
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultifunctional graphene oxide-bacteriophage based porous three-dimensional micro-nanocomposites†
Paolo
Passaretti
 *a,
Yiwei
Sun
b,
Inam
Khan
c,
Kieran
Chan
a,
Rania
Sabo
a,
Henry
White
d,
Timothy R.
Dafforn
e and
Pola Goldberg
Oppenheimer
*a
*a,
Yiwei
Sun
b,
Inam
Khan
c,
Kieran
Chan
a,
Rania
Sabo
a,
Henry
White
d,
Timothy R.
Dafforn
e and
Pola Goldberg
Oppenheimer
*a
aSchool of Chemical Engineering, University of Birmingham, Birmingham, B15 2TT, UK. E-mail: PXP561@student.bham.ac.uk; P.GoldbergOppenheimer@bham.ac.uk; Tel: +44 (0) 778 49 68 145 Tel: +44 (0) 121 41 45 287
bSchool of Physics and Astronomy, Queen Mary University of London, London, E1 4NS, UK
cSchool of Metallurgy and Materials, University of Birmingham, Birmingham, B15 2TT, UK
dBAE-Systems-Air Sector, Buckingham House, FPC 267, Filton, Bristol, BS34 7QW, UK
eSchool of Biosciences, University of Birmingham, Birmingham, B15 2TT, UK
First published on 28th June 2019
Abstract
Graphene, since its successful exfoliation and characterisation has been continuously drawing extensive research interests due to its potential for a broad range of applications ranging from energy, microelectronics, through polymer fillers and sensors to environmental and biomedical devices. Exploitation of its unique chemical and physical properties for the manufacturing of functional materials, requires careful structural control and scaling-up into three-dimensional morphologies. Here, a facile method is established to create and control the bottom-up self-assembly of graphene oxide nano-sheets via unprecedented integration with a highly versatile bio-ingredient, the filamentous bacteriophage M13, into hierarchical, three-dimensional, porous sponges of GraPhage13. This study explores the interplay of the GraPhage13 structure formation and studies the mechanisms that give rise to the controllable self-assembly. The straightforward fabrication of robust hierarchical micro-nano-architectures further lays a platform for applications in energy storage and conversion, catalysis and sensing.
Introduction
Carbon-based nanomaterials including graphene, graphene oxide (GO), carbon fibres and carbon nanotubes (CNTs) have been extensively studied for their potential exploitation in improving performance, functionality and durability of advanced materials.1 Therefore, they can be employed in a wide range of applications, ranging from composite materials with tailored functionality, through materials for electrochemical sensors and absorbers, to energy storage and conversion.1,2 Graphene is a two-dimensional (2D) material consisting of sp2-hybridized carbon atoms, exhibiting high-electrical and thermal conductivity as well as large surface area, high in-plane stiffness and ultra-thin thickness.1,3 Since the demonstration of the mechanical exfoliation for its produciton4 several alternative methods have been developed, including chemical vapour deposition (CVD),5–7 direct exfoliation of graphite1,8 and liquid phase exfoliation,9–11 with the latter method producing GO as an intermediate product,12,13 which can be reduced to partially restore the graphene structure (rGO).14–16 GO is comprised of several functional groups containing oxygen, namely hydroxyl which is epoxied to the top and bottom planes as well as carboxyl and carbonyl groups, located at the edges of the GO sheets (Fig. 1a).17,18 These functional groups render GO hydrophilic and therefore, make it easy to disperse in water, however, at the same time, they reduce its conductivity relative to graphene.1 Despite the continuous developments, it is still a considerable challenge to exploit the properties of graphene, GO and rGO for the fabrication of novel functional materials by assembling these 2D materials into three-dimensional (3D) architectures through facile and scalable procedures. Concurrently, however, the production of GO (rGO and graphene) sponges, which refers to foams, templates and aerogels, is a rapidly growing field of research.1 Such sponges are characterised by a 3D structure with, particularly low-density, porous network, large surface area and high conductivity.1 Several methods have been developed to assemble GO in a scalable manner into functional bulk materials, including, for instance, treatments at extreme conditions of high-temperature or high-pressure as well as in combination with polymers,19,20 DNA,21 proteins22 and peptides.1 Usually, the latter methods require the formation of a hydrogel, merely based on weak intermolecular interactions3,23–25 and subsequent drying processes, both crucial for the sponge formation.1 However, viral building blocks have not been previously exploited to fabricate GO micro-nano sponges.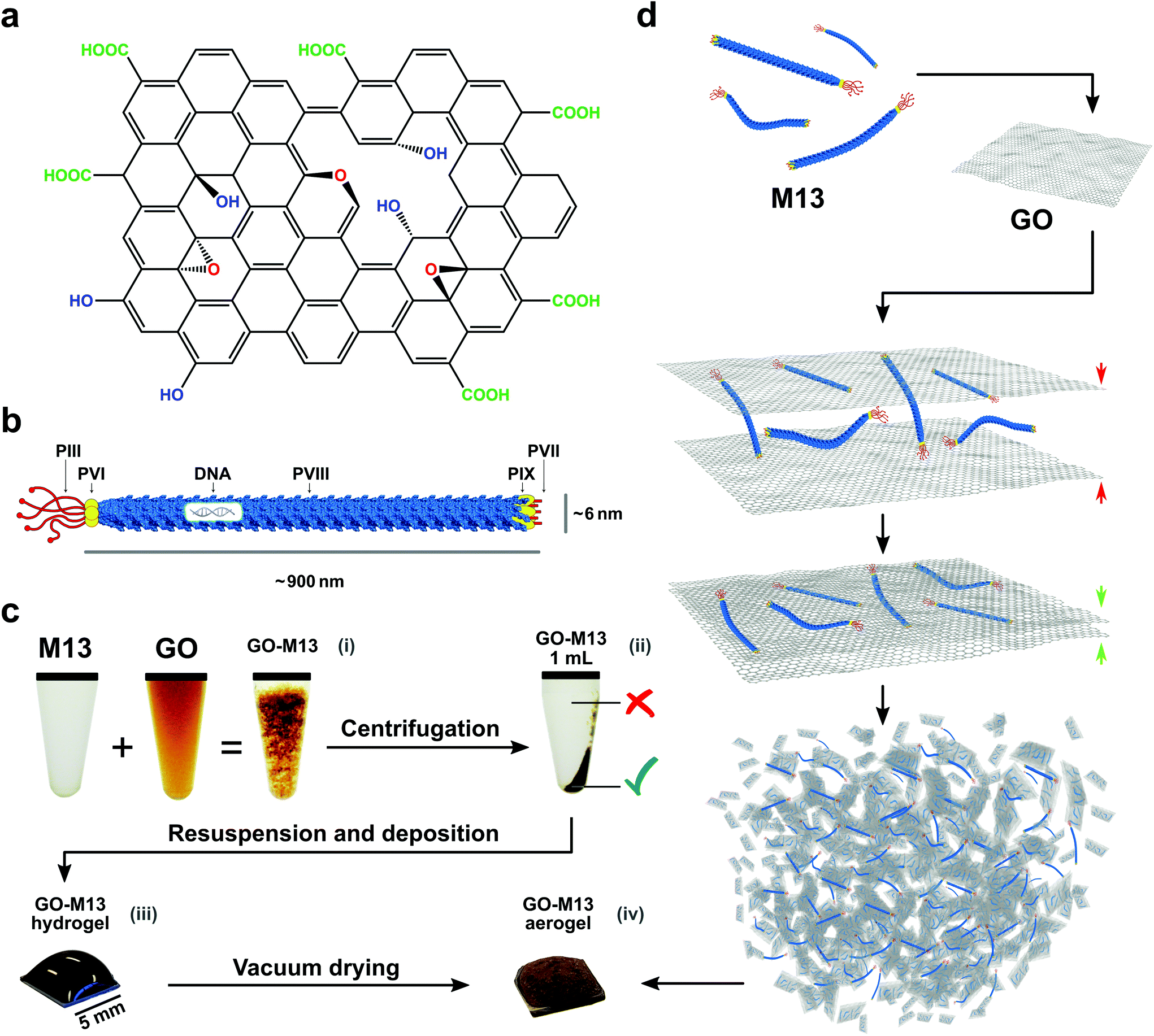 | ||
| Fig. 1 Schematic representation and pictures of the self-assembly mechanism. Schematics of: (a) graphene oxide and (b) M13 bacteriophage. (c) Overview of the fabrication process of the GraPhage13 including the four main steps: (i) assembly, (ii) precipitation, (iii) deposition and (iv) drying. (d) Tentative self-assembly mechanism of the GraPhage13, with M13 acting as an inter-linking agent for the GO nanosheets. | ||
In this work, we demonstrate a novel route for bottom-up self-assembly of GO into a 3D porous structure spanning the micro- and nanoscales, yielding a unique hierarchical GO sponge based on a synthetic biology approach, by exploiting the bacteriophage M13, acting as a cross-linker between the graphene nanosheets.
M13 is a filamentous bacteriophage that infects Escherichia coli (E. coli) strains showing the conjugative pilus. This virus establishes sustainable infections without killing the host, releasing new copies of itself to propagate the infection to the nearby E. coli cells.26,27 M13 phage has typical dimensions of approximately 880 nm in length and 6.6 nm in width, and it is composed of a circular, single-stranded DNA molecule (ssDNA), encoding ten genes encapsulated in a flexible rod-like shape capsid, consisting of 2700 copies of its major coat protein PVIII. At the ends, there are five copies of each minor coat protein, PIII and PVI on one side and PVII and PIX on the other (Fig. 1b).28–30
Our facile method to fabricate GO-M13 sponges (GraPhage13), is performed without the need of extreme conditions of temperature, pressure or pH rendering it an environmentally friendly and scalable material, further characterised by ultra-low-density and high-surface-area. The fabrication of GraPhage13 is based on four main steps: (i) assembly reaction in aqueous buffers, (ii) hydrogel separation through centrifugation, (iii) drop casting onto a silicon wafer and finally (iv) drying under vacuum (Fig. 1c and d). Furthermore, M13 can be functionalised through the many specific chemical groups on its surface with a broad range of functional molecules such as peptides, antibodies, proteins and enzymes.31–33 These hybrid micro-nano sponges, therefore have potential applications for the development of electrochemical energy storage and conversion devices, absorbers, sensors or composite scaffolds.1
Results and discussion
GraPhage13 hydrogel in water
Initially, GO and M13 were combined in deionised water (DIW), each at a concentration of 0.3 mg mL−1. Upon mixing of the two components, they immediately react forming an aggregate which is separated from the supernatant and subsequently deposited on the microtube bottom by centrifugation (Fig. 1c). To establish the optimal self-assembly conditions, we have studied the formation of the hydrogel by systematically analysing the ultraviolet-visible (UV-Vis) spectra of the supernatants after centrifugation (Fig. 2a–c). While the dispersed M13 was not producing a visible pellet post centrifugation (Fig. 2c), GO yielded a dark pellet with a clear supernatant (Fig. 2a). The precipitation of GO occurred due to its moderate instability in DIW, combined with the large size of the dispersed sheets and the presence of clusters of sheets.34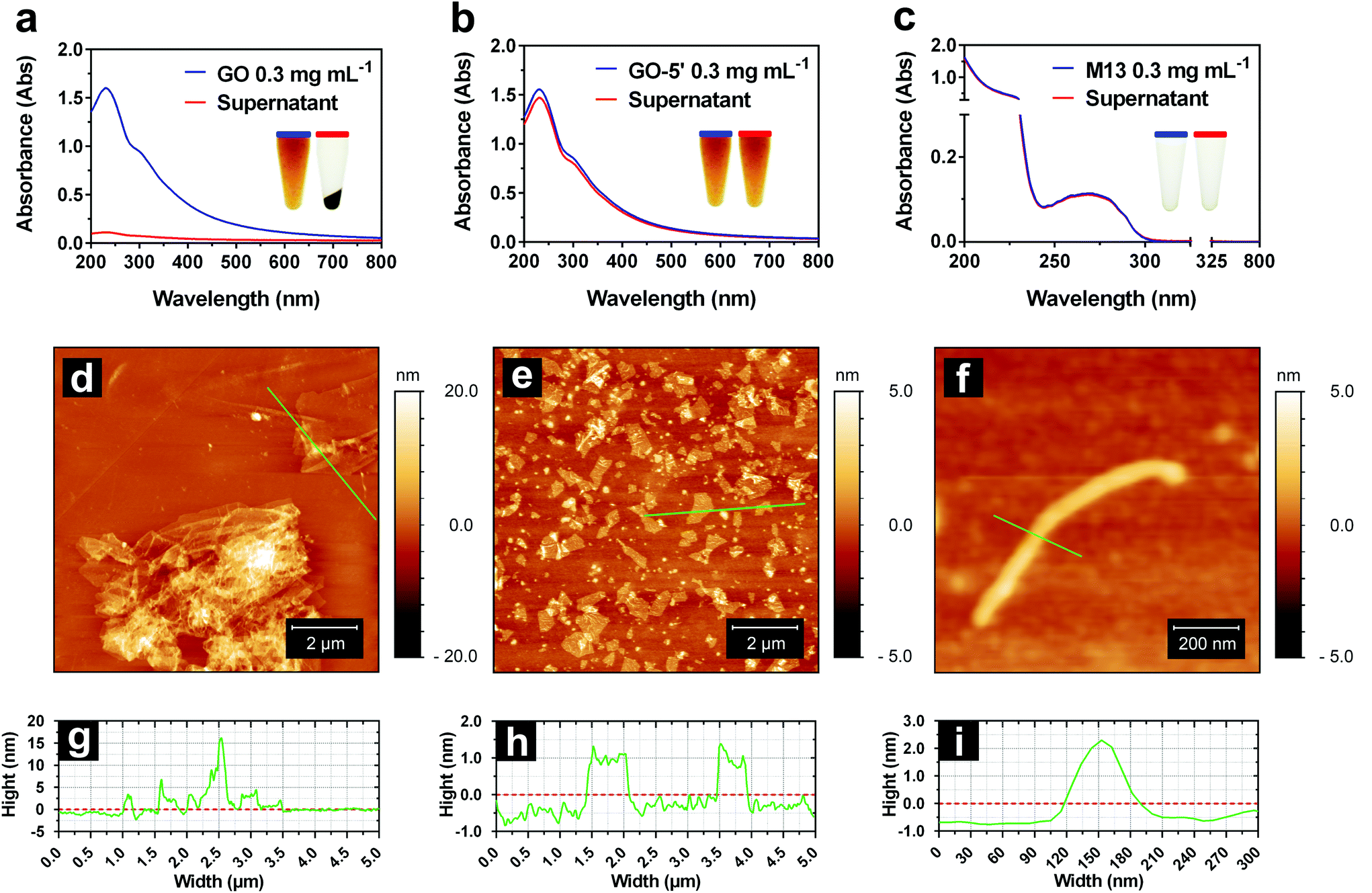 | ||
| Fig. 2 UV-Vis and AFM analyses of the individual components. UV-Vis spectra of (a) GO, (b) GO-5′ and (c) M13 dispersions in DIW (blue) with the corresponding supernatants (red). Insets: Photograph of each sample (left) and the corresponding supernatant (right). AFM height images of the (d) layered GO, (e) GO-5′ nanosheets and (f) individual M13 phage with the corresponding cross-sections (g–i). | ||
Under these conditions, it was not possible to distinguish between the GO precipitation due to the self-assembly processes or its clustering, therefore, hampering the spectrophotometric analysis of the hydrogel formation. To improve its stability, GO of 5 mg mL−1 from the stock solution, was sonicated for 5 min (GO-5′), obtaining a new stock. Moreover, the concentrations of the samples prepared by diluting both stocks (GO and GO-5′ of 5 mg mL−1), were estimated using calibration curves calculated by measuring the absorbance of serial dilutions (ESI Fig. S1†). The sonication allowed the separation of the layered flakes as well as obtaining smaller GO sheets, achieving an even dispersion (Fig. 2b).34
AFM height images (Fig. 2d–f) and the corresponding cross-sections (Fig. 2g–i) of the dispersed GO, GO-5′ and M13 bacteriophage clearly show that while the GO samples consist of large flakes and aggregates of heterogeneous thickness (Fig. 2d), the GO-5′ is predominantly composed of small, well-distributed nanosheets with a typical thickness of ∼1 nm (Fig. 2e).24,25 The M13 bacteriophages measure, on average, 880 μm in length, 60 μm in width and 3 μm in height (Fig. 2f). The apparent non-circular cross-section of the filamentous bacteriophage is due to a variety of conditions and in particular, the relative humidity of the sample, the applied force on the surface during the scan and tip convolution effects, caused by the curvature radius of the tip itself.35
UV-Vis spectra of GO exhibit a π → π* plasmon peak at 230 nm and a shoulder corresponding to the n → π* plasmon peak around 310 nm (Fig. 2a).36 The spectra of M13, show a high absorbance in the region between 200–230 nm, mostly due to the π → π* transitions in the peptide bonds37 and the typical peak at 269 nm of M13 bacteriophages which is due to the combination of the viral DNA and its major coat protein PVIII.38
The comparison of the absorbance intensities before and after the centrifugation enabled the stability of GO dispersion in DIW to be studied (Fig. 2a–c). It was found that only 7.05 ± 0.80% of the GO remained in the supernatant, while GO-5′, on the other hand, exhibited considerably better stability with a higher spectral absorbance intensity, corresponding to 94.51 ± 1.54% of the GO-5′ remaining within the supernatant. Also, 96.89 ± 6.60% of M13 remained dispersed in the supernatant after centrifugation (ESI Table S1 and Fig. S2†). Therefore, GO-5′ has been identified as the optimal GO preparation for the fabrication of GraPhage13.
To establish the optimal weight ratio between the two components for the formation of GraPhage13 hydrogel (GPH) self-assembly, GO was mixed with M13 in DIW at different weight ratios of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:1 and 1:2. GO concentration was kept constant at 0.3 mg mL−1 in all the samples, while M13 was of 0.15 mg mL−1 (GO-M1303015-5′), 0.3 mg mL−1 (GO-M130303-5′) and 0.6 mg mL−1 (GO-M130306-5′), respectively (Fig. 3).
:1, 1:1 and 1:2. GO concentration was kept constant at 0.3 mg mL−1 in all the samples, while M13 was of 0.15 mg mL−1 (GO-M1303015-5′), 0.3 mg mL−1 (GO-M130303-5′) and 0.6 mg mL−1 (GO-M130306-5′), respectively (Fig. 3).
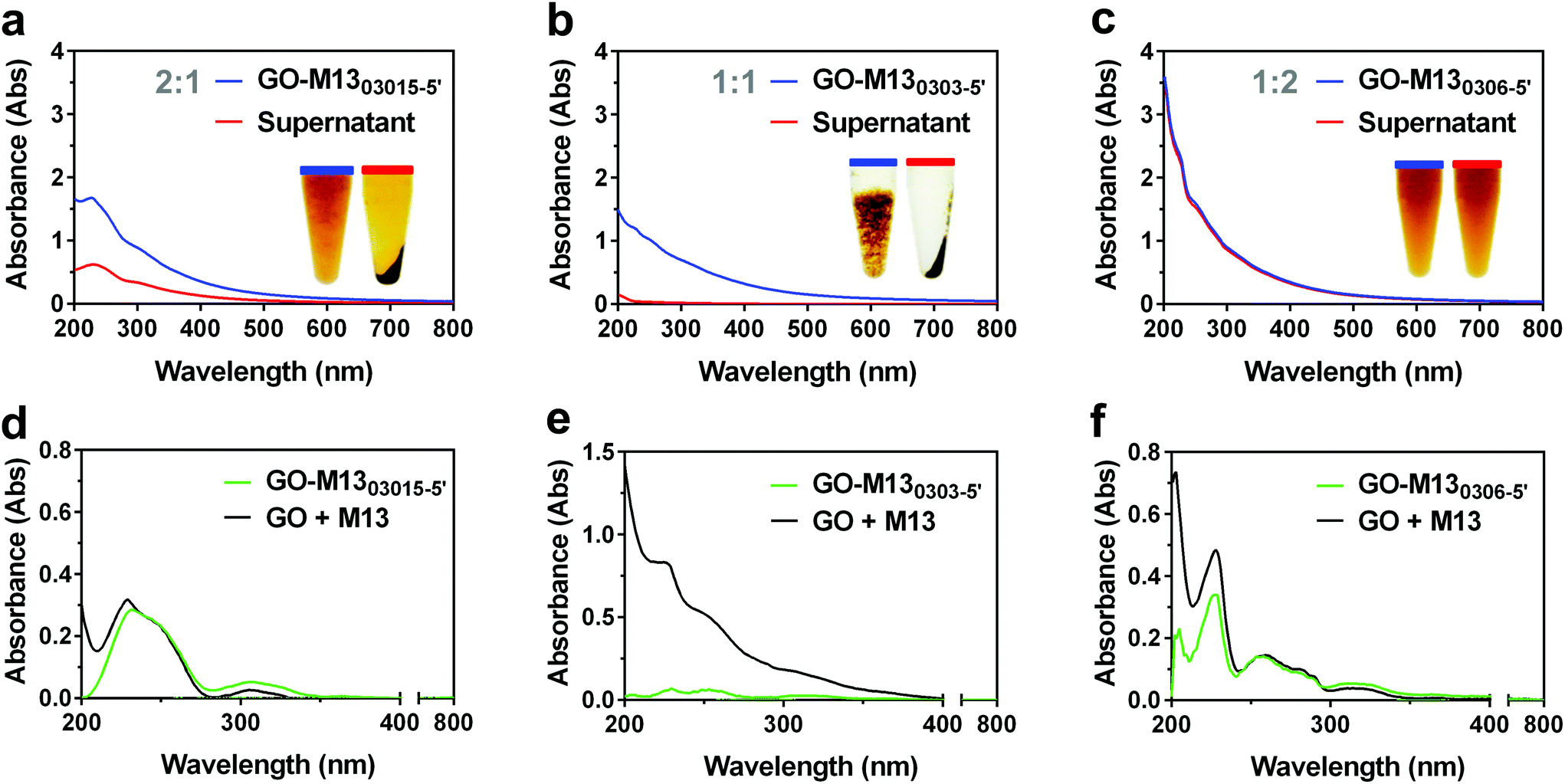 | ||
| Fig. 3 UV-Vis analysis of GO-M13 hydrogels. UV-Vis spectra of GPH at three different concentration ratios of (a) GO-M1303015-5′, (b) GO-M130303-5′ and (c) GO-M130306-5′. Inset: The corresponding photographs of each sample and its supernatant. (d–f) UV-Vis spectra of the three different wt/wt% ratios (green) in comparison to the theoretically calculated spectra, obtained by summing the values of the two components taken individually and at the same concentrations used to make the corresponding samples (black). | ||
GO-M1303015-5′ exhibited a minor aggregation and the characteristic spectrum of pure GO in solution, although, due to the presence of M13 in solution, with a slightly increased absorbance at 200 nm. Following the centrifugation, a dark pellet and a transparent light brown supernatant were formed indicating the presence of GO (Fig. 3a). GO-M130303-5′ exhibited an evident formation of the hydrogel (Fig. 3b), which is clearly separated from the supernatant with the corresponding spectrum showing a continuous increase of absorbance due to the macroaggregates of the hydrogel, responsible for the light scattering. The supernatant itself appeared transparent, and the lack of characteristic GO and M13 peaks in the UV-Vis spectrum, suggests the nearly complete precipitation of both components in the pellet. GO-M130306-5′ on the other hand, appeared clear with no apparent hydrogel formation and the corresponding spectrum exhibiting the fingerprint features of GO and M13 individually (Fig. 3c). Furthermore, following the centrifugation, only a small pellet was formed, and the supernatant spectrum was found to completely overlap with the spectrum of the GO-M130306-5′, suggesting that the two components did not interact, and they were still fully dispersed in the solution.
Given that the absorbance of two or more components in solution is derived by the sum of the individual absorbance values,39 the spectra of each sample were compared to the one obtained by the sum of their respective components, measured individually (Fig. 3d–f), to preliminary investigate the interaction between the two components. Interestingly, there was a reduction in the total absorbance in each case and in particular, for GO-M130303-5′ (Fig. 3e). Furthermore, this composition does not exhibit the characteristic peaks of absorbance of the individual GO and M13 components. In contrast, the GO-M130306-5′ spectrum almost entirely overlaps with the corresponding one obtained by the sum of the respective components (Fig. 3f), suggesting that GO and M13 were absorbing the light in the same manner as they would if they were freely dispersed in DIW. These results indicate that GO and M13 in the form of a hydrogel scatter the light, not exhibiting their typical absorption peaks.
GraPhage13 hydrogel in buffer
Two possible mechanisms can lie at the origin of the observed GPH self-assembly. The first is the result of the electrostatic forces acting between the negatively charged groups of GO and the positively charged groups of M13. The second mechanism relies on the non-covalent intramolecular interactions involving π-systems which may exist due to the increasing proximity between the two components and the progressive GO sheets stacking.3 We have, therefore, further experimentally examined which of the proposed mechanisms might be the dominant one and responsible for the assembly.The absence of the hydrogel formation in the case of GO-M130306-5′, whereas a pellet and an excess of M13 was expected in the supernatant (Fig. 3c), suggested that pH might play an important role in the GPH self-assembly. Subsequently, the effect of pH was systematically verified by adding set volumes of 1 M NaOH or HCl to the aqueous GO-M130303-5′ solution. While the excess of HCl led to hydrogel assembly, NaOH yielded a reverse outcome of disassembly. Moreover, the pH values of GO-M1303015-5′, GO-M130303-5′ and GO-M130306-5′ in DIW were measured and found to be 3.77 ± 0.02, 4.85 ± 0.02 and 5.7 ± 0.02, respectively (ESI Table S2†).
GO in DIW at a concentration of 0.3 mg mL−1 exhibited a pH of 3.49 ± 0.02, due to its acidic properties given by the presence of phenolic (pKa = 9.8) and carboxylic (pKa1 = 6.6 and pKa2 = 4.3) functional groups on its surface.40 These groups are responsible for its negative charge at pHs ranging from 2 to 12, and a stable dispersion can be typically achieved at pH ≥ 3.40,41 On the other hand, M13 in the same conditions showed a pH of 6.97 ± 0.06 and stability at pH ≥ 4.8. M13 is almost entirely comprised of its major coat proteins PVIII, which is made of 50 amino acids, and 13 of these, including the N- and the C-terminus contribute to the total protein charge (Fig. 4a). The C-terminus and the residues K40, K43, K44 and K48 face the internal cavity of the phage, stabilising the negative charge of the ssDNA during the capsid self-assembly mechanism.42 Moreover, Y21 and Y24 residues, mostly embedded in the protein structure, and exhibiting high pKa values (ESI Table S3†), do not contribute to the total charge of the virus. Therefore, just the N-terminus and the residues E2, D4, D5, K8 and E20 could be responsible for the M13's overall charge distributed on a surface area of ∼18300 nm2, calculated based on the assumption that the bacteriophage is of a shape of a regular cylinder (Fig. 4a).43,44
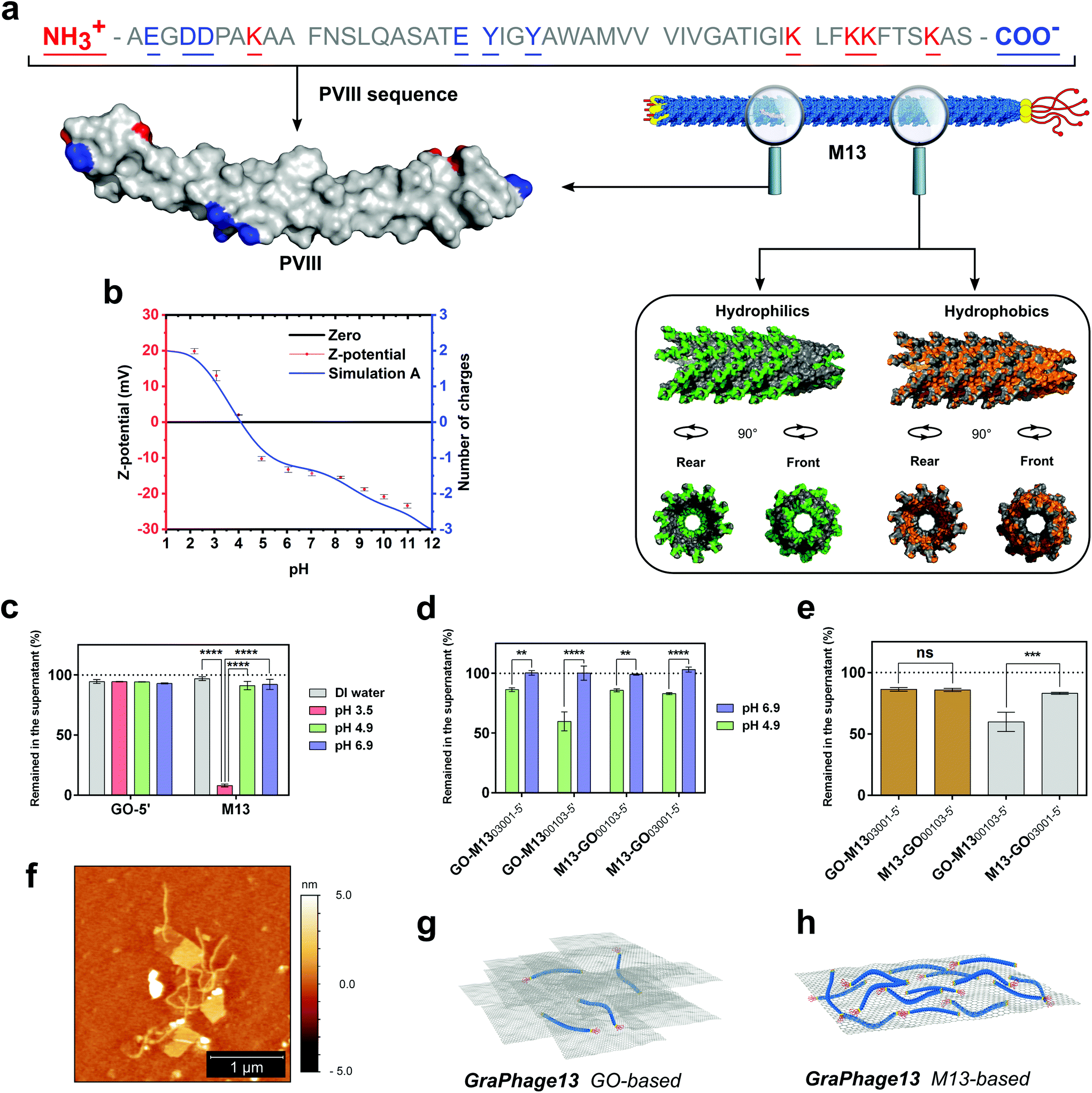 | ||
| Fig. 4 M13 PVIII structure analysis and GPHs characterisation. (a) M13 PVIII major coat protein sequence and 3D-structure (top left-hand side). On the right-hand side, a section of the phage structure made of 35 copies of the PVIII, assembled in the quaternary structure (PDB: 2MJZ).45 The hydrophilic surface (green) corresponds to the amino acids Arg, Lys, His, Glu, Asp, Asn, Gln, Thr, Ser and Cys, while the hydrophobic surface (orange) corresponds to the amino acids Ala, Gly, Val, Ile, Leu, Phe and Met. The cavity in two different orientations shows the location of the nucleic acid encapsulated within the M13. (b) The ζ-potential of M13 (red) at pH values ranging from 2 to 11 is overlapping the theoretically calculated values of M13's charge spectra (blue). (c) The amount of GO and M13 left in the supernatant after centrifugation at different buffer conditions, including the DIW and citrate buffer at pH 3.5, 4.9 and 6.9. (d) The comparison between four different GO-M13 formulations at pH 4.9 and 6.9 at which GO and M13 show self-assembly (green) as well as the respective control samples (blue), where there was no observed interaction taking place. (e) The amount of GO (brown) and M13 (grey) left in the supernatant after centrifugation of the four GO-M13 formulations in citrate buffer pH 4.9. (f) Representative AFM height image of the M13 bacteriophages interacting with GO. The unbound phage was washed with citrate buffer (10 mM, pH 4.9). Schematic representation of two different GraPhage13 hydrogel/aerogel compositions: (g) GO- and (h) M13-based. | ||
This hypothesis was further corroborated by measuring the ζ-potential of M13 and comparing it to the theoretically predicted values (Fig. 4b). The charge of each amino acid was assigned at each corresponding pH value, based on its pKa and following the Henderson–Hasselbalch equation.46 The theoretical values of the pKa of each amino acid were calculated by taking into consideration its chemical surrounding (ESI Table S3†). The isoelectric point (pI) of M13 was established as pH 4.1 from both the ζ-potential and the theoretical curves. Similar values were previously measured for fd bacteriophage, which has an identical, mature PVIII protein, excluding the substitution N35D.30,45,47
To establish the pH at which the assembly of GPH occurs, an acid titration, from pH 12 to 2, was performed. At pH 5.4, a minor aggregation started occurring, which further has been dissolved upon mixing followed by a more substantial non-soluble aggregation at pH ≤ 4.8. Notably, the self-assembly occurred at pH values where both components are stably dispersed in solution. The calculated ζ-potential and pH values indicate that the interaction between the two components is most probably due to the reduction of repulsion between the deprotonated carboxylic groups of the GO and the negatively charged amino acids present on M13, at the pH range from 5.4 to 4.8. Moreover, after the protonation of E20, the interaction between the positively charged N-terminus and K8 with the carboxylic groups on the GO can trigger the self-assembly process, further enabling the self-assembly of the GPH. Although this suggests that the electrostatic interactions dominate the self-assembly process, other noncovalent intramolecular interactions involving π-systems are not excluded.
The significant effect of pH on the interaction between the two components was further used to determine the optimal reaction conditions to form the GPH. Therefore, the stability of GO and M13 was tested in 10 mM citrate buffer (CB) at three specific values of pH 3.5, 4.9 and 6.9. UV-Vis analysis demonstrates that both components are stable at all the above-analysed pH conditions, except at pH 3.5, at which M13 has precipitated considerably due to its pI (Fig. 4c). Further UV-Vis analyses were performed to establish and quantify the assembly-ratio between the two components at optimal assembly conditions. GO was transferred into CB and mixed with a 30 times weight excess of M13 (GO-M1300103-5′) and reciprocally, M13 was mixed with 30 times excess of GO inside the buffer (M13-GO00103-5′). Moreover, two other samples were prepared by inverting the order of addition of the two components to evaluate if the mixing was affecting the reaction ratio (M13-GO03001-5′, GO-M1303001-5′). Due to the instability of M13 at pH ≤ 4.8, the self-assembly of GPH was carried-out at pH 4.9, which is slightly above its pI, and compared to the control solutions at pH 6.9, at which no interaction was expected to occur (ESI Fig. S3†).
The UV-Vis analyses further corroborate the significance of the pH on the interaction between the GO and M13 (Fig. 4d). Furthermore, the mixing appeared of significant importance (Fig. 4e) for the self-assembly process.
At optimal pH values for the self-assembly, it is possible to tune the reaction by varying the ratios of each component, to yield the two different assembly conditions called, GO- and M13-based (Fig. 4g and h), depending on which of the two is in excess. GO:M13 weight ratio ranges from 4:1 to 1:5, nevertheless, inverting the order of addition of the two components the M13-based hydrogel ratio can increase up to 1:12 (ESI Fig. S3 and Table S4†). These extreme assembly conditions are highly-appealing since they can potentially enable tailoring the properties and functional applications of GraPhage13 aerogel (GPA) such as porosity, surface area and stiffness.
GraPhage13 aerogel
Following the GPH assembly, the subsequent second step (ii) included the hydrogel separation (Fig. 1). Typically, the sample was centrifuged and 90% of the clear supernatant was discarded. After that, the pellet was re-suspended, yielding the final hydrogel behaving as a non-Newtonian fluid (ESI Fig. S4†) with a density of 1.005 ± 0.003 g cm−3.The drop casting (iii) and the drying (iv) steps included the deposition of 50 μL of the hydrogel onto a silicon wafer and the subsequent placing in a vacuum chamber. As the drying process progressed, caused by the vacuum evaporating the water, the hydrogel expanded into an extremely light 3D porous aerogel, showing ultra-low-density of 8.82 ± 0.03 mg cm−3 and high-surface area of 325.147 ± 34.307 m2 g−1 (ESI Fig. S5, Tables S5 and S6†).
Scanning electron microscopy (SEM) coupled with energy dispersive X-ray (EDX) spectroscopy was used to characterise the 3D morphology and composition of the formed GPA (Fig. 5), which exhibited a layered and porous 3D sponge-like structure with a random distribution of micro and nanopores (Fig. 5a–e). GO, on the other hand, exhibited a very fine and fragile structure (Fig. 5f), while the M13 appeared to be of an amorphous morphology with tangled fibres (Fig. 5g). The unique structure of the sponge-like GPA arises from the intertwined M13 nanowires, acting as threads in-between the thin GO nanosheets, forming the hierarchical 3D architectures with embedded microporous morphology. While GO was predominantly comprised of carbon and oxygen, M13 and GPA exhibited the presence of N, S, P, Na and Cl from the amino acids, DNA and traces of salts (Fig. 5i–k). The Si peak is due to the underlying supporting substrate on which the free-standing 3D sponge with an average thickness of ∼2 mm was generated. EDX analysis at three random locations across the macro-sponge yielded identical elemental composition throughout, indicating a homogeneous distribution of GO and M13 inside the 3D sponge (ESI Tables S7–9†). Raman spectroscopy was further carried out on the GO, M13 and GPA to quantify the effects of the interaction between GO and M13 in the sponge on the original bonds of each component. While Raman spectra of GO and GPA exhibited two clear peaks at 1350 cm−1 and 1600 cm−1 at the excitation wavelengths of 633 and 785 nm, accordingly (Fig. 6a), M13 exhibited three strong peaks at 1310, 1650 and 1590 cm−1 (Fig. 6a), agreeing with the literature.48 For the control sample comprised of stacked M13 on top of GO, with no sponge formation but of merely physical contact (PC) between the two components, Raman spectra exhibited highly similar fingerprints to those of GO and GPA (Fig. 6a).
 | ||
| Fig. 5 SEM-EDX analysis of GO-M13 aerogel and its components. Overview SEM images of (a–e) GO-M130303-5′ aerogel (GPA), (f) GO-5′, (g) M13 and (h) GPA, respectively. (Insets f–h) Images of the macro-samples with the corresponding EDX spectra (i–k). | ||
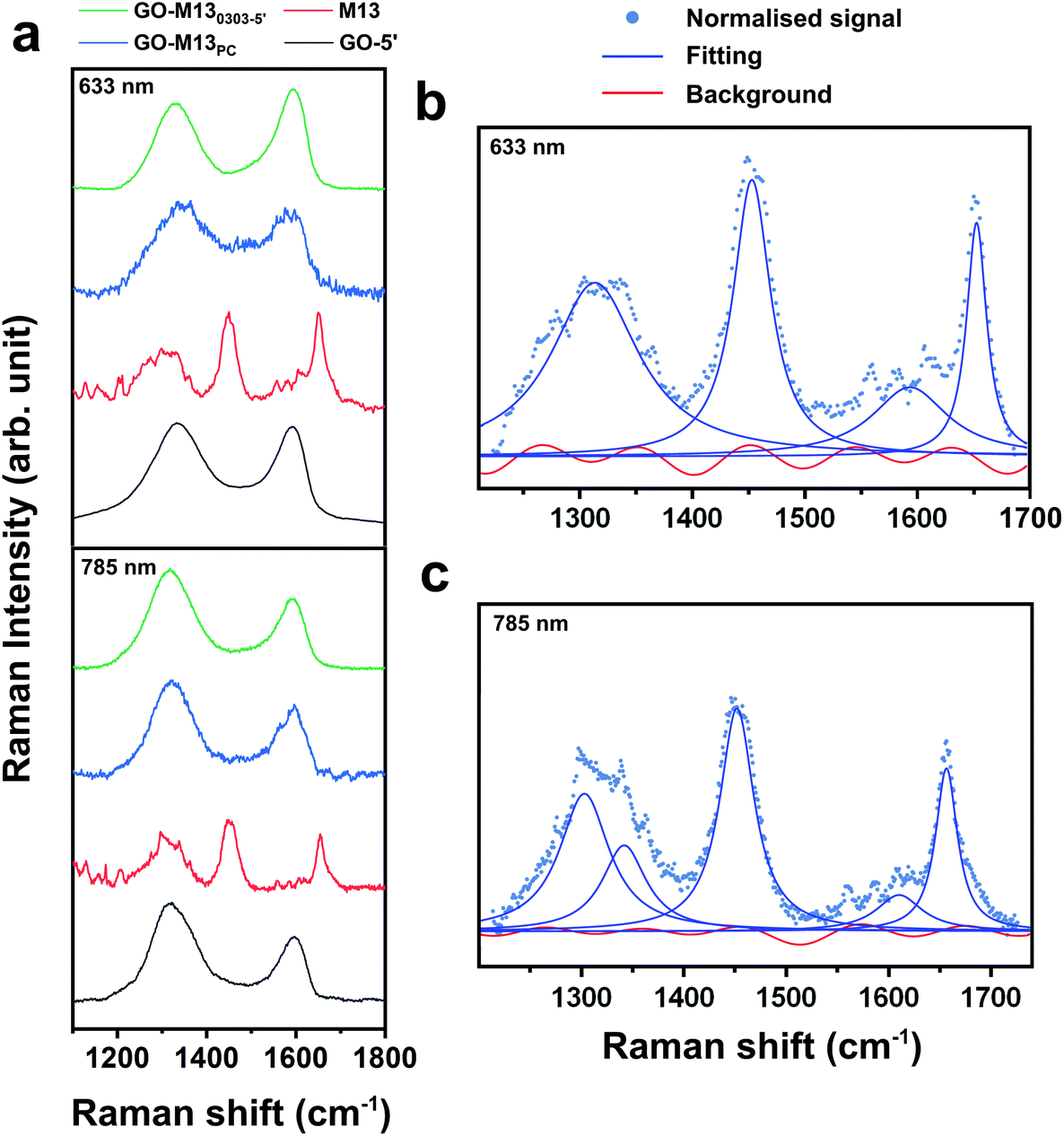 | ||
| Fig. 6 Raman spectroscopy of GPA and its components. (a) Representative Raman spectra of GO (black), M13 (red), GO-M13PC (blue) and GPA (green) at 633 and 785 nm excitation lasers. The best fit of the M13 spectra obtained at (b) 633 and (c) 785 nm, consisting of 4–5 Lorentzians for Raman peaks (blue) and Fourier series of 5–6 terms for the background (red), respectively for the 633 and the 785 nm excitation lasers. The spectra were flattened, and the background subtracted (blue dots). | ||
From Raman analysis, it is evident that the spectra of both GPA and GO-M13PC contain contribution mainly from GO rather than the M13. It was anticipated that due to the interaction between GO and M13 during the assembly of the sponge, two main peaks of GPA would be shifted, while those of GO-M13PC will not, indicating the modification of the GO bonds. Considering that all the spectra exhibited broad peaks with a large background, and by employing spectra fitting procedure, a subjective choice of the number of peaks and the background could affect the position of fitted peaks. Baseline-subtraction was not applied to the acquired spectra in Fig. 6a. Maximum likelihood estimation was employed to obtain the peak positions in the model (the peak shape, number of peaks, parameters of each peak and background) optimal for the acquired data ESI Tables S10–13.† Each spectrum was initially subtracted using a straight line for flattening, and subsequently, a corresponding number of Lorentzians and Fourier series of a range of terms were applied to give a fit for each spectrum (Fig. 6b and c).
Finally, by comparing the Bayesian information criteria (BIC) of the fits, the optimal fit was established for each spectrum. To note, no subjective input was provided to the BIC fitting, except setting two rational constraints: amplitudes of Lorentzians are positive, and coefficients of the Fourier series are comparatively small and, therefore, that they do not fit the peaks. The optimal fitting parameters of all the spectra are shown in ESI Tables S10–13.† The BIC fitting shows that the spectrum of the GO-M13PC consists of two peaks, which correspond to the unmodified sp2 (G mode) and sp3 (D mode) bonds of the GO.49 In the spectrum of the GPA, Raman G mode of GO splits, with approximately 30% upshifted and the rest remains unchanged. While the effect of charge transfer usually shifts the entire Raman peak, this observation can be interpreted as approximately 30% of the sp2 bond of GO stiffened by ∼2%, due to the interaction between M13 and GO. These results confirm that the interaction between GO and M13 in the fabricated sponge is strong enough to modify the original sp2 bonds.
Conclusions
This study introduces a novel, rapid and facile method to self-assemble and control GO into 3D hierarchical morphologies using the biological nanocomponent of the filamentous bacteriophage M13. The new graphene-viral sponge has been in-depth characterised, establishing the preferable conditions for the optimal self-assembly process, enabling the control over the stability as well as laying platform towards controllable pore dimensions. Investigating the formation mechanism of the hydrogel, signposted the electrostatic interaction as the driving force responsible for the self-assembly at specific pH conditions in solution. The porous structure of the micro-nano-composite exhibits ultra-low-density and high-surface-area with further advantages, compared with typical GO sponges, including the ease of assembly, low concentration of the reagents, scalability, environmental sustainability and low-production costs. GPA holds promise for numerous applications including for instance functional scaffolds, absorbers, filters, gas-sensors, chemical catalysis, supercapacitors and optoelectronics. Moreover, the M13 may provide a straightforward route for incorporating chemical functions by functionalisation along their longitudinal filament axis, rendering the GraPhage13 potentially useful for the development of rapid-response biological and chemical sensors for environmental and biomedical applications, where selective and targeted detection of biochemical molecules is crucial.Materials and methods
M13 propagation and purification
M13 bacteriophage (M13KE) was purchased from New England Biolabs® as double-stranded DNA (dsDNA) and transferred into One Shot™ TOP10F’ Chemically Competent E. coli (Thermo Fisher Scientific®) through heat shock. Subsequently, the phage was self-propagated in batch cultures using the above E. coli strain. For this preparation, 2 L of Nutrient Broth No. 2 (Thermo Fisher Scientific® Oxoid) were autoclaved. Once cooled down to room temperature, tetracycline was added to the final concentration of 5 μg mL−1. Subsequently, M13 and E. coli were inoculated and incubated at 37 °C, 150 rpm for 24 h. After the propagation, the culture was centrifuged two times at 8000 rpm (Beckman Coulter® JA 10, RCF = 11295g) and then the pellet containing E. coli was discarded. The supernatant containing M13 was subsequently incubated with PEG 6000 (Sigma-Aldrich®) and stirred for 90 min at 4 °C. Next, the supernatant/PEG solution was centrifuged at 10000 rpm (Beckman Coulter JA 10®, RCF = 17649g) and this time the supernatant was discarded. The obtained white pellet mainly consisting of M13, was resuspended in DIW. Finally, the concentration was determined with a UV-Vis spectrophotometer (Thermo Fisher Scientific® Evolution 600) measuring the absorbance at 269 nm and using an extinction coefficient (ε) of 3.84 ± 0.06 cm2 mg−1.29 The stock solution used for all the experiments was of a concentration of 5 mg mL−1.
GO stocks preparation
5 mg mL−1 GO liquid dispersion purchased from Graphene Supermarket® with a composition of Carbon and Oxygen at 79% and 20% respectively, was sonicated in glass vials using a probe sonicator (Fisherbrand™ Model 120 Sonic Dismembrator) at 70% power for 5 min (pulse 3 s ON and 9 s OFF). During the sonication, to avoid GO overheating, the vial was kept on ice.UV-Vis spectrophotometry
All samples were analysed in a 0.1 cm light path quartz cuvette with a UV-Vis spectrophotometer (Agilent® Cary 60 UV-Vis). The supernatant spectra were acquired after samples’ centrifugation in a benchtop centrifuge at 15000 rpm (SciSpin MICRO Centrifuge, RCF = 15100g) for 1 min.
Atomic force microscopy (AFM)
AFM (BRUKER Innova®) images were acquired in dry conditions on a P-type silicon wafer (Sigma-Aldrich®). Before depositing GO and GO-5′ onto the substrate, the latter was modified with (3-aminopropyl) triethoxysilane (APTES) (Sigma-Aldrich®). Then, the silicon was incubated for 30 min in a solution containing acetone and water with a volume ratio of 5:1 and 10 mM APTES.50 Subsequently, two different silicon-APTES wafers were soaked for 3 s in a solution of GO and GO-5′ respectively. Both solutions had a concentration of 1 μg mL−1. 20 μL of M13 25 μg mL−1 in DIW were drop-cast onto a ∼25 mm2 silicon wafer and dried at room temperature. The images were acquired in tapping mode with BRUKER® RTESPA-300 probe (T: 3.4 μm; L: 125 μm; W: 40 μm; f0: 300 kHz; k: 40 N m−1).
GO-M13 self-assembly
The GO-M130303-5′ hydrogel was immediately self-assembled after mixing GO and M13 in DIW. The final concentration of GO and M13 was 0.3 mg mL−1 each, within a final volume of 1 mL. Subsequently, the microtube was placed on an orbital shaker at 250 rpm for 15 min and centrifuged at 15000 rpm (SciSpin MICRO Centrifuge, RCF = 15100g) for 1 min. A P-type silicon wafer (Sigma-Aldrich®) was cleaved (∼25 mm2) and cleaned with a CO2 snow jet on a hot plate at a temperature of 250 °C. After the centrifugation, 900 μL of the supernatant was removed, and the pellet was well resuspended. 50 μL of the final hydrogel were deposited onto the Si substrate and finally, dried for 1 h in a vacuum chamber (Edwards® Rotary vacuum pump E2M2) until reaching a pressure of ∼90 Pa.
Zeta-potential
The ζ-potential of M13 1 mg mL−1 in 10 mM NaCl was analysed at pH ranging from pH 2 to 11 (Malvern Panalytical® Zetasizer Nano – ZS). The pH was measured with a Mettler Toledo FiveEasy™ equipped with a pH electrode InLab Micro.Rheology and hydrogel density
The viscosity and stress of GO-M130303-5′ were analysed using an HR-1 Discovery Hybrid Rheometer (TA Instruments®). A 75 μL sample was analysed with a TA geometry number 991437 (cone angle: 20°0′14′′, cone diameter 20 mm and truncation 55 μm) varying the shear rate from 0.1 to 1000 s−1. Moreover, the hydrogel density was predicted (1.003 mg cm−3) and calculated by weighing three samples of 1.0 mL on an analytical balance (OHAUS® Pioneer™ PA114C).
Scanning electron microscopy coupled with energy dispersive X-ray (SEM/EDX) spectroscopy
Before the image acquisition, all the samples were sputter coated with platinum for 4 min at 30 mA (Polaron® SC7640). The images and spectra were obtained with an XL30 ESEM FEG (Philips®) equipped with an INCA X-sight (Oxford Instruments®) at 15 kV.Volumetric mass density
To calculate the density of GPH, three microtubes containing 1 mL each of GO-M130303-5′ were systematically weighed on an analytical balance (OHAUS® Pioneer™ PA114C). Finally, the measured mass was correlated to the volume. Similarly, to calculate the density of GPA, three sponges were weighed individually using the inner balance of a Dynamic Vapor Sorption (DVS) apparatus (Surface Measurement Systems Ltd, DVS Advantage) under a controlled environment at 25 °C and 30% humidity. To correlate the obtained mass to the volume of the sponges, the three samples were photographed on a rotating stage at 0°, 90°, 180° and 270°, before and after drying, to compare the dimension of the hydrogel drop with the corresponding aerogel (ESI Fig. S11 and Table S9†). The results show that the area in pixels of the aerogel decreased by just 0.39% compared to the hydrogel. Therefore, to calculate the final density the weight of the three sponges was correlated to the volume of the hydrogel used (50 mm3).Brunauer, Emmett and Teller (BET)
The surface area was measured with a 3 Flex Physisorption (Micrometrics®) BET apparatus. The analysis was performed on GPA sponges prepared as described previously and dried overnight at 40 °C in a standard oven (Genlab Classic Oven) before the analysis.Raman spectroscopy
Non-polarised Raman spectra were obtained using a Renishaw inVia™ Qontor® Raman Microscope system with a confocal microscope (Leica® DM2500 equipped with a ×50 objective) at room temperature. Suitable edge filters for the 633 nm (RL633 Renishaw Class 3B HeNe) and 785 nm (RL785 Class 3B) laser were used as well as single 1200 grooves per mm grating for both lines, and the resolutions of the spectra were 1.5 cm−1 and 1.0 cm−1 at 633 nm and 785 nm, respectively. The laser power on the sample was kept below 5 mW for both lasers, to avoid laser-heating effects on the probed samples and the concomitant softening of their Raman peaks. Representative Raman spectra of GO, M13, GO-M13PC and GO-M130303-5′ on silicon substrates, were collected at 30, 70, 70 and 30 s at 633 nm and 10, 30, 20 and 30 s at 785 nm, enabling optimisation of the Raman signal while avoiding saturating of the detector.Software
All the figures were prepared using Inkscape. PyMOL® 1.7.x was used to highlight the hydrophilic, hydrophobic surfaces and polar contacts of the M13 PVIII major coat protein. Microsoft Office Excel, OriginLab 2018 and GraphPad PRISM® 6 were used for calculation and data plotting of UV-Vis spectra and AFM cross-sections. Error bars represent mean ± SD. Statistical significance was assessed performing 2-way ANOVA with a post hoc test of the relevant pairs of samples. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. The AFM images were prepared using Gwyddion®. Raman data was collected with the dedicated software (WiRE® 5.0), plotted and analysed using Wolfram Mathematica®.Author contributions
PP, PGO and TRD conceptualised the study and with HW guided it throughout and inputted to the experimental design and optimisation. PP with the assistance of IK, KC and RS carried out the experimental work and with YS and PGO analysed the results and wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge the Defence Science and Technology Laboratories (DSTLX-1000098511), and BAE-Systems (PO 97005101 and AIREF1701) for funding this research. PGO is a Royal Academy of Engineering (RAEng) Research Fellowship holder and would like to thank the RAEng for supporting this study.References
- V. Chabot, et al., A review of graphene and graphene oxide sponge: Material synthesis and applications to energy and the environment, Energy Environ. Sci., 2014, 7, 1564 RSC.
- X. Huang, et al., Graphene-based materials: Synthesis, characterization, properties, and applications, Small, 2011, 7, 1876–1902 CrossRef CAS PubMed.
- V. Georgakilas, et al., Functionalization of graphene: Covalent and non-covalent approaches, derivatives and applications, Chem. Rev., 2012, 112, 6156–6214 CrossRef CAS PubMed.
- K. S. Novoselov, Electric field effect in atomically thin carbon films, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- S. Bae, et al., Roll-to-roll production of 30-inch graphene films for transparent electrodes, Nat. Nanotechnol., 2010, 5, 574–578 CrossRef CAS PubMed.
- K. S. Kim, et al., Large-scale pattern growth of graphene films for stretchable transparent electrodes, Nature, 2009, 457, 706–710 CrossRef CAS PubMed.
- G. Eda, G. Fanchini and M. Chhowalla, Large-area ultrathin films of reduced graphene oxide as a transparent and flexible electronic material, Nat. Nanotechnol., 2008, 3, 270–274 CrossRef CAS PubMed.
- A. B. Bourlinos, V. Georgakilas, R. Zboril, T. A. Steriotis and A. K. Stubos, Liquid-Phase exfoliation of graphite towards solubilized graphenes, Small, 2009, 5, 1841–1845 CrossRef CAS PubMed.
- U. Khan, et al., Liquid-Phase exfoliation of graphite towards solubilized graphenes, Langmuir, 2011, 27, 9077–9082 CrossRef CAS PubMed.
- U. Khan, A. O'Neill, M. Lotya, S. De and J. N. Coleman, High-concentration solvent exfoliation of graphene, Small, 2010, 6, 864–871 CrossRef CAS PubMed.
- Y. Hernandez, et al., High-yield production of graphene by liquid-phase exfoliation of graphite, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS PubMed.
- D. C. Marcano, et al., Improved synthesis of graphene oxide, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, Preparation of graphitic oxide, J. Am. Chem. Soc., 1958, 80, 1339–1339 CrossRef CAS.
- C. Chen, M. Long, M. Xia, C. Zhang and W. Cai, Reduction of graphene oxide by an in-situ photoelectrochemical method in a dye-sensitized solar cell assembly, Nanoscale Res. Lett., 2012, 7, 101 CrossRef PubMed.
- H.-B. Zhang, et al., Vacuum-assisted synthesis of graphene from thermal exfoliation and reduction of graphite oxide, J. Mater. Chem., 2011, 21, 5392 RSC.
- Z.-J. Fan, et al., Facile synthesis of graphene nanosheets via Fe reduction of exfoliated graphite oxide, ACS Nano, 2011, 5, 191–198 CrossRef CAS PubMed.
- H. He, J. Klinowski, M. Forster and A. Lerf, A new structural model for graphite oxide, Chem. Phys. Lett., 1998, 287, 53–56 CrossRef CAS.
- A. Lerf, H. He, M. Forster and J. Klinowski, Structure of graphite oxide revisited, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- H.-P. Cong, P. Wang and S.-H. Yu, Stretchable and self-healing graphene oxide–polymer composite hydrogels: a dual-network design, Chem. Mater., 2013, 25, 3357–3362 CrossRef CAS.
- A. Sahu, W. Choi II and G. Tae, A stimuli-sensitive injectable graphene oxide composite hydrogel, Chem. Commun., 2012, 48, 5820 RSC.
- Y. Xu, Q. Wu, Y. Sun, H. Bai and G. Shi, Three-dimensional self-assembly of graphene oxide and DNA into multifunctional hydrogels, ACS Nano, 2010, 4, 7358–7362 CrossRef CAS PubMed.
- M. Ardini, et al., Supramolecular self-assembly of graphene oxide and metal nanoparticles into stacked multilayers by means of a multitasking protein ring, Nanoscale, 2016, 8, 6739–6753 RSC.
- Y. Zhang, C. Wu, S. Guo and J. Zhang, Interactions of graphene and graphene oxide with proteins and peptides, Nanotechnol. Rev., 2013, 2, 27–45 CAS.
- F. Zhang, et al., Horseradish peroxidase immobilized on graphene oxide Physical properties and applications in phenolic compound removal.pdf, J. Phys. Chem. C, 2010, 114, 8469–8473 CrossRef CAS.
- J. Zhang, et al., Graphene oxide as a matrix for enzyme immobilization, Langmuir, 2010, 26, 6083–6085 CrossRef CAS PubMed.
- P. H. Hofschneider, E. coli K 12-BAKTERIOPHAGEN I Untersuchungen über ‘kleine’ E. coli K 12 Bakteriophagen, Z. Naturforschg, 1963, 18, 203–210 Search PubMed.
- D. A. Marvin and B. Hohn, Filamentous bacterial viruses, Bacteriol. Rev., 1969, 33, 172–209 CAS.
- T. J. Henry and D. Pratt, The proteins of bacteriophage M13, Proc. Natl. Acad. Sci. U. S. A., 1969, 62, 800–807 CrossRef CAS PubMed.
- S. A. Berkowitz and L. A. Day, Mass, length, composition and structure of the filamentous bacterial virus fd, J. Mol. Biol., 1976, 102, 531–547 CrossRef CAS PubMed.
- P. M. G. F. van Wezenbeek, T. J. M. Hulsebos and J. G. G. Schoenmakers, Nucleotide sequence of the filamentous bacteriophage M13 DNA genome: comparison with phage fd, Gene, 1980, 11, 129–148 CrossRef CAS PubMed.
- G. P. Smith and V. A. Petrenko, Phage Display, Chem. Rev., 1997, 97, 391–410 CrossRef CAS PubMed.
- W. J. Chung, D. Y. Lee and S. Y. Yoo, Chemical modulation of m13 bacteriophage and its functional opportunities for nanomedicine, Int. J. Nanomed., 2014, 9, 5825–5836 Search PubMed.
- K. Mohan and G. A. Weiss, Chemically Modifying Viruses for Diverse Applications, ACS Chem. Biol., 2016, 11, 1167–1179 CrossRef CAS PubMed.
- S. Ye and J. Feng, The effect of sonication treatment of graphene oxide on the mechanical properties of the assembled films, RSC Adv., 2016, 6, 39681–39687 RSC.
- X. Ji, J. Oh, a. K. Dunker and K. W. Hipps, Effects of relative humidity and applied force on atomic force microscopy images of the filamentous phage fd, Ultramicroscopy, 1998, 72, 165–176 CrossRef CAS PubMed.
- Q. Lai, S. Zhu, X. Luo, M. Zou and S. Huang, Ultraviolet-visible spectroscopy of graphene oxides, AIP Adv., 2012, 2, 3–8 Search PubMed.
- A. R. Goldfarb, L. J. Saidel and E. Mosovich, The Ultraviolet Absorption Spectra of Proteins, J. Biol. Chem., 1951, 193(193), 397–404 CAS.
- L. A. Day and R. L. Wiseman, A Comparison of DNA Packaging in the Virions of fd, Xf, and Pf1, Cold Spring Harb. Monogr. Arch., 1978, 8, 605–625 Search PubMed.
- W. Mäntele and E. Deniz, UV–VIS absorption spectroscopy: Lambert-Beer reloaded, Spectrochim. Acta, Part A, 2017, 173, 965–968 CrossRef PubMed.
- B. Konkena and S. Vasudevan, Understanding aqueous dispersibility of graphene oxide and reduced graphene oxide through pKa measurements, J. Phys. Chem. Lett., 2012, 3, 867–872 CrossRef CAS PubMed.
- D. Li, M. B. Müller, S. Gilje, R. B. Kaner and G. G. Wallace, Processable aqueous dispersions of graphene nanosheets, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- T. A. Roth, G. A. Weiss, C. Eigenbrot and S. S. Sidhu, A minimized M13 coat protein defines the requirements for assembly into the bacteriophage particle, J. Mol. Biol., 2002, 322, 357–367 CrossRef CAS PubMed.
- K. Zimmermann, H. Hagedorn, C. C. Heuck, M. Hinrichsen and H. Ludwig, The ionic properties of the filamentous bacteriophages Pf1 and fd, J. Biol. Chem., 1986, 261, 1653–1655 CAS.
- N. F. Steinmetz and M. Manchester, Viral nanoparticles: tools for materials science and biomedicine, Pan Stanford, 2011 Search PubMed.
- O. Morag, N. G. Sgourakis, D. Baker and A. Goldbourt, The NMR–Rosetta capsid model of M13 bacteriophage reveals a quadrupled hydrophobic packing epitope, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 971–976 CrossRef CAS PubMed.
- J. N. Aronson, The Henderson-Hasselbalch Equation Revisited, Biochem. Mol. Biol. Educ., 1983, 11, 68 CAS.
- Y. M. Lee, et al., Nanomesh-Structured Ultrathin Membranes Harnessing the Unidirectional Alignment of Viruses on a Graphene-Oxide Film, Adv. Mater., 2014, 26, 3899–3904 CrossRef CAS PubMed.
- K. L. Aubrey and G. J. Thomas, Raman spectroscopy of filamentous bacteriophage Ff (fd, M13, f1) incorporating specifically-deuterated alanine and tryptophan side chains. Assignments and structural interpretation, Biophys. J., 1991, 60, 1337–1349 CrossRef CAS PubMed.
- F. Tuinstra and J. L. Koenig, Raman spectrum of graphite, J. Chem. Phys., 1970, 53, 1126–1130 CrossRef CAS.
- J. Ou, et al., Tribology study of reduced graphene oxide sheets on silicon substrate synthesized via covalent assembly, Langmuir, 2010, 26, 15830–15836 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures and discussion. See DOI: 10.1039/c9nr03670a |
| This journal is © The Royal Society of Chemistry 2019 |