
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVersatile route to core–shell reinforced network nanostructures†
Pascal
Rusch
a,
Fabian
Niemeyer
ac,
Denis
Pluta
ac,
Björn
Schremmer
a,
Franziska
Lübkemann
a,
Marina
Rosebrock
a,
Malte
Schäfer
bc,
Mandy
Jahns
b,
Peter
Behrens
 bcd and
Nadja C.
Bigall
*acd
bcd and
Nadja C.
Bigall
*acd
aInstitute of Physical Chemistry and Electrochemistry, Leibniz Universität Hannover, Callinstraße 3A, 30167 Hannover, Germany. E-mail: nadja.bigall@pci.uni-hannover.de
bInstitute for Inorganic Chemistry, Leibniz Universität Hannover, Callinstraße 9, 30167 Hannover, Germany
cLaboratory of Nano and Quantum Engineering, Leibniz Universität Hannover, Schneiderberg 39, 30167 Hannover, Germany
dCluster of Excellency PhoenixD (Photonics, Optics, and Engineering – Innovation Across Disciplines), Hannover, Germany
First published on 22nd July 2019
Abstract
In this work we present the generation of new core–shell network nanostructures of macroscopic dimensionality by a two-step process analogous to the seeded-growth method in colloidal nanoparticle modification. The nanoparticle-based core network is assembled first and in a separate second step it is coated with a continuous metal oxide shell by sol–gel methods. The interparticle contact of the nanoparticles comprising the core network is kept intact throughout the process. By analyzing the local elemental distribution, the shells can be shown to be homogeneous over the macroscopic network monolith. The shell network can be used to considerably reinforce the mechanical strength of the final core–shell network structure.
Introduction
After the introduction of aerogels in 1931 by Kistler1 and the optimistic outlook to extend the list of materials from which aerogels can be generated, it still took the better part of a century until a method was used to generate nanocrystal-based aerogel networks in 2005.2,3 This method to form randomly oriented network structures out of pre-formed colloidal nanoparticles offers higher customizability as it allows the fine tuning of the individual building blocks by modern colloid chemical routes as opposed to the one-step generation of particles and networks in molecular routes of conventional sol–gel chemistry. In recent years the number of nanoparticle materials that were converted into such macroscopic and porous network structures steadily increased4–7 and was expanded to noble metals8 as well as a number of intricate particles provided by the well advanced chemical synthesis of nanoparticles.9–13 The procedure usually includes the controlled destabilization of a colloidally stable nanoparticle solution, often by attacking the stabilizing ligand shell of the particle, e.g., by oxidation agents. More recently different methods were described based on connecting the particles – sometimes also reversibly – via the interaction of ligands, organic and inorganic, with added cations.14,15 Also methods completely free of chemical interactions but rather based on physical procedures have been employed.16 Still, as the catalogue of new nanoparticles grows and the number of these converted into self-supporting 3D networks increases, generally this innovation stops once a network is indeed generated.In this work we want to show that the network formation alone does not need to be the last step, but instead some of the modification procedures developed for colloidal nanoparticles can – under the right conditions – be applied to a nanoparticle network substrate as well. Network structures build up from metal nanoparticle building blocks have shown very promising results as catalysts in the oxygen evolution reaction17,18 as well as fuel cell applications.19 Similarly semiconductor nanoparticle based networks were employed as photocatalysts.20 Our group recently demonstrated a semiconductor network based sensor also showing the higher sensitivity of the network compared to plain nanoparticles.21 One drawback of the monolithic network structures is their poor mechanical stability which is widely regarded as the point inhibiting application of these networks.22,23 Here, we propose the method of post-assembly shell growth as a new way to increase mechanical stability and thereby resolve one major problem holding self-supporting nanoparticle network structures back from their potential applications. For example, in recent studies of the catalytic activity of nanoparticle aerogels, it was found that either the networks had to be broken apart and dispersed24 or the reaction conditions needed to be changed drastically13 to accommodate this problem. It has recently been shown that it is possible to grow metallic shells around nanoparticle-based networks by electrochemical deposition.25 Our method also entails another modification step after the initial nanoparticle network formation but using wet-chemical methods. The method employed is similar to a seeded growth mechanism in colloid chemistry, where first nanocrystal seeds are synthesized and subsequently a different compound can be grown onto the seeds under conditions of heterogeneous nucleation only. In our method, however, instead of the seed nanoparticle we employ a seed nanoparticle network, as schematically shown in Fig. 1. More specifically, we propose the growth of metal oxide shells around the whole NANOparticle-based NETwork Structure (NANONETS) (consisting of interconnected nanocrystal building blocks) using sol–gel processes. The method presented in this work is applicable for different substrates as shown by the shell growth onto the semiconductor NANONETS as well as metallic ones. It can also be used to grow not only silica shells but also titania alike (also see Fig. 2). This indicates the versatility of our approach.
 | ||
| Fig. 1 Schematic depiction of the two-step process employed: first, the assembly of individual nanocrystal building blocks into a porous network structure and second the modification of this network by shell growth. | ||
 | ||
| Fig. 2 Different material combination of post-gelation modified networks. (a) CdSe/CdS nanorod core network with a silica shell, (b) CdSe/CdS nanorod core network with a titania shell and (c) Au–Ag mixed core network with a silica shell. | ||
Contrary to other methods already published in the literature we do not embed particles in a gel matrix made of the metal oxide.26–29 Instead the original network is made up of the nanoparticle based “core” network which is encased in a second “shell” network applying a post-assembly coating step. The crystal-to-crystal contact between the nanoparticles in the “core” network is still present in the newly formed structure, as schematically illustrated in Fig. 1.
Experimental
Chemicals used
Tri-n-octylphosphine oxide (TOPO, 99%), 3-mercaptopropionic acid (MPA, 99%), potassium hydroxide (KOH, >85%), hexane (99%), toluene (99.7%), methanol (99.8%), sulfur (S, 99.98%), ammonia (NH3, 28.0–30.0% aqueous solution), titanium(IV) butoxide (97%), sodium borohydride (98%), (3-aminopropyl)-trimethoxysilane (APTMS, 97%) and tetraethyl orthosilicate (TEOS, 99.999%) were purchased from Sigma-Aldrich. Acetone (99.5%) and hydrogen peroxide (35% aqueous solution) were purchased from Honeywell. Cadmium oxide (CdO, 99.998%), hydrogen tetrachloroaurate trihydrate (99.99%), silver nitrate (99.9%) and selenium (Se, 99.999%) were purchased from Alfa Aesar. Hexylphosphonic acid (HPA, >99%) and octadecylphosphonic acid (ODPA, >99%) were purchased from PCI Synthesis. Tri-n-octylphosphine (TOP, 97%) and trisodium citrate dihydrate (99.0%) were purchased from ABCR. Acetylacetone (acac, 99%) was purchased from Merck. Ethanol (99.8%) was purchased from Roth. All chemicals were used as received.Synthesis procedures
The CdSe/CdS nanorods used for network formation were synthesized according to the literature procedure30 and assembled as described in earlier studies of our group.12 The whole process will be described briefly here.The nanorods in solution (750 μL) were precipitated by the addition of methanol and centrifuged at 5000 rcf. The supernatant was discarded and the particles were dissolved in 10 mL of hexane. To this solution 242 μL of MPA was added as well as 10 mL of a 0.1 M solution of KOH in methanol. The mixture is shaken for 24 hours during which the now MPA capped nanorods precipitated. The particles were separated from the remaining phase transfer solution by centrifugation and could be dissolved in 0.1 M aqueous KOH.
The coating of networks with titania was performed accordingly using titanium n-butoxide as a metal oxide precursor. As the hydrolysation of titanium alkoxides is much faster than the reaction of the equivalent silicon compounds acetylacetonate was added in this case to slow down the reaction by complexing the titanium cation. The amount of acetylacetonate was chosen in relation to the amount of titania precursor added.
The coating of noble metal nanoparticle based networks was performed in an identical manner. In this case the addition of a linker, namely, APTMS, at a 0.1 ratio compared to the noble metal concentration could be used to improve the uniformity of the shells.
Characterization
SEM samples were prepared by finely slicing off a part of the macroscopic network structure with a scalpel. This section was stuck to the adhesive carbon film and measured. The SEM used was a JEOL JSM 6700F field emission scanning electron microscope with an acceleration voltage of 2 kV and a secondary electron detector at a working distance of 8 mm. In the SEM, elemental analysis was performed using an Oxford Instruments INCA 300 energy dispersive X-ray spectrometer with the SEM operating at 10 kV at a working distance of 15 mm.
Results and discussion
For the functionalization of nanoparticles with a silica shell, a plethora of methods is described in the literature which are mostly based on the Stöber process.35 Due to the substrate not being a nanoparticle in solution but rather a macroscopic body (consisting of a network of nanocrystals), methods involving microemulsions36,37 could not be used. We therefore opted for a synthesis that is similar to the direct coating methods reported,38,39 with much simpler conditions than the microemulsion routes in terms of the solvent system since no surfactants are employed, see the Experimental section for details. This procedure could be used with small adaptions for the growth of titania shells as well as for the growth of silica on noble metal NANONETSs. The successful modification of the NANONETS with a metal oxide shell can be observed by transmission electron microscopy (TEM) as shown in Fig. 2.For the pristine semiconductor NANONETS a smooth surface of the NRs forming the network is visible (Fig. 3b) in the TEM measurements. With increasing amounts of metal oxide precursor added to the structure there is no optical difference between the dried network structures as can be seen in the photographs under UV- and daylight (Fig. 3a). In electron microscopy an increase in surface roughness is visible due to small domains of silica forming on the nanocrystal surfaces (see Fig. 3c and d). In these cases, the difference in electronic TEM contrast between silica and the NRs is unfortunately not sufficient to make the new shell clearly distinguishable from cadmium sulfide. With higher amounts of silica precursor and consequently thicker shells, the core network and the surrounding silica shell become easily discernable (Fig. 3e). The porous structure of the network is visible in the SEM (Fig. 4d) and could also be measured by krypton physisorption. The surface area derived from the BET evaluation is 100 m2 g−1 for the unmodified network which is comparable to the ones reported for similar structures.11,40 After the modification the BET surface areas increase considerably to 160–180 m2 g−1 potentially due to microporosity in the silica shell itself as well as the higher mechanical stability, preventing the network from damage during processing. It also indicates that the modification does not block the porosity of the network (see Fig. S11†).
 | ||
| Fig. 3 Core–shell network structures prepared with different ratios of silica precursors relative to the cadmium content of the core network. (a) Photographs of the monolithic dried networks under ambient (top) and UV (bottom) light with increasing amounts of silica precursor added from left to right (starting at 0 to 10/1). TEM micrographs of networks modified with different silica/cadmium ratios, (b) 0/1, (c) 1/1, (d) 2/1 and (e) 10/1. Arrows are used to indicate some of the recognizable domains of silica growth on the building block surfaces. | ||
 | ||
| Fig. 4 Analysis of local elemental distribution to evaluate the homogeneity of the silica shells. (a) Schematic depiction of the SEM sample in relation to the whole network body and (b) photograph of a dried network monolith under UV light to demonstrate the shape analogy. (c) Large scale SEM micrograph of the network slice modified with an initial Si/Cd ratio of 10/1, depicting the measurement positions for EDX (assembled from two separate pictures) and (d) higher resolution SEM image of the same network structure displaying the porous structure. (e) Si/Cd ratio found by EDX measurements at various measurement positions for initial Si/Cd ratios of 10/1 in grey and 2/1 in red. | ||
The successful modification with a homogeneous silica shell is clearly visible in the nanometer size regime. However, this only shows the modification in a random spot test. Hence, to investigate the homogeneity of the modification on a much larger scale, scanning electron microscopy (SEM) with elemental analysis by energy-dispersive X-ray spectroscopy (EDX) is employed. The elemental distribution is measured at different positions of a section of the modified NANONETS. These samples are prepared by slicing a thin piece off the body of the network structure (possible for samples with Si/Cd ratios larger than 1) without further modification of the sample (schematically shown in Fig. 4a). It should be noted that this is to the best of our knowledge the first time that the dried network of nanoparticles can be cut easily by simply employing a scalpel, since “conventional” nanocrystal network structures are either far too brittle or far too deformable for such a preparation method. By our post-assembly silica growth route, it is possible to perform structural investigations of nanocrystal network structures to a greater extent. Here, we have investigated the ratio of Si/Cd at various points in the NANONETS (center, top, bottom, and outer areas) in order to determine the homogeneity of our silica shell growth route. The SEM images show the highly porous network structure expected for these types of nanoparticle assemblies (Fig. 4d). The elemental measurements illustrate the silica-shell modification as well as its homogeneity. In the case of low silica amounts (Si/Cd < 1/1) the NANONETS breaks uncontrollably during sample preparation and random measurement positions were chosen (see the ESI†). Still, the elemental measurements fit well within our assumption of a largely homogeneous silica shell growth as discussed in the ESI.† For higher amounts of silica the NANONETS can be sliced in a controlled way and so the elemental distribution can be examined from the surface of the NANONETS down to the center. As an example, the measurement positions for one sample are marked in Fig. 3c on an overview (for positions on other samples see Fig. S4–S6† for the corresponding EDX spectra). In the case of NANONETSs modified with an initial (that is, as inserted during synthesis) ratio of 2/1 of Si/Cd, this initial ratio is retained for the surface part of the final structure, while in the center the ratio drops to 1.64/1. When an initial ratio of 10/1 of Si/Cd is used, the ratio measured from the surface of the NANONETS is 7.5/1 (measured at the position “outside”) going down to 4.8/1 inside the structure (measured at the position “center”). This gradient in the Si/Cd ratio exemplifies the strong influence of the transport limitation inside of the network pores, so that the modification is therefore not totally uniform for the whole of the macroscopic body of the NANONETS. On the other hand, it also shows that the modification is – while not completely uniform – present over the whole macroscopic network. It needs to be taken into account that transport is a major obstacle in the attempt of transferring nanoparticle modification methods in colloidal solution onto nanoparticle network substrates. Considering that the size regime of the distance that the silica precursors have to travel is increased from nanometers in a colloid to millimeters in such types of networks, the silica shells produced by this method throughout the structure are remarkably homogeneous.
The pH-value and concentration of water are key parameters for the Stöber synthesis. The standard parameters for this work have been adapted from literature procedures for the functionalization of noble metal nanoparticles with a silica shell in solution.41 The parameters are then varied to optimize the functionalization for a NANONETS substrate. The amount of water and ammonia is varied from no addition of the reactant to a tenfold excess in the case of ammonia or a fivefold excess in the case of water compared to the standard values adapted from the literature. All other parameters are kept constant. From the TEM micrographs of the NANONETS the clear necessity of both water and ammonia for the modification process can be derived. If neither water nor ammonia is added (Fig. S2a†) the shell growth is highly inhomogeneous even on a low size scale. Silica forms probably by reaction with residual water in the pores or from the atmosphere, but it does not form a clearly defined shell around the network. Instead some pores are filled with silica, while no silica is visible in other places. If only 30% ammonia solution is added but no additional water, the results are quite similar (Fig. S2b†). In this case a silica gel seems to form encasing the original NANONETS. The pores of the original structure are filled with the silica material. If water is added but no ammonia, the growth of silica also appears to be largely inhomogeneous (Fig. S3a†) with thicker silica shells visible at certain spots and no shell visible at other places. As soon as both reactants, water and ammonia, are added, the desired structure of the original NANONETS is surrounded by a continuous silica shell without any visible side nucleation. While the amount of ammonia does not seem to have a pronounced influence for the volumes investigated as long as it is present (Fig. S3†), water concentrations between 2 and 10 mmol mL−1 yield the most homogeneous and pronounced silica shells (Fig. S2†).
It was also investigated if it was possible to transfer these reaction conditions while changing either the core network material or the shell material. To this end similar conditions were applied while using titanium butoxide instead of tetraethyl orthosilicate (Fig. 1c). With this it was possible to grow titania shells in an equal manner (also see Fig. S7†). Slight adjustments in this procedure are needed such as the addition of acetylacetone (acac). acac acts as a chelating ligand which considerably decreases the reactivity of the precursor. This counteracts the effect that titania alkoxides are in general much more reactive compared to silica alkoxides as has been described for similar processes in colloidal particle modification (also see Fig. S8†).42,43 When examining the local elemental distribution by SEM-EDX as described for the silica shells earlier this influence is clearly visible. Without the addition of acac the homogeneous nucleation of titania outside of the network is immediately visible even by the eye. If the amount of acac in relation to the amount of titania precursor is increased the ratio of Ti/Cd measured inside the network increases due to the suppression of homogeneous nucleation. Also the homogeneity increases, indicated by the small difference in the Ti/Cd ratio measured at different spots on the gel. However, if very high amounts of acac are added the titania shell formation inside the gel is suppressed and there is almost no titania grown in this instance. A ratio of 2/1 acac/Ti-precursor seems to yield the most promising results in this study. The BET surface area of this titania modified network is ca. 140 m2 g−1 which is still higher than that of the unmodified networks (see Fig. S11†).
Furthermore, it was possible to use networks assembled from a mixture of gold and silver nanoparticles to grow a noble metal core silica shell network. Electron micrograms of these are shown in Fig. 5. The nanoparticles assemble into a chain-like interconnected highly porous network structure. In higher magnifications the silica shell is clearly visible homogeneously surrounding the whole continuous network (Fig. 5b). Measuring the elemental distribution at high magnifications is very difficult as the networks tend to move during the measurement. Still in the HAADF-STEM the shell is visible as a slightly brighter shell surrounding the noble metal chains. In the EDX signals of all three elements of interest (Au, Ag and Si) are detected along the chains of the network (Fig. 5c and d). The physisorption measurements revealed a BET surface area of the pristine Au–Ag nanoparticle networks of 25 m2 g−1 which is similar to the values reported in the literature.8 As was the case for semiconductor nanoparticle based networks this surface area increases considerably with the silica modification to 105 m2 g−1 (see Fig. S11†). This hints at the large inner surface of the porous silica shell. Similar to the growth of titania shells in this instance it was also needed to minimally adjust the experimental procedure. The addition of low amounts of a silica linker (here: 3-aminopropyl-trimethoxy-silane, at a 0.1 ratio compared to the amount of silica precursor used) was needed to grow homogeneous shells (also see Fig. S9† for details). Without the addition of a linker the shell growth is not homogeneous and limited to randomly dispersed spots on the network structure.
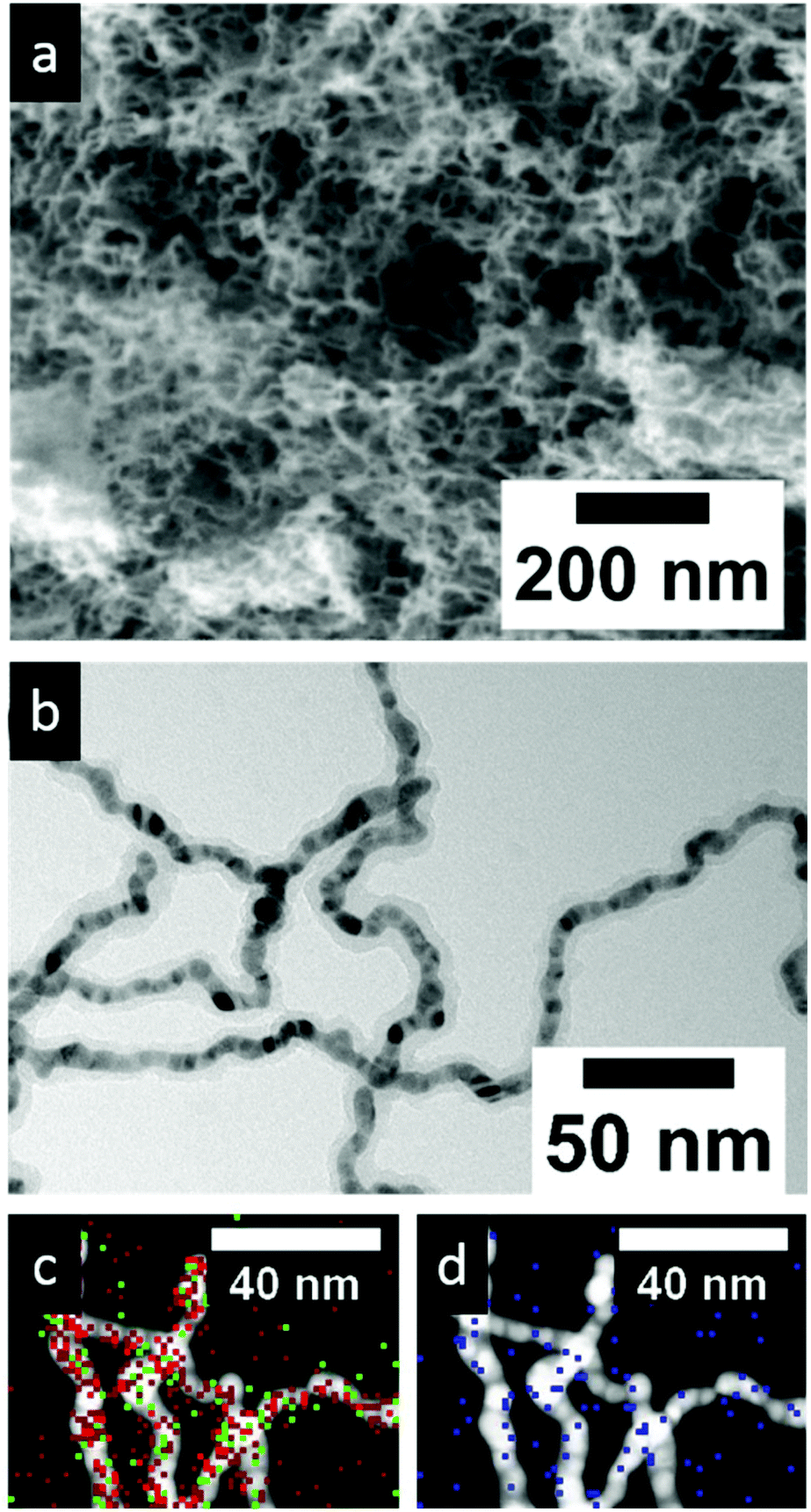 | ||
| Fig. 5 Network consisting of a mixture of Au and Ag nanoparticles with a silica shell grown around the network in (a) SEM, (b) BF-TEM and (c, d) HAADF-STEM with the overlaying EDX measurement. Red color indicates Au, green Ag and blue Si. | ||
The silica shell modification was expected to considerably increase the resistivity of the NANONETS towards mechanical stress as the network is essentially reinforced by the shell. Although the mechanical instability of nanoparticle networks is often a drawback it is seldom investigated or even mentioned in the literature. In fact the studies of mechanical properties have been reported only for organic aerogels,44,45 carbon aerogels46,47 or silica aerogels48,49 and recently for other metal–oxide aerogels.50 In this work a compression test was employed to evaluate the mechanical stability of silica shell modified and unmodified NANONETSs. As the dimensions of the NANONETS are small, and as the forces the samples are subjected to are also very small, it is not possible to actually measure the Young's modulus (which is usually done in the area of elastic deformation at low strain levels). Still, information about the relative mechanical stability can be obtained by the measurements performed. The measurements of the unmodified NANONETS show a drastic drop in stress, which occurs when a large part of the structure breaks and is a signature of its brittleness.
This is not observed in the case of silica modified networks (see Fig. 6) within the measurement range investigated. In a more practical context this increased mechanical resistance can also be observed when the dried NANONETS is added back to a solvent, i.e., water. The unmodified NANONETS falls apart in a few seconds into a multitude of small particles (Fig. S10†), which we attribute to the occurrence of capillary forces. In contrast, the silica shell modified NANONETS sinks to the bottom of the container after some time while still remaining largely in one piece (also see Fig. S10†). This result further emphasizes the benefit of post-assembly silica shell growth for nanoparticle aerogels, since it enables rehydration while keeping the advantageous properties of the nanocrystal networks in the macroscopic body.
 | ||
| Fig. 6 (a) Mechanical measurement of dried silica shell modified and unmodified CdSe/CdS NR networks by compression tests. The photographs (b) show a silica shell modified network structure (Si/Cd 10/1) at various points of the measurement. | ||
Conclusions
In summary, we have presented a novel approach to the metal oxide shell modification of the NANONETS which leaves the initial network and the interparticle crystal-to-crystal contact intact. To our knowledge this marks the first time that the possibility of modifying self-supporting nanocrystal networks by a sol–gel post-assembly modification with a thin inorganic shell has been described. This process leads to a new, formerly unknown structure. This method was employed to successfully coat nanostructures of different compositions ranging from the semiconducting to metallic ones and is therefore highly versatile. It could also be expanded to the growth of titania instead of silica. The integrity of the nanocrystal network could be confirmed by electron microscopy measurements and the homogeneity of the silica shell was examined by local elemental analysis and found to be highly uniform over the whole body for all modifications performed. It could be shown that the modification with silica shells severely increases the mechanical stability of the structures, which can now be cut into pieces without breaking. All of this opens up a plethora of new possible applications of nanocrystal network assemblies in, e.g., electrochemistry or catalysis where the network structures would need to be rehydrated with an electrolyte while maintaining the network properties. Due to the increased mechanical stability the fabrication of flow-through reactors can be imagined as well as the introduction of external stirring to facilitate mixing. All this can be done while retaining the monolithic structure, leading to an easy separation. As the particles forming the core network are still connected the networks should be able to conduct generated charge carriers, potentially improving the efficiency of the catalyst. Also the transfer of this method to other catalytically active metal oxides could prove interesting.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Armin Feldhoff and Juergen Caro for the use of the SEM, as well as the Laboratory of Nano and Quantum Engineering for the use of the TEM and their support. The authors would also like to thank the REBIRTH cluster of excellence for access to the mechanical testing experiment. The project leading to these results received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No. 714429). N. C. B. furthermore acknowledges funding from German Federal Ministry of Education and Research (BMBF) within the framework of NanoMatFutur (support code 03X5525). This project was in part funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy within the Cluster of Excellence PhoenixD (EXC 2122). M. S. is grateful for a fellowship from the PhD programme “Hannover School for Nanotechnology-Sensors” of the LNQE, funded by the state of lower Saxony.Notes and references
- S. S. Kistler, Nature, 1931, 127, 741–741 CrossRef CAS.
- J. L. Mohanan, I. U. Arachchige and S. L. Brock, Science, 2005, 307, 397–400 CAS.
- I. U. Arachchige, J. L. Mohanan and S. L. Brock, Chem. Mater., 2005, 17, 6644–6650 CrossRef CAS.
- K. K. Kalebaila, D. G. Georgiev and S. L. Brock, J. Non-Cryst. Solids, 2006, 352, 232–240 CrossRef CAS.
- A. Hitihami-Mudiyanselage, K. Senevirathne and S. L. Brock, ACS Nano, 2013, 7, 1163–1170 CrossRef CAS PubMed.
- S. Ganguly and S. L. Brock, J. Mater. Chem., 2011, 21, 8800 RSC.
- N. Gaponik, A. Wolf, R. Marx, V. Lesnyak, K. Schilling and A. Eychmüller, Adv. Mater., 2008, 20, 4257–4262 CrossRef CAS.
- N. C. Bigall, A.-K. Herrmann, M. Vogel, M. Rose, P. Simon, W. Carrillo-Cabrera, D. Dorfs, S. Kaskel, N. Gaponik and A. Eychmüller, Angew. Chem., Int. Ed., 2009, 48, 9731–9734 CrossRef CAS PubMed.
- H. Yu and S. L. Brock, ACS Nano, 2008, 2, 1563–1570 CrossRef CAS PubMed.
- I. U. Arachchige and S. L. Brock, J. Am. Chem. Soc., 2007, 129, 1840–1841 CrossRef CAS PubMed.
- S. Naskar, J. F. Miethe, S. Sanchez-Paradinas, N. Schmidt, K. Kanthasamy, P. Behrens, H. Pfnür and N. C. Bigall, Chem. Mater., 2016, 28, 2089–2099 CrossRef CAS.
- S. Sanchez-Paradinas, D. Dorfs, S. Friebe, A. Freytag, A. Wolf and N. C. Bigall, Adv. Mater., 2015, 27, 6152–6156 CrossRef CAS PubMed.
- S. Naskar, A. Freytag, J. Deutsch, N. Wendt, P. Behrens, A. Köckritz and N. C. Bigall, Chem. Mater., 2017, 29, 9208–9217 CrossRef CAS.
- V. Lesnyak, S. V. Voitekhovich, P. N. Gaponik, N. Gaponik and A. Eychmüller, ACS Nano, 2010, 4, 4090–4096 CrossRef CAS PubMed.
- V. Sayevich, B. Cai, A. Benad, D. Haubold, L. Sonntag, N. Gaponik, V. Lesnyak and A. Eychmüller, Angew. Chem., Int. Ed., 2016, 55, 6334–6338 CrossRef CAS PubMed.
- A. Freytag, S. Sánchez-Paradinas, S. Naskar, N. Wendt, M. Colombo, G. Pugliese, J. Poppe, C. Demirci, I. Kretschmer, D. W. Bahnemann, P. Behrens and N. C. Bigall, Angew. Chem., Int. Ed., 2016, 55, 1200–1203 CrossRef CAS PubMed.
- W. Liu, A.-K. Herrmann, D. Geiger, L. Borchardt, F. Simon, S. Kaskel, N. Gaponik and A. Eychmüller, Angew. Chem., Int. Ed., 2012, 51, 5743–5747 CrossRef CAS PubMed.
- W. Liu, P. Rodriguez, L. Borchardt, A. Foelske, J. Yuan, A.-K. Herrmann, D. Geiger, Z. Zheng, S. Kaskel, N. Gaponik, R. Kötz, T. J. Schmidt and A. Eychmüller, Angew. Chem., Int. Ed., 2013, 52, 9849–9852 CrossRef CAS PubMed.
- S. Henning, H. Ishikawa, L. Kühn, J. Herranz, E. Müller, A. Eychmüller and T. J. Schmidt, Angew. Chem., Int. Ed., 2017, 56, 10707–10710 CrossRef CAS PubMed.
- L. Korala, J. R. Germain, E. Chen, I. R. Pala, D. Li and S. L. Brock, Inorg. Chem. Front., 2017, 4, 1451–1457 RSC.
- A. Schlosser, L. C. Meyer, F. Lübkemann, J. F. Miethe and N. C. Bigall, Phys. Chem. Chem. Phys., 2019, 21, 9002–9012 RSC.
- B. Cai, V. Sayevich, N. Gaponik and A. Eychmüller, Adv. Mater., 2018, 30, 1707518 CrossRef PubMed.
- W. Wan, R. Zhang, M. Ma and Y. Zhou, J. Mater. Chem. A, 2018, 6, 754–775 RSC.
- D. Wen, W. Liu, D. Haubold, C. Zhu, M. Oschatz, M. Holzschuh, A. Wolf, F. Simon, S. Kaskel and A. Eychmüller, ACS Nano, 2016, 10, 2559–2567 CrossRef CAS PubMed.
- B. Cai, R. Hübner, K. Sasaki, Y. Zhang, D. Su, C. Ziegler, M. B. Vukmirovic, B. Rellinghaus, R. R. Adzic and A. Eychmüller, Angew. Chem., Int. Ed., 2018, 57, 2963–2966 CrossRef CAS PubMed.
- S. Jun, J. Lee and E. Jang, ACS Nano, 2013, 7, 1472–1477 CrossRef CAS PubMed.
- Z. Li, L. Kong, S. Huang and L. Li, Angew. Chem., Int. Ed., 2017, 56, 8134–8138 CrossRef CAS PubMed.
- B. Belache, Y. Khelfaoui, M. Bououdina, T. Souier and W. Cai, Mater. Sci. Semicond. Process., 2018, 76, 42–49 CrossRef CAS.
- L. G. Tartuci, L. F. T. Domingos, J. Bettini, K. O. Vieira, E. Raphael, B. R. C. Vale, J. L. Ferrari and M. A. Schiavon, J. Nanopart. Res., 2017, 19, 250 CrossRef.
- L. Carbone, C. Nobile, M. De Giorgi, F. Della Sala, G. Morello, P. Pompa, M. Hytch, E. Snoeck, A. Fiore, I. R. Franchini, M. Nadasan, A. F. Silvestre, L. Chiodo, S. Kudera, R. Cingolani, R. Krahne and L. Manna, Nano Lett., 2007, 7, 2942–2950 CrossRef CAS PubMed.
- W. W. Yu, L. Qu, W. Guo and X. Peng, Chem. Mater., 2003, 15, 2854–2860 CrossRef CAS.
- T. Kodanek, H. M. Banbela, S. Naskar, P. Adel, N. C. Bigall and D. Dorfs, Nanoscale, 2015, 7, 19300–19309 RSC.
- H. G. Bagaria, E. T. Ada, M. Shamsuzzoha, D. E. Nikles and D. T. Johnson, Langmuir, 2006, 22, 7732–7737 CrossRef CAS PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- X. Tang, E. Kröger, A. Nielsen, C. Strelow, A. Mews and T. Kipp, Langmuir, 2017, 33, 5253–5260 CrossRef CAS PubMed.
- F. Pietra, R. J. A. van Dijk - Moes, X. Ke, S. Bals, G. Van Tendeloo, C. de Mello Donega and D. Vanmaekelbergh, Chem. Mater., 2013, 25, 3427–3434 CrossRef CAS.
- E. Mine, A. Yamada, Y. Kobayashi, M. Konno and L. M. Liz-Marzán, J. Colloid Interface Sci., 2003, 264, 385–390 CrossRef CAS PubMed.
- Y. Kobayashi, H. Katakami, E. Mine, D. Nagao, M. Konno and L. M. Liz-Marzán, J. Colloid Interface Sci., 2005, 283, 392–396 CrossRef CAS PubMed.
- H. Yu and S. L. Brock, ACS Nano, 2008, 2, 1563–1570 CrossRef CAS PubMed.
- E. Mine, A. Yamada, Y. Kobayashi, M. Konno and L. M. Liz-Marzán, J. Colloid Interface Sci., 2003, 264, 385–390 CrossRef CAS.
- S. Lee, K. Lee, W. D. Kim, S. Lee, D. J. Shin and D. C. Lee, J. Phys. Chem. C, 2014, 118, 23627–23634 CrossRef CAS.
- X. Zhang, H. Sun, X. Tao and X. Zhou, RSC Adv., 2014, 4, 31313–31317 RSC.
- K. Kanamori, M. Aizawa, K. Nakanishi and T. Hanada, Adv. Mater., 2007, 19, 1589–1593 CrossRef CAS.
- H. Sehaqui, M. Salajková, Q. Zhou and L. A. Berglund, Soft Matter, 2010, 6, 1824 RSC.
- C. Zhu, T. Y.-J. Han, E. B. Duoss, A. M. Golobic, J. D. Kuntz, C. M. Spadaccini and M. A. Worsley, Nat. Commun., 2015, 6, 6962 CrossRef CAS PubMed.
- H.-W. Liang, Q.-F. Guan, L.-F. Chen, Z. Zhu, W.-J. Zhang and S.-H. Yu, Angew. Chem., Int. Ed., 2012, 51, 5101–5105 CrossRef CAS PubMed.
- J. P. Randall, M. A. B. Meador and S. C. Jana, ACS Appl. Mater. Interfaces, 2011, 3, 613–626 CrossRef CAS PubMed.
- K. E. Parmenter and F. Milstein, J. Non-Cryst. Solids, 1998, 223, 179–189 CrossRef CAS.
- A. Benad, F. Jürries, B. Vetter, B. Klemmed, R. Hübner, C. Leyens and A. Eychmüller, Chem. Mater., 2018, 30, 145–152 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Rehydration experiments, further TEM and SEM measurements. See DOI: 10.1039/c9nr03645h |
| This journal is © The Royal Society of Chemistry 2019 |