
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Shape-persistent porous organic cage supported palladium nanoparticles as heterogeneous catalytic materials†
Shan
Jiang
 *a,
Harrison J.
Cox
a,
Evangelos I.
Papaioannou
b,
Chenyang
Tang
b,
Huiyu
Liu
a,
Billy J.
Murdoch‡
c,
Emma K.
Gibson
de,
Ian S.
Metcalfe
b,
John S. O.
Evans
a and
Simon K.
Beaumont
*a
*a,
Harrison J.
Cox
a,
Evangelos I.
Papaioannou
b,
Chenyang
Tang
b,
Huiyu
Liu
a,
Billy J.
Murdoch‡
c,
Emma K.
Gibson
de,
Ian S.
Metcalfe
b,
John S. O.
Evans
a and
Simon K.
Beaumont
*a
aDepartment of Chemistry, Science Site, Durham University, South Road, Durham DH1 3LE, UK. E-mail: shan.jiang@durham.ac.uk; simon.beaumont@durham.ac.uk
bSchool of Engineering, Newcastle University, Newcastle-upon-Tyne, NE1 7RU, UK
cNational EPSRC XPS Users’ Service (NEXUS), School of Engineering, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK
dSchool of Chemistry, University of Glasgow, University Avenue, Glasgow, G12 8QQ, UK
eUK Catalysis Hub, Research Complex at Harwell, Oxfordshire, OX11 0FA, UK
First published on 26th July 2019
Abstract
Porous Organic Cages (POCs) are an emerging class of self-assembling, porous materials with novel properties. They offer a key advantage over other porous materials in permitting facile solution processing and re-assembly. The combination of POCs with metal nanoparticles (NPs) unlocks applications in the area of catalysis. In this context, POCs can function as both the template of ultra-small NPs and a porous, but reprocessable, heterogeneous catalyst support. Here, we demonstrate the synthesis of ultra-small Pd NPs with an imine linked POC known as ‘CC3’, and show that hydrogen gas can be used to form metallic NPs at ∼200 °C without the reduction of the organic cage (and the accompanying, unwanted loss of crystallinity). The resulting materials are characterized using a range of techniques (including powder diffraction, scanning transmission electron microscopy and synchrotron X-ray absorption spectroscopy) and shown to be recrystallizable following dissolution in organic solvent. Their catalytic efficacy is demonstrated using the widely studied carbon monoxide oxidation reaction. This demonstration paves the way for using ultra-small NPs synthesized with POCs as solution-processable, self-assembling porous catalytic materials.
Introduction
Porous materials such as zeolites, metal organic frameworks (MOFs),1 covalent organic frameworks (COFs)2 and porous polymers3 attract considerable attention for applications such as gas storage, separations, sensing and catalysis.4 These materials can be rationally designed to exhibit desirable properties such as porosity and selectivity.5 The synthetic diversity of this class of materials permits the incorporation of numerous and highly diverse chemical functionalities in the pore structure.6 When employed as catalysts or catalyst supports, porous materials’ high surface areas, tuneable porosity and chemical functionality can enhance performance, as well as improving separation and recovery relative to homogeneous catalysts.7 Many industrial catalysts comprise nanoscale metal nanoparticles (NPs) on inorganic porous materials such as high surface area alumina or fumed-silica.8 Metal NPs confined in well-defined pore structures, such as metal organic frameworks (MOFs), have recently begun to be investigated as heterogeneous catalysts.9 The pore cavities have the potential to confine the growth of metal NPs, affording excellent particle size control and, in turn, improving the surface area and possibly selectivity and stability of the catalyst. Additionally, synergistic interactions between the metal catalyst and porous support have been suggested to enhance catalytic performance.10However, taking advantage of porous materials in this manner, and ensuring good control of the size, composition, spatial distribution, confinement and accessibility of ultra-small metal NPs in the pore structures remains challenging.10
Self-assembly of discrete, shape-persistent cage molecules – known as porous organic cages (POCs) – is a rapidly expanding field of study.11 These materials differ from other porous materials in that they comprise individual molecular building blocks, the cages, which assemble into a macroscopic structure. This is in clear contrast to zeolites or MOFs that are typically synthesized as an extended structure. This decoupling of assembly from synthesis is a transformative difference, because it enables solution processing of POC-derived materials: they can be readily dissolved in solvents to their molecular form, then re-assembled as solids, membranes or thin films.12 Such materials are already of widespread interest for applications such as gas storage, gas separation and molecular recognition.5 The use of POCs to control the synthesis and assembly of ultra-small metal NPs is therefore highly appealing as a route to make ultra-small NPs, but also to enable facile solution processing compared to other porous material templates.10
‘Mix and match’ assembly strategies using more than one cage molecule type can permit the fabrication of both porous organic alloys,13 and functional composites.14 The possibility of including multiple components in these self-assembling materials unlocks further possibilities for controlled and tuneable fabrication of bimetallic or multicomponent systems in the context of nanoparticle and catalyst syntheses.
To date, there are only a few attempts to fabricate ultra-small metal NPs using POCs as stabilizing and templating agents.15–20 These have allowed metal NPs to be homogenized in solution or used as a heterogeneous catalyst for a range of liquid phase reactions (Suzuki coupling,18 nitrophenol hydrogenation,16 cyanation of aryl halides,17 amine borane decomposition,16 photoreduction of nitroarene to azobenzenes20) and have demonstrated the potential of organic cage molecules to template metal NPs with uniform size. However, the two types of organic cage systems employed in these examples are either not crystalline/prone to self-assembly or contain imine linkages that are reduced to amines by NaBH4 (a reducing agent being necessary to reduce the metal salt precursor). This conversion of the imines to amines is known to lead to the POCs losing their shape persistence, crystallinity and porosity.21 It therefore remains an important challenge to prepare metal NPs templated by shape persistent POCs, without loss of their crystallinity. It is their crystallinity that is crucial for applications requiring facile processability and re-assembly of these highly ordered materials.
Here, we describe a simple and efficient synthetic approach that enables production of metal NPs with controlled size (around 1.6 nm) using the shape-persistent POC CC3 (Fig. 1(a)). This is achieved with the material and NPs retaining the ability to self-assemble into porous crystalline form, demonstrating their potential for easy post-synthesis processing.
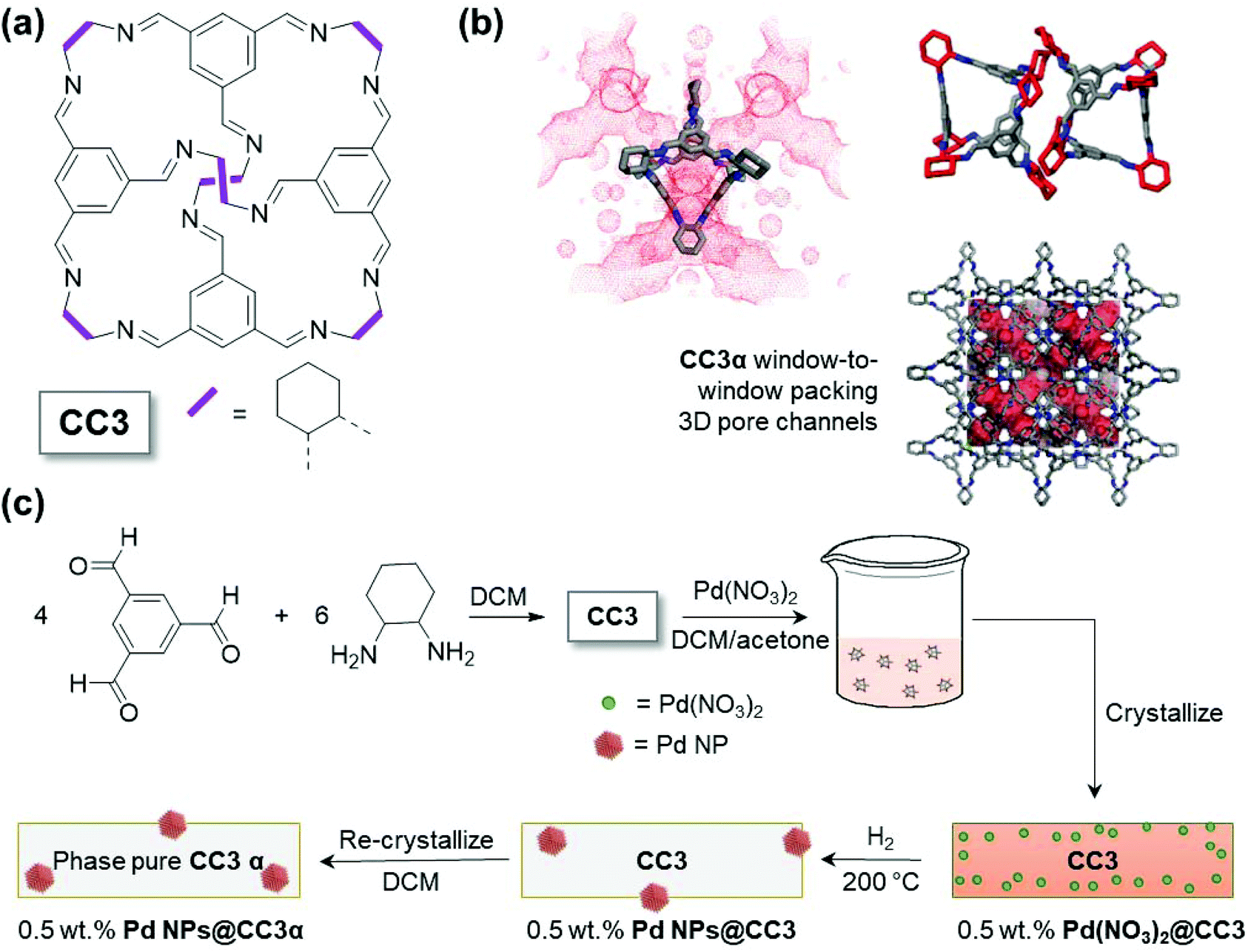 | ||
| Fig. 1 (a) Molecular structure of porous organic cage – CC3. (b) Crystal structure depictions of thermodynamic CC3α polymorph to show the pore network connectivity around one molecule (the accessible pore structure is seen from the connolly surfaces in red generated using a probe radius of 1.82 Å), the local cage packing motif, and the wider crystal structure. (c) Schematic representation of the overall synthesis of Pd NPs@CC3 material (DCM = dichloromethane, rectangles are the CC3 cage material, the 0.5 wt% Pd(NO3)2@CC3 is presumed to contain cages that incorporate Pd2+ species). | ||
Results and discussion
As can be seen from Fig. 1(a), CC3 is an imine linked cage with a tetrahedral shape. The cage has four windows allowing access for guest species to the internal void. When crystallized in the absence of guest species, the thermodynamically most stable polymorph, denoted CC3α, is formed. CC3α shows a window-to-window cage-packing motif, which results in an interconnected three dimensional (3D) pore network as shown in Fig. 1(b). The pores comprise both the internal void of the cage but also extrinsic voids that result from inefficient packing of the cages, leaving spaces between the CC3 molecules. The limiting pore size that will govern access for guest species to this pore network can, however, be approximated by the cage windows. In this work, Pd(NO3)2 was consequently selected as the metal precursor because, in contrast to larger metal precursors, the size of one Pd(NO3)2 complex means it is likely to fit into the CC3 molecule (Fig. S1†), via these cage windows. The initial encapsulation of Pd(NO3)2 inside the cage cavities in solution was anticipated to be important in enabling the metal to become distributed within the porous solid upon crystallization. By way of contrast, similar experiments with the larger Pd(acac)2 metal precursor, which is not expected to fit within the cage cavities, produced a material which exhibited very poor Pd particle size control (demonstrating the critical importance of the size of the Pd(NO3)2 relative to the size of the cage molecule cavity, Fig. S2†vs.Fig. 3).The synthesis of the Pd NPs@CC3 is summarized schematically in Fig. 1(c). First, CC3 was prepared using previously reported methodology (the [4 + 6] cycloimination reaction of 1,3,5-triformylbenzene with (1R,2R)-1,2-diaminocyclohexane).11 The successful synthesis of CC3 was confirmed by 1H and 13C NMR (Fig. S3†). Next, the Pd(NO3)2 metal precursor was incorporated into CC3 in solution, and subsequently crystallized together to form Pd(NO3)2@CC3, before reducing in H2 gas to form ∼1.6 nm metal NPs (Pd NPs@CC3). Gas-phase H2 reduction of the metal salt impregnated crystalline cages enables formation of metal NPs without hydrogenation of the imine linkages (still seen by NMR, Fig. S4†), in contrast to liquid phase reduction with NaBH4 reported in previous studies.15–19 The Pd NPs@CC3 materials were shown to be catalytically competent in the oxidation of carbon monoxide, a widely used reaction for the benchmarking of conventional heterogeneous catalysts.22 The Pd NPs@CC3 material could also be reproducibly recrystallized as a single (cubic) polymorph in which the (XRD silent) NPs appear to be held within the bulk of CC3 particles. We note that the absence of reflections for Pd metal in the XRD pattern is a confirmation that significant amounts of larger size particles are not present; for the small (<2 nm) particles observed by TEM, peak broadening effects mean that scattering from Pd would not be observable from the data, particularly at the low loadings present.23 The inclusion of the Pd NPs within the crystalline POC matrix is an interesting observation, recrystallization normally being used as a purification procedure – possible reasons for this behaviour will be discussed later in the section. The recrystallization in the final step is conducted in DCM, conditions under which the Pd-free CC3 is known to be fully dissolved such that crystallisation into other polymorphs is also possible.24 Overall this procedure yields size controlled, ultra-small NPs in a high quality POC matrix.
Pd(NO3)2@CC3 crystals (light red) were obtained after slow evaporation of the solvent (the slow crystallization being known to produce pure CC3 crystals with few defects).25,26 The Pd loadings, which ranged from 0.5 to 2.0 wt%, were verified by Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES, Table S2†).
Having crystallized the Pd precursor together with CC3 (Pd(NO3)2@CC3), we then examined the most suitable conditions to conduct the reduction under H2 to yield ultra-small metal NPs (Pd NPs@CC3), without chemical damage to the cage structure itself. Thermogravimetric analysis (TGA) was employed to follow the changes that occurred on heating Pd(NO3)2@CC3 samples in both an inert atmosphere and 10% hydrogen (Fig. 2(a)). In N2 only small changes, possibly due to residual solvent loss are seen below 300 °C, at which temperature decomposition occurs. In 10% H2/N2, however, mass loss that exceeds that for the sample in N2 was seen to have an onset as low as 150 °C, with eventual complete decomposition at ∼400 °C. The H2-reduction temperature being substantially below the temperature of decomposition in N2 mirrors the behaviour already known for bulk Pd(NO3)2.27 To investigate the process, two samples were prepared by reduction for 1 h at either 150 or 200 °C. The color of the samples (Fig. S5†) shows a change has occurred with the original light orange-red color changing to yellow and then dark grey on reduction at 150 and 200 °C, respectively.
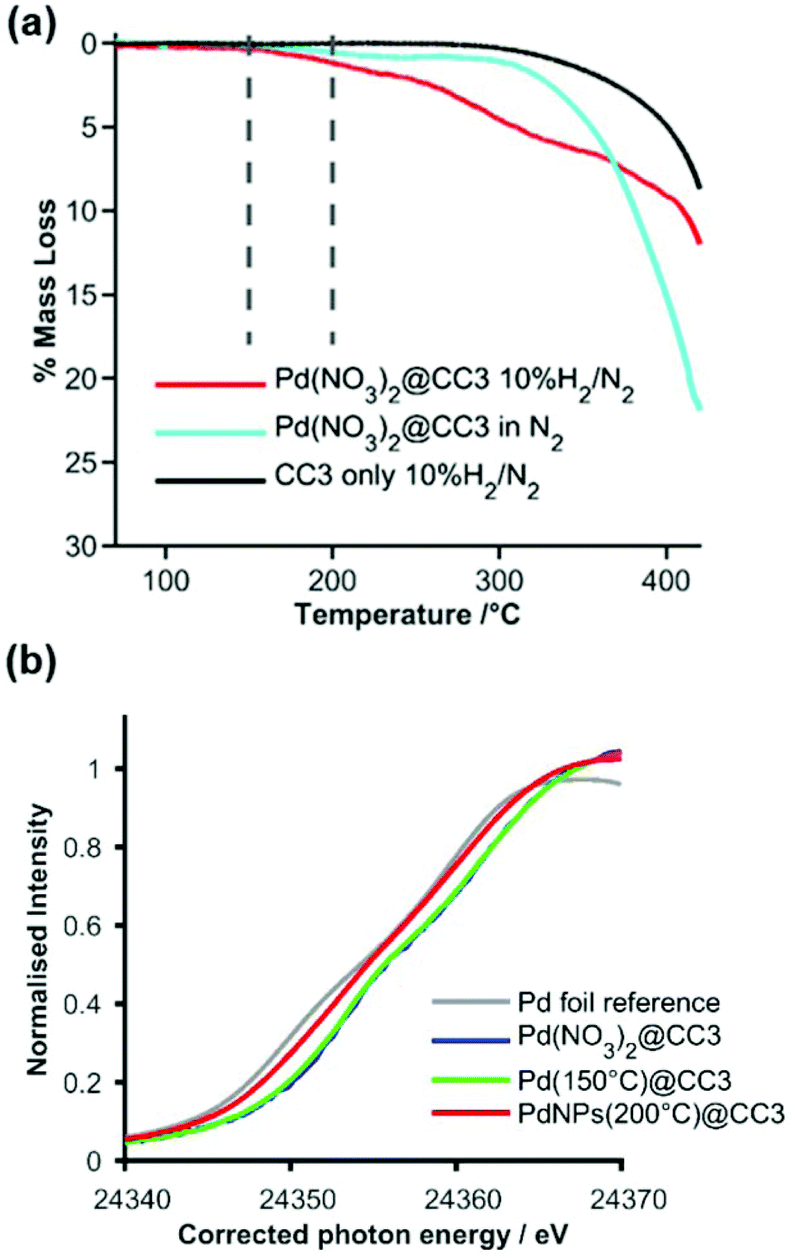 | ||
| Fig. 2 (a) TGA mass loss profiles in either inert (nitrogen, pale blue) or reducing (H2/N2, red) gas for Pd(NO3)2@CC3, and in H2/N2 for CC3 (black); vertical dashed lines show the temperature (150 and 200 °C) at which reduced samples were prepared. (b) Normalized XANES spectra expanded around the Pd K-edge for Pd reference foil (grey), Pd(NO3)2@CC3 before reduction (blue – behind green) and Pd NPs@CC3 after reduction in H2 at 150 °C (green) and 200 °C (red). The photon energy scale has been corrected by alignment of concurrently measured Pd foil data. Full spectra are shown in Fig. S6.† | ||
X-ray Absorption Spectroscopy (XAS) was used to monitor the reduction process more carefully, confirming little change at 150 °C but a change to the metallic Pd0 oxidation state after reduction at 200 °C. The expanded near edge XAS region (Fig. 2(b)) clearly shows a shift in edge energy between the 150 °C reduced sample (which is similar to the unreduced sample) and the 200 °C reduced sample, which is very similar to a metallic Pd foil reference sample (full spectra are shown in Fig. S6†). Analysis of the Extended-edge X-ray Absorption Fine Structure (EXAFS) spectra obtained from Pd(NO3)2@CC3 or the 150 °C reduced sample indicated no significant Pd–Pd interactions in the first co-ordination sphere (Fig. S7–9, Tables S4 and 5†), while the sample reduced at 200 °C (Pd NPs@CC3) showed an average first shell Pd–Pd co-ordination number of around 5 palladium atoms per Pd scatterer. This is indicative of ultra-small NPs around 1 nm in size (even allowing for different possible particle morphologies28).
Scanning electron microscopy (SEM) of the Pd NPs@CC3 (reduced at 200 °C) shows the octahedral shape of the Pd containing organic crystals is preserved with respect to their Pd-free counterparts, which also exhibit an octahedral microscopic mophology (Fig. S10†). Higher resolution imaging using Scanning Transmission Electron Microscopy (STEM) is shown in Fig. 3(a–d). Small near monodisperse Pd NPs can be seen with a mean diameter and corresponding size distribution of 1.6 ± 0.3 nm. No NPs were seen for reduction at lower temperature. The size determined from STEM imaging is broadly in agreement with the analysis of the EXAFS spectra, any small difference could either result from the tendency of microscopy to overestimate the size due to the fact larger particles are easier to see, or else to the fact a small amount of residual Pd2+ remains as isolated sites and lowers the co-ordination number derived from XAS. If the latter, only a very small fraction of isolated Pd2+ can remain based on XANES oxidation state analysis. STEM images of a higher Pd loading sample (Fig. S11†) indicate the size of metal NPs does not change significantly from that present in the 0.5 wt% sample.
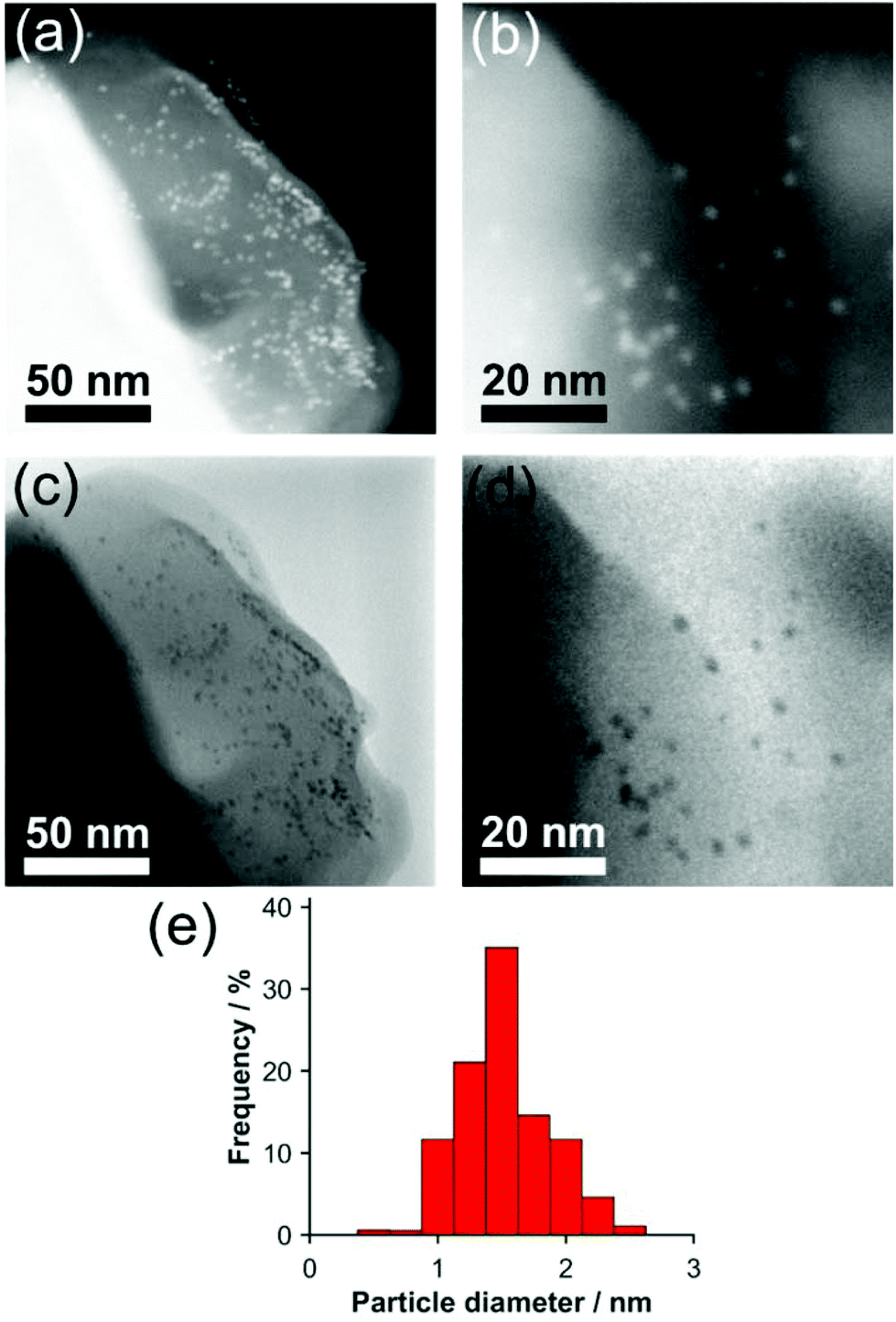 | ||
| Fig. 3 (a and b) dark field and (c and d) bright field STEM images of 0.5 wt% Pd NPs@CC3. (e) Size histogram of Pd NP diameters. | ||
The STEM images show the NPs are embedded within the POC crystal matrix. If NPs were present only at the surface of the POC crystals, when projected into 2D by the TEM most particles would appear decorating only the crystal edges (with apparent particle densities lower on the top and bottom faces), as seen for NPs on the surface of polystyrene spheres.29 This is not the case for Pd NPs@CC3 (Fig. 3), which exhibit an even spatial distribution in the STEM images, providing evidence of incorporation in the material rather than merely on its surface.
Gas sorption analysis was performed on CC3 and 0.5 wt% Pd NPs@CC3 as shown in Fig. S12.† The CC3 material was found to have a Brunauer–Emmett–Teller (BET) surface area of 400 m2 g−1, consistent with the reported value of 409 m2 g−1 for highly crystalline CC3.26 0.5 wt% Pd NPs@CC3 was found to have a similar Type-I N2 sorption isotherm with a BET surface area of 350 m2 g−1. Pd NPs@CC3 also had a total adsorption of 4.7 mmol g−1 N2 at 77 K and a relative pressure (P/P0) of 0.998, compared to 5.5 mmol g−1 for pure CC3. The key point is the 0.5 wt% Pd NPs@CC3 material's porosity doesn't substantially decrease compared to that of the CC3 parent material, meaning that the porosity necessary for reagent access during catalysis is preserved. Any slight decrease in accessible surface area could be indicative of pores blocked by the Pd NPs, but since the decrease is only small this should not be over-interpreted.
A number of attempts were made to identify interactions between either the Pd salt and the cage functional groups in Pd(NO3)2@CC3 or Pd NPs and the cage in Pd NPs@CC3, but were not successful. Neither 1H NMR (Fig. S4†), nor infrared spectroscopy (Fig. S13†) showed any discernable differences in the cage structure before or after reduction as a result of incorporating Pd, possibly because of the low ratio of Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CC3 (1 Pd atom:20 cage molecules). The similarity of the organic structures confirm the cages have remained intact and are not reduced (the imine linkages therefore appear likely too far away or too hindered for hydrogenation to be catalyzed by the nascent Pd NPs during reduction). There are also no major differences discernible by powder X-ray diffraction (PXRD) (Fig. 4 and S14†) between CC3 and the samples in which Pd is present (before or after reduction). Notably, there is no evidence for Pd(NO3)2 forming an independent precipitated phase. The PXRD pattern of the pure CC3 cage material before incorporation of Pd shows that the crystals possess cubic F4132 symmetry with a ≈ 25 Å, in good agreement with the reported single crystal structure of CC3α (Fig. 4 and Fig. S14†).
:CC3 (1 Pd atom:20 cage molecules). The similarity of the organic structures confirm the cages have remained intact and are not reduced (the imine linkages therefore appear likely too far away or too hindered for hydrogenation to be catalyzed by the nascent Pd NPs during reduction). There are also no major differences discernible by powder X-ray diffraction (PXRD) (Fig. 4 and S14†) between CC3 and the samples in which Pd is present (before or after reduction). Notably, there is no evidence for Pd(NO3)2 forming an independent precipitated phase. The PXRD pattern of the pure CC3 cage material before incorporation of Pd shows that the crystals possess cubic F4132 symmetry with a ≈ 25 Å, in good agreement with the reported single crystal structure of CC3α (Fig. 4 and Fig. S14†).
 | ||
| Fig. 4 PXRD patterns for CC3 (black), desolvated 0.5 wt% Pd(NO3)2@CC3 (red), 0.5 wt% Pd NPs@CC3 after H2 reduction at 200 °C for 1 hour (blue), and recrystallization of 0.5 wt% Pd NPs@CC3 in DCM to form pure CC3α phase (cyan). Yellow arrows denote additional reflection at 7.6°. | ||
The only change seen on going from CC3 to Pd(NO3)2@CC3 is that the PXRD pattern of Pd(NO3)2@CC3 contains some weak additional reflections – most notably at 7.6°2θ, although changes at 11° and 19.5°2θ can be identified in a Pawley fit to the data.30 The presence of a feature as low as 7.6°2θ points to a unit cell with at least one dimension greater than 11 Å, and therefore the possible presence of a second polymorph of the organic cage material. These features are seen to persist in the structure of the Pd NPs@CC3 material after gas phase reduction. The crystal packing motif adopted by CC3 is (like many compounds) known to be sensitive to surface functionalization or guest species (including solvents) present during their assembly.24 In addition to the CC3α form, CC3 has also been shown to form a second polymorph, CC3β (space group P3), and several gas or solvent inclusion crystals, such as (CC3).(Et2O)3.CH2Cl2.24 This low concentration component likely results from similar inclusion crystals of either the Pd(NO3)2 or the directed inclusion of other compounds (e.g. solvent) present due to difference in ionic strength. The additional features are not present if the CC3 is subject to the same DCM/acetone recrystallization in the absence of Pd(NO3)2, Fig. S15.† This second phase was only formed in low quantities; attempts to load substantially more palladium resulted in less crystallinity and attempts at single crystal analysis showed the crystals were unstable (evidenced by a rapid color change) in intense X-rays, hence the precise identification of this second polymorph has remained elusive. As will be seen below this does not alter the use or potential applications since the material (cages and NPs) can be recrystallized easily from DCM into the usual CC3α motif, Fig. 4.
The observation that only a single phase (CC3α) is formed when Pd NPs@CC3 is dissolved in DCM and recrystallized (Fig. 4 & S14†) is consistent with the fact that on reduction to form NPs, the Pd species do not interact with a sufficient number of cage molecules to influence the packing motif adopted (for hypothetically 100 Pd atom NPs at 0.5 wt% Pd loading there is one NP for every ∼1900 cage units). The ability to recrystallize the NPs dispersed within the CC3 material is crucial for applications. Since STEM images of Pd NPs@CC3 were obtained after dissolving and effectively recrystallizing the sample by evaporation on the TEM grid, this confirms the NPs remain dispersed throughout the material upon solution processing. It is interesting that recrystallization is typically employed as a purification procedure but here the NPs clearly become embedded in the CC3 material. This can be rationalised by the very high surface free energies of the Pd NPs being stabilised by (at the very least) van der Waals interactions with the CC3 (in the absence of any other stabilising agent), and thus remaining bound to CC3 molecules.31 Speculatively, the CC3 molecules already attached to the Pd NPs during crystallisation can then act as a ‘Trojan horse’ ligand to mimic the expected supramolecular interactions during crystal epitaxy, as found for the inclusion of NPs with appropriate ligands in liquid crystals.32
A number of reports have argued or postulated the NPs form within the cages,15,18–20 their relative size range (about 1.3–1.9 nm) makes this option unrealistic given the small size of the cavity (diameter ∼0.8 nm); this is also true for the pore cavity between two cage molecules with window-to-window packing (again ∼0.8 nm). This is illustrated in Fig. S16.† Instead, given the similarity of the particle size with the size of the cage (max diameter = 2 nm), it more likely becomes a guest/defect occupying the space of one cage molecule within the porous crystalline structure (given that by TEM they are not strongly clustered or only at the surface of the material, Fig. 3). The role of the cages is therefore in the controlled assembly of the ultra-small NPs and their subsequent external stabilization, rather than as conventional templates.
CO oxidation was used as a benchmark reaction, to demonstrate the use of Pd NPs@CC3 materials as catalysts. Fig. 5 shows light-off data (conversion as a function of increasing temperature) for the CO/O2 reaction conducted in a fixed packed-bed reactor over the temperature range 100–200 °C. In order to measure the rate of reaction under gradientless conditions the reactor was operated under conditions of differential CO conversion (<20%). The CO oxidation reaction catalyzed by the 0.5 wt% Pd NPs@CC3 catalyst proceeds at ∼20 mmolCO2 molPd−1 s−1 at 200 °C (∼6% CO conversion). Additionally, Fig. 5 shows points for a typical reference Pd/Al2O3 catalyst and two literature catalysts.33,34 It can be seen that the Pd NPs@CC3 exhibit broadly comparable catalytic activity to other Pd catalysts (normalised to Pd amount).§ However, the other catalysts shown are not as amenable to solution phase processing techniques as Pd NPs@CC3, but once assembled are fixed in form.
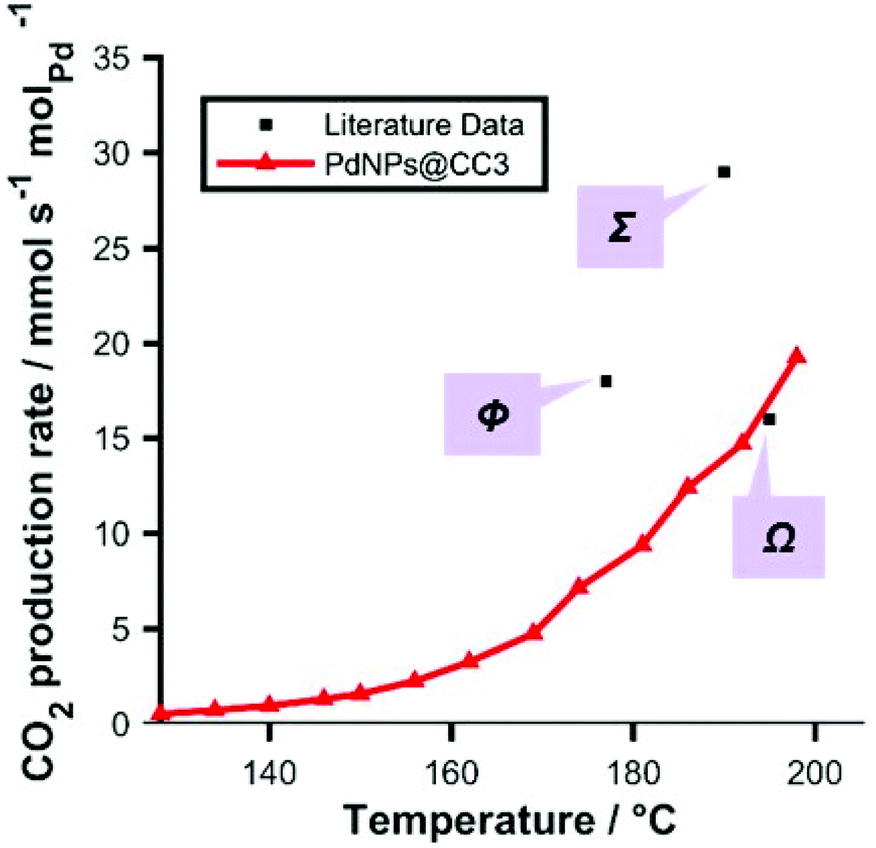 | ||
| Fig. 5 Light-off data: red: CO oxidation rate as a function of temperature catalyzed by Pd NPs@CC3 (<10% conversion; 3% CO, 15% O2, balance He, total flow 60 mL min−1 (NPT); ambient pressure; 71.5 mg of 0.5 wt% Pd catalyst). Black: reference/literature data (all measured at <20% conversion) for typical Pd catalysts: Ω, Pd triptycene based microporous polymer;33Σ, Pd/SiO2 (literature);34Φ, Pd/Al2O3 commercial reference sample (Alfa Aesar) (as a sample of similar particle size could not be sourced, a sample with around 3 m2 g−1 metal surface area was employed—about double the geometric surface area assuming spherical particles in the CC3 derived sample—and so the data point for this sample only was normalized for known relative surface area to aid in comparison of this sample with our system) tested in same apparatus. | ||
The stability of the catalyst during reaction was also demonstrated by repeat cycling of the material in temperature (5 times, Fig. S17†), which shows very little change and is consistent with both TGA in air (Fig. S18†) and variable temperature XRD (Fig. S19†), both of which show stability above these temperatures. In addition to using low conversions that avoid a significant change in concentration along the reactor bed, a series of experiments at varying flow rates were conducted confirming the absence of mass transfer effects along the reactor, which if present could lead to erroneous catalytic performance data (Fig. S20†).
Conclusions
We have developed a synthetic approach that enables controlled assembly of metal NPs and their incorporation with shape persistent POC materials. The Pd NP containing material remains soluble and recrystallizable after NP incorporation. The cage molecules act as a stabilizer and effective catalyst support for the ultra-small Pd NPs produced (∼1.6 nm as determined by STEM). A range of techniques (including PXRD, TGA, XAS) was used to give detailed insight into the synthesis of ultra-small Pd NPs@CC3via gas phase reduction in hydrogen. The resulting Pd-embedded porous organic material has been shown to be an effective CO oxidation catalyst. In contrast to other recent reports of organic cage templated NPs, liquid phase reduction of the organic cage was avoided by use of the gas-phase reductant H2 for the first time, preventing loss of cage bonding that inhibits crystal formation/assembly. This proof-of-concept demonstration paves the way for taking advantage of the solution processability and facile directed assembly of porous organic cage materials to assemble and process ultra-small nanoparticle catalysts.Experimental
Synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, 12H), 7.91 (s, Ar–H, 12H), 3.35 (m, CHN, 12H), 1.9–1.90 (m, –CH2, 48H) ppm. 13C {1H} NMR (100 MHz, CDCl3) δ 159.1, 136.6, 129.5, 74.7, 33.0, 24.4 ppm. ASAP: m/z 1117.7 [M]+ (100%).)
N, 12H), 7.91 (s, Ar–H, 12H), 3.35 (m, CHN, 12H), 1.9–1.90 (m, –CH2, 48H) ppm. 13C {1H} NMR (100 MHz, CDCl3) δ 159.1, 136.6, 129.5, 74.7, 33.0, 24.4 ppm. ASAP: m/z 1117.7 [M]+ (100%).)
Characterization
350 eV). Each sample (sufficient that the absorption coefficient jump at the edge was satisfactory for transmission measurements) was prepared by pressing finely ground powders into self-supporting disks using an IR die press and then supporting on Kapton tape. These were employed for transmission XAS measurements with pure Pd metal foil measured concurrently between a second and third ionisation chamber, enabling X-ray energy calibration and data alignment. Further details of the analysis of data processing methods are given in the ESI.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
SJ was funded by the UK Catalysis Hub, which is kindly thanked for resources and support provided via membership of the UK Catalysis Hub Consortium and funded by the UK Engineering and Physical Sciences Research Council (EPSRC, Grants EP/K014706/2, EP/K014668/1, EP/K014854/1, EP/K014714/1 and EP/M013219/1). The authors acknowledge additional EPSRC funding, grant EP/P007767/1. The authors gratefully acknowledge Stewart Clark (DU) for access to the Materials Studio software, the DU GJ Russell Microscopy Facility for access to SEM and TEM, Budhika Mendis (DU) for assistance with STEM measurements, and Thomas Chamberlain for helpful discussions. The authors wish to acknowledge the Diamond Light Source for provision of beamtime through the Catalysis BAG (SP15151-4) and are grateful for the support of the B18 beamline scientists. BM acknowledges the UK National EPSRC XPS Users’ Service, an EPSRC Mid-Range Facility.Notes and references
- G. Ferey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, eeal1585 CrossRef PubMed.
- R. Dawson, A. I. Cooper and D. J. Adams, Prog. Polym. Sci., 2012, 37, 530–563 CrossRef CAS.
- M. E. Davis, Nature, 2002, 417, 813 CrossRef CAS PubMed.
- A. G. Slater and A. I. Cooper, Science, 2015, 348, aaa8075 CrossRef PubMed.
- J. Long, W. Yang, J. Hai-Long and X. Qiang, Adv. Mater., 2017, 1703663 Search PubMed.
- L. Jiao, Y. Wang, H.-L. Jiang and Q. Xu, Adv. Mater., 2018, 30, 1703663 CrossRef PubMed.
- J. E. Bailie, G. J. Hutchings and S. O'Leary, in Encyclopedia of Materials: Science and Technology, ed. K. H. J. Buschow, R. W. Cahn, M. C. Flemings, B. Ilschner, E. J. Kramer, S. Mahajan and P. Veyssière, Elsevier, Oxford, 2001, pp. 8986–8990 Search PubMed.
- M. Meilikhov, K. Yusenko, D. Esken, S. Turner, G. Van Tendeloo and R. A. Fischer, Eur. J. Inorg. Chem., 2010, 2010, 3701–3714 CrossRef.
- Q.-L. Zhu and Q. Xu, Chem., 2016, 1, 220–245 CAS.
- T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973–978 CrossRef CAS PubMed.
- Q. Song, S. Jiang, T. Hasell, M. Liu, S. Sun, A. K. Cheetham, E. Sivaniah and A. I. Cooper, Adv. Mater., 2016, 28, 2629–2637 CrossRef CAS PubMed.
- T. Hasell, S. Y. Chong, M. Schmidtmann, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2012, 51, 7154–7157 CrossRef CAS PubMed.
- A. F. Bushell, P. M. Budd, M. P. Attfield, J. T. A. Jones, T. Hasell, A. I. Cooper, P. Bernardo, F. Bazzarelli, G. Clarizia and J. C. Jansen, Angew. Chem., Int. Ed., 2013, 52, 1253–1256 CrossRef CAS PubMed.
- X. Yang, J.-K. Sun, M. Kitta, H. Pang and Q. Xu, Nat. Catal., 2018, 1, 214–220 CrossRef CAS.
- J.-K. Sun, W.-W. Zhan, T. Akita and Q. Xu, J. Am. Chem. Soc., 2015, 137, 7063–7066 CrossRef CAS PubMed.
- B. Mondal, K. Acharyya, P. Howlader and P. S. Mukherjee, J. Am. Chem. Soc., 2016, 138, 1709–1716 CrossRef CAS PubMed.
- L. Qiu, R. McCaffrey, Y. Jin, Y. Gong, Y. Hu, H. Sun, W. Park and W. Zhang, Chem. Sci., 2018, 9, 676–680 RSC.
- R. McCaffrey, H. Long, Y. Jin, A. Sanders, W. Park and W. Zhang, J. Am. Chem. Soc., 2014, 136, 1782–1785 CrossRef CAS PubMed.
- G.-J. Chen, W.-L. Xin, J.-S. Wang, J.-Y. Cheng and Y.-B. Dong, Chem. Commun., 2019, 55, 3586–3589 RSC.
- M. Liu, M. A. Little, K. E. Jelfs, J. T. A. Jones, M. Schmidtmann, S. Y. Chong, T. Hasell and A. I. Cooper, J. Am. Chem. Soc., 2014, 136, 7583–7586 CrossRef CAS PubMed.
- H.-J. Freund, G. Meijer, M. Scheffler, R. Schlögl and M. Wolf, Angew. Chem., Int. Ed., 2011, 50, 10064–10094 CrossRef CAS PubMed.
- K. R. Beyerlein, R. L. Snyder and P. Scardi, J. Appl. Crystallogr., 2011, 44, 945–953 CrossRef CAS.
- M. A. Little, S. Y. Chong, M. Schmidtmann, T. Hasell and A. I. Cooper, Chem. Commun., 2014, 50, 9465–9468 RSC.
- S. Jiang, Q. Song, A. Massey, S. Y. Chong, L. Chen, S. Sun, T. Hasell, R. Raval, E. Sivaniah, A. K. Cheetham and A. I. Cooper, Angew. Chem., Int. Ed., 2017, 56, 9391–9395 CrossRef CAS PubMed.
- T. Hasell, S. Y. Chong, K. E. Jelfs, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2012, 134, 588–598 CrossRef CAS PubMed.
- S. Yuvaraj, L. Fan-Yuan, C. Tsong-Huei and Y. Chuin-Tih, J. Phys. Chem. B, 2003, 107, 1044–1047 CrossRef CAS.
- A. M. Beale and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2010, 12, 5562–5574 RSC.
- W. Shi, Y. Sahoo, M. T. Swihart and P. N. Prasad, Langmuir, 2005, 21, 1610–1617 CrossRef CAS PubMed.
- G. Pawley, J. Appl. Crystallogr., 1981, 14, 357–361 CrossRef CAS.
- F. J. Heiligtag and M. Niederberger, Mater. Today, 2013, 16, 262–271 CrossRef CAS.
- H. K. Bisoyi and S. Kumar, Chem. Soc. Rev., 2011, 40, 306–319 RSC.
- Q. Liang, J. Liu, Y. Wei, Z. Zhao and M. J. MacLachlan, Chem. Commun., 2013, 49, 8928–8930 RSC.
- A. Siani, O. S. Alexeev, G. Lafaye and M. D. Amiridis, J. Catal., 2009, 266, 26–38 CrossRef CAS.
- Y. Yang, K. M. Saoud, V. Abdelsayed, G. Glaspell, S. Deevi and M. S. El-Shall, Catal. Commun., 2006, 7, 281–284 CrossRef CAS.
- M. Haruta, S. Tsubota, T. Kobayashi, H. Kageyama, M. J. Genet and B. Delmon, J. Catal., 1993, 144, 175–192 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional synthetic and XAS methodological details; structural models; TEM, STEM, SEM NMR, IR, XAS, N2 porosimetry, PXRD, TGA and catalytic data. See DOI: 10.1039/c9nr04553h |
| ‡ Present address B.M.: School of Science, RMIT University, VIC 3001, Melbourne, Australia. |
| § It should be noted that Pd with reducible oxides, e.g. Ceria,35 and Au based systems36 have much lower light off temperatures, but this is due to a different mechanism at play. Pure Pd catalysts are known to be strongly inhibited by absorption of CO, which blocks oxygen access, below around 150 °C. |
| This journal is © The Royal Society of Chemistry 2019 |