
High photoluminescence of shortwave infrared-emitting anisotropic surface charged gold nanoclusters†

Abstract
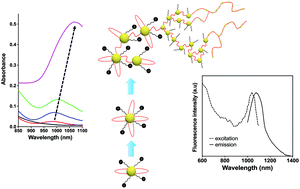
Incorporating anisotropic surface charges on atomically precise gold nanoclusters (Au NCs) led to a strong absorption in the near-infrared region and could enable the formation of self-assembled Au NCs exhibiting an intense absorption band at ∼1000 nm. This surface modification showed a striking enhancement of the photoluminescence in the Shortwave Infrared (SWIR) region with a quantum yield as high as 6.1% in water.
- This article is part of the themed collection: Editor’s Choice: Controlling anisotropy in nanomaterials
 Please wait while we load your content...
Please wait while we load your content...

