
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational design of nanosystems for simultaneous drug delivery and photodynamic therapy by quantum mechanical modeling†
Moloud
Kaviani
 and
Cristiana
Di Valentin
*
and
Cristiana
Di Valentin
*
Dipartimento di Scienza dei Materiali, Università di Milano-Bicocca, via R. Cozzi 55, 20125 Milano, Italy. E-mail: cristiana.divalentin@unimib.it
First published on 12th August 2019
Abstract
Drug delivery systems are based on reversible interactions between carriers and drugs. Spacers are often introduced to tailor the type of interaction and to keep drugs intact. Here, we model a drug delivery system based on a functionalized curved TiO2 nanoparticle of realistic size (700 atoms – 2.2 nm) by the neurotransmitter dopamine to carry the anticancer chemotherapeutic agent doxorubicin (DOX). The multiscale quantum chemical study aims at unraveling the nature and mechanism of the interactions between the components and the electronic properties of the composite system. We simulate the temperature effect through molecular dynamics runs of thermal annealing. Dopamine binds preferentially to low coordinated Ti sites on the nanoparticle through dissociated bidentate and chelate modes involving the diol groups. DOX is tethered by H-bonds, π–π stacking, dipole–dipole interactions and dispersion forces. Comparing different coverage densities of the spacer on the nanoparticle surface, we assess the best conditions for an effective drug transport and release: only at full coverage, DOX does not slip among the dopamine molecules to reach the nanoparticle surface, which is crucial to avoid the formation of stable coordinative bonds with under-coordinated Ti atoms. Finally, given the strong absorption properties and fluorescence of DOX and of the TiO2 photocatalyst, we model the effect of light irradiation through excited state calculations to localize excitons and to follow the charge carrier's life path. This fundamental study on the nature and mechanism of drug/carrier interaction provides a solid ground for the rational design of new experimental protocols for a more efficient drug transport and release and its combination with photodynamic therapy.
1. Introduction
Doxorubicin (DOX) is one of the most effective anticancer chemotherapy drugs.1 It is classified as an “anthracycline antibiotic” and is widely used to treat different types of cancer, such as breast cancer, bladder cancer, Kaposi's sarcoma, acute lymphocytic leukemia, lymphoma, etc.2 DOX can suppress the growth of cancer cells by blocking the Type II topoisomerase enzyme that is needed for them to divide and grow. Moreover, doxorubicin has strong absorption and fluorescence in the visible spectral region, which is a valuable tool in tracking of DOX-containing conjugates by optical techniques.3–5 However, its clinical use has been limited by few factors, such as cardiotoxicity, which forces the treatment to become dose-dependent6 and its low specificity.7–9Therefore, delivery vehicles involving multifunctional components are needed to overcome these issues. Depending on the choice they will be responsive to different stimuli, such as pH,10 heat,11 light12 or intra-cellular enzymes.13 Nanoparticles (NPs) have been used as carriers for drug delivery,14–16 resulting in high drug loading17,18 and prolonged in vivo circulation time. Due to their size, NPs can easily penetrate into tissues and accumulate in tumor cells.19,20 Moreover, imaging or therapeutic functions can be integrated, making them promising multifunctional nanoplatforms for both diagnosis and cure.21–24
In drug delivery systems, one of the crucial factors that influences therapeutic efficiency is the drug loading mode. Non-covalent complexation (H-bonding, dispersion forces, electrostatic interactions) and covalent conjugation are the two most common approaches. Non-covalent complexation not only keeps the drug in its pristine state, but also elevates the therapeutic activity by increasing cellular internalization.25,26 The therapeutic activity of a covalently conjugated drug is commonly compromised27,28 because the conjugation might alter the original chemical structure of the drug. Also, conjugation may prevent effective drug release. For example, Qin et al.29 showed that the non-covalent loading of DOX to TETT (N-(tri-methoxysilylpropyl) ethylene diamine triacetic acid, trisodium salt) functionalized TiO2 NPs exhibited a greater cytotoxicity than free DOX, in contrast to the corresponding covalent conjugated system that resulted in an observed decreased cytotoxicity and antitumor activity.
To hamper the direct interaction between the drug and the NP, one can use bifunctional linkers that guarantee the needed space and allow controlling the binding strength through the proper choice of the two functional groups used to anchor the surface and to tether the drug.30 One of the most extensively utilized bifunctional linkers for direct conjugation to metal oxide NP surfaces is the neurotransmitter dopamine (4-(2-aminoethyl)benzene-1,2-diol). It can directly bind to the undercoordinated surface metal atoms by its enediol portion through coordination bonds, while the primary amine remains exposed to the surrounding environment, imparting water dispersibility and acting as a potential handle for biomolecules.
In addition to dopamine, polydopamine (PDA) is also one of the most used materials to coat NPs. It can be obtained by polymerization of dopamine in weakly basic environments.31–36 The simplicity in the preparation of PDA virtually on any material surfaces, such as noble metals (Au, Ag, Pt, and Pd), oxides (TiO2, non-crystalline SiO2, crystalline SiO2 (quartz) Al2O3, and Nb2O5), polymers, magnetite nanoparticles37etc., enables it to be a versatile coating. Moreover, the competitive advantages of using PDA functionalization lie in its convenience and good chemical reactivity for post-modification.38
Dopamine contains phenyl, amino and hydroxyl groups, which provides active surfaces for loading the aromatic chemotherapy drugs such as DOX via hydrogen bonding and π–π stacking interactions.34,35,39 DOX release is pH-dependent. For example, the drug release by Fe3O4@PDA-PEG-EGFR-DOX NPs (PEG: polyethylene glycol and EGFR: epidermal growth factor receptor) was examined at pH values 5.0 and 7.4. It was reported that the amount of DOX released at pH 5.0 was approximately four times higher than that at pH 7.4. A possible rationalization is that the protonation of the amino group in DOX at a lower pH value enhanced its hydrophilicity and triggered the drug release.35
TiO2 nanoparticles (NPs) are one of the most produced and widely used inorganic semiconducting metal oxide nanomaterials and they have recently emerged as excellent candidates for biomedical applications, due to their unique photocatalytic properties, excellent biocompatibility, low toxicity40 for both humans and the environment and low cost and high chemical stability.41,42 The most relevant biomedical applications of TiO2 is in photodynamic therapy for cancer treatment,43–46 being an excellent ROS (reactive oxygen species) generator under light irradiation, but it could also be used as a drug delivery system,46 for cell imaging,47–49 biosensors, antimicrobial and bactericidal action and genetic engineering.41,50
In previous studies,51,52 we have shown and discussed why curved TiO2 nanoparticles are more suitable than flat (101) TiO2 surfaces for biomedical applications, in agreement with experimental results.42 The main reasons are that curved NPs expose many low coordinated sites53 that can strongly anchor functionalizing linkers and that the high density of linkers on curved NPs prevents their bending towards the surface, which makes them more available for tethering bioactive molecules.
In this work, we present a quantum mechanical investigation of the interplay between a dopamine-functionalized curved TiO2 NP and doxorubicin to gain insight into the crucial aspects of the drug transport and delivery processes. For this study, we consider realistically sized TiO2 NPs of 700 atoms (2.2 nm).54 These spherical NPs are similar to those used in many experimental studies of NP + dopamine complexation.43,55–60 We investigate different coverage regimes of dopamine molecules on the surface to determine how they affect the DOX binding mode, binding energy and electronic properties. We also perform molecular dynamics runs and observe how the DOX and dopamine molecules rearrange on the surface during a simulated annealing process. Finally, we consider the effect of light irradiation on the DOX@DOPAMINE@TiO2-NP triad system, to establish its effectiveness in the separation of photoinduced charge carriers.61
All the calculations for the low coverage regime are with both the hybrid density functional theory (DFT-B3LYP)62,63 and self-consistent-charge density functional tight-binding (SCC-DFTB or hereon shortened to DFTB)64,65 methods, which allow assessing the accuracy of DFTB in the description of both structural and electronic properties of these complex systems. The higher coverage regime has been investigated by means of DFTB static and dynamic calculations, except for the electronic properties that were analyzed through single-point DFT calculations on the DFTB geometries for a correct description of the TiO2 band gap.
2. Computational details
As mentioned in the introduction, in this work we have used two levels of theory: density functional theory (DFT) and self-consistent charge density functional tight-binding (SCC-DFTB). Both methods have been employed for geometry optimization and electronic structure calculations. For performing molecular dynamics (MD), we only used the SCC-DFTB approach, as detailed at the end of this section.The DFT calculations have been carried out with the GAUSSIAN16 code66 and the CRYSTAL17 simulation code.67,68 The GAUSSIAN16 code has been used only for the free DOX molecule (section 3.1) at the B3LYP 6-311+G** level of theory. Its geometry was relaxed at both the equilibrium structure of the S0, S1 and T1 electronic states using ground state DFT calculations, the time-dependent density functional theory (TD-DFT) scheme and spin constrained DFT calculations, respectively. The solvent effect was included by means of the Polarizable Continuum Model (PCM), using the integral equation formalism variant (IEFPCM).69,70 Our results are in good agreement with theoretical and experimental studies reported in ref. 71 and 72.
In the CRYSTAL17 calculation, the Kohn–Sham orbitals are expanded in Gaussian-type orbitals. The all-electron basis sets are Ti 86-4111(d41) and O 8-4111(d1) for the oxygens of TiO2; H 5-111(p1), C 6-31111 (d1), O 8-41111 (d1) and N 6-311(d1) have been employed for hydrogen, carbon, oxygen and nitrogen of the adsorbed molecules. We used the hybrid functional B3LYP, for a more accurate description of the TiO2 band gap73 and we applied the a posteriori correction by Grimme (D*) to include dispersion forces.74,75 The cut-off limits in the evaluation of Coulomb and exchange series/sums appearing in the SCF equation were set to 10−7 for Coulomb overlap tolerance, 10−7 for Coulomb penetration tolerance, 10−7 for exchange overlap tolerance, 10−9 for exchange pseudo-overlap in the direct space, and 10−30 for exchange pseudo-overlap in the reciprocal space. The condition for the SCF convergence was set to 10−6 a.u. on the total energy difference between two subsequence cycles. The equilibrium structure is determined by using a quasi-Newton algorithm with a BFGS Hessian updating scheme.76 Geometry optimization was performed without any symmetry constraint; forces were relaxed to be less than 4.5 × 10−4 au and displacements to be less than 1.8 × 10−3 au.
For all the SCC-DFTB calculations we used the DFTB + open source package.77 We employed the MATORG parameterization set78 for the pairwise interaction of the atoms of both TiO2 and adsorbed molecules. The description of the hydrogen bonding has been further improved with the inclusion of the empirical HBD correction (ζ = 4).79,80 For geometry relaxations, the threshold for the convergence of the self-consistent charge (SCC) procedure was set to 10−6 charge au and forces were relaxed to be less than 10−4 au.
The anatase TiO2 spherical nanoparticle (NP) model used throughout this work has been designed through global optimization with a simulated annealing process at the SCC-DFTB level of theory in our previous work.54 The stoichiometry of the model is (TiO2)22310H2O and it is characterized by an equivalent diameter of 2.2 nm. The nanoparticle has been treated as a large isolated molecule in the vacuum without any periodic boundary condition.
The total adsorption energy of the dopamine molecule on the spherical nanoparticle has been defined as:
| ΔEDOPAads = ETot − (ENP + EDOPA) |
The total adsorption energy for DOX molecules on the dopamine-functionalized spherical nanoparticle has been defined as:
| ΔEDOXads = ETot − (ENP+nDOPA + EDOX) |
Total energy values do not include any entropic term and therefore the adsorption energy does not include the entropy loss following molecular adsorption on the NP surface, which would reduce the overall energy gain.
For the triplet excited states, obtained by spin constraint calculations, we compared hybrid DFT (B3LYP) and SCC-DFTB results with the on-site Coulomb correction81 (DFTB+U) method with an effective U–J value of 3.5 eV.82
Born–Oppenheimer MD simulations at the DFTB level of theory for the functionalized NPs were performed within the canonical ensemble (NVT). The Newton equations of motion were integrated with the velocity Verlet algorithm, and a relative small time step of 0.5 fs was used to ensure reversibility. During the molecular dynamic simulation, the temperature has been kept constant at 160 K by using the Nosé–Hoover thermostat (time constant of 0.04 ps) and the system has been let to evolve for 12 ps.
3. Results and discussion
3.1. Doxorubicin (DOX) molecule
To screen the different possible conformations of DOX molecules (Fig. 1a), we performed DFTB MD simulations. Statistical sampling has been enhanced by running five separate simulations, where different random seeds for velocity assignment were considered. The MD runs at 1 atm and 300 K for 100 ps. The optimized structures and relative energies in the singlet ground state (S0), at both DFTB and DFT levels of theory, are all reported in Fig. S1.† The most stable configuration, whose schematic drawing along with the relevant OH distances and along with the DFT (B3LYP) HOMO and LUMO plots is shown in Fig. 1a and b, will be used as reference in the following of this work. The HOMO is mainly concentrated on the π system of the aromatic ring and on the hydroxyl group, while in the LUMO the electron cloud is more shifted toward the keto groups. The HOMO–LUMO gap is 2.82 eV.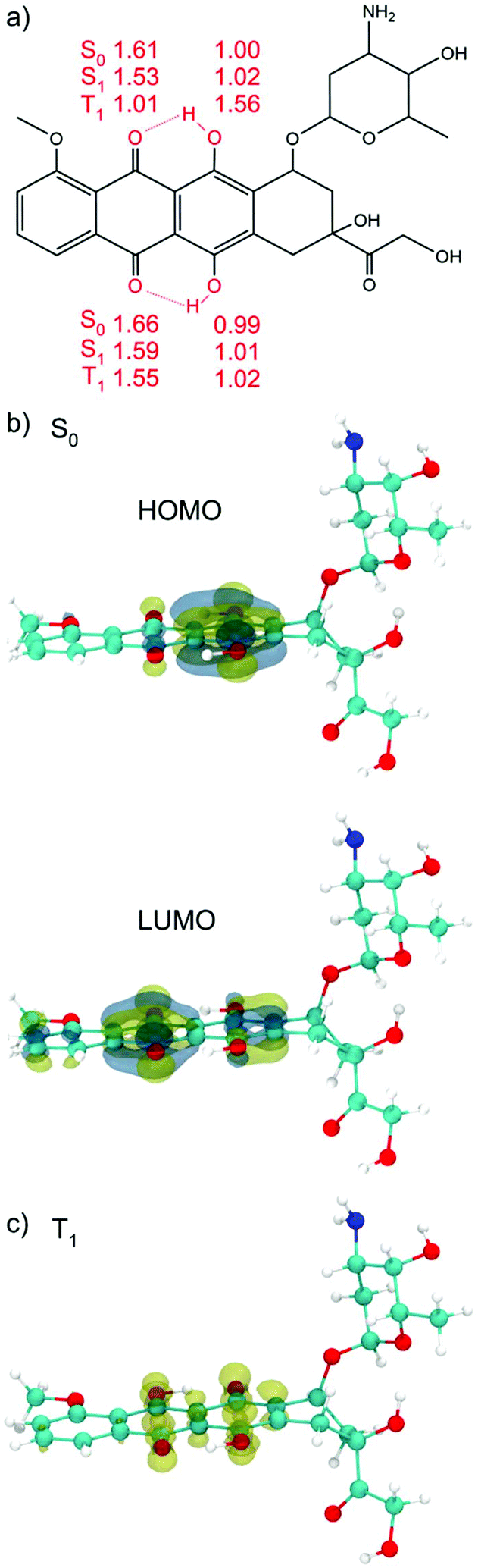 | ||
| Fig. 1 (a) Schematic drawing of the DOX chemical formula. The relevant OH and OH⋯O distances for the S0, S1 and T1 DFT (B3LYP) optimized structures in vacuum are reported in Å. The sketch resembles the S0 position of the H atom. (b) DFT (B3LYP) HOMO and LUMO plots for the most stable S0 conformation. The HOMO–LUMO gap is 2.82 eV and (c) DFT (B3LYP) spin density plot for the T1 optimized structure. C atoms are in cyan, H atoms are in white, the N atom is in blue and O atoms are in red. | ||
Hydroxyl groups can serve as proton donors, and carbonyl oxygen as proton acceptors. In the DOX molecule, with the proton donor and acceptor in such close proximity, the “excited state intra-molecular proton transfer” (ESIPT) may occur upon light irradiation.83 When a vertical electronic excitation occurs, the charge redistribution makes the proton donor more acidic and the proton acceptor more basic. In the S0 state (vacuum) the O–H bond lengths of the hydroxyl groups on the phenyl rings of DOX are 1.00 and 0.99 Å and the distances between hydrogens of the hydroxyl groups and the oxygens of the keto group are 1.61 and 1.66 Å (see Fig. 1a), respectively.
In the optimized excited S1 state, obtained by full atomic relaxation in the TD-DFT scheme (for details see the computational section), these bond lengths become 1.02, 1.01, 1.53 and 1.59 Å, respectively. The OH groups on one DOX phenyl ring are slightly elongated but still intact. On the other hand, the H⋯O distances for the oxygen of the keto group on the nearby six-member ring are shortened (see Fig. 1a), which indicates that these two intra-molecular H-bonds are strengthened. This distortion from the S0 to the S1 geometry suggests a propensity for the proton transfer upon excitation. This has been extensively studied in the literature for DOX and other molecules.71,72,84–89
The lowest DOX excited triplet state (T1) was also computed and fully relaxed by spin constrained calculations (see Fig. 1c). In the T1 optimized structure, we observe the complete proton transfer from the hydroxyl group to the keto oxygen, mentioned above. The energy difference between S0 and T1 is +1.29 eV.
As a further analysis, we have characterized the vertical S0 → S1 excitation and the S1 → S0 emission processes by TD-DFT in different nonpolar and polar solvents (see Table S1†) (for further technical details, see the computational section) and compared them with existing data from previous experimental and computational studies.71,72 Calculations can correctly describe the relative shifts in absorption and emission due to different solvents, although they are rather small. Even absolute values are quite close to experimental data. We do not expect the solvent effect to play a crucial role even when the molecule is loaded on the functionalized NP surface.
3.2. DOX loaded on dopamine-functionalized TiO2 nanoparticles
In this section, we analyze the adsorption of DOX molecules on the dopamine-functionalized 2.2 nm spherical NP surface at different coverage densities. In a previous study, we extensively investigated the adsorption modes of dopamine on the surface of this TiO2 nanoparticle.51,52 We have shown that while the bidentate catechol portion of dopamine binds to the oxide surface through coordinate bonds, in agreement with infrared studies,54,58 the primary amine could remain exposed to the surrounding environment providing a hook to tether biomolecules or drugs (DOX here). However, we also showed that, especially at low coverage, the molecule could bend towards the NP and establish either a hydrogen or a coordinate bond with the surface atoms. When the coordinate bond (–H2N⋯Ti) is formed, the molecule inclination is very high, with the phenyl ring almost parallel to the surface. Clearly, in such cases the amino group is not available for tethering of DOX. Therefore, for the sake of the present work, we will consider only dopamine adsorption configurations (four), where the –NH2 functional group of the dopamine is free, pointing towards the vacuum, or weakly interacting with the TiO2 surface through H-bonds. For such configurations, we will investigate DOX adsorption structures, binding energies and electronic properties of the composite systems, in different dopamine coverage regimes (low in section 3.2.1, medium in section 3.2.2 and high in section 3.2.3) and upon thermal treatment. Furthermore, in section 3.4, we will analyze the effect of light irradiation in terms of vertical excitation (exciton formation), atomic relaxation in the excited state (exciton self-trapping) and charge carrier dynamic evolution.In Fig. S2† we present all the configurations that have been considered in this work. We compare bond lengths and adsorption energies per molecule (ΔEDOPAads) as obtained after full geometry relaxation at DFTB and, only for NRD and SRD, also at DFT (B3LYP-D*) level of theory. The SRD configuration (Fig. S2c†) is the most stable one, as a result of the dopamine bending towards the surface, making two hydrogen bonds between the ethyl-amino group and the surface of the NP. In contrast, SLU (Fig. S2d†) is the least stable configuration since the dopamine stands up toward the vacuum, with no interaction between the molecule and the NP surface. This is in-line with our previous work on the adsorption of dopamine on the spherical NP.51 In addition, we note that details of the DFTB structures to some extent differ from DFT ones, especially as regards the H-bond distances; however, the general features of the different configurations and of relative adsorption energies are consistent with DFT.
As a next step, DOX is placed on top of the dopamine–functionalized NP models, as shown in Fig. 2. The configurations are labeled as: DOX@X, where X is the dopamine + NP structure considered. In all cases, the starting geometry for the atomic relaxation was conceived with a H-bond between the N of the ethyl-amino functional group of the dopamine and the hydrogen of the hydroxyl groups at the extreme of the aliphatic part of the DOX (see Fig. 1a).
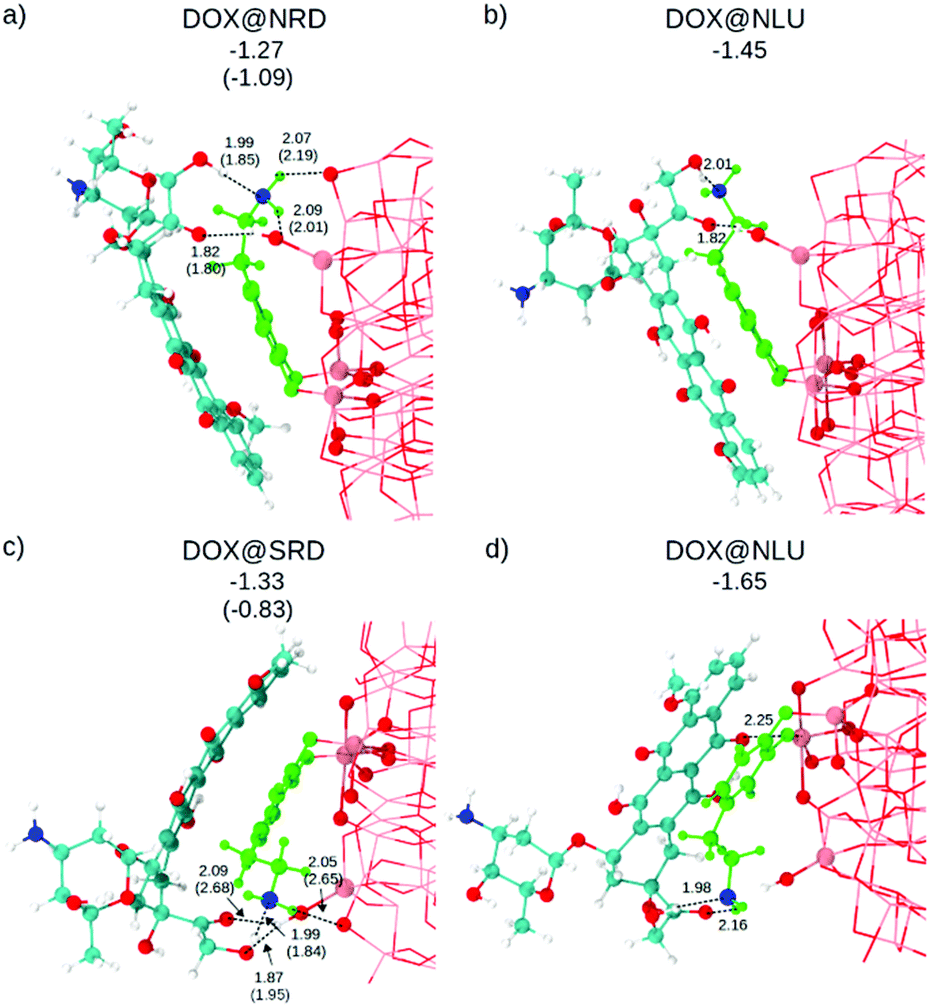 | ||
| Fig. 2 Side view of the optimized structures after DFTB (DFT) calculations. For more clarity, the atoms of the dopamine are colored in light green except for the N atom that is in blue. DOX C atoms are in cyan, H atoms are in white and the N atom is in blue. O atoms are in red and Ti atoms of the NP are in pink. Relevant distances are reported in Å in proximity of the dashed lines. Adsorption energies are in eV. | ||
The optimized structures and the relative DFTB and DFT (in parenthesis) adsorption energies (ΔEDOXads) are shown in Fig. 2. Except for the Ti5c⋯OKETO (2.25 Å) coordinate bond in DOX@SLU (Fig. 2d), all the other newly established bonds are H-bonds. As a consequence of the higher strength of the coordinate bond compared to the H-bond, this structure is the most favored among those considered (see adsorption energies in Fig. 2).
Geometry optimization at the hybrid DFT (B3LYP) level of theory for these structures is extremely expensive due to their large size; therefore, we performed it only for DOX@NRD and DOX@SRD. We considered these two specific cases, because here the DOX molecules do not make any coordinate bonds to the surface, the dopamine molecule establishes hydrogen bond(s) with the surface of the NP and there is π–π stacking between the DOX and the dopamine molecules. These hydrophobic interactions are crucial to reduce the solubility of DOX, and its premature loss, at least under basic conditions.4
3.2.1.a. Electronic properties. The electronic structure of these configurations has been calculated in three ways: (i) with DFTB (see Fig. S3†), (ii) performing a single-point DFT calculation on the optimized DFTB geometry (DFT/DFTB) and (iii) with DFT on the DFT optimized geometries (see Fig. 3). By comparing the results, we can assess whether DFTB is adequately robust to describe the electronic properties of these complex systems.
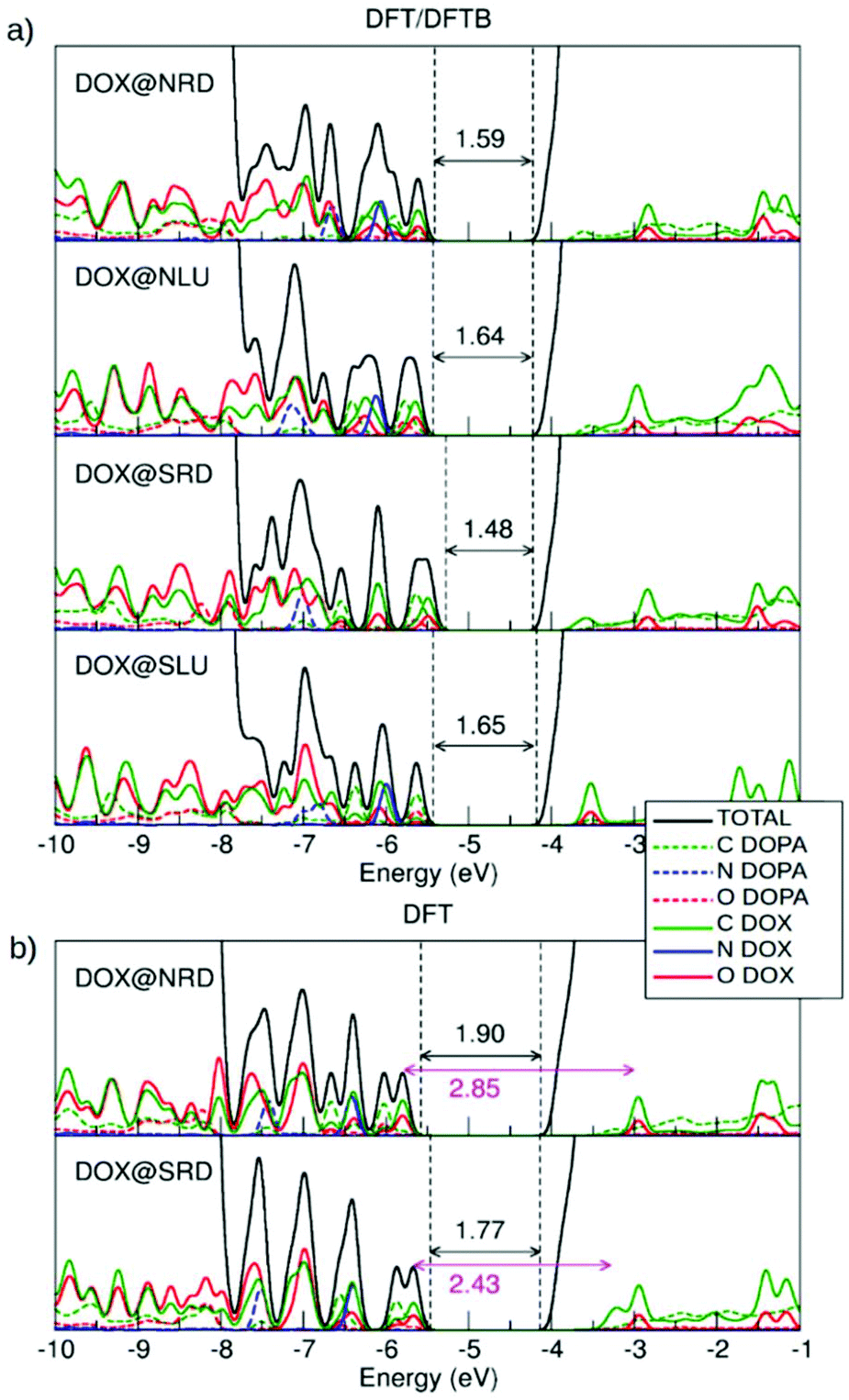 | ||
| Fig. 3 Total (DOS) and projected (PDOS) density of states calculated by (a) DFT/DFTB and (b) DFT calculations. C is represented in green, N is in blue and O is in red. The values for the HOMO–LUMO gap are reported with double-head arrows on each plot for DFT/DFTB and DFT calculations. | ||
The DOS by DFTB (Fig. S3†) is characterized by the presence of four mid-gap states. Starting from top of the valence band, there are two states associated with DOX and one with dopamine. There is also one DOX empty state below the bottom of the conduction band (LUMO). On the other hand, in both the DFT/DFTB (Fig. 3a) and DFT (Fig. 3b) calculations, we can identify many more states above the top of the valence band from ∼−7.7 eV (three states are associated with dopamine adsorbed in the bidentate mode on the surface of the spherical nanoparticle as explained in ref. 51. Therefore, one can see that DFTB results are not sufficiently reliable as regards the description of the electronic properties of these systems. We also note that the band gap value for the bare NP is underestimated by DFTB (3.11 eV) compared to DFT (4.13 eV).
For the free isolated DOX, nine states are observed above the −7.7 eV energy threshold, which is analogous to what was observed for DOX@NRD, DOX@NLU and DOX@SRD in Fig. 3a, because, in those configurations, DOX does not interact directly with the surface of the spherical nanoparticle but only with the dopamine. Differently, we note that in DOX@SLU, there are fewer states in the band gap due to the stabilizing effect of the coordinate bond. In this configuration, HOMO is associated with the O and C of the dopamine molecule, while for all the other ones, where there is no interaction between DOX and the NP surface, the HOMO is associated with O and C of the DOX molecule.
In the DOX@NRD configuration, the energy difference between the HOMO (localized on DOX oxygen and carbon atoms) and the empty state of the DOX inside the conduction band is around 2.85 eV (see the double headed arrow in Fig. 3b) which is very close to the HOMO–LUMO gap for the free DOX molecule (2.82 eV, see section 3.1). The position of the DOX states is further modified in DOX@SRD, due to a larger π–π interaction than in DOX@NRD, leading to a HOMO–LUMO gap value reduction to 2.43 eV. Consequently, we expect an appreciable absorption red-shift.
Thus, we may conclude that, although DFTB methods are not sufficiently accurate for the description of the electronic properties, DFT calculations on DFTB geometries correctly resemble DFT calculations on DFT optimized ones, based on the comparison between Fig. 3a and b. On these grounds, we will use the same combined approach for larger systems below.
At low dopamine coverage, however, the DOX molecule tends to slip towards the NP surface forming some coordinative bonds, as we have observed during some short MD runs (see Fig. S4 in the ESI†).
Therefore, in order to avoid these undesirable interactions, we increased the coverage density of dopamine molecules on the NP, to create a barrier for the DOX to reach and get in contact with the NP surface. In particular, we adsorbed a cluster of seven dopamine molecules on a portion of the NP surface. To achieve a very high density, we adsorbed them in a mixture of bidentate (two) and chelated (five) modes.51
We used MD and simulated thermal annealing to explore the potential energy surface and to obtain stable configurations. We started from structures a-, b- and c-DOX@7DOPA in Fig. S5,† where no direct interaction between DOX and the NP surface is present and, then, we investigated the temperature effect on the structures. The optimized geometries after performing MD are shown in Fig. 4. The adsorption energy values (ΔEDOXads) are −3.77, 1.69 and −1.43 eV, respectively. There is a huge energy stabilization for a-DOX@7DOPA after MD (compare with Fig. S5†), whose origin will be discussed below. In Fig. 4 and Fig. S5,† the dopamine molecules are color coded as follows: dopamine 1 green (bidentate), dopamine 2 tan (chelated), dopamine 3 ice-blue (chelated), dopamine 4 orange (chelated), dopamine 5 pink (chelated), dopamine 6 yellow (chelated) and dopamine 7 is in red (bidentate).
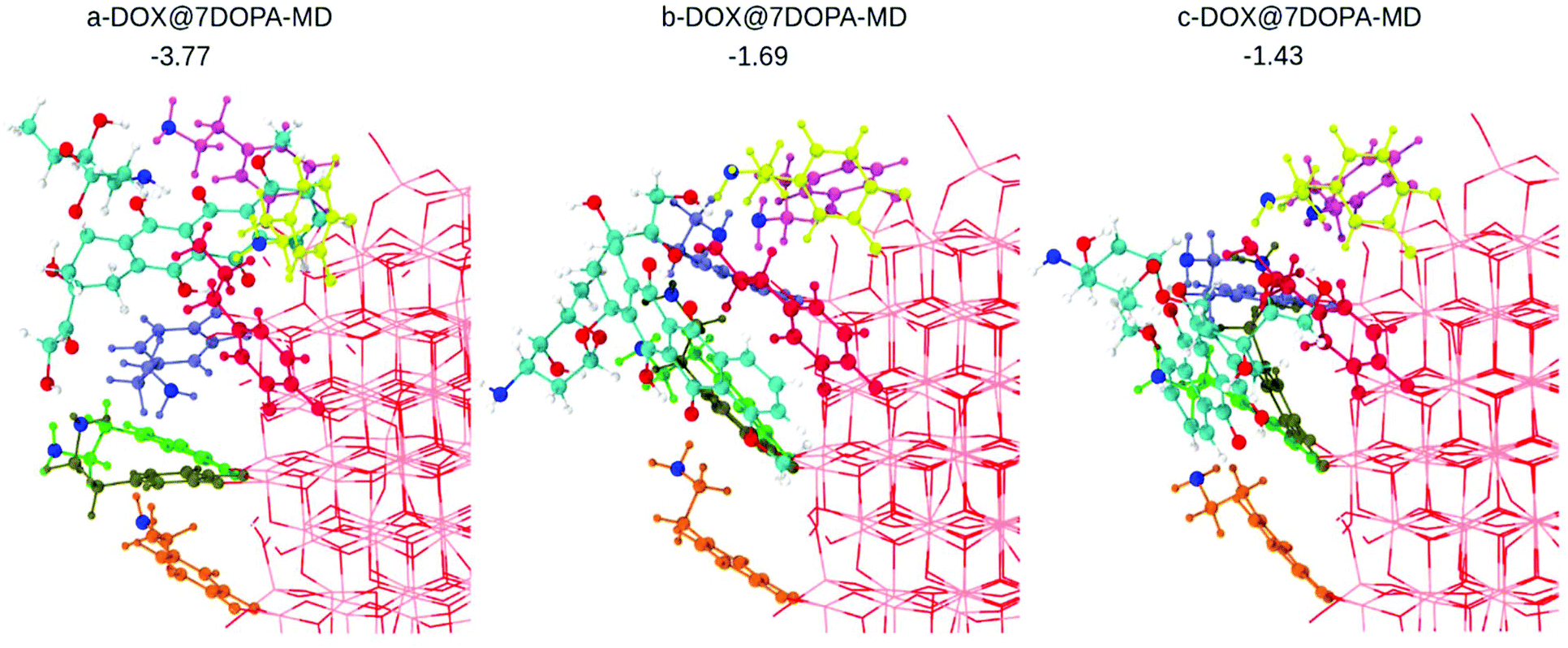 | ||
| Fig. 4 Optimized structures after performing MD on the corresponding configurations shown in Fig. S4a–c,† as obtained by DFTB calculations. The dopamine molecules adsorbed on the surface of the NP are color coded as explained in the text. In addition, all the N atoms of the –NH2 groups from the dopamine molecules and the DOX molecule are in blue. DOX C atoms are in cyan, H atoms are in white and the N atom is in blue. O atoms are in red and Ti atoms of the NP are in pink. Adsorption energies are in eV. | ||
In the case of b- and c-DOX@7DOPA-MD, there is no interaction between the DOX and the NP surface. We also note that all of the existing interactions, either DOX/dopamine, dopamine/NP or dopamine/dopamine, are of H-bond type (see Table S3†) with the addition, of some π–π stacking interactions between the red dopamine and DOX in b-DOX@7DOPA-MD.
Differently, in the case of a-DOX@7DOPA-MD, the DOX slipped into the void between the red, pink and ice-blue dopamine molecules during the simulated annealing. Here we observed the formation of a coordinate bond between DOX and the NP surface (OKETO⋯Ti4c of 2.24 Å) in addition to the typical H-bonds seen in the other configurations (b- and c-) and a considerable π–π stacking interaction between pink dopamine and DOX (see Table S3†). We attribute the enhanced adsorption energy (−3.77 eV) computed for this structure to the established OKETO⋯Ti4c bond (estimated to be about −0.95 eV from a model calculation of a formaldehyde molecule adsorbed on the same site), to large π–π stacking and dispersion interactions as a consequence of the close packing of DOX among the dopamine molecules.
We have also plotted the density of states (DOS) for the three optimized structures after MD. As discussed in section 3.2.1.a, the DOS is obtained by the combined DFT/DFTB approach, thus performing a single-point DFT calculation on the DFTB geometry. The results are shown in Fig. 5 and should be compared to Fig. 3. Having more dopamine molecules on the surface causes the appearance of new peaks both inside the band gap and at the top of the valence band. The HOMO–LUMO gap value decreases with respect to that for the low coverage in Fig. 3. The highest occupied peak is now made of dopamine states, in contrast to the low coverage where it was arising from the DOX states.
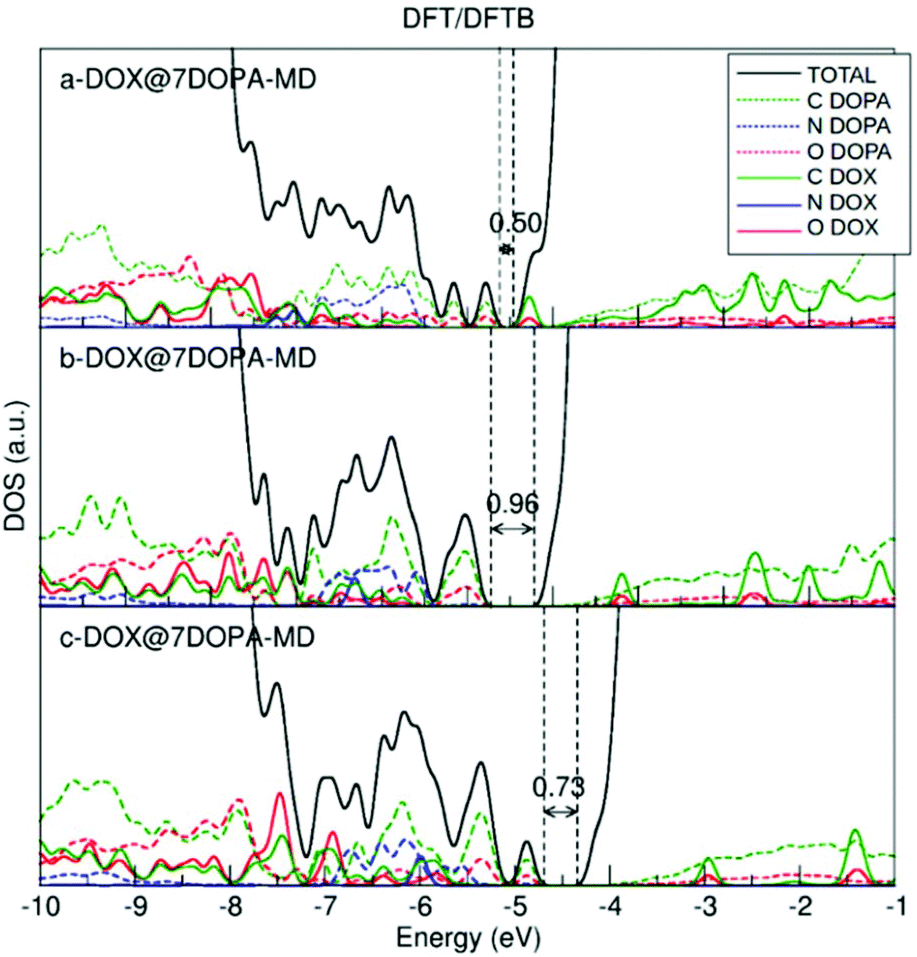 | ||
| Fig. 5 DFT total (DOS) and projected (PDOS) density of states for the DFTB optimized DOX@7DOPA-MD structures after performing MD, shown in Fig. 4. | ||
A DOX molecule was then placed on this highly covered NP in different relative positions, as shown in Fig. S7† after atomic relaxation (a-, b-, c- and d-DOX@46DOPA). However, some simulated annealing is mandatory to explore the potential energy surface of these very large systems. This was carried out by performing DFTB MD runs at 160 K for 12 ps. The corresponding optimized structures after performing MD are shown in Fig. 6 (a-, b-, c- and d-DOX@46DOPA-MD).
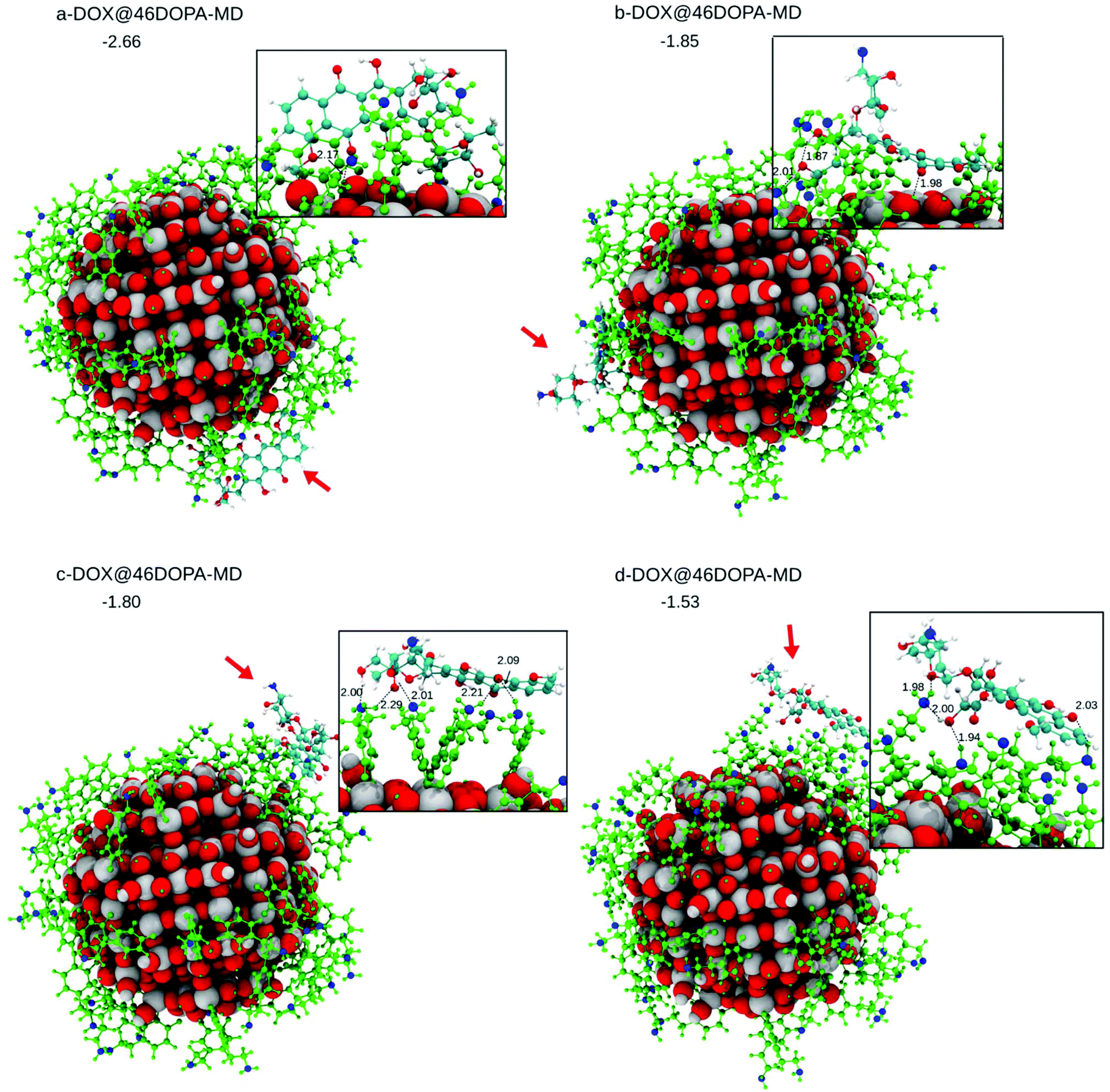 | ||
| Fig. 6 (a–d) Four optimized DFTB model structures of DOX on the curved TiO2 nanoparticle functionalized by 46 dopamine molecules (DOX@46DOPA-MD), after performing MD. In the inset, the DOX fragment is magnified. Red arrows indicate the position of the DOX on the dopamine-functionalized TiO2 NP. For the sake of clarity, all of the 46 dopamine molecules on the surface of the NP are colored in green except for the N atoms that are in blue. DOX C atoms are in cyan, H atoms are in white and the N atom is in blue. O atoms are in red and Ti atoms of the NP are in pink. Relevant distances are reported in Å in proximity of the dashed lines. Adsorption energies are in eV. | ||
In c-DOX@46DOPA-MD and d-DOX@46DOPA-MD, DOX has no direct interaction with the surface of the NP but only some H-bonds with the dopamine molecules are established, where the dopamine NH2 groups play both the role of H-bond donors or acceptors towards the carbonyl O or OH groups in DOX, respectively. For the d-DOX@46DOPA-MD structure, we have additionally heated up the system until 300 K, followed by full atomic relaxation (see Fig. S8†), to assess whether the optimized structure after MD at 160 K was in a deep stable minimum or not. Indeed, we only observe some molecular rearrangement of the H-bond network and a tiny stabilization of about 0.3 eV but no formation of any coordinative bonds with the undercoordinated Ti atoms on the NP surface.
In a-DOX@46DOPA-MD and b-DOX@46DOPA-MD, we observe that the DOX molecule has slipped into some voids among the dopamine molecules and succeeded in reaching the NP surface, although no coordinative bonds are formed. In configuration a-DOX@46DOPA-MD the –NH2 group of the DOX makes a H-bond with a surface OH (2.00 Å) in addition to the H-bond between the DOX keto group and another surface OH (OKETO⋯HOTiNP: 2.17 Å). In configuration b-DOX@46DOPA-MD the keto group of the DOX molecule makes a H-bond with a surface OH group (OKETO⋯HOTiNP: 1.94 Å).
The statistics of DOX configurations on the highly covered NP by dopamine molecules we have reached is not very large. However, we can clearly state that some configurations exist where, even after some temperature annealing simulation, no contact between the DOX molecule and the NP surface is observed but only H-bonds with the decorating dopamine molecules. Those configurations are the most effective for a successful drug delivery process because a pH lowering effect, commonly observed in cancer cells, will not affect much the overall interaction of the DOX with the drug carrier systems but will cause the protonation of the NH2 group in DOX increasing its solubility in the physiological media for a consequent efficient release.
Therefore, based on the results of this and of the previous section, we must conclude that only full dopamine coverage on the TiO2 nanoparticle is efficacious for the drug delivery process. A mid-coverage dopamine coating will leave enough space for DOX to interact with the NP surface by some relatively strong coordinate bonds that are detrimental for its release.
3.3. Effect of light irradiation
In this last section, we will analyze the effect of light irradiation on the DOX@DOPAMINE@TiO2-NP system to investigate the life path of photoexcited excitons and to establish the effectiveness of this triad in the separation of photoinduced charge carriers. We take as a model system DOX@NRD from section 3.2.1 (see Fig. 2a), although we also consider DOX@SRD in the ESI (Fig. S10†). We simulate the excitation by computing first the vertically excited state, in the Franck–Condon approximation, from the S0 to the T1 state through a spin-constrained DFT (B3LYP) calculation. Then, we allow for full atomic relaxation in the T1 spin state. We obtain two optimized T1 structures that are shown in Fig. 7a and b. These two solutions are degenerate in energy (0.02 eV difference).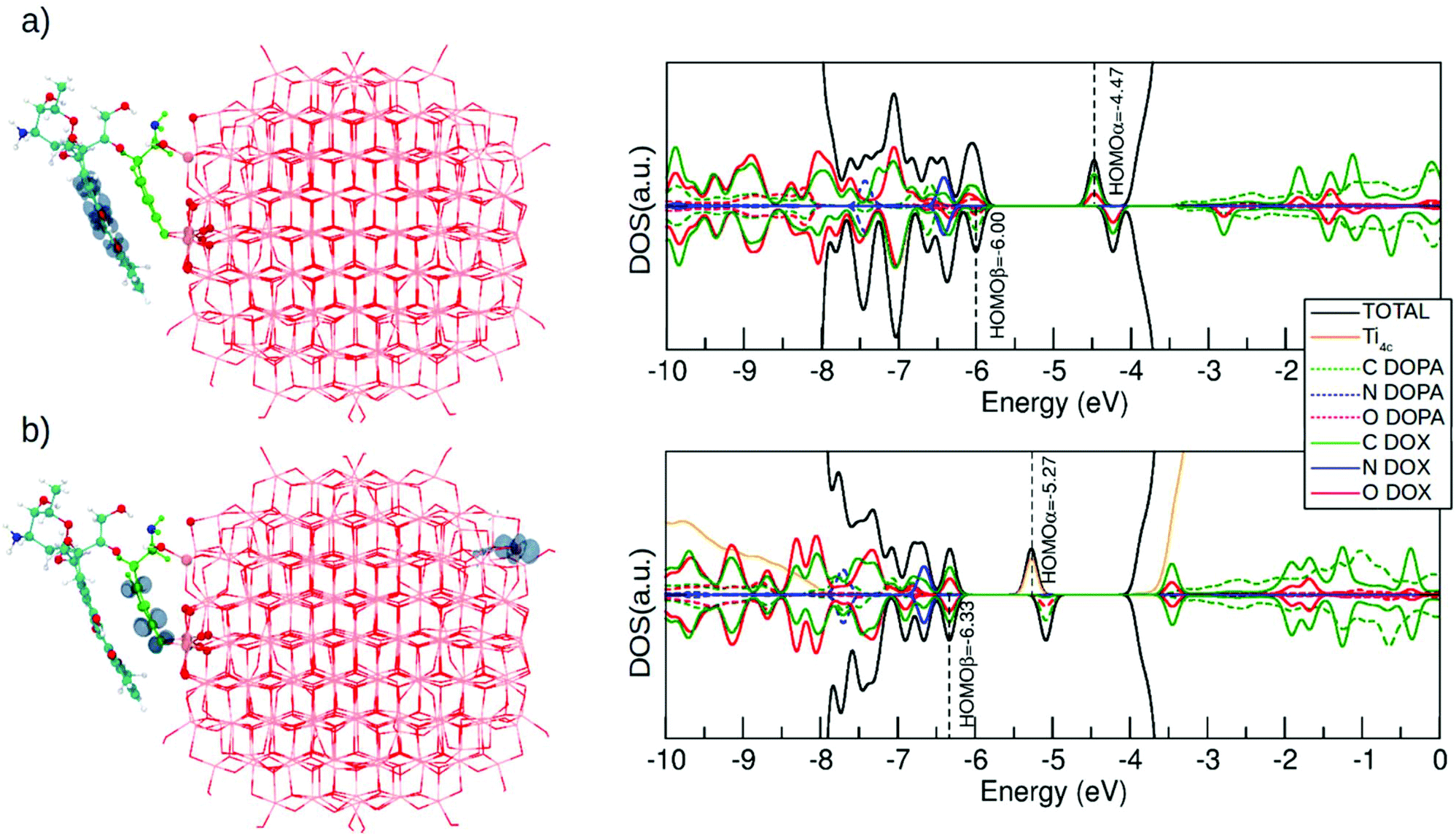 | ||
| Fig. 7 Right panel shows the spin density plots and the left panel shows the total (DOS) and projected (PDOS) density of states (DFT/DFTB) for the DOX@NRD configuration at the T1 state where (a) electrons and holes are localized on the DOX molecule and (b) the electron is localized on the Ti4c and the hole is localized on the dopamine molecule. Corresponding DOS and PDOS for the vertically excited state and for the DOX@SRD are displayed in Fig. S11.† For clarity, the atoms of the dopamine are colored in light green except for the N atom that is in blue. DOX C atoms are in cyan, H atoms are in white and the N atom is in blue. O atoms are in red and Ti atoms of the NP are in pink. | ||
In the first optimized T1 structure shown in Fig. 7a, both the photoexcited electron and hole are localized on the DOX molecule (see the spin plot). We also observe an intramolecular proton transfer, totally analogous to that for the T1 optimized state of the isolated DOX molecule in section 3.1 (see Fig. 1b). In the ground S0 state, the H–O bonds in the DOX are 1.00 Å (see the numbers in the parenthesis in Fig. S10a†), while in the T1 excited state one of the H–O bonds is so much stretched that it leads to the proton transfer to the O atom of the keto group (see Fig. S10a†).
To assess the performance of DFTB with respect to this type of calculation in the excited state, we show the density plot obtained by the DFTB+U method in Fig. S12a.† Spin density localization associated with the photoexcited exciton is very similar to what obtained with DFT calculations.
The DFT (B3LYP) total energy difference for an isolated DOX molecule between S0 and T1 (ΔE(T1–S0)) states, in their respective relaxed structures, is 1.32 eV. When DOX is adsorbed on dopamine/TiO2 NP (DOX@NRD), the ΔE(T1–S0) is only reduced by −0.01 eV (1.31 eV) (by −0.09 eV for DOX@SRD). This agrees very well with the DOX HOMO–LUMO gaps (2.82 vs. 2.85 eV) when free or DOX@NRD, reported in Fig. 1 and in Fig. 3b and discussed in section 3.2.1.
The density of states for the T1 optimized structure in Fig. 7a is shown on its right side. This should be compared with that for the S0 state for DOX@NRD in Fig. 3b. We observe that the photoexcited electron (HOMOα) is about 0.5 eV below the bottom of the conduction band in a DOX state involving C and O atoms. This state was empty in the ground state and lied about 1 eV above the bottom of the conduction band in Fig. 3b (DOX@NRD).
In the second optimized T1 structure shown in Fig. 7b, the photoexcited hole is localized on the dopamine molecule, whereas the photoexcited electron is trapped on a surface Ti atom far apart from the adsorption site. We observe some bond elongation for the Ti–O bonds at the Ti3+ trapping site and some C–C and Ti–O bind elongation in the hole hosting dopamine molecule. The DOX binding energy to such excited dopamine@TiO2 NP is approximately the same as that of the corresponding non-excited system in Fig. 2 (−1.11 eV vs. −1.09 eV). The DOS clearly shows that the photoexcited electron in the Ti d state is about 1.3 eV below the bottom of the conduction band, thus much deeper than in the previous case of Fig. 7a.
Although these two optimized T1 states are essentially at the same energy, one may expect to selectively excite to one or the other by using a different light frequency for irradiation. The energy to vertically excite an electron to the bottom of the conduction band is clearly much lower than the energy to excite an electron from the DOX occupied states to the DOX unoccupied ones. This is corroborated by the energy differences reported in the DOS for DOX@NRD in Fig. 3b.
Finally, we also investigate the light irradiation effect at dopamine mid coverage for structure a-DOX@7DOPA shown in Fig. 4. Given the size of the system, and the successful assessment of the DFTB+U method on the low coverage case, we used such an approach to compute the T1 excited state, by means of a spin-constrained atomic optimization (see Fig. S12b†). Here, the photoexcited electron becomes trapped at a surface Ti site, whereas the hole is localized on the dopamine. We could not succeed in the DOX photoexcitation at mid coverage density. The explanation for this is that, at this dopamine coverage density, the highest occupied states are all dopamine states (see DOS in Fig. 5a), with no contribution from the DOX molecule, as in the case of low coverage (compare with Fig. 3b).
4. Conclusions
In this work, we have investigated the anticancer drug doxorubicin loading on a functionalized curved semiconducting oxide nanoparticle. A titanium dioxide spherical nanoparticle of realistic size (2.2 nm with 700 atoms) was used as a light-responsive drug carrier. However, to reduce its direct interaction with DOX, a spacer or linker is anchored to the surface that is capable of tethering DOX, without altering its pristine chemical structure, through weak interactions, such as H-bonds, π–π stacking, dispersion forces, and dipole–dipole interactions. Dopamine, a catechol derivative molecule, with an additional amino group was chosen since it is a bifunctional linker with a high affinity towards oxide surfaces.The main objective is to assess, by means of accurate density functional based calculations, what is the most effective coverage density of the bifunctional linker on the nanoparticle surface to limit the interaction of DOX with the oxide surface for an effective transport but also for an effective release. We also aim at gaining insight into the electronic properties of these composite systems and to investigate the effect of vis-light irradiation.
We have considered three coverage densities of the linker: low (1 dopamine), medium (7 dopamines) and high (46 dopamines). At low coverage, we have performed both DFTB and DFT (B3LYP) calculations to assess the reliability of DFTB. We proved that DFT optimized structures are good, whereas electronic properties must be obtained with a single point DFT calculation on the DFTB geometry (DFT/DFTB) to achieve a satisfactory accuracy. At mid and high coverage, we have used DFTB to perform some thermal annealing simulations and full atomic relaxation. Electronic properties were studied with the DFT/DFTB approach.
When DOX is loaded on the dopamine-functionalized TiO2 NP at low coverage, we observe that DOX tends to sandwich over a bent dopamine molecule that lies almost flat on the oxide surface. The electronic structure indicates a very large reduction of the HOMO–LUMO gap between 1.5 and 1.7 eV due to several states in the gap arising from both dopamine and DOX molecules. The topmost states are assigned to DOX.
In the mid coverage regime, DOX is found to slip into the voids between the dopamine molecules that still have several degrees of freedom to move since they are not fully packed. In this way, DOX can reach the oxide surface and establish some rather strong coordinate bonds with the undercoordinated surface Ti ions.
Only at high coverage of dopamine, DOX molecules do not have any contact with the oxide surface, even after a reasonably long thermal simulated annealing. Under these conditions, DOX can be efficiently transported by the carrier but also successfully delivered because only weak hydrophobic interactions (H-bonding, dispersion forces and π–π stacking) are established that guarantee low DOX solubility at basic pH and efficient release at acidic pH.
Another relevant outcome of this study is the insight into the vis-light irradiation effect on this complex composite or triad systems (DOX@DOPAMINE@TiO2-NP). Free DOX absorbs in the vis-region at 480 nm. TD-DFT calculations accurately reproduce this observation and shift arising from the use of different solvents. When DOX is loaded on the dopamine-functionalized TiO2, its HOMO–LUMO states are only slightly rigidly shifted but the difference in energy is almost unchanged (2.85 vs. 2.82 eV in free DOX). However, the LUMO of DOX is deep in the conduction band; thus the lowest unoccupied states are TiO2 states (i.e. Ti 3d). When the system is excited and relaxed in a triplet exciton, two solutions can be obtained: one is essentially an excited DOX molecule adsorbed on the dopamine-functionalized TiO2 NP; the other presents a photoexcited electron trapped at a low-coordinated surface Ti ion and the hole localized on the dopamine molecule. Although the two solutions are essentially degenerate, they could be selectively excited, depending on the final purpose (photodynamic therapy or imaging), by using a different light frequency of irradiation, since the energy to vertically excite an electron to the bottom of the conduction band (1.90 eV) is much lower than the energy to excite an electron from the DOX occupied to the DOX unoccupied states (2.85 eV).
This computational study unravels fundamental aspects of drug interaction with their carriers. A future development of this work will be the investigation of the drug loading capacity of this drug delivery system and of the pH effect due to the environment. However, given the increasing size, we will probably have to resort to lower level theories, such as force-field methods. A comprehensive computational understanding of these systems will be useful to develop new experimental protocols for a more efficient drug transport and release in combination with photodynamic therapy.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Lorenzo Ferraro for his technical help, and to Costanza Ronchi, Martina Datteo and Daniele Selli for fruitful discussions. The project has received funding from the European Research Council (ERC) under the European Union's HORIZON2020 research and innovation programme (ERC Grant Agreement No. [647020]).References
- F. Arcamone, Doxorubicin: Anticancer Antibiotics, Academic Press, San Diego, 1982 Search PubMed.
- P. K. Singal and N. Iliskovic, N. Engl. J. Med., 1998, 339, 900–905 CrossRef CAS PubMed.
- L. Angeloni, G. Smulevich and M. Marzocchi, Spectrochim. Acta, Part A, 1982, 38, 213–217 CrossRef.
- Z. Liu, X. Sun, N. Nakayama-Ratchford and H. Dai, ACS Nano, 2007, 1, 50–56 CrossRef CAS PubMed.
- E. Heister, V. Neves, C. Tilmaciu, K. Lipert, V. S. Beltran, H. M. Coley, S. R. P. Silva and J. McFadden, Carbon, 2009, 47, 2152–2160 CrossRef CAS.
- O. Tacar, P. Sriamornsak and C. R. Dass, J. Pharm. Pharmacol., 2013, 65, 157–170 CrossRef CAS PubMed.
- F. Wang, Y. C. Wang, S. Dou, M. H. Xiong, T. M. Sun and J. Wang, ACS Nano, 2011, 5, 3679–3692 CrossRef CAS PubMed.
- J. Prados, C. Melguizo, R. Ortiz, C. Velez, P. J. Alvarez, J. L. Arias, M. A. Ruiz, V. Gallardo and A. Aranega, Anti-Cancer Agents Med. Chem., 2012, 12, 1058–1070 CrossRef CAS PubMed.
- P. Singh, R. Sharma, K. McElhanon, C. D. Allen, J. K. Megyesi, H. Bene and S. P. Singh, Free Radicals Biol. Med., 2015, 86, 90–101 CrossRef CAS PubMed.
- D. Ren, F. Kratz and S. W. Wang, Small, 2011, 7, 1051–1060 CrossRef CAS PubMed.
- G. Kong, R. D. Braun and M. W. Dewhirst, Cancer Res., 2000, 60, 4440–4445 CAS.
- Y. Shamay, L. Adar, G. Ashkenasy and A. David, Biomaterials, 2011, 32, 1377–1386 CrossRef CAS PubMed.
- G. M. Dubowchik, R. A. Firestone, L. Padilla, D. Willner, S. J. Hofstead, K. Mosure, J. O. Knipe, S. J. Lasch and P. A. Trail, Bioconjugate Chem., 2002, 13, 855–869 CrossRef CAS PubMed.
- E. K. Lim, T. Kim, S. Paik, S. Haam, Y. M. Huh and K. Lee, Chem. Rev., 2015, 115, 327–394 CrossRef CAS PubMed.
- K. Sun, J. Wang, J. Zhang, M. Hua, C. Liu and T. Chen, Int. J. Biol. Macromol., 2011, 49, 173–180 CrossRef CAS PubMed.
- K. E. Sapsford, W. R. Algar, L. Berti, K. B. Gemmill, B. J. Casey, E. Oh, M. H. Stewart and I. L. Medintz, Chem. Rev., 2013, 113, 1904–2074 CrossRef CAS PubMed.
- K. Y. Win and S. S. Feng, Biomaterials, 2006, 27, 2285–2291 CrossRef CAS PubMed.
- W. H. De Jong and P. J. A. Borm, Int. J. Nanomed., 2008, 3, 133–149 CrossRef CAS.
- Y. R. Zhang, R. Lin, H. J. Li, W. L. He, J. Z. Du and J. Wang, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2019, 11, e1519 Search PubMed.
- S. Poh, V. Chelvam and P. S. Low, Nanomedicine, 2015, 10, 1439–1449 CrossRef CAS PubMed.
- M. Li, Z. Luo and Y. Zhao, Chem. Mater., 2018, 30, 25–53 CrossRef CAS.
- L. Hajba and A. Guttman, Biotechnol. Adv., 2016, 34, 354–361 CrossRef CAS PubMed.
- J. Li, Sci. Bull., 2015, 60, 488–490 CrossRef CAS.
- J. H. Lee and Y. Yeo, Chem. Eng. Sci., 2015, 125, 75–84 CrossRef CAS PubMed.
- M. T. Morgan, Y. Nakanishi, D. J. Kroll, A. P. Griset, M. A. Carnahan, M. Wathier, N. H. Oberlies, G. Manikumar, M. C. Wani and M. W. Grinstaff, Cancer Res., 2006, 66, 11913–11921 CrossRef CAS PubMed.
- Y. Cheng, A. C. Samia, J. D. Meyers, I. Panagopoulos, B. Fei and C. Burda, J. Am. Chem. Soc., 2008, 130, 10643–10647 CrossRef CAS PubMed.
- G. Wu, R. F. Barth, W. Yang, S. Kawabata, L. Zhang and K. Green-Church, Mol. Cancer Ther., 2006, 5, 52–59 CrossRef CAS PubMed.
- X. Yang, H. Hong, J. J. Grailer, I. J. Rowland, A. Javadi, S. A. Hurley, Y. Xiao, Y. Yang, Y. Zhang, R. J. Nickles, W. Cai, A. A. Steeber and S. Gong, Biomaterials, 2011, 32, 4151–4160 CrossRef CAS PubMed.
- Y. Qin, L. Sun, X. Li, Q. Cao, H. Wang, X. Tang and L. Ye, J. Mater. Chem., 2011, 21, 18003–18010 RSC.
- S. P. Pujari, L. Scheres, A. T. Marcelis and H. Zuilhof, Angew. Chem., Int. Ed., 2014, 53, 6322–6356 CrossRef CAS PubMed.
- R. Mrówczyński, J. Jurga-Stopa, R. Markiewicz, E. L. Coy, S. Jurga and A. Woźniak, RSC Adv., 2016, 6, 5936–5943 RSC.
- C. Bi, R. Jiang, X. He, L. Chen and Y. Zhang, RSC Adv., 2015, 5, 59408–59416 RSC.
- Y. Dai, D. Yang, D. Yu, C. Cao, Q. Wang, S. Xie, L. Shen, W. Feng and F. Li, ACS Appl. Mater. Interfaces, 2017, 9, 26674–26683 CrossRef CAS PubMed.
- F. Liu, X. He, Z. Lei, L. Liu, J. Zhang, H. You, H. Zhang and Z. Wang, Adv. Healthcare Mater., 2015, 4, 559–568 CrossRef CAS PubMed.
- X. Mu, F. Zhang, C. Kong, H. Zhang, W. Zhang, R. Ge, Y. Liu and J. Jiang, Int. J. Nanomed., 2017, 12, 2899–2911 CrossRef CAS PubMed.
- J. Xi, L. Da, C. Yang, R. Chen, L. Gao, L. Fan and J. Han, Int. J. Nanomed., 2017, 12, 3331–3345 CrossRef CAS PubMed.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- J. Cui, Y. Yan, G. K. Such, K. Liang, C. J. Ochs, A. Postma and F. Caruso, Biomacromolecules, 2012, 13, 2225–2228 CrossRef CAS PubMed.
- D. R. Dreyer, D. J. Miller, B. D. Freeman, D. R. Paul and C. W. Bielawski, Langmuir, 2012, 28, 6428–6435 CrossRef CAS PubMed.
- H. Shi, R. Magaye, V. Castranova and J. Zhao, Part. Fibre Toxicol., 2013, 10, 15 CrossRef CAS PubMed.
- Z. F. Yin, L. Wu, H. G. Yang and Y. H. Su, Phys. Chem. Chem. Phys., 2013, 15, 4844–4858 RSC.
- T. Rajh, N. M. Dimitrijevic, M. Bissonnette, T. Koritarov and V. Konda, Chem. Rev., 2014, 114, 10177–10216 CrossRef CAS PubMed.
- E. A. Rozhkova, I. Ulasov, B. Lai, N. M. Dimitrijevic, M. S. Lesniak and T. Rajh, Nano Lett., 2009, 9, 3337–3342 CrossRef CAS PubMed.
- N. Kotagiri, G. P. Sudlow, W. J. Akers and S. Achilefu, Nat. Nanotechnol., 2015, 10, 370–379 CrossRef CAS PubMed.
- D. Duan, H. Liu, Y. Xu, Y. Han, M. Xu, Z. Zhang and Z. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 5278–5286 CrossRef CAS PubMed.
- N. K. Shrestha, J. M. Macak, F. Schmidt-Stein, R. Hahn, C. T. Mierke, B. Fabry and P. Schmuki, Angew. Chem., Int. Ed., 2009, 48, 969–972 CrossRef CAS PubMed.
- K. T. Thurn, T. Paunesku, A. Wu, E. M. B. Brown, B. Lai, S. Vogt, J. Maser, M. Aslam, V. Dravid, R. Bergan and G. E. Woloschak, Small, 2009, 5, 1318–1325 CrossRef CAS PubMed.
- P. J. Endres, T. Paunesku, S. Vogt, T. J. Meade and G. E. Woloschak, J. Am. Chem. Soc., 2007, 129, 15760–15761 CrossRef CAS PubMed.
- K. Brown, T. Thurn, L. Xin, W. Liu, R. Bazak, S. Chen, B. Lai, S. Vogt, C. Jacobsen and G. E. Woloschak, Nano Res., 2018, 11, 464–476 CrossRef CAS PubMed.
- E. Katz and I. Willner, Angew. Chem., Int. Ed., 2004, 43, 6042–6108 CrossRef CAS.
- C. Ronchi, M. Datteo, M. Kaviani, D. Selli and C. Di Valentin, J. Phys. Chem. C, 2019, 123, 10130–10144 CrossRef CAS.
- C. Ronchi, D. Selli, W. Pipornpong and C. Di Valentin, J. Phys. Chem. C, 2019, 123, 7682–7695 CrossRef CAS PubMed.
- G. Fazio, L. Ferrighi and C. Di Valentin, J. Phys. Chem. C, 2015, 119, 20735–20746 CrossRef CAS.
- D. Selli, G. Fazio and C. Di Valentin, J. Chem. Phys., 2017, 147, 164701 CrossRef PubMed.
- E. Rozhkova, I. Ulasov, D.-H. Kim, N. Dimitrijevic, V. Novosad, S. Bader, M. Lesniak and T. Rajh, Int. J. Nanosci., 2011, 10, 899–908 CrossRef CAS PubMed.
- T. Rajh, L. Chen, K. Lukas, T. Liu, M. Thurnauer and D. Tiede, J. Phys. Chem. B, 2002, 106, 10543–10552 CrossRef CAS.
- J. Liu, L. de la Garza, L. Zhang, N. M. Dimitrijevic, X. Zuo, D. M. Tiede and T. Rajh, Chem. Phys., 2007, 339, 154–163 CrossRef CAS.
- M. B. Radoičić, I. A. Janković, V. N. Despotović, D. V. Šojić, T. D. Savić, Z. V. Šaponjić and B. F. Abramović, Appl. Catal., B, 2013, 138, 122–127 CrossRef.
- L. D. L. Garza, Z. V. Saponjic, N. M. Dimitrijevic, M. C. Thurnauer and T. Rajh, J. Phys. Chem. B, 2006, 110, 680–686 CrossRef PubMed.
- M. Niederberger, G. Garnweitner, F. Krumeich, R. Nesper, H. Cölfen and M. Antonietti, Chem. Mater., 2004, 16, 1202–1208 CrossRef CAS.
- N. M. Dimitrijevic, L. D. L. Garza and T. Rajh, Int. J. Mod. Phys. B, 2009, 23, 473–491 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–788 CrossRef CAS PubMed.
- T. Frauenheim, G. Seifert, M. Elstner, Z. Hajnal, G. Jungnickel, D. Porezag, S. Suhai and R. Scholz, Phys. Status Solidi, 2000, 217, 41–62 CrossRef CAS.
- M. Elstner and G. Seifert, Philos. Trans. R. Soc., A, 2014, 372, 20120483 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, et al., Gaussian16, revision B.01, Gaussian, Inc, Wallingford, CT, 2016 Search PubMed.
- A. Erba, J. Baima, I. Bush, R. Orlando and R. Dovesi, J. Chem. Theory Comput., 2017, 13, 5019–5027 CrossRef CAS PubMed.
- R. Dovesi, V. R. Saunders, C. Roetti, R. Olando, C. M. Zicovich-Wilson, F. Pascale, B. Civalleri, K. Doll, N. M. Harrison, I. J. Bush, et al., CRYSTAL17 User's Manual, University of Torino, Torino, Italy, 2017 Search PubMed.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- J.-L. Pascual-ahuir, E. Silla and I. Tunon, J. Comput. Chem., 1994, 15, 1127–1138 CrossRef CAS.
- M. Jia, X. Song, Q. Zhang and D. A. Yang, J. Cluster Sci., 2018, 29, 673–678 CrossRef CAS.
- D. K. Rana, S. Dhar, A. Sarkar and S. C. Bhattacharya, J. Phys. Chem. A, 2011, 115, 9169–9179 CrossRef CAS PubMed.
- F. De Angelis, C. Di Valentin, S. Fantacci, A. Vittadini and A. Selloni, Chem. Rev., 2014, 114, 9708–9753 CrossRef CAS PubMed.
- B. Civalleri, C. M. Zicovich-Wilson, L. Valenzano and P. Ugliengo, CrystEngComm, 2008, 10, 405–410 RSC.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- B. Civalleri, P. D'Arco, R. Orlando, V. Saunders and R. Dovesi, Chem. Phys. Lett., 2001, 348, 131–138 CrossRef CAS.
- B. Aradi, B. Hourahine and T. Frauenheim, J. Phys. Chem. A, 2007, 111, 5678–5684 CrossRef CAS PubMed.
- D. Selli, G. Fazio, G. Seifert and C. Di Valentin, J. Chem. Theory Comput., 2017, 13, 3862–3873 CrossRef CAS PubMed.
- H. Hu, Z. Lu, M. Elstner, J. Hermans and W. Yang, J. Phys. Chem. A, 2007, 111, 5685–5691 CrossRef CAS PubMed.
- M. Elstner, Theor. Chem. Acc., 2006, 116, 316–325 Search PubMed.
- B. Hourahine, S. Sanna, B. Aradi, C. Köhler, T. Niehaus and T. Frauenheim, J. Phys. Chem. A, 2007, 111, 5671–5677 CrossRef CAS PubMed.
- A. Tilocca and A. Selloni, J. Phys. Chem. C, 2012, 116, 9114–9121 CrossRef CAS.
- J. Seo, S. Kim and S. Y. Park, J. Am. Chem. Soc., 2004, 126, 11154–11155 CrossRef CAS PubMed.
- M. H. V. Huynh and T. Meyer, Chem. Rev., 2007, 107, 5004–5064 CrossRef CAS PubMed.
- J. Zhao, X. Liu and Y. Zheng, J. Phys. Chem. A, 2017, 121, 4002–4008 CrossRef CAS PubMed.
- L. Poudel, A. M. Wen, R. H. French, V. A. Parsegian, R. Podgornik, N. F. Steinmetz and W. Y. Ching, ChemPhysChem, 2015, 16, 1451–1460 CrossRef CAS PubMed.
- W. Yang and X. Chen, Phys. Chem. Chem. Phys., 2014, 16, 4242–4250 RSC.
- D. Spry and M. Fayer, J. Phys. Chem. A, 2007, 127, 204501 CrossRef CAS PubMed.
- X. Y. Liu, J. F. Zhao and Y. J. Zheng, RSC Adv., 2017, 7, 51318–51323 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: DFTB and DFT optimized structure of DOX before and after performing MD; spectroscopic Parameters of DOX; adsorption configurations and energies for one dopamine molecule on the surface of the TiO2 NP; DOS(PDOS) calculated by DFTB for DOX@1DOPA structures; DFTB optimized structure of DOX@SRD after performing MD; DFTB optimized structures of 7DOPA and DOX@7DOPA; interactions between DOX/Dopamine, dopamine/dopamine and Dopamone/NP; DOS(PDOS) calculated by DFT/DFTB for 7DOPA and DOX@7DOPA configurations; DFTB optimized structures of DOX@46DOPA; DFTB optimized structure of d-DOX@46DOPA after performing MD at 300 K; DOS(PDOS) for DOX@46DOPA calculated by DFT/DFTB; DFT optimized structures of triplet exciton and spin density plots for DOX@NRD and DOX@SRD; DOS(PDOS) for the vertical and adiabatic calculations of the DOX@NRD and DOX@SRD obtained by DFT. DFTB+U spin density plots using for DOX@NRD and for b-DOX@7DOPA-MD. See DOI: 10.1039/c9nr03763b |
| This journal is © The Royal Society of Chemistry 2019 |