
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantification of DNA damage induced repair focus formation via super-resolution dSTORM localization microscopy†
Dániel
Varga‡
 a,
Hajnalka
Majoros‡
bc,
Zsuzsanna
Ujfaludi
bc,
Miklós
Erdélyi
*a and
Tibor
Pankotai
*bc
a,
Hajnalka
Majoros‡
bc,
Zsuzsanna
Ujfaludi
bc,
Miklós
Erdélyi
*a and
Tibor
Pankotai
*bc
aUniversity of Szeged, Department of Optics and Quantum Electronics, Hungary. E-mail: erdelyim@physx.u-szeged.hu
bUniversity of Szeged, Department of Biochemistry and Molecular Biology, Hungary. E-mail: pankotai@bio.u-szeged.hu; pankotai.tibor@stoma.szote.u-szeged.hu
cUniversity of Szeged, Department of Oral Biology and Experimental Dental Research, Hungary
First published on 1st July 2019
Abstract
In eukaryotic cells, each process, in which DNA is involved, should take place in the context of a chromatin structure. DNA double-strand breaks (DSBs) are one of the most deleterious lesions often leading to chromosomal rearrangement. In response to environmental stresses, cells have developed repair mechanisms to eliminate the DSBs. Upon DSB induction, several factors play roles in chromatin relaxation by catalysing the appropriate histone posttranslational modification (PTM) steps, therefore promoting the access of the repair factors to the DSBs. Among these PTMs, the phosphorylation of the histone variant H2AX at its Ser139 residue (also known as γH2AX) could be observed at the break sites. The structure of a DNA double-strand break induced repair focus has to be organized during the repair as it contributes to the accessibility of specific repair proteins to the damaged site. Our aim was to develop a quantitative approach to analyse the morphology of single repair foci by super-resolution dSTORM microscopy to gain insight into chromatin organization in DNA repair. We have established a specific dSTORM measurement process by developing a new analytical algorithm for gaining quantitative information about chromatin morphology and repair foci topology at an individual γH2AX enriched repair focus. Using this method we quantified single repair foci to show the distribution of γH2AX. The image of individual γH2AX referred to as the Single target Molecule response scatter Plot (SMPlot) was obtained by using high lateral resolution dSTORM images. Determination of the average localization numbers in an SMPlot was one of the key steps of quantitative dSTORM. A repair focus is made up of nanofoci. Such a substructure of repair foci can only be resolved and detected with super-resolution microscopy. Determination of the number of γH2AXs in the nanofoci was another key step of quantitative dSTORM. Additionally, based on our new analysis method, we were able to show the number of nucleosomes in each nanofocus that could allow us to define the possible chromatin structure and the nucleosome density around the break sites. This method is one of the first demonstrations of a single-cell based quantitative measurement of a discrete repair focus, which could provide new opportunities to categorize the spatial organization of nanofoci by parametric determination of topological similarity.
Introduction
The DNA in the nucleus is constantly targeted by different damaging agents derived from both endogenous and exogenous sources. DNA double-strand breaks (DSBs) are the most deleterious lesions, and therefore they have to be repaired as quickly and efficiently as possible to prevent chromosomal loss and translocation. Since DSBs affect DNA integrity simultaneously with the recruitment of early DNA repair factors, a DNA damage response (DDR) is activated in the cells, which can arrest the cell cycle.1,2 For efficient DDR activation, different DSB sensors are required to activate chromatin reorganization and recruitment of downstream repair proteins that can eventually accomplish the efficient repair process.3 Recent studies have already shown that DNA damage can lead to immediate chromatin relaxation around the site of damage.4–6 One of the first steps of DSB induced chromatin reorganization is the phosphorylation of the histone variant H2AX at its S139 residue (called γH2AX) in the proximity of the damaged site.4,5,7,8 The γH2AX enriched chromosomal locus, considered as the repair focus, marks the damage site to initiate the recruitment of further repair proteins required for the process and performance of the repair.9,10 Several factors, such as the cell cycle state, functional activity of genes, break position along the DNA sequence, temporal state of DNA compaction, number of simultaneously occurring DSBs, etc., have been known to influence this process, thereby assigning the fate of the cell.11,12 The γH2AX signal detection is regularly used to visualize and quantify the extent of DSBs and to follow the DNA repair kinetics.Several techniques have been applied to follow the changes in γH2AX signal intensity. Chromatin immunoprecipitation studies have revealed that the γH2AX signal shows asymmetrical distribution around the damage site with lower density at the transcribed regions.13–20 It has already been shown that both the chromatin topology and state are crucial for the organization of the recruitment of repair proteins. A recent study based on chromosome conformation capture (4C) experiments has highlighted the complexity of genome re-organization, including megabase range associations between certain chromosomal regions as well as smaller genomic interactions, which involve kilobase-length DNA segments.21,22 Additionally, optical methods, such as conventional confocal microscopy, have also been regularly used for mapping the spatial distribution of DSBs. Due to the lateral resolution of these methods, the DDR signal can be detected in maximum 300 nm resolution. By using these techniques, it was shown that the γH2AX signal distributes up to a megabase around the damaged site8 generating DSB repair foci (repair foci) with a typical feature size of half a micron, which is just above the resolution limit of traditional fluorescence microscopes.
High resolution imaging based datasets of repair focus structures have already been published and demonstrated via single molecule localization methods (SMLM),23–28 structured illumination microscopy (SIM),26,29,30 and stimulated emission depletion (STED)29–31 super-resolution methods. These images can be further evaluated by using cluster analysis, and both the spatial distribution and the geometrical parameters of repair foci and even nanofoci can be determined.32,33 Single molecule localization methods (SMLM), such as dSTORM, provide the highest lateral resolution among optical methods and open the way for imaging biological structures in the sub-20 nm regime.34–36 SMLM determines the positions of individual molecules, which are used to create the final image.37–39 Different SMLM techniques such as PALM, PAINT and dSTORM have been proposed for quantitative analysis. However, taking into account the advantages and the limitations of the labelling procedures we opted to use the dSTORM technique.40–46 Although dSTORM separates the nanofoci of the repair focus, quantitative evaluation of the images has still been challenging because the number of detected localizations (Nlocalizations) generated by a single-labelled histone had remained unknown. Therefore, for quantitative analysis, the number of localizations per labelled γH2AX histone (number of localizations in a SMPlot) has to be statistically determined.
Here we provide insight into γH2AX distribution in nanometre resolution by using a super-resolution dSTORM microscopy technique applied either on U2OS cells exposed to neocarzinostatin treatment or on AsiSI endonuclease-expressing DIvA cells.18,47 By surpassing the limitation of classical confocal microscopy, super-resolution dSTORM microscopy possesses high prospecting capacity, which allows us to enlarge complex structures at γH2AX-enriched chromatin regions in 20 nm resolution. With this technique we measured the DDR profiles of several genomic regions and gained temporal, functional and structural insights into the damaged chromatin units evolved during the DSB repair. By means of dSTORM, in accord with the already published data, we observed that the number of repair foci increased following DSB induction.48,49 In addition, we demonstrated that the sizes of these repair foci were extended under DSB formation and we could also provide a higher resolution of the repair foci spatial organization using our new statistical approaches. This article preconcerts the nano-scale organization of the repair foci, which could highlight the spatial localization of the nanofocus structure and quantitative measurements of these repair centres.
Results
The experimental system and the determination of the parameters used in dSTORM
The typical size of a repair focus is about half micron, just above the resolution limit of traditional fluorescence microscopes. However, at a high density of the DNA breaks when individual repair foci overlap and form larger clusters the traditional imaging methods cannot be utilized. In such cases optical super-resolution microscopy is required to distinguish individual repair foci and reveal their sub-structures containing 20–60 nm nanofoci.50 In order to prove this, we generated DNA DSBs by applying neocarzinostatin and 4-OHT treatment to U2OS and DIvA cells, respectively.14,47 We quantified the DSB triggered repair foci formation 2 hours following the break induction by labelling H2AX S139 phosphorylated sites with fluorophore-conjugated antibody. We generated traditional EPI fluorescence and high resolution dSTORM images from the nuclei of non-treated and treated cells (Fig. 1A vs. B). By performing dSTORM, we observed an increase in the number and distribution of repair foci following DSB induction. At a higher resolution of individual repair foci, we could identify nanofoci that we used in our quantitative analysis (Fig. 1C).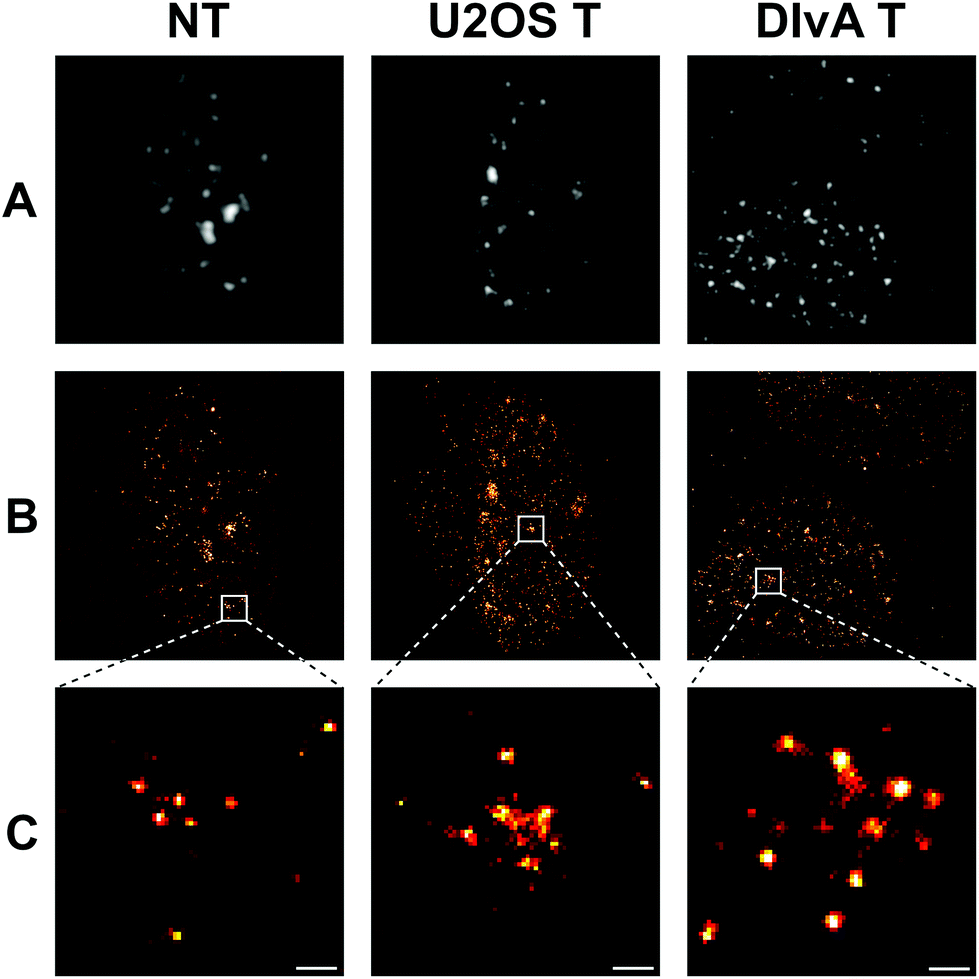 | ||
| Fig. 1 Traditional EPI fluorescence (A) and dSTORM super-resolved (B) images of nuclei of nontreated (NT) and treated (via NCS and 4-OHT) U2OS and DIvA cells, respectively. Magnified dSTORM images (C) of the selected individual repair foci. Scale bar represents 200 nm. | ||
Spatial distribution of repair foci within cells
In contrast to traditional optical microscopy images, in which the separation of individual repair foci is a great challenge and their size can only be quantified by their intensity values, cluster-analysis dSTORM images pave the way for quantitative evaluation. Quantitative functions of repair foci, such as their spatial density variation and their area distribution, were evaluated by means of 2D density-based spatial cluster analysis (DBSCAN). This algorithm requires two input parameters: a minimum number of points that form a cluster (Ncore) and the maximum distance between two adjacent points (ε).51 For the elimination of non-specific labelling and imprecise, out-of-focus localization, Ncore and ε were set to 8 and 50 nm during the calculations, respectively. Fig. 2A, C and E show a typical dSTORM super-resolved image of non-treated (NT) and treated (T) (with NCS and 4-OHT) U2OS and DIvA cells. The cluster-analysis images of the selected cells are also shown (Fig. 2B, D and F). In order to efficiently reveal the repair focus distribution pattern inside the nucleus, the cluster analysis module was implemented into our rainSTORM localization software (for details see Materials and methods).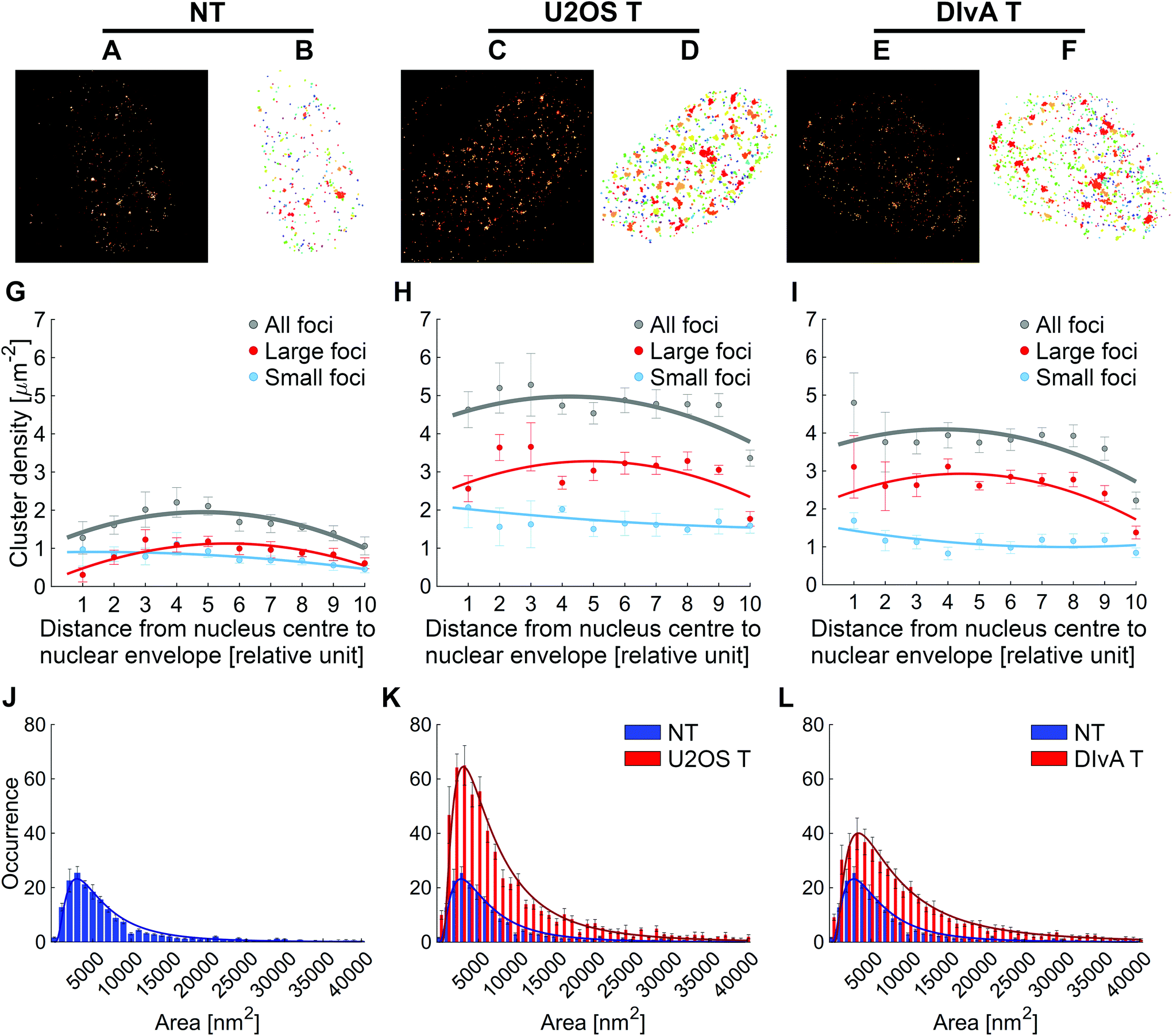 | ||
| Fig. 2 dSTORM (A, C, and E) and cluster-analysis (B, D, and F) images of untreated (A, B) and treated U2OS (C, D) and DIvA (E, F) cells. The average repair focus density functions are indicated in grey (G, H and I). The density function calculated for small (<5000 nm2) and large (>5000 nm2) repair foci are also shown in blue and red, respectively. Comparative histograms of the area distribution of repair focus sizes are presented for untreated (J), NCS treated U2OS (K) and DIvA cells (L). | ||
The algorithm isolated and quantified each repair focus based on their area and spatial distribution inside the nucleus. While Fig. 2A–F show single but typical nuclei, Fig. 2G–L depict the evaluation results of several, 5 untreated, 4 treated U2OS and 6 treated DIvA nuclei, respectively. By using the algorithm, discrete nanofoci were analysed by quantifying the γH2AX tag-pair distances. These defined nanofoci are different from those detected by standard microscopy showing the sub-structures of molecular arrangements (Fig. 2B, D and F).
It was reported that in euchromatic milieu DNA breaks could be repaired more effectively since these breaks do not need to be repositioned outside of the heterochromatic domain for the successful repair.52 In order to detect the distribution of the γH2AX enriched repair foci within the nucleus we applied cluster recognition, and we compared their position to the centre of the nucleus determined by a computer algorithm. This position was used as a central point to plot individual repair foci by modelling their localization as a circular shell. The spatial density of repair foci show a non-linear distribution (grey dots and trend lines in Fig. 2G, H and I). Following DSB induction, in comparison to the control nuclei, we could detect almost 3-times more repair foci formation in the treated U2OS and DivA cells (grey dots and trend lines in Fig. 2H and I vs. G), respectively. The amount of the measured density increases from the periphery towards the centre of the nucleus due to the different chromatin organizations. For further quantitative analysis of the repair foci, we sorted them into two classes based on their size (indicated in red and blue in Fig. 2G–I). While in the nuclei of untreated cells, both populations have similar and regular density distribution, in the treated cells the number of large repair foci (>5000 nm2) was found to be 2 times more than the number of small (<5000 nm2) ones (Fig. 2H and I red vs. blue lines). Therefore, the larger repair foci could appear upon DNA damage induction, and it could differ from the repair foci induced by endogenous DNA damage.
Finally, for deep evaluation, several individual repair foci of each treatment category were chosen. For that, the sizes of clusters associated with the repair foci were categorized by their area, and their distribution was presented in histograms shown in Fig. 2J, K and L. The measured distributions could be fitted with lognormal curves.37 In control cells, the expected area of the calculated surface was found to be 2950 nm2, and this value was only slightly changed in the treated U2OS and DivA cells (2850 nm2 and 3150 nm2). However, the mean values of the calculated distributions were increased by 18% (8750 nm2) and 55% (11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 550 nm2) in the treated cells compared to the untreated ones (7450 nm2), since the normalized occurrence of the large-sized repair foci was enriched following DSB induction. The presented data reveal that this algorithm can also be used to separate the γH2AX background, i.e. endogenous versus DSB induced signals (Fig. 2J vs. K and L). These data suggest that DSBs induced by either neocarzinostatin or 4-OHT could result in elevated γH2AX histone enriched repair foci both in number and size.
550 nm2) in the treated cells compared to the untreated ones (7450 nm2), since the normalized occurrence of the large-sized repair foci was enriched following DSB induction. The presented data reveal that this algorithm can also be used to separate the γH2AX background, i.e. endogenous versus DSB induced signals (Fig. 2J vs. K and L). These data suggest that DSBs induced by either neocarzinostatin or 4-OHT could result in elevated γH2AX histone enriched repair foci both in number and size.
The evaluation of spatial distribution of repair foci is based on the cluster analysis of the raw dSTORM images, in which the pixel value represents the number of accepted localizations. However, the number of localizations (Nlocalizations) generated by a single labelled histone strongly depends on the lifetime of the fluorescence ON state (Nlifetime), the labelling density (Nlabelling) and the number of reactivation cycles of the applied dye molecules (Nactivation). Due to multiple localizations, the accepted ones belonging to the same target molecule form an SMPlot, the size of which depends on the localization precision.
| Nlocalizations = Nlifetime × Nlabelling × Nactivation |
Segmentation and quantitative evaluation of SMPlots are required to determine the response function of dSTORM imaging, in other words, the number of localizations belonging to a single labelled histone molecule. Being aware of this response function the size and spatial distribution of the captured repair foci can be statistically evaluated.
Trajectory fitting of individual blinking events (Nlifetime)
In dSTORM, the fluorescence dye molecules are stochastically switched between their OFF (no fluorescence), ON (fluorescence) and bleached states. The occupation of these states can be controlled by a special switching buffer and data acquisition (laser power etc.) parameters.53 The lifetime of the ON state strongly depends on the biological sample and the local chemical environment.50 Ideally, the lifetime of the ON state is in accordance with the exposure time, and the captured photons emitted by a single dye molecule can be visualized on a single image frame. However, the detector is not triggered, and the lifetime of the ON state is not constant. As a result, the same dye molecule can be captured on sequential frames and the trajectory length of single emitted fluorescence shows an exponential decay (Fig. 3A). A trajectory fitting module was built into the rainSTORM localization software that can realign these sections.54 This resulted in less but more precise detection of localizations (Fig. 3B). Labelling density, buffer conditions and image acquisition parameters were set to minimize the possibility of spatial and temporal overlap of individual PSFs (Point Spread Function), hence single Gaussian fitting could be used throughout this work. Fig. 3 shows the dSTORM (Fig. 3C and D) and the cluster analysis images (Fig. 3E and F) of a repair focus before and after trajectory fitting, respectively. The trajectory fitted image reveals more structural details, and consequently provides a more appropriate data source for cluster analysis (Fig. 3D and F).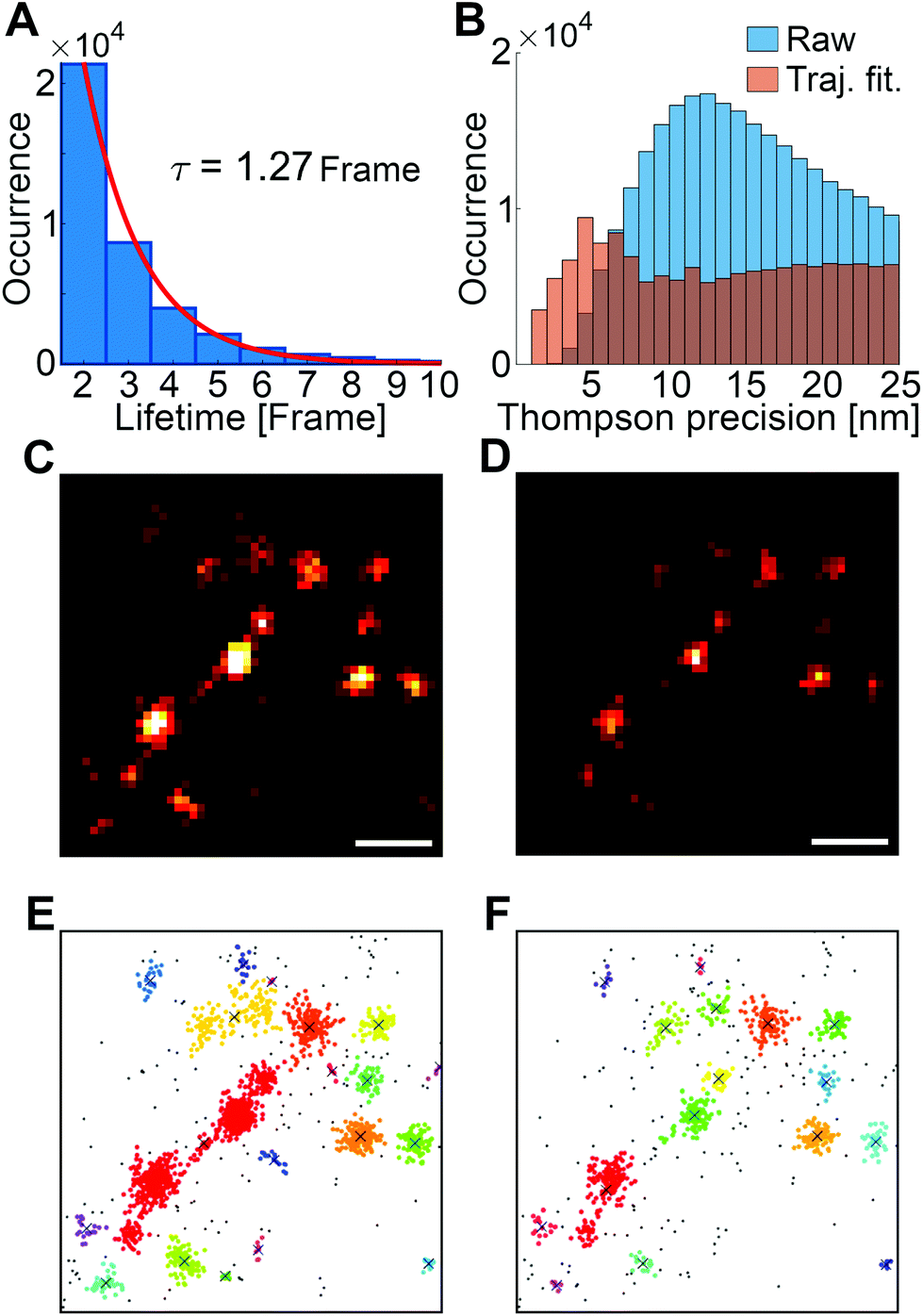 | ||
| Fig. 3 The histogram represents the lifetime of the ON state (A). Trajectory fitting precise localizations (orange) and the original (raw) distribution (blue) are represented in B. 2D original (C) and trajectory fitted dSTORM image (D) of the same repair focus, respectively. Scale bar is 200 nm. Graphical representation of cluster analysis of a repair focus before (E) and after (F) trajectory fitting, respectively. The different colours indicate different nanofoci within a repair focus. | ||
Number of blinkings per individual SMPlot (Nlabelling × Nactivation)
Generally, during immunostaining techniques, proteins are recognized by primary and fluorophore-conjugated secondary antibodies (Fig. 4A). However, the number of molecules, taking part in the labelling procedure and then in super-resolution imaging, strongly depend on the actual biological sample (number of epitopes etc.) and the local environment (pH, permeability etc.). During our measurements, due to the steric hindrance of the nucleosomes, a single primary antibody can bind to the target γH2AX molecule. In addition, our measurements also support the already published data that the stoichiometry of the primary and secondary antibody is not equal (i.e. IgG), since the 2nd antibody could recognize two epitope surfaces on the first antibody binding.55 Consequently, a single γH2AX molecule is labelled by one primary and one or two secondary antibodies. The number of dye molecules per secondary antibody was set to four, based on consultations with the manufacturer. In conclusion, we used a model in which a single target histone molecule is labelled either by 4 or 8 fluorescent dye molecules, therefore the number of dye molecules per γH2AX molecule is| Nlabelling = Nprimary × Nsecondary × Ndye, |
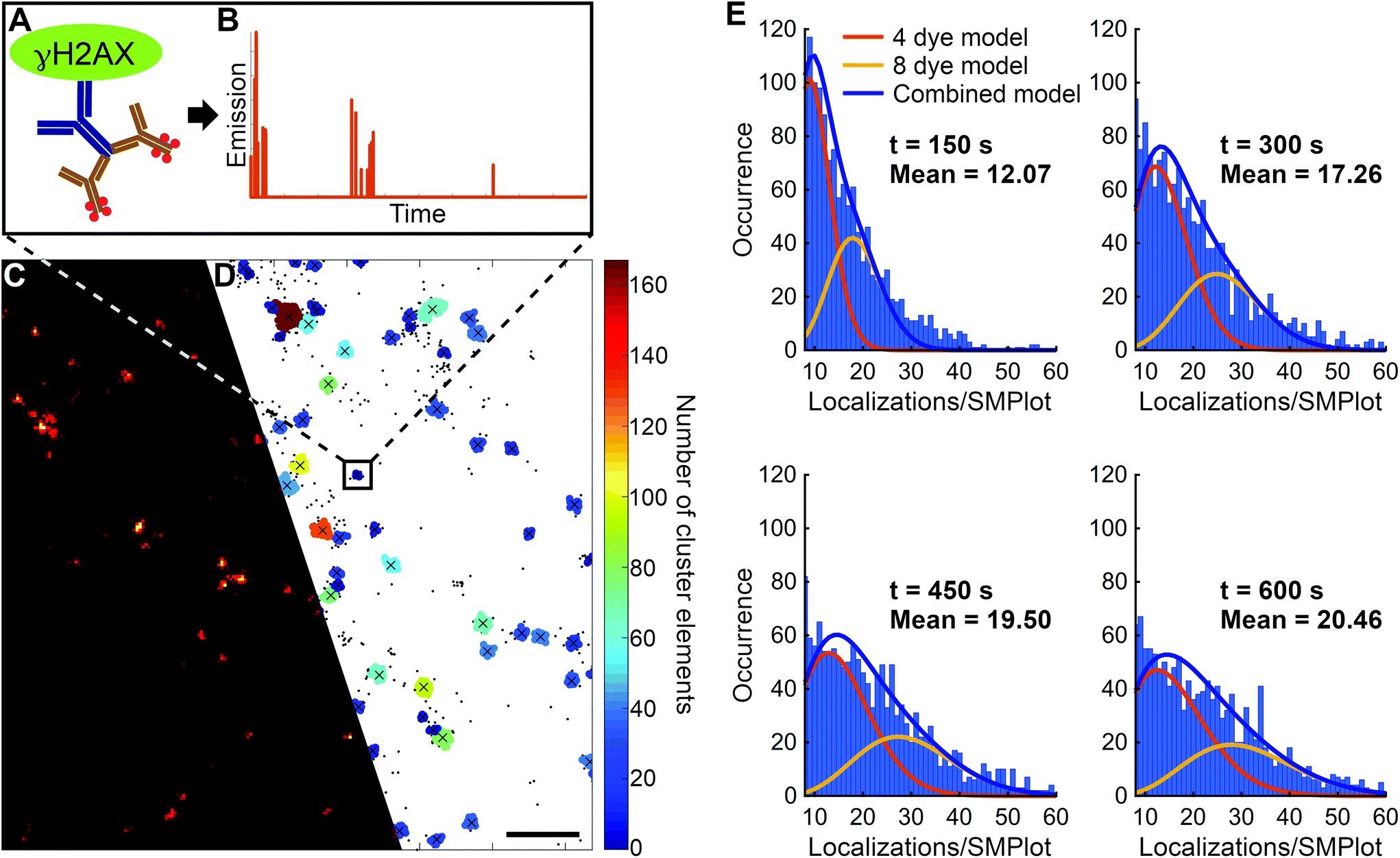 | ||
| Fig. 4 Schematic representation of the binding of first and secondary antibodies to γH2AX (A) and their frame indexes are shown (B). Super-resolved dSTORM image before (C) and after (D) cluster analysis. Images obtained during dSTORM (C) and the SMPlots were selected via cluster analysis (D) and their histogram was applied to determine the ratio of labelling via 4 and 8 dye molecules (E) and the response function. Scale bar represents 500 nm. | ||
Additionally, this ratio also depends on the duration of the measurement, since photobleaching plays a central role in the model. In the dSTORM technique dye molecules can be switched ON and OFF several times before they are finally bleached. It was shown that in a three-state switching model the number of switching cycles follow Poisson and geometrical distributions in short (kblt ≪ 1) and long (kblt ≫ 1) data acquisition times, respectively.46 However, the typical number of switching cycles (Nactivation) has already been published for several dyes,56 and it should be determined more specifically, since it strongly depends on the sample, the buffer conditions and the data acquisition parameters. Based on the evaluation of the fitted curves (Fig. 4E) it can be realized that a measurement time longer than 500 s (>20000 image frame with 30 ms exposure time) was required for adequate statistical data analysis. This evaluation proved that under the measurement conditions detailed above on average 20 localizations belonged to a single γH2AX molecule, i.e. the response function (number of localizations in SMPlot) of the system was found to be 20 localizations per target molecule.
Quantitative analysis of single DBS repair foci
Based on the statistically given response function we could determine the number of labelled γH2AX in the individual repair foci both in the untreated and in the treated cells. dSTORM images and their cluster maps of three randomly selected cells with three typical repair foci are shown in Fig. 5(A–I). In the treated cells, the densities of repair foci were found to be 2.8- and 2.2-times higher compared to the untreated U2OS control cells (Fig. 5D and G vs. A). In these cells, in which DSBs were induced, an increased number of γH2AX could be observed within the repair focus compared to the control cells. The histograms obtained from the quantitative measurements are shown in Fig. 5M–O. The distribution of cluster sizes based on their γH2AX histone numbers follow the same kinetics shown in Fig. 2 (Fig. 2J–L). Consequently, the number of γH2AX molecules within a repair focus is linearly proportional to its area in untreated (397 ± 7 γH2AX per nm2), treated U2OS (427 ± 3 γH2AX per nm2), and treated DivA (412 ± 4 γH2AX per nm2) cells shown in Fig. 5J, K and L, respectively. | ||
| Fig. 5 Super-resolved dSTORM images of the entire nuclei of untreated (A) and treated U2OS (D) and DIvA (G) cells. Three typical repair foci were selected (B, E and H) and cluster analysed (C, F, and I). The number of γH2AX histones as a function of the area of the repair focus are depicted and fitted by a linear curve (J, K, and L) based on the evaluation of 5 untreated, 4 treated U2OS and 6 treated DIvA cells. Histograms of the γH2AX histone number/focus (M, N and O) using the same data show a similar distribution to the area distribution. | ||
The major advantage of our algorithm is that it allows the schematic representation of the individual localization of repair foci and we could apply a topological analysis of the captured images. By using the parameters (localization, primary and secondary antibody numbers, fluorophores, etc.) determined in our measurements, we could mathematically analyse the topology of nanofoci within a repair focus. Each blinking event was measured, quantified and following the calculations the number of independent nanofoci were plotted into two-dimensional complexes (Fig. 5A–I). In the representation process each point which localized in proximity (N = 8 and ε = 50 nm) were considered to belong to the same repair focus. The described plots of each condition (U2OS control, NCS treated U2OS and 4-OHT treated DivA cells) are shown in Fig. 5C, F and I. These characterization studies allow a compact and illustrative visualization of specific nanofoci. Based on this plot we could tag the point structures with barcodes, which provides novel possibilities of analysing and categorizing the number of nanofoci structures in cell nuclei (Fig. 5J–L). These representations demonstrate that the endogenous and induced DSBs are covered by approximately 10–50 H2AX S139 phosphorylated histones (Fig. 5M–O).57,58
Discussion
DNA double-strand breaks are one of the most harmful DNA lesions since the dsDNA strand loses its integrity and the improper association of these broken DNA strands could lead to chromosomal rearrangements. During the DSB repair, the chromatin structure is rearranged and H2AX S139 phosphorylation rapidly appears around the damage sites.5,6,24 These steps allow the efficient recruitment of the repair factors to the damaged DNA regions and affect the choice between the DNA repair pathways. For examining the DSB-induced chromatin changes, confocal microscopy-based techniques are used in most of the studies, although over the last few years the high-throughput chromosome conformation capture technique (4C) and single cell microscopy were utilized to gain detailed insights about the protein interactions and cascades involved in the different repair pathways.31,59–63 A more detailed overview has raised more questions, which could be answered only at a single-cell level: how the different DNA repair pathways are chosen and how individual repair proteins are regulated to access the DNA repair site. Answering these questions requires a better resolution, most favourably in a single molecule detection level deeply into the mechanistic organization of the orchestrated repair focus. For single molecule detection, the 200–300 nm resolution, which is the limitation of conventional microscopy would not provide sufficiently detailed image resolution. Recently, G. Legube's laboratory has published detailed information about the chromatin organization of DNA repair foci by using 4C.59 However, the limitation of the technique is that it shows the average of a given repair focus by combining the data obtained from a large population of cells. Recent applications of electron-microscopy and super-resolution light microscopy24,30,31,61–63 have demonstrated that it is feasible to study single molecular arrangements within a repair focus. By improving the resolution of microscopy and data evaluation of structures on the meso- and nano-scale level, the search for the best-suited analysis parameters and potentially useful classification criteria of repair foci and damaged chromatin sites has become important.In this study, we addressed the nano-scale resolution of a single repair focus by the quantitative dSTORM technique in order to reveal the structure of γH2AX containing repair foci within the nuclear environment. For this, we quantified numerous parameters, such as the number of fluorophores, and primary and secondary antibodies, which could bind to a single target molecule, etc. and we applied these parameters to evaluate the images by using dSTORM based image processing. Using this unconventional procedure we provided 20 nm resolution imaging followed by cluster analysis of various repair foci. However, the quantitative dSTORM technique has been used for studying cellular events, such as cytoskeleton formation in the cytoplasm, and in our study it has been utilized for the first time to study cellular events in the nucleus. By the data we obtained from our quantitative measurements it is one of the first demonstrations of the deep structure of a DNA repair focus, in which a single nucleosome resolution has been obtained together with the nanofoci organization. Our data suggest a looping mechanism, in which approximately twenty S139 phosphorylated H2AX histones are included within a single chromatin sub-domain. Based on the literature data and our experimental results we could estimate that a H2AX S139 phosphorylated histone could be located in one out of ten or twenty nucleosomes, which suggest that a single nanofocus is localized within an approximately 40–50 kb DNA region.14,30,57,58,64
Additionally, another important finding of our study is that a single repair focus contains approximately 10 units of γH2AX enriched nanofoci. However, we could not determine whether it is a single DSB or several broken DNA regions are associated with one repair focus. Nanofoci spatially distribute in the nuclear space according to a pattern that is dependent on the progression of DDR. This pattern recapitulates the previously described repair kinetics, underlying an euchromatin-to-heterochromatin repair trend since it was shown that heterochromatin regions require further structural remodelling before specific DNA repair proteins could assess those regions. These data highlight another mechanism, in which the complex DNA breaks could be associated with repair centres for efficient DNA repair. This question could be answered in the future by using our quantitative dSTORM method.
In conclusion, we could show that dSTORM is the most adequate tool for deep investigation of repair focus formation. We believe that nowadays this is the most appropriate procedure for quantitative analyses of the structural changes of a single repair focus in individual cells at nano-scale resolution. The measurements and the procedure we applied in our study allow ultra-resolution insights into structures and architectures, offering new perspectives for further understanding the mechanisms of chromatin function in DNA repair.
Materials and methods
Trajectory fitting algorithms
The exposure time in localization microscopy is matched to the ON state lifetime of individual molecules. However, due to the stochastic feature of the blinking process, a single fluorescent molecule is typically captured in several sequential frames. Trajectory fitting is an inbuilt algorithm in the rainSTORM localization software that links together photons emitted by the same dye molecule. Localizations on sequential frames which are closer to each other than a preliminary defined acceptance radius are assumed to belong to the same fluorescence dye molecule. As a result, the code calculates the weighted localization coordinates taking into consideration the captured photon numbers. Therefore, the higher the localization precision, the higher the weight factor.54Determination of cluster density
A Matlab code was written to determine the spatial density of repair foci inside the nuclei using localization data provided by rainSTORM. First the selected nucleus was segmented with a simple and irregular Npolygon-sided (Npolygon ≈ 100) polygon using the sum image of the captured frames. The centre of the polygon65 was calculated and connected to all the vertices of the polygon, and all these lines were segmented into ten equal parts (nine division points). In the next step, ten polygons were formed by the nth division points of each line. Repair foci inside the ith, but outside the (i − 1)th polygons were counted and the normalized area repair focus density was calculated in each.Implementation of 2D/3D DBSCAN into rainSTORM
A DBSCAN based cluster analysis module was implemented into the rainSTORM program. After the reconstruction of the high resolution (SupRes) image the user can select a region using the box tracking tool, and set the two cluster analysis parameters (Ncore, ε). The program plots and saves data for further evaluation and visualization. Larger areas (entire nuclei etc.) can also be selected, but the code automatically segments them into smaller regions to avoid computation fails. After cluster analysis is performed for all sub-regions, the code saves the merged data.Experimental determination of bleach rate
The number of cumulative localizations (Ncumulative) as a function of time follow an exponential curve the decay of which is proportional to the bleach rate (kbl):| Ncumulative = N0(1 − e−kblt), |
Statistics of Ndye on independently switching fluorophores
Fluorescence switching was described by a three-state (ON, OFF and bleached) model. The probability of detecting n blinkings of Ndye fluorophores iswhere xi gives the blinking number of the i-th molecule and P1(m) is the probability of m blinkings of a single fluorophore.46 Due to the assumption that single γH2AX molecules were labelled by 4 or 8 fluorophores, the overall probability was given as the linear combination of the probabilities

| P4,8(m) = a1·P4(m1) + a2·P8(m2), |
Cell lines, media and culture conditions
DIvA cells were cultured at 37 °C in DMEM (Dulbecco's Modified Eagle's Medium, 4.5 g l−1 glucose, supplemented with L-pyruvate; Lonza,) supplemented with 10% foetal bovine serum (Lonza), 4 mM L-glutamine (Sigma-Aldrich), 1 mM puromycin (Gibco) and 1% antibiotics (Lonza). U2OS osteosarcoma cells were cultured at 37 °C in DMEM (Dulbecco's Modified Eagle's Medium; Lonza) supplemented with 10% foetal bovine serum (Lonza), 4 mM L-glutamine (Sigma-Aldrich) and 1% antibiotics (Sigma-Aldrich).Both cell lines were grown under standard conditions.
The U2OS cell line was purchased from ATCC, and DIvA cells were provided by G. Legube. All experimental protocols were approved by the guidelines of the University of Szeged and the Medical Research Council.
Neocarzinostatin (NCS) treatment
U2OS cells were treated with 5 ng ml−1 concentration of neocarzinostatin and incubated for 15 minutes. Following the treatment, the cells were washed with PBS (phosphate-buffered saline) and incubated in a culture medium for 2 hours.4-Hydroxytamoxifen (4-OHT) treatment
DIvA cells were treated with a 1 μM concentration of 4-OHT and incubated for 2 hours for the nuclear transport of AsiSI endonuclease. Following the treatment, the cells were washed with PBS and then immunostained.Immunocytochemistry
The cells were washed with PBS and then incubated with CSK buffer 3 times for 3 minutes and once for half minute [10 mM Hepes pH 7.0 (Sigma-Aldrich), 100 mM sucrose (Sigma-Aldrich), 3 mM MgCl2 (Sigma-Aldrich), 0.7% Triton X-100 (Sigma-Aldrich), 0.3 mg ml−1 RNase A (Roche)]. The cells were washed twice with PBS, and then fixed with 4% formaldehyde (Sigma-Aldrich) for 10 minutes. The cells were permeabilized with 0.2% Triton X-100/PBS for 5 minutes. After washing steps, the cells were blocked with 5% BSA (Sigma-Aldrich) in PBST [0.1% Tween 20 (Sigma-Aldrich) in PBS], supplemented with the GAR HRP antibody in 1:2000 dilution for 20 minutes. The cells were washed with PBST, and then incubated with primary antibodies diluted in 1% BSA/PBST:anti-γH2AX (Abcam, ab2893) in 1:400 dilution. After washing steps, the following secondary antibody was used: GAR Alexa 647 (Abcam, ab150091) in 1:1500 dilution. After several washing steps with PBST the experiments were conducted after the addition of imaging buffer, which is an aqueous solution diluted in PBS containing an enzymatic oxygen scavenging system GluOx (2000 U ml−1 glucose-oxidase (Sigma-Aldrich), 40000 U ml−1 catalase (Sigma-Aldrich), 25 mM potassium chloride (Sigma-Aldrich), 22 mM tris(hydroxymethyl)aminomethane (Sigma-Aldrich), 4 mM tris(2-carboxyethyl)phosphine (TCEP) (Sigma-Aldrich)) with 4% (w/v) glucose (Sigma-Aldrich) and 100 mM β-mercaptoethylamine (MEA) (Sigma-Aldrich). The final pH was set to 6.0–8.5.66–68
dSTORM microscopy
We used a Nikon Eclipse Ti-E frame with a Nikon CFI Apochromat TIRF objective (NA 1.49, 100× magnification, oil immersion) for imaging. EPI-fluorescence illumination was applied at an excitation wavelength of 647 nm (2RU-VFL-P-300-647-B1, 300 mW, MPB Communications Ltd). A filter set from Semrock was used in the microscope (Di03-R405/488/561/635-t1-25x36BrightLine® quad-edge quad-edge super-resolution/TIRF dichroic beamsplitter and FF01-446/523/600/677-25BrightLine® quad-band bandpass filter). An Andor iXon3 DU897 EMCCD camera was used for image acquisition (pixel size: 16 μm) with the following acquisition parameters: 30 ms exposure time, EM gain of 100, temperature of −75 °C.Author contributions
Conceived the project and designed the experiments: T.P., M.E., H.M., D.V., and Zs.U. Performed the experiments: H.M. and D.V. Analysed the data: H.M., D.V., Zs.U., T.P., and M.E. Contributed reagents/materials/analysis tools: M.E. and T.P. Wrote the paper: T.P., M.E., H.M., D.V., and Zs.U.Funding
This research was supported by a grant from the National Research, Development and Innovation Office grants GINOP-2.3.2-15-2016-00020, GINOP-2.3.2-15-2016-00036, the Hungarian Brain Research Program (2017-1.2.1-NKP-2017-00002), the EU-funded Hungarian Grant EFOP-3.6.1-16-2016-00008, and the Tempus Foundation.Conflicts of interest
No potential conflicts of interest were disclosed.Acknowledgements
We thank Nikolett Barbara Boros, Zoltán Gábor Páhi, Imre M. Boros and Evi Soutoglou for discussion and revisions. We also thank Ivett Berzsenyi and Elvira Czvik Ponyeczkiné for technical assistance.References
- A. Ciccia and S. J. Elledge, Mol. Cell, 2010, 40, 179–204 CrossRef CAS.
- S. P. Jackson and J. Bartek, Nature, 2009, 461, 1071–1078 CrossRef CAS PubMed.
- A. N. Blackford and S. P. Jackson, Mol. Cell, 2017, 66, 801–817 CrossRef CAS.
- G. Du, G. A. Drexler, W. Friedland, C. Greubel, V. Hable, R. Krucken, A. Kugler, L. Tonelli, A. A. Friedl and G. Dollinger, Radiat. Res., 2011, 176, 706–715 CrossRef CAS PubMed.
- B. Jakob, J. Splinter, S. Conrad, K. O. Voss, D. Zink, M. Durante, M. Lobrich and G. Taucher-Scholz, Nucleic Acids Res., 2011, 39, 6489–6499 CrossRef CAS.
- Y. Zhang, G. Mate, P. Muller, S. Hillebrandt, M. Krufczik, M. Bach, R. Kaufmann, M. Hausmann and D. W. Heermann, PLoS One, 2015, 10, e0128555 CrossRef PubMed.
- N. F. Lowndes and G. W. Toh, Curr. Biol., 2005, 15, R99–R102 CrossRef CAS.
- V. Turinetto and C. Giachino, Nucleic Acids Res., 2015, 43, 2489–2498 CrossRef CAS.
- E. P. Rogakou, C. Boon, C. Redon and W. M. Bonner, J. Cell Biol., 1999, 146, 905–916 CrossRef CAS.
- E. P. Rogakou, D. R. Pilch, A. H. Orr, V. S. Ivanova and W. M. Bonner, J. Biol. Chem., 1998, 273, 5858–5868 CrossRef CAS PubMed.
- R. Ceccaldi, B. Rondinelli and A. D. D'Andrea, Trends Cell Biol., 2016, 26, 52–64 CrossRef CAS.
- M. Falk, M. Hausmann, E. Lukasova, A. Biswas, G. Hildenbrand, M. Davidkova, E. Krasavin, Z. Kleibl, I. Falkova, L. Jezkova, L. Stefancikova, J. Sevcik, M. Hofer, A. Bacikova, P. Matula, A. Boreyko, J. Vachelova, A. Michaelidesova and S. Kozubek, Crit. Rev. Eukaryotic Gene Expression, 2014, 24, 205–223 CrossRef CAS.
- E. Berkovich, R. J. Monnat Jr. and M. B. Kastan, Nat. Cell Biol., 2007, 9, 683–690 CrossRef CAS.
- P. Caron, F. Aymard, J. S. Iacovoni, S. Briois, Y. Canitrot, B. Bugler, L. Massip, A. Losada and G. Legube, PLoS Genet., 2012, 8, e1002460 CrossRef CAS.
- J. S. Iacovoni, P. Caron, I. Lassadi, E. Nicolas, L. Massip, D. Trouche and G. Legube, EMBO J., 2010, 29, 1446–1457 CrossRef CAS.
- J. A. Kim, M. Kruhlak, F. Dotiwala, A. Nussenzweig and J. E. Haber, J. Cell Biol., 2007, 178, 209–218 CrossRef CAS.
- A. Meier, H. Fiegler, P. Munoz, P. Ellis, D. Rigler, C. Langford, M. A. Blasco, N. Carter and S. P. Jackson, EMBO J., 2007, 26, 2707–2718 CrossRef CAS.
- T. Pankotai, C. Bonhomme, D. Chen and E. Soutoglou, Nat. Struct. Mol. Biol., 2012, 19, 276–282 CrossRef CAS.
- V. Savic, K. B. Sanborn, J. S. Orange and C. H. Bassing, Cell Cycle, 2009, 8, 3285–3290 CrossRef CAS.
- R. Shroff, A. Arbel-Eden, D. Pilch, G. Ira, W. M. Bonner, J. H. Petrini, J. E. Haber and M. Lichten, Curr. Biol., 2004, 14, 1703–1711 CrossRef CAS.
- B. D. Pope, T. Ryba, V. Dileep, F. Yue, W. Wu, O. Denas, D. L. Vera, Y. Wang, R. S. Hansen, T. K. Canfield, R. E. Thurman, Y. Cheng, G. Gulsoy, J. H. Dennis, M. P. Snyder, J. A. Stamatoyannopoulos, J. Taylor, R. C. Hardison, T. Kahveci, B. Ren and D. M. Gilbert, Nature, 2014, 515, 402–405 CrossRef CAS.
- S. S. Rao, M. H. Huntley, N. C. Durand, E. K. Stamenova, I. D. Bochkov, J. T. Robinson, A. L. Sanborn, I. Machol, A. D. Omer, E. S. Lander and E. L. Aiden, Cell, 2014, 159, 1665–1680 CrossRef CAS.
- M. J. Grant, M. S. Loftus, A. P. Stoja, D. H. Kedes and M. M. Smith, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4992–4997 CrossRef CAS.
- M. Hausmann, E. Wagner, J. H. Lee, G. Schrock, W. Schaufler, M. Krufczik, F. Papenfuss, M. Port, F. Bestvater and H. Scherthan, Nanoscale, 2018, 10, 4320–4331 RSC.
- M. Krufczik, A. Sievers, A. Hausmann, J. H. Lee, G. Hildenbrand, W. Schaufler and M. Hausmann, Int. J. Mol. Sci., 2017, 18 DOI:10.3390/ijms18051005.
- R. Lopez Perez, G. Best, N. H. Nicolay, C. Greubel, S. Rossberger, J. Reindl, G. Dollinger, K. J. Weber, C. Cremer and P. E. Huber, FASEB J., 2016, 30, 2767–2776 CrossRef.
- H. Ma, R. Fu, J. Xu and Y. Liu, Sci. Rep., 2017, 7, 1542 CrossRef.
- K. Prakash, D. Fournier, S. Redl, G. Best, M. Borsos, V. K. Tiwari, K. Tachibana-Konwalski, R. F. Ketting, S. H. Parekh, C. Cremer and U. J. Birk, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 14635–14640 CrossRef CAS.
- S. Britton, J. Coates and S. P. Jackson, J. Cell Biol., 2013, 202, 579–595 CrossRef CAS.
- F. Natale, A. Rapp, W. Yu, A. Maiser, H. Harz, A. Scholl, S. Grulich, T. Anton, D. Horl, W. Chen, M. Durante, G. Taucher-Scholz, H. Leonhardt and M. C. Cardoso, Nat. Commun., 2017, 8, 15760 CrossRef CAS.
- J. Reindl, S. Girst, D. W. Walsh, C. Greubel, B. Schwarz, C. Siebenwirth, G. A. Drexler, A. A. Friedl and G. Dollinger, Sci. Rep., 2017, 7, 40616 CrossRef CAS.
- E. Bobkova, D. Depes, J.-H. Lee, L. Jezkova, I. Falkova, E. Pagacova, O. Kopecna, M. Zadneprianetc, A. Bacikova and E. Kulikova, Int. J. Mol. Sci., 2018, 19, 3713 CrossRef.
- A. Hofmann, M. Krufczik, D. Heermann and M. Hausmann, Int. J. Mol. Sci., 2018, 19, 2263 CrossRef.
- J. Folling, M. Bossi, H. Bock, R. Medda, C. A. Wurm, B. Hein, S. Jakobs, C. Eggeling and S. W. Hell, Nat. Methods, 2008, 5, 943–945 CrossRef.
- M. J. Rust, M. Bates and X. Zhuang, Nat. Methods, 2006, 3, 793–795 CrossRef CAS.
- M. Sauer, J. Cell Sci., 2013, 126, 3505–3513 CrossRef CAS PubMed.
- E. Betzig, G. H. Patterson, R. Sougrat, O. W. Lindwasser, S. Olenych, J. S. Bonifacino, M. W. Davidson, J. Lippincott-Schwartz and H. F. Hess, Science, 2006, 313, 1642–1645 CrossRef CAS PubMed.
- B. Huang, W. Wang, M. Bates and X. Zhuang, Science, 2008, 319, 810–813 CrossRef CAS.
- W. E. Moerner, Angew. Chem., Int. Ed., 2015, 54, 8067–8093 CrossRef CAS.
- A. M. Bittel, A. Nickerson, I. S. Saldivar, N. J. Dolman, X. Nan and S. L. Gibbs, Sci. Rep., 2016, 6, 29687 CrossRef CAS.
- F. Fricke, J. Beaudouin, R. Eils and M. Heilemann, Sci. Rep., 2015, 5, 14072 CrossRef.
- K. S. Grussmayer, A. Kurz and D. P. Herten, ChemPhysChem, 2014, 15, 734–742 CrossRef CAS.
- R. Jungmann, M. S. Avendano, M. Dai, J. B. Woehrstein, S. S. Agasti, Z. Feiger, A. Rodal and P. Yin, Nat. Methods, 2016, 13, 439–442 CrossRef CAS.
- C. L. Kruger, M. T. Zeuner, G. S. Cottrell, D. Widera and M. Heilemann, Sci. Signaling, 2017, 10 DOI:10.1126/scisignal.aan1308.
- S. H. Lee, J. Y. Shin, A. Lee and C. Bustamante, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17436–17441 CrossRef CAS.
- R. P. Nieuwenhuizen, M. Bates, A. Szymborska, K. A. Lidke, B. Rieger and S. Stallinga, PLoS One, 2015, 10, e0127989 CrossRef.
- P. Caron, J. Choudjaye, T. Clouaire, B. Bugler, V. Daburon, M. Aguirrebengoa, T. Mangeat, J. S. Iacovoni, A. Álvarez-Quilón and F. Cortés-Ledesma, Cell Rep., 2015, 13, 1598–1609 CrossRef CAS.
- A. Kalousi, A. S. Hoffbeck, P. N. Selemenakis, J. Pinder, K. I. Savage, K. K. Khanna, L. Brino, G. Dellaire, V. G. Gorgoulis and E. Soutoglou, Cell Rep., 2015, 11, 149–163 CrossRef CAS.
- C. Lemaitre, A. Grabarz, K. Tsouroula, L. Andronov, A. Furst, T. Pankotai, V. Heyer, M. Rogier, K. M. Attwood, P. Kessler, G. Dellaire, B. Klaholz, B. Reina-San-Martin and E. Soutoglou, Genes Dev., 2014, 28, 2450–2463 CrossRef.
- D. Sisario, S. Memmel, S. Doose, J. Neubauer, H. Zimmermann, M. Flentje, C. S. Djuzenova, M. Sauer and V. L. Sukhorukov, FASEB J., 2018, 32(12), 6469–6477 CrossRef CAS.
- M. Ester, H.-P. Kriegel, J. Sander and X. Xu, Presented in part at the Proceedings of the Second International Conference on Knowledge Discovery and Data Mining, Portland, Oregon, 1996.
- A. Janssen, G. A. Breuer, E. K. Brinkman, A. I. van der Meulen, S. V. Borden, B. van Steensel, R. S. Bindra, J. R. LaRocque and G. H. Karpen, Genes Dev., 2016, 30, 1645–1657 CrossRef CAS PubMed.
- L. Nahidiazar, A. V. Agronskaia, J. Broertjes, B. van den Broek and K. Jalink, PLoS One, 2016, 11, e0158884 CrossRef.
- A. O. I. Group, rainSTORM user guide, http://titan.physx.u-szeged.hu/~adoptim/?page_id=582%20?, (accessed 30 Mar, 2019).
- C. R. Bauer, Methods Mol. Biol., 2014, 1131, 543–548 CrossRef CAS.
- G. T. Dempsey, J. C. Vaughan, K. H. Chen, M. Bates and X. Zhuang, Nat. Methods, 2011, 8, 1027–1036 CrossRef CAS.
- L. Vian, A. Pękowska, S. S. P. Rao, K. R. Kieffer-Kwon, S. Jung, L. Baranello, S. C. Huang, L. El Khattabi, M. Dose, N. Pruett, A. L. Sanborn, A. Canela, Y. Maman, A. Oksanen, W. Resch, X. Li, B. Lee, A. L. Kovalchuk, Z. Tang, S. Nelson, M. Di Pierro, R. R. Cheng, I. Machol, B. G. St Hilaire, N. C. Durand, M. S. Shamim, E. K. Stamenova, J. N. Onuchic, Y. Ruan, A. Nussenzweig, D. Levens, E. L. Aiden and R. Casellas, Cell, 2018, 173, 1165–1178 CrossRef CAS.
- W. Xiang, M. J. Roberti, J. K. Heriche, S. Huet, S. Alexander and J. Ellenberg, J. Cell Biol., 2018, 217, 1973–1984 CrossRef CAS.
- F. Aymard, M. Aguirrebengoa, E. Guillou, B. M. Javierre, B. Bugler, C. Arnould, V. Rocher, J. S. Iacovoni, A. Biernacka, M. Skrzypczak, K. Ginalski, M. Rowicka, P. Fraser and G. Legube, Nat. Struct. Mol. Biol., 2017, 24, 353–361 CrossRef CAS.
- B. Bintu, L. J. Mateo, J. H. Su, N. A. Sinnott-Armstrong, M. Parker, S. Kinrot, K. Yamaya, A. N. Boettiger and X. Zhuang, Science, 2018, 362 DOI:10.1126/science.aau1783.
- M. Bach, C. Savini, M. Krufczik, C. Cremer, F. Rosl and M. Hausmann, Int. J. Mol. Sci., 2017, 18 DOI:10.3390/ijms18081726.
- J. P. Eberle, A. Rapp, M. Krufczik, M. Eryilmaz, M. Gunkel, H. Erfle and M. Hausmann, Methods Mol. Biol., 2017, 1663, 1–13 CrossRef CAS.
- M. Eryilmaz, E. Schmitt, M. Krufczik, F. Theda, J. H. Lee, C. Cremer, F. Bestvater, W. Schaufler, M. Hausmann and G. Hildenbrand, Cancers, 2018, 10 Search PubMed.
- J. Bewersdorf, B. T. Bennett and K. L. Knight, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 18137–18142 CrossRef CAS.
- P. Bose and G. Toussaint, Computing the constrained Euclidean, geodesic and link centre of a simple polygon with applications, Proceedings of CG International'96. IEEE, 1996.
- P. Tinnefeld, C. Eggeling and S. W. Hell, Far-Field Optical Nanoscopy, 2015 Search PubMed.
- P. Schafer, S. van de Linde, J. Lehmann, M. Sauer and S. Doose, Anal. Chem., 2013, 85, 3393–3400 CrossRef.
- S. van de Linde, A. Loschberger, T. Klein, M. Heidbreder, S. Wolter, M. Heilemann and M. Sauer, Nat. Protoc., 2011, 6, 991–1009 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr03696b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |