
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mixed dysprosium-lanthanide nitride clusterfullerenes DyM2N@C80-Ih and Dy2MN@C80-Ih (M = Gd, Er, Tm, and Lu): synthesis, molecular structure, and quantum motion of the endohedral nitrogen atom†
C.
Schlesier
,
F.
Liu
 *,
V.
Dubrovin
,
L.
Spree
,
B.
Büchner
,
S. M.
Avdoshenko
* and
A. A.
Popov
*
*,
V.
Dubrovin
,
L.
Spree
,
B.
Büchner
,
S. M.
Avdoshenko
* and
A. A.
Popov
*
Leibniz Institute for Solid State and Materials Research (IFW Dresden), Helmholtzstrasse 20, 01069 Dresden, Germany. E-mail: f.liu@ifw-dresden.de; s.avdoshenko@ifw-dresden.de; a.popov@ifw-dresden.de
First published on 13th June 2019
Abstract
Systematic exploration of the synthesis of mixed-metal Dy–M nitride clusterfullerenes (NCFs, M = Gd, Er, Tm, Lu) is performed, and the impact of the second metal on the relative yield is evaluated. We demonstrate that the ionic radius of the metal appears to be the main factor allowing explanation of the relative yields in Dy–M mixed-metal systems with M = Sc, Lu, Er, and Gd. At the same time, Dy–Tm NCFs show anomalously low yields, which is not consistent with the relatively small ionic radius of Tm3+ but can be explained by the high third ionization potential of Tm. Complete separation of Dy–Gd and Dy–Er, as well as partial separation of Dy–Lu M3N@C80 nitride clusterfullerenes, is accomplished by recycling HPLC. The molecular structures of DyGd2N@C80 and DyEr2N@C80 are analyzed by means of single-crystal X-ray diffraction. A remarkable ordering of mixed-metal nitride clusters is found despite similar size and electronic properties of the metals. Possible pyramidalization of the nitride clusters in these and other nitride clusterfullerenes is critically analyzed with the help of DFT calculations and reconstruction of the nitrogen inversion barrier in M3N@C80 molecules is performed. Although a double-well potential with a pyramidal cluster structure is found to be common for most of them, the small size of the inversion barrier often leads to an apparent planar structure of the cluster. This situation is found for those M3N@C80 molecules in which the energy of the lowest vibrational level exceeds that of the inversion barrier, including Dy3N@C80 and DyEr2N@C80. The genuine pyramidal structure can be observed by X-ray diffraction only when the lowest vibrational level is below the inversion barrier, such as those found in Gd3N@C80 and DyGd2N@C80. The quantum nature of molecular vibrations becomes especially apparent when the size of the inversion barrier is comparable to the energy of the lowest vibrational levels.
Introduction
Nitride clusterfullerenes (NCFs), i.e. metallofullerenes with endohedral M3N clusters, are the most abundant and versatile family of endohedral clusterfullerenes.1 Since the discovery of the first NCF, Sc3N@C80, in 1999,2 a large variety of NCFs with various fullerene cage sizes and different metal ions (M = Sc, Y, or lanthanides) have been synthesized.3 In the M3N cluster, three M3+ ions are located at the vertices of a triangle with the nitride ion N3− in its center. The cluster transfers six electrons to the surrounding carbon cage. The fullerene C80-Ih with a particularly stable hexaanion is the most suitable host for the endohedral species donating six electrons to the cage,4 and for M = Sc, Y, and lanthanides from Gd to Lu the M3N@C80-Ih NCFs are obtained with the highest yield among other cage sizes. However, when the cluster's size is incommensurate with the C80-Ih cage, as is observed for the early lanthanides from La to Nd with large ionic radii, the distribution of the fullerene sizes shifts to larger cages and the total NCF yield is decreased substantially.5 For Gd and Tb, although Gd3N@C80-Ih and Tb3N@C80-Ih are still the most abundantly produced Gd- and Tb-NCFs, the large size of the nitride cluster leads to substantial inner strain, which is partially compensated via a considerable pyramidalization of the M3N cluster inside the C80-Ih cage.3d,6 For smaller metals, the M3N cluster in M3N@C80-Ih is planar or nearly planar.3d,6a Thus, the balance between the ionic radius of the metal and the fullerene size is an important factor influencing the yield and molecular structure of NCFs.1b,c,6bCombining metals of different sizes within one NCF molecule, which would be then dubbed “mixed-metal NCF”, is an efficient approach to vary the cluster size and hence alter the product distribution of NCFs. As the ionic radius of Sc is considerably smaller than those of lanthanides, Sc has often been used in the synthesis of binary mixed-metal NCFs. Sc–Y,7 Sc–Ti,8 and Sc–V,9 all Sc–lanthanide binary systems forming NCFs, and even two ternary-metal NCFs, ScYErN@C80 and DyErScN@C80, have been produced.10 A number of non-Sc mixed-metal NCFs have also been synthesized, including Ti–Y,11 Ce–Y,10e,f Ce–Lu,12 Gd–Ho,13 Gd–Lu,13,14 Ho–Y,15 Ho–Lu,14,15 and Lu–Y.16 In addition to the alteration of yields and product distribution in comparison with corresponding homometallic NCFs, mixed-metal NCFs may also exhibit different chemical,7,17 electrochemical,10e,f,12a and magnetic properties.15,18
The structural diversity of nitride clusters offers a possibility to tune the magnetism of lanthanide-NCFs. The nitride ion in the center of the M3N cluster bears a large negative charge (the formal charge is −3e; the QTAIM analysis gives the value of ca −1.7e).19 Besides, NCF molecules feature rather short lanthanide–nitrogen bonds. The combination of these two factors leads to a large negative charge from a nitride ion in close proximity to a lanthanide and creates a strong axial ligand field (LF) in NCFs, which results in an easy-axis magnetic anisotropy for Ce, Pr, Nd, Tb, Dy, and Ho, and an easy-plane anisotropy for Er and Tm ions.10iAb initio calculations performed for Dy-NCFs predict very large LF splitting of Dy3+ total momentum (J) states in the range of 1500 cm−1,20 and all three DyxSc3−xN@C80-Ih NCFs (x = 1–3) were found to be single molecule magnets.18a,21 However, due to the different cluster compositions and intramolecular Dy⋯Dy interactions, DySc2N@C80-Ih, Dy2ScN@C80-Ih, and Dy3N@C80-Ih exhibit substantially different magnetic properties, which shows that combining Dy with other metals in nitride clusterfullerenes may lead to even more interesting magnetic phenomena, and further exploration of Dy-based mixed-metal NCFs is therefore an important task.
In this work we focus on the binary Dy–metal NCFs with a C80-Ih cage (Fig. 1) and study the influence of the non-Dy metals in the cluster on the synthesis and molecular structure of mixed-metal NCFs, keeping in mind further studies of their magnetic properties, which will be reported elsewhere. As a partner for Dy we have chosen the following elements: Lu as the lanthanide with the smallest ionic radius (Shannon's ionic radius22R3+ = 0.86 Å); Gd as the largest lanthanide (R3+ = 0.94 Å), still favoring the formation of M3N@C80-Ih; and Er (R3+ = 0.89 Å) and Tm (R3+ = 0.88 Å) as lanthanides with intermediate sizes. Besides, these lanthanides have different magnetic states in NCFs,10i and hence their combination with Dy may lead to diverse magnetic properties of mixed-metal NCFs. In the following, we first present a systematic analysis of the influence of the metal on the relative yields of mixed-metal Dy NCFs, and then an analysis of how a combination of Dy with lanthanide ions of different sizes affects the molecular structure and in particular the shape of the trimetal-nitride cluster. In the end we demonstrate how quantum vibrational effects dramatically affect the outcome of the diffraction experiments.
 | ||
| Fig. 1 DyM2N@C80 (a) and Dy2MN@C80 (b) molecules based on the C80-Ih carbon cage isomer. Dy is depicted in green, the lanthanide M in dark orange, N in blue and carbon atoms in light gray. | ||
Synthesis
The synthesis of all mixed-metal EMFs in this work was performed following the same protocol. Graphite rods were core-drilled and filled with a mixture of Dy/M (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio), graphite, and melamine as a source of nitrogen. The molar ratio Dy:M:N:C was kept at 1:1:10:15. The rods were evaporated under a He atmosphere (200 mbar) using a current of 100 A. Current phases were 30–60 s long, followed by a cooling period. As only the cathode is evaporated in the arc-discharge, the polarity of the rods was changed between the phases to ensure evaporation of both electrodes. The soot produced was collected, washed with acetone and extracted with CS2 for 20 hours. Then the CS2 was evaporated, the fullerene extract was redissolved in toluene, characterized by analytical HPLC and LDI mass-spectrometry, and then separated by multi-step HPLC as described below.
:1 molar ratio), graphite, and melamine as a source of nitrogen. The molar ratio Dy:M:N:C was kept at 1:1:10:15. The rods were evaporated under a He atmosphere (200 mbar) using a current of 100 A. Current phases were 30–60 s long, followed by a cooling period. As only the cathode is evaporated in the arc-discharge, the polarity of the rods was changed between the phases to ensure evaporation of both electrodes. The soot produced was collected, washed with acetone and extracted with CS2 for 20 hours. Then the CS2 was evaporated, the fullerene extract was redissolved in toluene, characterized by analytical HPLC and LDI mass-spectrometry, and then separated by multi-step HPLC as described below.
Comparison of the binary Dy–M NCF systems
Fig. 2 compares chromatograms of the raw fullerene extracts obtained for Dy–Gd, Dy–Er, Dy–Tm, and Dy–Lu systems. For all systems, chromatograms are dominated by a strong peak with a retention time near 32 min, which based on our previous experience with NCF synthesis is assigned to the mixture of DyxM3−xN@C80-Ih NCFs (x = 0–3). The peak next to it with a retention time of ca. 34 min is assigned to NCFs with a C80-D5h cage, whereas the peaks at longer retention times are NCFs with larger cages. Note that the relative yield of NCFs with larger cages is the highest in the Dy–Gd system. Nonetheless, as discussed in the introduction, NCFs with C80-Ih cages are produced with the highest yield, and in the following we will focus only on this cage isomer. Unless otherwise stated, from here on we will omit the cage symmetry designation and assume C80 to be C80-Ih.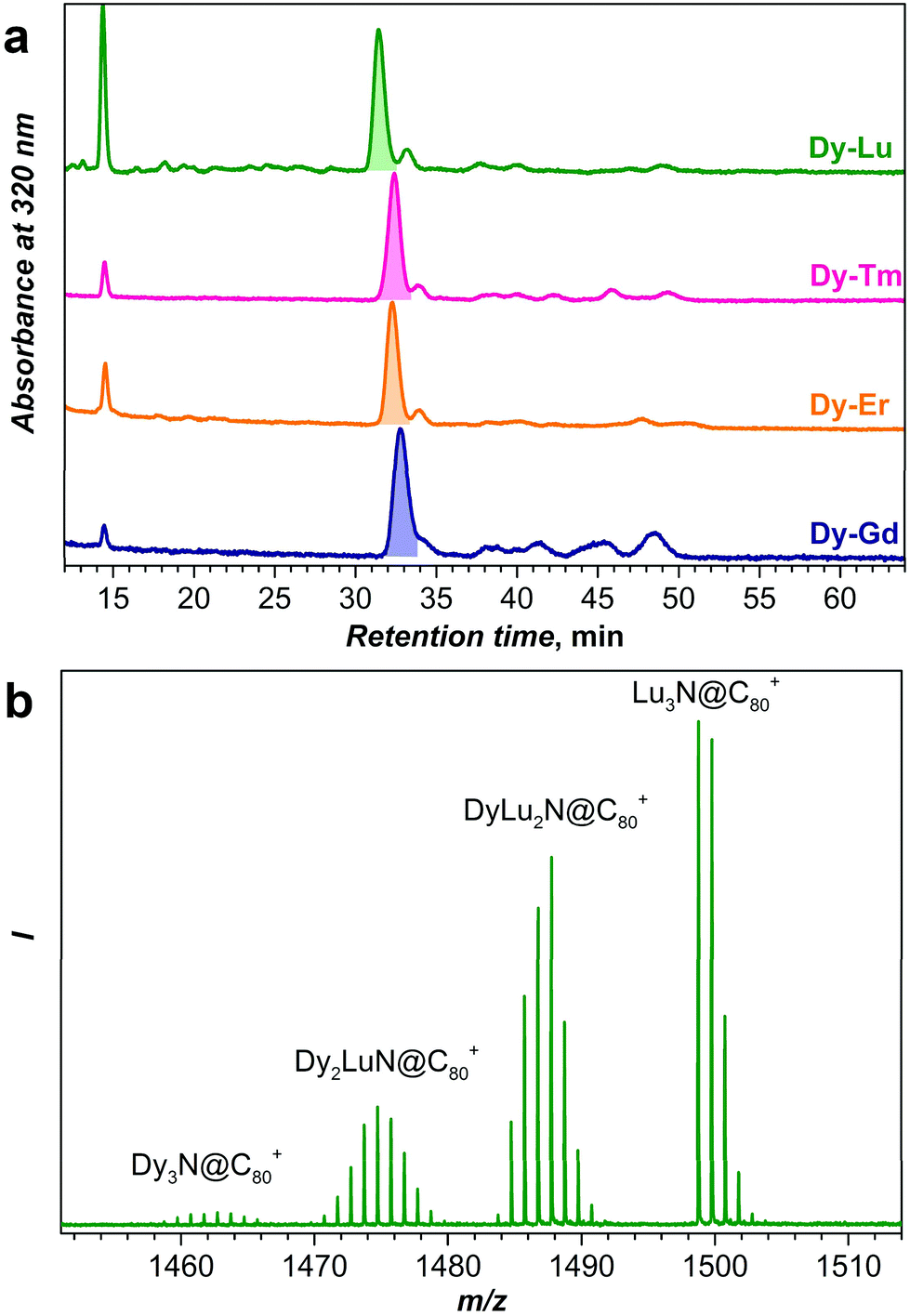 | ||
| Fig. 2 (a) HPLC chromatograms of raw fullerene extracts obtained in the Dy–Lu, Dy–Er, Dy–Tm, and Dy–Gd NCF syntheses (analytical BuckyPrep column, toluene as the eluent, 40 °C, 1.6 mL min−1). The sharp peak at 14–15 min is attributed to C70, the main NCF fractions comprising DyxM3−xN@C80-Ih (x = 0–3) eluted at 31–34 min (shaded with the corresponding color). (b) LDI mass-spectrum of the DyxLu3−xN@C80 (x = 0–3) fraction measured in the positive ion mode. | ||
For an equimolar mixture of Dy and a metal M in the starting material (i.e. the Dy:M molar ratio is 1:1), the ratio of Dy3N:Dy2MN:DyM2N:M3N clusters in the synthesized NCF mixture should be 1:3:3:1 given that the product ratio follows binominal statistical distribution without a bias for any of the metals. However, the studies of mixed-metal EMFs showed that the ratio of the cluster composition rarely follows the expected statistical distribution.2,10e,h,23 The use of an array of metals in combination with Dy in this work enables a systematic study of this phenomenon. As a method for fast analysis of the sample composition, here we use laser-desorption ionization mass-spectrometry (LDI-MS). Although LDI-MS can hardly be applied for a quantitative estimation of an arbitrary fullerene mixture because of different ionization efficiencies, it is known that all M3N@C80 NCFs have almost identical oxidation potentials independent of the metals in the cluster,1d and hence similar ionization efficiency in the positive ion mode can be expected for all M3N@C80 molecules studied in this work. Thus, the analysis of the LDI-MS intensities should give if not the quantitative estimation of the relative yield in each mixed-metal system, but at least a trend in the values.
Fig. 2b and 3 show mass-spectra of the M3N@C80 fraction for each Dy–M system. As Dy and Lu have sufficiently different atomic masses, the peaks of all four DyxLu3−xN@C80 (x = 0–3) species can be easily distinguished in the mass spectrum (Fig. 2b). For other Dy–M systems, the relatively broad isotopic distribution and closer atomic masses result in overlapping spectral patterns, which still can be used for the estimation of the sample composition by matching with theoretical isotopic distributions for the involved species (Fig. 3 and S1†). The relative yield of a given DyxM3−xN@C80 compound in the Dy–M mixed system referred to the yield of Dy3N@C80 is then computed as a ratio of the total LDI-MS intensities of DyxM3−xN@C80 to Dy3N@C80 (Table S1†). The relative yield of Dy3N@C80 in each system is then considered to be equal to 1. The relative yields estimated from mass-spectral data are plotted in Fig. 4 along with the data on the Dy–Sc system studied in our group earlier. A strong deviation from the binominal statistical distribution (1:3:3:1) is found in all binary systems. Note that the following discussion is based on the relative yields of NCFs in mixed-metal systems. The absolute yield of Dy3N@C80 varies in different mixed-metal systems, and hence Fig. 4 and the data in Table S1† do not give an estimation of the total yields of NCFs. We do observe a general trend that the absolute yield of Dy3N@C80 in the mixed-metal systems with a small second metal (Sc or Lu) is considerably lower than the absolute yield of Dy3N@C80 in a mixed-metal system with bigger metals (such as Gd), but a precise estimation of the absolute yields is not possible due to the small amount of EMFs produced by arc-discharge synthesis and is not discussed further.
 | ||
| Fig. 3 LDI mass-spectra of the DyxM3−xN@C80-Ih (x = 0–3) fractions measured in positive ion mode: (a) M = Gd; (b) M = Er; and (c) M = Tm. Each panel contains overlaid calculated mass-spectra of individual DyxM3−xN@C80 molecules (bottom) and a comparison of the experimental and calculated mass-spectra of the whole fraction (top). For the sake of a clearer comparison experimental mass-spectra are shifted to higher masses by 0.3 a.m.u. | ||
 | ||
| Fig. 4 Relative yields of DyxM3−xN@C80-Ih NCFs (M = Sc, Lu, Er, Tm and Gd) in each mixed-metal system determined from LDI-MS intensities and normalized to the intensity of Dy3N@C80-Ih (note that the data are plotted in a logarithmic scale). Also shown are the yields expected for the binominal statistical distribution of metals in the nitride cluster (1:3:3:1). Numerical data used to prepare this plot can be found in Table S1 in the ESI.† Note that the absolute yield of Dy3N@C80 varies in different mixed-metal systems. | ||
A clear tendency of the relative yield variation with the size of the metal is seen in Fig. 4. Sc with the smallest ionic radius gives the highest relative yields of Dy2MN@C80, DyM2N@C80, and M3N@C80. The ratio of Dy2ScN and DySc2N is 0.5 instead of the expected 1, and the ratio of DySc2N and Sc3N is 1.5 instead of 3. Clearly, under identical conditions, Sc is much more predisposed to the formation of M3N@C80 than Dy. In the Dy–Lu and Dy–Er systems the relative yields of DyM2N@C80 and M3N@C80 are also much higher than the expected 3 to 1, respectively, but smaller than those for Sc analogs. The decrease of the relative yield of mixed-metal species is in line with the increase of the ionic radii from Sc (0.75 Å) to Lu (0.86 Å) to Er (0.89 Å). For Gd (0.94 Å), whose ionic radius is higher than that of Dy (0.91 Å), the situation is reversed, and the relative yields of Dy2GdN, DyGd2N, and Gd3N species decrease with the number of Gd ions in the cluster. Thus, a simple conclusion might be drawn based on these data: the metals with an ionic radius smaller than Dy are more favorable for NCF formation, whereas the metals with a bigger ionic radius are less predisposed to NCF formation. However, the data for the Dy–Tm system shows that the ionic radius is not the only factor affecting the yield of NCFs.
With an ionic radius of 0.88 Å, Tm3+ is slightly smaller than Er3+ and considerably smaller than Dy3+, but the distribution of Dy–Tm NCFs points to the higher preference for Dy versus Tm for NCF formation. The relative yields of Dy2TmN@C80 and DyTm2N@C80 are even smaller than those of Dy2GdN@C80 and DyGd2N@C80. We suggest that the reason for the anomalously low yield of Dy–Tm NCFs lies in the electronic properties of Tm, particularly in its third ionization potential. It is known that in monometallofullerenes some lanthanides are divalent, whereas others adopt a trivalent state. Shinohara et al. demonstrated that the valence states of lanthanides in EMFs correlate well with their third ionization potentials (IP3, see also Fig. S2†).24 The metals with a large IP3 exceeding 23 eV, namely Sm, Eu, Tm, and Yb, donate only two electrons to the fullerene cage and remain divalent, whereas IP3 values of La, Ce, Pr, Nd, Gd, Tb, Dy, Ho, Er, and Lu are below the threshold of 23 eV. These lanthanides are ionized by a hosting fullerene cage to their trivalent state. Furthermore, the lanthanides with an IP3 below 23 eV form nitride clusterfullerenes (in which the valence state of the metal is also trivalent), whereas clusterfullerenes of Sm, Eu, and Yb are not known. Tm is the only lanthanide able to adopt a variable valence state in EMFs, a divalent state in monometallofullerenes and a trivalent state in NCFs.25 Presumably, the high IP3 value of Tm still takes its toll in NCF formation by lowering the synthetic yields of Dy2TmN@C80, DyTm2N@C80, and Tm3N@C80.
As the yield of Dy–Tm NCFs is too low, further accumulation and separation of DyxM3−xN@C80 NCFs were only performed for M = Gd, Er, and Lu. DyxM3−xN@C80 NCFs with the same fullerene cage show very similar retention behavior, and their separation requires several steps of recycling HPLC. Fig. 5 shows plots of recycling chromatograms for all three mixtures obtained for the main DyxM3−xN@C80 fractions highlighted in Fig. 2 at the first step (Fig. S3–S8† show further separation details). The Dy–Gd system offers the most straightforward separation among the three. Clear resolution into four components (with Dy3N, Dy2GdN, DyGd2N, and Gd3N clusters) can be seen after 16 cycles. For the DyxEr3−xN@C80 mixture, a similar level of separation requires at least twice more cycles (Fig. 5 and S5†), which further complicates the separation not only because of the increased time demands, but also because of the overlap of the fastest fractions at the nth cycle with the slowest fractions at the (n−1)th cycle. Surprisingly, the DyxLu3−xN@C80 mixture proved to be the most difficult to separate despite the largest difference in the ionic radii of Dy3+ and Lu3+. Whereas the resolution of the Dy3N@C80 and Dy2LuN@C80 HPLC peaks is possible after 25 and 40 cycles, respectively (Fig. 5 and S7†), the retention times of Lu3N@C80 and DyLu2N@C80 are so close that their separation cannot be accomplished even after 70 cycles (Fig. S7 and 8†).
 | ||
| Fig. 5 Recycling HPLC chromatograms of DyxM3−xN@C80 mixtures (x = 0–3) for M = Lu, Er, and Gd (semipreparative BuckyPrep column, toluene as the eluent, 25 °C, flow rate: 1.0 mL min−1). Insets show an enlarged fragment of the chromatogram obtained at the 22nd cycle (Dy–Lu and Dy–Er system) or 16th cycle (Dy–Gd system). | ||
In summary, multistep recycling HPLC allowed the isolation of Dy2GdN@C80, DyGd2N@C80, DyEr2N@C80, and Dy2ErN@C80 in high compositional purity exceeding 90–95% based on mass-spectroscopic analysis (see Fig. S4, S6, and S8†). The retention behavior of Dy2LuN@C80, DyLu2N@C80 and Lu3N@C80 was found to be too similar to offer efficient separation without a substantial loss of the material. Dy2LuN@C80 could be separated to ca. 90% purity (the sample also contained ca. 10% DyLu2N@C80 and ca. 3% Lu3N@C80), whereas the separation of DyLu2N@C80 from Lu3N@C80 is hardly possible and was aborted when the relative content of the two NCFs in the sample was ca. 1:1.35. Note that earlier we have had similar difficulties with the separation of the HoxLu3−xN@C80 NCFs.15
Spectroscopic characterization
Since the absorption of UV and visible light by EMFs are dominated by the π → π* transition of the fullerene cage, UV-Vis absorption spectra of M3N@C80 NCFs are known to be very similar irrespective of the endohedral M3N cluster, unless the latter also includes Sc atoms, in which case the differences between spectra are more pronounced.1d,10g,k,16,23 Indeed, UV-Vis absorption spectra of the Dy–Gd and Dy–Er M3N@C80 families also follow this trend and have an almost identical appearance with absorptions near 325, 410, and 555 nm and 3–4 weak features in the long-wavelength range at 620–720 nm (Fig. 6). However, more detailed analysis of the lowest-energy part of the spectra shows a small but systematic variation of the peak energies. Upon going from Gd3N@C80 to Dy3N@C80 and then to Er3N@C80via corresponding mixed-metal counterparts (i.e. with a gradual decrease of the cluster size), absorption energies shift to shorter wavelengths (higher energies) from 708/675 nm in Gd3N to 695/665 nm in Er3N@C80 (Fig. 6, inset). Although the HOMO and LUMO of non-Sc M3N@C80-Ih NCFs are mainly localized on the fullerene cage, a small contribution of the metal cluster to the LUMO was also established,26 which is presumably responsible for the blue-shift of the lowest energy transitions with the decrease of the cluster size. | ||
| Fig. 6 Overlaid UV-Vis absorption spectra of isolated M3N@C80 NCFs measured in toluene solution at room temperature. The inset shows an expansion of the low-energy part; the spectra are offset for easier comparison. | ||
Single-crystal analysis and molecular structures
The molecular structures of DyEr2N@C80 and DyGd2N@C80 were established by single-crystal X-ray diffraction using synchrotron radiation at BESSY II of the Helmholtz-Zentrum Berlin.27 Single crystals suitable for diffraction analysis were obtained by co-crystallization of DyEr2N@C80 and DyGd2N@C80 with NiII(OEP) (OEP = octaethylporphyrin). The DyM2N@C80·NiII(OEP) fragments of DyGd2N@C80·NiII(OEP)·2(C6H6) and DyEr2N@C80·NiII(OEP)·2(C6H6) crystals are shown in Fig. 7. As in many other M3N@C80·NiII(OEP) co-crystals, both fullerene cages and NiII(OEP) molecules are well ordered with their nearest cage carbon–Ni contacts of 2.81–2.82 Å.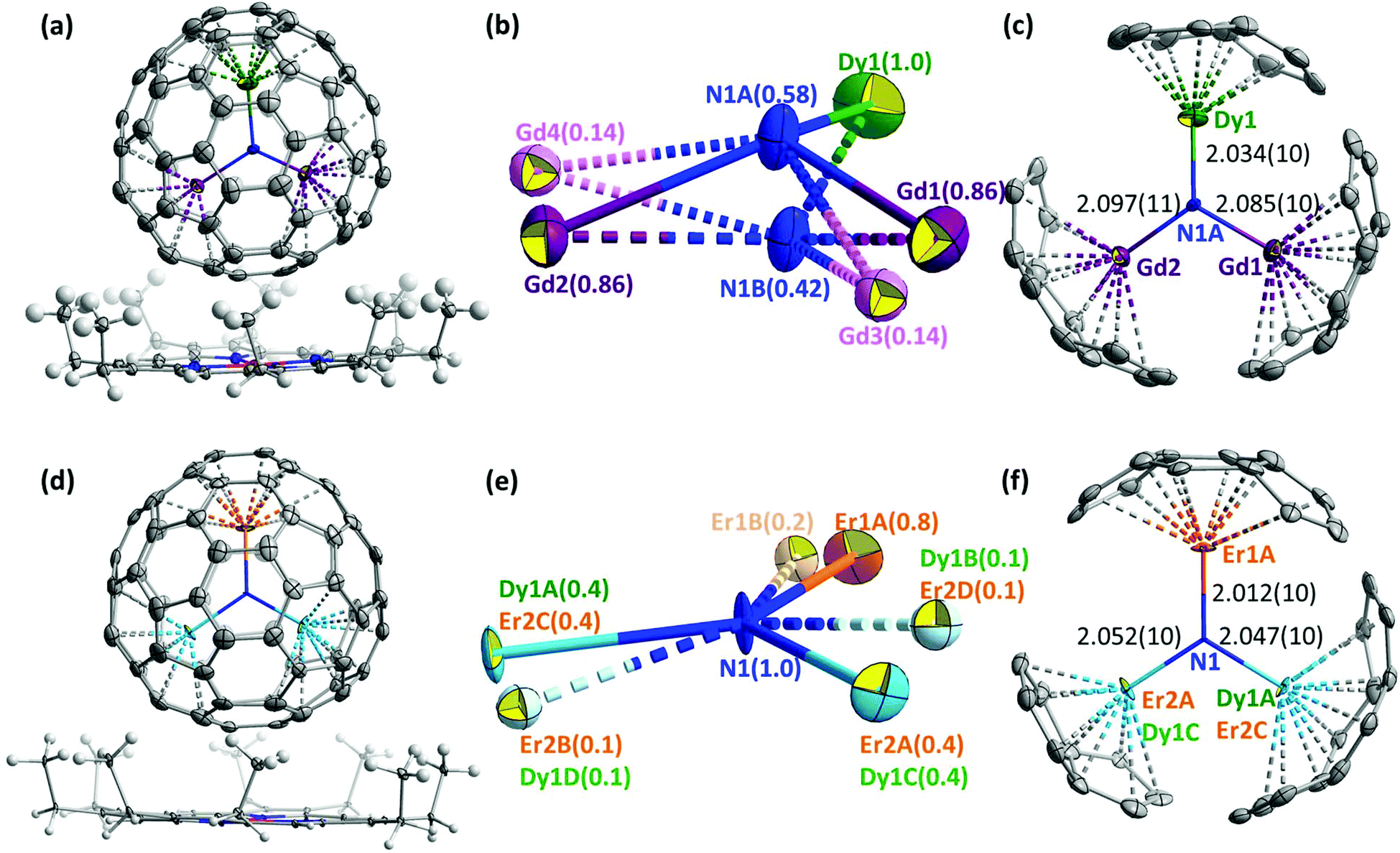 | ||
| Fig. 7 (a) Crystal structure of DyGd2N@C80·NiII(OEP)·2(C6H6). Solvent molecules are omitted for clarity and only the main site of the DyGd2N cluster is shown; (b) all sites of the DyGd2N cluster in the crystal; the site occupancies are noted in parentheses; (c) the main DyGd2N site and coordinated fragment of the fullerene cage; also shown are metal–nitrogen bond lengths (in Å). (d) Crystal structure of DyEr2N@C80·NiII(OEP)·2(C6H6). Solvent molecules are omitted for clarity and only site A of the DyEr2N cluster is shown. (e) All sites of the DyEr2N cluster in the crystal; the site occupancies are noted in parentheses and (f) the DyEr2N site A and coordinated cage carbons and metal–nitrogen bond lengths (in Å). The displacement parameters are shown at the 30% probability level. | ||
In the icosahedral C80-Ih cage the M3N cluster has many energetically equivalent positions, which result in the free rotation of the cluster inside the cage in solution and may lead to disorder in the crystalline phase; even stronger disorder may be expected for the mixed-metal NCFs due to the overlap of different metal positions in the cluster. The unique advantage of co-crystallization with NiII(OEP) for NCFs is that not only the fullerene cage, but also the endohedral cluster is often well ordered in the crystal.6b,10c,j The plane of the M3N cluster is usually perpendicular to the porphyrin plane of the NiII(OEP) molecule, two metals face the NiII(OEP)-coordinated part of the cage, and the third metal is oriented towards the opposite side of the fullerene cage (Fig. 7). In the MSc2N@C80·NiII(OEP) and M2ScN@C80·NiII(OEP) co-crystals, the position farthest from the porphyrin is always adopted by the larger lanthanide, whereas one or two Sc atoms are oriented closer to the NiII(OEP). Likewise, in the CeLu2N@C80·NiII(OEP)·2C7H8 crystal (CeLu2N@C80-Ih is the only non-Sc mixed-metal NCF studied by single-crystal X-ray diffraction before this work), the larger Ce atom is located further away from the NiII(OEP), and smaller Lu atoms are located closer to the porphyrin.12b We have recently studied the ordering of the endohedral cluster in Sc3N@C80 and YSc2N@C80 by NiII(OEP) theoretically and found that the ordering can be explained by simple electrostatic consideration based on the distribution of the electrostatic potential in the molecules.28 The importance of the electrostatic potential distribution for co-crystallization with NiII(OEP) has also been reported for some other empty and endohedral fullerenes.29 Furthermore, the study of YSc2N@C80 showed that different metals (Y and Sc in that case) cause slightly different distribution of electrostatic potentials outside the fullerene cage, which may lead to a further alignment of the mixed-metal clusters.
Although in the DyM2N@C80 NCFs studied in this work the difference of the ionic sizes of metals is not as pronounced as in the already reported mixed-metal NCFs, co-crystallization with NiII(OEP) still results in remarkable ordering of the nitride clusters. In DyGd2N@C80·NiII(OEP), the single Dy ion is fully ordered and is located remotely from the NiII(OEP), whereas Gd atoms are refined to two sites with occupancies of 0.86 and 0.14 (Fig. 7). Two sites with occupancies of 0.58 and 0.42 located at a distance of 0.91(2) Å from each other are refined for nitrogen. For the main site of the DyGd2N cluster shown in Fig. 7c, the bond lengths are 2.034(10), 2.085(10), and 2.097(11) Å for Dy1–N1A, Gd1–N1A, and Gd2–N1A, respectively, and the bond angles at nitrogen N1A are: ∠Dy1N1AGd1 = 116.8(5)°, ∠Dy1N1AGd2 = 118.0(5)°, and ∠Gd1N1AGd2 = 111.1(5)° with a sum of 345.9°. The geometry of the minor site of the DyGd2N cluster shown in Fig. S9b† has a small deviation from the major site geometry. Splitting of the nitrogen positions and deviation of the sum of ∠MNM’ angles from 360° indicate that the DyGd2N cluster inside the C80-Ih cage is pyramidal.
In DyEr2N@C80·NiII(OEP), two sites of DyEr2 are well recognized with occupancies of 0.8 and 0.2. The Er1 atom is located far from the NiII(OEP). The Er2 and Dy1 atoms are overlapped and face the porphyrin-coordinated site of the cage. Thus, in both DyGd2N@C80·NiII(OEP) and DyEr2N@C80·NiII(OEP) crystals, the position farthest from the porphyrin is occupied by the smaller lanthanide (Dy or Er), in contrast to the situation described earlier for MSc2N,10c,d,j,21b,30 M2ScN,10j,21c and CeLu2N12b clusters.
The nitrogen in the DyEr2N cluster is nearly located on the plane of DyEr2 with an out of plane displacement of 0.05(1) Å. The nitrogen ellipsoid is strongly elongated perpendicular to the DyEr2 plane. Due to the overlap of Dy1 and Er2, estimation of the Dy–N bond distance cannot be very accurate. The bond lengths are 2.012(10), 2.047(10), and 2.052(10) Å for Er1A–N1, Dy1A/Er2C–N1, and Er2A/Dy1C–N1, respectively. Thus, the average of Dy1–N and Er2–N bond lengths is longer than the Er1–N distance in agreement with the larger ionic radius of Dy. The bond angles at the nitrogen site are: ∠Dy1A/Er2CN1Er1A = 123.1(5)°, ∠Dy1A/Er2CN1Er2A/Dy1C = 115.1(5)°, and ∠Er1AN1Er2A/Dy1C = 121.6(5)°, with a sum of 360°.
Note that Dy/Gd or Dy/Er are crystallographically rather similar and it is hardly possible to distinguish them unambiguously. For DyGd2N@C80, our assignment shown in Fig. 7b is based on the higher electron density at the Dy1 site when compared to both Gd sites. Besides, the shorter Dy–N and longer Gd–N bond distances determined with this assignment (Fig. 7c) are in good agreement with the larger ionic radius of Gd3+ in comparison with that of Dy3+. Likewise, assignment of the Er1(A/B) site in DyEr2N@C80 is also based on the higher electron density at that position and on the reasonable distribution of the metal–nitrogen bond lengths following the expected shorter Er–N and longer Dy–N bonds (Fig. 7f). However, current experimental data do not allow us to exclude some overlap (in the range of 10%) between Gd and Dy positions in DyGd2N@C80 or a further overlap between Er1 and Dy in DyEr2N@C80.
The pronounced pyramidalization of the DyGd2N cluster and the nearly planar shape of the DyEr2N cluster are in line with the crystallographic studies of other M3N@C80 molecules.6b The shape of the nitride cluster in NCFs depends on the match between the size of the cluster and the cage size. The most stable form of the cluster is planar, and it is realized in those NCFs which offer sufficient inner space for the cluster. If the cage size is fixed as in the M3N@C80 series considered here, the increase of the metal size leads to the inner strain of the structure. The geometrical parameters of the M3N cluster have a certain flexibility, and the M–C and M–N bonds may become shorter in comparison with their optimal values, to maintain the planarity of the cluster. However, when the planarity of the cluster requires shortening of the M–N and M–C bonds beyond a certain threshold, the nitrogen atom is pushed out of the M3 plane and the cluster adopts a pyramidal shape instead.
Table 1 summarizes the Gd–N, Dy–N, Er–N, and Lu–N bond lengths in different M3N@C80 NCFs determined by single-crystal X-ray diffraction studies. The average M–N bond length in Lu3N@C80, Er3N@C80, and Dy3N@C80 is in the range of 2.04–2.05 Å, although the ionic radii of Dy3+ and Lu3+ are different by 0.05 Å. The Er3N and Lu3N clusters are nearly planar, whereas for Dy3N the experimental data allow two alternative interpretations: one suggests a planar cluster and a single nitrogen site, in which the thermal ellipsoid is strongly elongated in the out-of-plane direction.31 A more recent interpretation of the same diffraction data suggests splitting of the nitrogen position between two sites leading to a pyramidal Dy3N cluster.6b In Gd3N@C80, a further increase of the metal size results in an average Gd–N bond length of 2.08 Å, and the Gd3N cluster has to adopt a pyramidal configuration in M3N@C80.6a
| Gd–N | Dy–N | Er–N | Lu–N | |
|---|---|---|---|---|
| a Shannon radii for the lanthanide 3+ ions in a six-coordinate environment from the study in ref. 22. b “av” stands for an averaged M–N bond length over two or three M–N bonds. c The bond length is strongly underestimated because of the disorder in the cluster and near overlap with the Sc site. d The value is likely to be an average of the Dy–N and Er–N bond lengths. | ||||
| R(M3+)a | 0.94 | 0.91 | 0.89 | 0.86 |
| X-ray diffraction | ||||
| M 3 N | Gd 3 N (ref. 6a) | Dy 3 N (ref. 6b and 31) | Er 3 N (ref. 6b) | Lu 3 N |
| 2.038(8) | 2.004(8) | 2.046(3) | 2.050 (av) (ref. 34) | |
| 2.085(4) | 2.068(6) | 2.059(3) | 2.032 (av) (ref. 35) | |
| 2.117(5) | 2.055(7) | 2.028(3) | 2.041 (av) (ref. 3c) | |
| 2.080 (av) | 2.042 (av) | 2.044 (av) | ||
| MSc 2 N | GdSc 2 N (ref. 10j) | DySc 2 N (ref. 21b) | ErSc 2 N (ref. 30) | |
| 2.149(10) | 2.096(6) | 2.089(9) | ||
| M 2 ScN | Gd 2 ScN (ref. 10j) | Dy 2 ScN (ref. 21c) | ||
| 2.072(3) | 2.078(6) | |||
| 2.102(3) | 1.965(6) (overlap Sc) | |||
| MM′ 2 N | DyGd 2 N | DyGd 2 N | DyEr 2 N | CeLu 2 N (ref. 12b) |
| 2.085(10) | 2.034(10) | 2.012(10) | 2.016(7) | |
| 2.097(11) | DyEr 2 N | 2.047(10)d | 2.061(8) | |
| 2.047(10)d | 2.052(10)d | 2.038 (av) | ||
| 2.052(10)d | ||||
| DFT calculations | ||||
| 2.090 (Gd3N) | 2.063 (Dy3N) | 2.055 (Er3N) | 2.051 (Lu3N) | |
| 2.090 (DyGd2N) | 2.072 (Dy2LaN) | 2.049 (DyEr2N) | 2.036 (DyLu2N) | |
| 2.089 (Dy2GdN) | 2.063 (Dy2GdN) | 2.044 (Dy2ErN) | 2.020 (Dy2LuN) | |
| 2.064 (Dy2YN) | ||||
| 2.064 (Dy2ErN) | ||||
| 2.073 (Dy2LuN) | ||||
| 2.106 (Dy2ScN) | ||||
| 2.076 (DyLa2N) | ||||
| 2.064 (DyGd2N) | ||||
| 2.067 (DyY2N) | ||||
| 2.070 (DyEr2N) | ||||
| 2.090 (DyLu2N) | ||||
| 2.156 (DySc2N) | ||||
Note that Dy–N bonds of 2.0–2.1 Å determined for Dy-containing NCFs are much shorter than typical Dy–N bond lengths. Fig. 8 shows the distribution of 5369 Dy–N bond lengths in molecular Dy compounds retrieved from the Cambridge Structural Database (CSD). The shortest non-fullerene Dy–N bond reported to date is 2.14 Å long,32 whereas in a majority of such compounds the Dy–N bond lengths are in the range of 2.3–2.7 Å.33 The short Dy–N bond length is an indication of significant strain experienced by lanthanide M3N clusters encapsulated inside the C80 fullerene cage.
 | ||
| Fig. 8 Distribution of 5369 Dy–N bond lengths in molecular Dy compounds deposited in the Cambridge Structural Database. The range of Dy–N bond lengths in Dy-based M3N@C80 NCFs is highlighted in light red. The inset shows an expansion of the data in the short distance range; the abscissa scale for the inset and the main figure is the same. | ||
Mixed-metal nitride clusters allow redistribution of the bond lengths and hence a partial release of the strain. Geometrical parameters of MSc2N and M2ScN clusters (M = Gd, Dy, Er) clearly illustrate this effect. M–N bonds in MSc2N@C80 NCFs are considerably longer than the M–N bonds in the corresponding M3N@C80 molecules (Table 1). In M2ScN@C80 (M = Gd, Dy) the M–N bond lengths become shorter than those in MSc2N clusters and approach the values found in M3N@C80 molecules. Yet, both MSc2N and M2ScN clusters are planar for all lanthanides studied so far.10c,d,j,21b,c,30 In DyGd2N@C80, the length of the Gd–N bonds is comparable to those in Gd3N, and the Dy–N bond is even somewhat shorter than that in Dy3N@C80. Thus, both Gd–N and Dy–N bonds reached their shortest threshold values, and the DyGd2N cluster has to become pyramidal to release the strain further. For DyEr2N the situation is less obvious because of the overlapped Dy/Er positions, which make the bond length estimation unreliable. For the well-modeled Er1 atom in DyEr2N@C80, the Er1A–N bond is noticeably shorter than that in Er3N@C80, which indicates that the Dy–N bond can release its strain by elongation at the expense of the shortening of the Er–N bonds.
As a measure of the pyramidalization of the M3N cluster it is convenient to use the displacement of the nitrogen atom from the metal plane (Table 2). For instance, in Gd3N@C80 and Tb3N@C80 nitrogen atoms are displaced above/below the M3 plane by 0.522/0.463 and 0.453/0.405 Å, respectively (the nitrogen atom in these molecules is split between two sites, hence two different values).3d,6a In DyGd2N@C80 this displacement distance is 0.456/0.452 Å, somewhat smaller than that in Gd3N but larger than that in the Tb3N cluster. If the split-nitrogen model of the Dy3N@C80 data is adopted, the out-of-plane displacement of nitrogen in Dy3N would be 0.26 Å (here the value is calculated as a half of the distance between two nitrogen sites). I.e., pyramidalization is decreased considerably from Gd3N over Tb3N to Dy3N.
| M3N@C80-Iha | X-ray, Å | DFT, Å | γ M3N, cm−1 | l z , Å |
|---|---|---|---|---|
| a The molecules are listed in the descending order of their DFT-predicted pyramidalization (displacement of nitrogen from the metal plane). b l z is computed as a square root of l2 in eqn (1), in which ν and μ are substituted with the DFT vibrational frequency of the γM3N mode and atomic mass of nitrogen, respectively. c Single nitrogen site. d Nitrogen is split between two sites and pyramidalization is computed as a half of the distance between two nitrogen sites. e In the pyrrolidine monoadduct. | ||||
| DyLa2N@C80 | — | 0.820 | ||
| Dy2LaN@C80 | — | 0.593 | 159 | 0.096 |
| Gd3N@C80 | 0.522(8)/0.463(14) (ref. 6a) | 0.468 | 143 | 0.104 |
| DyGd2N@C80 | 0.456(19)/0.452(12) | 0.396 | ||
| Tb3N@C80 | 0.453(4)/0.405(7) (ref. 3d) | 0.365 | 128 | 0.114 |
| CeLu2N@C80 | 0.349(9)/0.324(9) (ref. 12b) | 0.352 | 114 | 0.125 |
| Dy2GdN@C80 | — | 0.311 | ||
| Dy3N@C80 | 0.068(11) (ref. 31)c | 0.203 | 72 | 0.187 |
| 0.26 (ref. 6b)d | ||||
| Dy2YN@C80 | — | 0.125 | 86 | 0.159 |
| DyY2N@C80 | — | 0.087 | ||
| Dy2ErN@C80 | — | 0.086 | ||
| Ho3N@C80 | 0.058(9)c (ref. 6b) | 0.068 | 51 | 0.259 |
| 0.090d | ||||
| Y3N@C80 | 0.129(6) (ref. 38)e | 0.059 | 74 | 0.183 |
| DyEr2N@C80 | 0.050(11) | 0.057 | ||
| Dy2LuN@C80 | — | 0.046 | ||
| Er3N@C80 | 0.025(3)c (ref. 6b) | 0.020 | 97 | 0.143 |
| 0.064d | ||||
| Dy2ScN@C80 | 0.116(8) (ref. 21c) | 0.020 | 133 | 0.110 |
| DyLu2N@C80 | — | 0.015 | 93 | 0.149 |
| DySc2N@C80 | 0.003(12) (ref. 21b) | 0.014 | 211 | 0.079 |
| Lu3N@C80 | 0.0953(1) (ref. 34) | 0.007 | 145 | 0.103 |
| 0.01(2) (ref. 35) | ||||
| 0.001(8) (ref. 3c) | ||||
For a deeper understanding of these phenomena, we also studied the pyramidalization of the nitride cluster in a series of lanthanide and mixed-lanthanide M3N@C80 molecules theoretically. DFT calculations were performed at the PBE-D level with a plane-wave basis set and corresponding projector augmented-wave potentials, treating 4f-electrons as a part of the core as implemented in the VASP 5.0 package.36 The computed out-of-plane displacements of nitrogen for Gd3N@C80, DyGd2N@C80, and Tb3N@C80 are 0.468, 0.396, and 0.365 Å respectively. Notably, for the midsize cluster in Dy3N@C80 calculations predict the value of 0.203 Å, but already in DyEr2N@C80 the cluster is nearly planar, with the nitrogen displacement amounting to only 0.057 Å (compare to the experimental result of 0.05(1) Å). Similarly, for Dy2GdN@C80 and Dy2ErN@C80 the calculation predicts a pyramidal cluster with a displacement parameter of 0.311 and 0.086 Å, respectively. Finally, nearly planar cluster geometries are also found for Er3N@C80 (0.020 Å) and Lu3N@C80 (0.007 Å). These computational results are in agreement with the trends predicted in other theoretical studies of M3N@C80 molecules.13,37 Overall, DFT calculations support well the experimental observations of the strong cluster pyramidalization for Gd3N and DyGd2N, a moderately pyramidal cluster geometry in Dy3N@C80, and a nearly planar cluster in DyEr2N@C80. However, as we show below, a direct comparison of X-ray data and results of DFT calculations may be misleading if the real physical quantities behind diffraction data are not considered correctly.
Cluster pyramidalization and vibrational displacements
Structural analysis of NCFs such as that given in the previous section or in numerous publications on this subject (see ref. 6b for a systematic analysis of M3N@C80 structures) implicitly assumes that X-ray diffraction provides the ground state geometry of the molecule. Strictly speaking, this is not entirely correct. Thermal motion of atoms and population of excited vibrational levels may significantly distort the observed structure, especially if the potential energy surface is rather shallow. Since these effects may affect the interpretation of the diffraction data substantially, we decided to analyze in depth how vibrational degrees of freedom may influence the structure of the nitride cluster.Harmonic approximation
DFT-based vibrational analysis of M3N@C80 NCF molecules (M = Y, Gd–Er, Table 2) shows that vibrations of the nitrogen atom in these molecules are well separated from vibrational displacements of the other atoms. Three degrees of freedom of the nitrogen atom produce two vibrational modes of the M3N cluster. The displacement of the nitrogen perpendicular to the plane of metals forms the cluster deformation mode (γM3N), whereas the in-plane motion constitutes the two-fold degenerate antisymmetric M–N stretching mode (νasM–N). The νasM–N mode occurs at frequencies of 700–750 cm−1 and is easily detectable in IR spectra due to its medium-strong intensity.31,39 The out-of-plane oscillation frequency predicted in the harmonic approximation depends strongly on the metal size and ranges from 256 cm−1 in Sc3N@C80 (exp. value is 236 cm−1, ref. 40) over 145 cm−1 in Lu3N@C80 and down to 51 cm−1 in Ho3N@C80, for which DFT predicts the lowest γM3N frequency in the whole M3N@C80 series (Table 2). The frequency variation from Sc to Lu and Ho reflects the trend of the M3N cluster to flatten its potential energy surface along the pyramidalization coordinate with the increase of the metal size. When the metal size increases beyond Ho, the cluster adopts a pyramidal shape with a double-well energy potential, and the calculated γM3N frequency increases again from 72 cm−1 in Dy3N@C80 to 128 cm−1 in Tb3N@C80 and 143 cm−1 in Gd3N@C80.The large difference of the γM3N and νasM–N vibrational frequencies results in a more pronounced out-of-plane displacement of the nitrogen atom in M3N@C80, leading to an elongation of the nitrogen thermal ellipsoid in the out-of-plane direction. In the harmonic approximation, which implies that the energy is a quadratic function of atomic displacements, the thermal displacements can be calculated as the root-mean-square vibrational amplitudes of a quantum harmonic oscillator:41
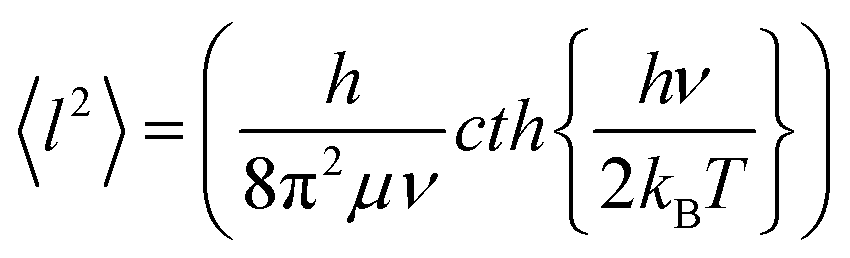 | (1) |
To summarize, rather large vibrational displacements of the nitrogen atom may potentially lead to erroneous conclusions on the perceived cluster geometry. When the cluster is on the edge between planar and pyramidal shapes, the potential energy surface is flat, and the vibrational frequency of the out-of-plane deformation is low. This defines the large vibrational amplitudes of ca. 0.2 Å and partial localization of the probability density above and below the metal plane. The thermal ellipsoid of nitrogen should then be strongly elongated, and the question whether nitrogen should be treated as a single site (“planar cluster”) or split into two sites (“pyramidal cluster”) may become very ambiguous.
Beyond harmonic approximation
Simple harmonic approximation already points to the potentially complicated contribution of vibrational motion to the experimental molecular structure. But flat energy potentials and large vibrational amplitudes also indicate that the harmonic approximation may not be adequate by itself. A more reliable description of the system requires explicit consideration of the energy potential along the pyramidalization coordinate. With the use of DFT, we obtained potentials for five M3N@C80 molecules by optimizing their structures with the nitrogen atom fixed at different distances above and below the plane of metal atoms (Fig. 9). | ||
| Fig. 9 (a) Molecular structure of M3N@C80 with a pyramidal cluster and definition of the axis z used in the analysis; an equivalent position of the nitrogen atom below the plane of metal atoms is semitransparent. (b) Probability density for finding the nitrogen atom at a certain position along the z axis in different M3N@C80 molecules computed for T = 100 K as the temperature often used in X-ray diffraction studies of EMFs. | ||
For the strongly pyramidal Gd3N cluster in Gd3N@C80, the energy profile (E(z)) has a classical double-well potential shape with the minima for the nitrogen atom at 0.45 Å above and below the Gd3 plane (Fig. 10). Quite common in many fields of analytical physics, the double-well potentials are well developed and understood since the studies of pyramidal inversion in NH3 and related molecules.42 The E(z) maximum at z = 0 Å, i.e. the pyramidal inversion barrier, is only 177 cm−1 in Gd3N@C80. The symmetrized ab initio potential profile (dots in Fig. 10) can be described well using an analytical function E(z) = Az4 − Bz2 (solid lines in Fig. 10). After fitting A and B parameters the Schrödinger equation is solved numerically using this analytical potential and a particle mass equal to that of a nitrogen atom (14 Da), which gives a quasi-degenerate ground state for Gd3N@C80 with the symmetric and antisymmetric state energies of 71.2 and 73.2 cm−1, respectively, and an inversion splitting of 2 cm−1. The probability to find the nitrogen atom at a certain z-position (defined as a square of the wavefunction |Ψn2| for the nth state) has two maxima closely coinciding with the potential minima. Thus, the pyramidal shape of the Gd3N cluster is well defined since the zeroth (|0〉) and first (|1〉) vibrational levels lie within the potential wells. However, already the second energy level at 174.5 cm−1 almost coincides with the inversion barrier, and |Ψ22| has a broad maximum at z = 0. At higher energy, the structure of the vibrational levels resembles that of a system with a harmonic potential, although the levels are not equidistant.
 | ||
| Fig. 10 Potential energy profiles (top row) and probability densities at different temperatures (bottom row) for the pyramidalization of the M3N cluster in M3N@C80 molecules with different cluster compositions and sizes. In potential energy profiles dots are results of DFT calculations, solid colored lines are fits of DFT results with the Az4 − Bz2 function, red dashed vertical lines mark positions of the minima of the potential, dashed horizontal lines are vibrational levels obtained by the solution of the Schrödinger equation and gray solid lines are probability densities for each vibrational level (i.e. |Ψn2| function for the nth level). For the sake of comparison probability densities for different M3N@C80 molecules in the bottom row are shown on one scale. | ||
The experimentally observable positions of the nitrogen (e.g., from diffraction data) correspond to the maxima of the total probability density, which is temperature dependent because of the variation of thermal populations of the energy levels. The probability density at a temperature T can be obtained as a sum 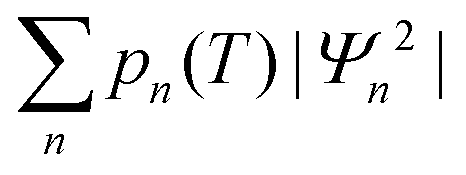 , where pn(T) are weight factors determined from Boltzmann distribution,
, where pn(T) are weight factors determined from Boltzmann distribution,  . For Gd3N@C80 we find a well-behaving double maximum picture (Fig. 9b and 10) with a slight temperature variation of the maximum position and a considerable broadening of the distribution. Yet two maxima are clearly seen up to room temperature ensuring that X-ray diffraction data give a correct description of the pyramidal Gd3N cluster.
. For Gd3N@C80 we find a well-behaving double maximum picture (Fig. 9b and 10) with a slight temperature variation of the maximum position and a considerable broadening of the distribution. Yet two maxima are clearly seen up to room temperature ensuring that X-ray diffraction data give a correct description of the pyramidal Gd3N cluster.
A double-well energy profile with an inversion barrier of only 69 cm−1 is found for DyGd2N@C80. The decrease of the barrier caused by the reduced size of the M3N cluster leads to a considerable change in the structure of the vibrational levels in comparison with those in Gd3N@C80. The ground vibrational level of DyGd2N@C80 is found at 45.3 cm−1, which lies within the double-well. However, the |Ψ02| function has a complex shape with two broad and strongly overlapping peaks, whose maxima are shifted from the positions of the potential minima to much smaller z values. Thus, if X-ray diffraction would be performed at helium temperature, an apparent pyramidalization deduced from diffraction data would be underestimated or even missed because the probability minimum at z = 0 is rather shallow. The first excited vibrational level of DyGd2N@C80 is located at 63.7 cm−1. Note that the quasi-degeneracy of the |0〉 and |1〉 levels observed in Gd3N@C80 is lifted here. The |Ψ12| distribution has a more common shape with two well-defined maxima near the positions corresponding to the minima of the energy profile. Considerable alteration of the shape of the |Ψ02| and |Ψ12| functions and a comparably small energy difference between the levels lead to pronounced changes in the probability density even after a modest temperature increase from 5 to 40 K: the maxima become sharper and shift further away from each other, whereas the minimum between them becomes more pronounced. Thus, the temperature increase will result in rather counterintuitive changes in the diffraction data as positions of nitrogen will be better defined at a higher temperature than near 0 K. The second and higher excited vibrational levels of DyGd2N@C80 resemble the levels of a system with a single energy minimum. A temperature increase above 40 K leads to a gradual shift of the probability maxima to higher |z| values and broadening of the probability density.
The decrease of the cluster size to that of Dy3N results in a conceptual change of the form of the potential energy profile. Although its double-well nature with the minima near |z| = 0.2 Å is formally preserved, the inversion barrier is reduced to a small value of 1.5 cm−1, which is already comparable to the numerical precision of the computational approach. The ground vibrational level is found at 25.3 cm−1, considerably higher than the inversion barrier, and |Ψ02| has a single maximum at z = 0. Essentially the structure of vibrational levels in Dy3N@C80 is that of a system with a single well potential. Substantial anharmonicity of the potential, which has ∼z4 dependence, leads to a non-equidistant spacing of the levels. The probability density for the nitrogen position in Dy3N@C80 has a single-peak shape at all temperatures. The peak is rather broad and gets flattened at higher temperatures. Thus, one can expect that the diffraction data for Dy3N should show a single nitrogen position with an anomalously elongated thermal ellipsoid as was indeed reported in ref. 31.
A situation similar to Dy3N is also predicted for the DyEr2N and Lu3N clusters. The pyramidalization of the cluster and inversion barrier in DyEr2N are even smaller than those in Dy3N. In Lu3N@C80, a single energy minimum is found. The potential is not harmonic even for the relatively small Lu3N cluster and for both molecules it is well described by a quartic function with only a small contribution of z2. A decrease of the cluster size results in a narrowing of the potential well and an increase of the distance between vibrational levels. Accordingly, the probability density with the maximum at z = 0 becomes narrower with the decrease of the cluster size. Thus, diffraction data should give a planar structure of the M3N cluster with a gradual decrease of the elongation of the nitrogen ellipsoid from Dy3N over DyEr2N to Lu3N.
Fig. 11 compares probability densities, obtained from computational studies of DyGd2N@C80 and DyEr2N@C80, to the difference in the electron density near the position of the nitrogen, obtained by subtracting the density reconstructed from the refined positions of all atoms except nitrogen from the experimental density determined by X-ray diffraction. Good agreement between experiment and theory can be observed. For DyGd2N@C80, an elongated density profile with two well-defined maxima can be distinguished and the maxima are close to the maxima in the probability density function. For DyEr2N@C80, a single density lobe is seen and its spatial extension along the z-axis corresponds to the peak shape of the probability density.
 | ||
| Fig. 11 Difference electron density maps for the nitrogen atom in DyEr2N@C80 (top) and DyGd2N@C80 (bottom). The calculated density was built using refined positions and structure factors of all atoms except nitrogen. 2D maps of the density are overlaid with the probability density function for the nitrogen atom at the z-coordinate computed for 100 K (the same as in Fig. 9). For the sake of comparison the color scale and the scale for probability density are the same for both molecules. | ||
To summarize, the analysis of the potential energy profile along the cluster pyramidalization coordinate shows that precise structure determination of the M3N cluster from diffraction data may be complicated. Calculations show a gradual development of the double-well potential with increasing metal size, which means that the pyramidalization of the M3N cluster should increase gradually as long as the metal size is larger than that of Lu. However, if the inversion barrier is below the lowest vibrational level, the probability density has a single maximum (Fig. 9b and 10). Diffraction methods (which do not restore the shape of the potential but determine high-probability atomic positions) should then give the planar shape of the cluster even when the energy profile has two minima. At the same time, the thermal ellipsoid of nitrogen should be strongly elongated in the direction perpendicular to the metal plane. Description with single or split nitrogen positions may then give similar R-factors. A true pyramidal shape of the cluster can be obtained when at least one vibrational level of the system lies below the inversion barrier. But even in this case, a mismatch between the shape of the potential and probability density function, i.e. when the maxima in the latter do not correspond to the minima in the former, can be rather strong, as found here for the DyGd2N cluster, and pyramidalization deduced from diffraction data should exhibit considerable temperature dependence. Only when two or more vibrational levels are below the inversion barrier, as found for Gd3N@C80, a stable pyramidal shape of the cluster with moderate temperature dependence can be observed by X-ray diffraction. Needless to say, additional disorder of the fullerene cage and metal positions will further complicate the situation. Thus, the molecular structure elucidation of endohedral fullerenes with labile endohedral species at a single temperature may give ambiguous results. Variable temperature studies with careful analysis of thermal motion are needed to address this problem.43
In the end we would like to note that parameters of the double-well potentials found in this work for M3N@C80 molecules with large endohedral clusters may be of use for design of the sources of terahertz radiation or even masers. The very first two-state maser system designed back in 195444 utilized ammonia gas and operated at a frequency of 24.0 GHz corresponding to an inversion splitting of 0.8 cm−1 in an NH3 molecule. The inversion barrier of 177 cm−1 in Gd3N@C80 is almost an order of magnitude smaller than the insertion barrier of 2020 cm−1 in NH3, but the vibration effective mass of 14 Da is an order of magnitude greater in Gd3N@C80 against 1 Da for ammonia. Thus, the transmission coefficient for the quantum tunneling defined as  in the Wentzel–Kramers–Brillouin approximation is of the same order for both molecules and hence similar quantum dynamics can be expected for them. Without going into deep detail at this moment, we just highlight that the Gd3N@C80 system may be driven and modulated by an external electric field in the same way and fashion as is possible for the ammonia system and hence Gd3N@C80 is a prospective candidate for a two-state maser system with an operating frequency of 60.0 GHz (inversion splitting: 2 cm−1). A combination of other lanthanides can be used to tune the shape of the double-well potential and hence the inversion splitting and operating frequency. Differently from the ammonia gas, in the case of the fullerene crystal the M3N units are encapsulated in carbon cages and hence are well-separated from each other and isolated from the environment. Thus, the M3N-based maser combines the simplicity of the ammonia maser (two-state system; no need for pumping as the energy split is small; a lack of spontaneous emission) with higher efficiency in amplification and higher photon densities due to the solid state density of the M3N units in the crystal.45
in the Wentzel–Kramers–Brillouin approximation is of the same order for both molecules and hence similar quantum dynamics can be expected for them. Without going into deep detail at this moment, we just highlight that the Gd3N@C80 system may be driven and modulated by an external electric field in the same way and fashion as is possible for the ammonia system and hence Gd3N@C80 is a prospective candidate for a two-state maser system with an operating frequency of 60.0 GHz (inversion splitting: 2 cm−1). A combination of other lanthanides can be used to tune the shape of the double-well potential and hence the inversion splitting and operating frequency. Differently from the ammonia gas, in the case of the fullerene crystal the M3N units are encapsulated in carbon cages and hence are well-separated from each other and isolated from the environment. Thus, the M3N-based maser combines the simplicity of the ammonia maser (two-state system; no need for pumping as the energy split is small; a lack of spontaneous emission) with higher efficiency in amplification and higher photon densities due to the solid state density of the M3N units in the crystal.45
Conclusions
In this work we have undertaken a systematic exploration of various Dy-lanthanide combinations in the synthesis of mixed-metal nitride clusterfullerenes. A variation of the size of the metal combined with Dy offered a possibility to gradually tune the size of the nitride cluster and study the influence of this factor on the synthesis and structure. The ionic radius of the metal employed in the synthesis is shown to be the main factor affecting the relative yield of the majority of DyxM3−xN@C80 species. Yet, the electronic properties of metals such as the third ionization potential, also play an important role and may cause completely unexpected distribution of products, such as that demonstrated in this work for the Dy–Tm system.A single-crystal X-ray diffraction study of the molecular structures of DyGd2N@C80 and DyEr2N@C80 along with DFT computation for a broad range of DyxM3−xN@C80 molecules offered a possibility to analyze how the variation of the size of the nitride cluster affects its shape. DFT calculations showed that a gradual increase of the metal size leads to the variation of the cluster shape from a planar triangle when the cluster is relatively small to a trigonal pyramid with gradually increasing height when the metal size increases. For pyramidal clusters the energy profile along the pyramidalization coordinate has a double-well shape. At the same time, calculations show that the inversion barrier for the pyramidal inversion in the M3N cluster is quite small. This situation results in a fundamental problem for structure determination rooted in the quantum nature of molecular vibrations: if the energy of the zeroth vibrational level is higher than the inversion barrier, the apparent structure of the pyramidal M3N cluster as perceived from diffraction data will be planar. Particularly interesting is the situation when the inversion barrier is comparable to the lowest-energy vibrational levels. In this case, quantum effects may reveal themselves as the strong variation of the probability distribution with temperature and potentially may be detected by variable-temperature X-ray diffraction experiments or spectroscopic studies. Finally, we propose that the double-well potential and inversion splitting of the levels for larger M3N clusters have a number of parameters which make them prospective candidates for terahertz light applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from the European Union's Horizon 2020 research and innovation programme, European Research Council (grant agreement No 648295 to A. A. P.) and Marie Skłodowska-Curie action (grant agreement No. 748635 to S. M. A.) and the Deutsche Forschungsgemeinschaft (grants PO 1602/4-1 and PO 1602/5-1 to A. A. P.). Diffraction data have been collected on BL14.3 at the BESSY II electron storage ring operated by the Helmholtz-Zentrum Berlin; we would particularly like to acknowledge the help and support of Manfred Weiss and his group members during the experiments at BESSY II. Computational resources were provided by the Center for High Performance Computing at the TU Dresden. We appreciate the technical support with computational resources in IFW Dresden by Ulrike Nitzsche. Sandra Schiemenz is acknowledged for the help with spectroscopic measurements.References
- (a) J. Zhang, S. Stevenson and H. C. Dorn, Acc. Chem. Res., 2013, 46, 1548 CrossRef CAS PubMed; (b) L. Dunsch and S. Yang, Small, 2007, 3, 1298 CrossRef CAS PubMed; (c) M. N. Chaur, F. Melin, A. L. Ortiz and L. Echegoyen, Angew. Chem., Int. Ed., 2009, 48, 7514 CrossRef CAS PubMed; (d) A. A. Popov, S. Yang and L. Dunsch, Chem. Rev., 2013, 113, 5989 CrossRef CAS PubMed; (e) S. Yang, T. Wei and F. Jin, Chem. Soc. Rev., 2017, 46, 5005 RSC.
- S. Stevenson, G. Rice, T. Glass, K. Harich, F. Cromer, M. R. Jordan, J. Craft, E. Hadju, R. Bible, M. M. Olmstead, K. Maitra and A. J. Fisher, et al. , Nature, 1999, 401, 55 CrossRef CAS.
- (a) S. F. Yang and L. Dunsch, J. Phys. Chem. B, 2005, 109, 12320 CrossRef CAS PubMed; (b) M. Krause, J. Wong and L. Dunsch, Chem. – Eur. J., 2005, 11, 706 CrossRef CAS PubMed; (c) W. Shen, L. B. Bao, S. Hu, X. Gao, Y. Xie, X. Gao, W. Huang and X. Lu, Chem. – Eur. J., 2018, 24, 16692 CrossRef CAS PubMed; (d) T. M. Zuo, C. M. Beavers, J. C. Duchamp, A. Campbell, H. C. Dorn, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2007, 129, 2035 CrossRef CAS PubMed; (e) L. Dunsch, M. Krause, J. Noack and P. Georgi, J. Phys. Chem. Solids, 2004, 65, 309 CrossRef CAS; (f) W. Fu, L. Xu, H. Azurmendi, J. Ge, T. Fuhrer, T. Zuo, J. Reid, C. Shu, K. Harich and H. C. Dorn, J. Am. Chem. Soc., 2009, 131, 11762 CrossRef CAS PubMed; (g) M. N. Chaur, A. J. Athans and L. Echegoyen, Tetrahedron, 2008, 64, 11387 CrossRef CAS.
- (a) A. A. Popov and L. Dunsch, J. Am. Chem. Soc., 2007, 129, 11835 CrossRef CAS PubMed; (b) A. Rodriguez-Fortea, N. Alegret, A. L. Balch and J. M. Poblet, Nat. Chem., 2010, 2, 955 CrossRef CAS PubMed; (c) K. Nakao, N. Kurita and M. Fujita, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 11415 CrossRef CAS PubMed.
- (a) F. Melin, M. N. Chaur, S. Engmann, B. Elliott, A. Kumbhar, A. J. Athans and L. Echegoyen, Angew. Chem., Int. Ed., 2007, 46, 9032 CrossRef CAS PubMed; (b) M. N. Chaur, F. Melin, B. Elliott, A. Kumbhar, A. J. Athans and L. Echegoyen, Chem. – Eur. J., 2008, 14, 4594 CrossRef CAS PubMed; (c) M. N. Chaur, F. Melin, J. Ashby, A. Kumbhar, A. M. Rao and L. Echegoyen, Chem. – Eur. J., 2008, 14, 8213 CrossRef CAS PubMed.
- (a) S. Stevenson, J. P. Phillips, J. E. Reid, M. M. Olmstead, S. P. Rath and A. L. Balch, Chem. Commun., 2004, 2814 RSC; (b) M. M. Olmstead, T. Zuo, H. C. Dorn, T. Li and A. L. Balch, Inorg. Chim. Acta, 2017, 468, 321 CrossRef CAS.
- N. Chen, L. Z. Fan, K. Tan, Y. Q. Wu, C. Y. Shu, X. Lu and C. R. Wang, J. Phys. Chem. C, 2007, 111, 11823 CrossRef CAS.
- S. Yang, C. Chen, A. Popov, W. Zhang, F. Liu and L. Dunsch, Chem. Commun., 2009, 6391 RSC.
- T. Wei, S. Wang, X. Lu, Y. Tan, J. Huang, F. Liu, Q. Li, S. Xie and S. Yang, J. Am. Chem. Soc., 2016, 138, 207 CrossRef CAS PubMed.
- (a) N. Chen, E. Y. Zhang and C. R. Wang, J. Phys. Chem. B, 2006, 110, 13322 CrossRef CAS PubMed; (b) Y. Zhang, K. B. Ghiassi, Q. Deng, N. A. Samoylova, M. M. Olmstead, A. L. Balch and A. A. Popov, Angew. Chem., Int. Ed., 2015, 52, 495 Search PubMed; (c) S. Stevenson, C. B. Rose, J. S. Maslenikova, J. R. Villarreal, M. A. Mackey, B. Q. Mercado, K. Chen, M. M. Olmstead and A. L. Balch, Inorg. Chem., 2012, 51, 13096 CrossRef CAS PubMed; (d) X. L. Wang, T. M. Zuo, M. M. Olmstead, J. C. Duchamp, T. E. Glass, F. Cromer, A. L. Balch and H. C. Dorn, J. Am. Chem. Soc., 2006, 128, 8884 CrossRef CAS PubMed; (e) Y. Zhang, A. A. Popov and L. Dunsch, Nanoscale, 2014, 6, 1038 RSC; (f) Y. Zhang, S. Schiemenz, A. A. Popov and L. Dunsch, J. Phys. Chem. Lett., 2013, 4, 2404 CrossRef CAS; (g) S. Yang, A. A. Popov, C. Chen and L. Dunsch, J. Phys. Chem. C, 2009, 113, 7616 CrossRef CAS; (h) A. L. Svitova, A. A. Popov and L. Dunsch, Inorg. Chem., 2013, 52, 3368 CrossRef CAS PubMed; (i) Y. Zhang, D. Krylov, M. Rosenkranz, S. Schiemenz and A. A. Popov, Chem. Sci., 2015, 6, 2328 RSC; (j) S. Stevenson, C. Chancellor, H. M. Lee, M. M. Olmstead and A. L. Balch, Inorg. Chem., 2008, 47, 1420 CrossRef CAS PubMed; (k) Y. Zhang, A. A. Popov, S. Schiemenz and L. Dunsch, Chem. – Eur. J., 2012, 18, 9691 CrossRef CAS PubMed; (l) R. M. Macfarlane, D. S. Bethune, S. Stevenson and H. C. Dorn, Chem. Phys. Lett., 2001, 343, 229 CrossRef CAS; (m) M. Nie, J. Xiong, C. Zhao, H. Meng, K. Zhang, Y. Han, J. Li, B. Wang, L. Feng, C. Wang and T. Wang, Nano Res., 2019, 12, 1727 CrossRef.
- C. Chen, F. Liu, S. Li, N. Wang, A. A. Popov, M. Jiao, T. Wei, Q. Li, L. Dunsch and S. Yang, Inorg. Chem., 2012, 51, 3039 CrossRef CAS PubMed.
- (a) L. Zhang, A. A. Popov, S. Yang, S. Klod, P. Rapta and L. Dunsch, Phys. Chem. Chem. Phys., 2010, 12, 7840 RSC; (b) S. Stevenson, H. R. Thompson, K. D. Arvola, K. B. Ghiassi, M. M. Olmstead and A. L. Balch, Chem. – Eur. J., 2015, 21, 10362 CrossRef CAS PubMed.
- Z. Zhang, Y. Liu, P. Han, S. Zhuang, T. Wang, S. Luo and B. Xu, ChemPhysChem, 2015, 16, 295 CrossRef CAS PubMed.
- E. B. Iezzi, J. C. Duchamp, K. R. Fletcher, T. E. Glass and H. C. Dorn, Nano Lett., 2002, 2, 1187 CrossRef CAS.
- Y. Zhang, D. Krylov, S. Schiemenz, M. Rosenkranz, R. Westerstrom, J. Dreiser, T. Greber, B. Buchner and A. A. Popov, Nanoscale, 2014, 6, 11431 RSC.
- S. Yang, A. A. Popov and L. Dunsch, Angew. Chem., Int. Ed., 2008, 47, 8196 CrossRef CAS PubMed.
- N. Chen, E. Y. Zhang, K. Tan, C. R. Wang and X. Lu, Org. Lett., 2007, 9, 2011 CrossRef CAS PubMed.
- (a) R. Westerström, J. Dreiser, C. Piamonteze, M. Muntwiler, S. Weyeneth, K. Krämer, S.-X. Liu, S. Decurtins, A. Popov, S. Yang, L. Dunsch and T. Greber, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 060406 CrossRef; (b) A. L. Svitova, Y. Krupskaya, N. Samoylova, R. Kraus, J. Geck, L. Dunsch and A. A. Popov, Dalton Trans., 2014, 43, 7387 RSC; (c) L. Spree and A. A. Popov, Dalton Trans., 2019, 48, 2861 RSC.
- A. A. Popov and L. Dunsch, Chem. – Eur. J., 2009, 15, 9707 CrossRef CAS PubMed.
- (a) V. Vieru, L. Ungur and L. F. Chibotaru, J. Phys. Chem. Lett., 2013, 4, 3565 CrossRef CAS; (b) F. Cimpoesu, N. Dragoe, H. Ramanantoanina, W. Urland and C. Daul, Phys. Chem. Chem. Phys., 2014, 16, 11337 RSC; (c) C.-H. Chen, D. S. Krylov, S. M. Avdoshenko, F. Liu, L. Spree, R. Yadav, A. Alvertis, L. Hozoi, K. Nenkov, A. Kostanyan, T. Greber and A. U. B. Wolter, et al. , Chem. Sci., 2017, 8, 6451 RSC.
- (a) R. Westerström, J. Dreiser, C. Piamonteze, M. Muntwiler, S. Weyeneth, H. Brune, S. Rusponi, F. Nolting, A. Popov, S. Yang, L. Dunsch and T. Greber, J. Am. Chem. Soc., 2012, 134, 9840 CrossRef PubMed; (b) D. Krylov, F. Liu, A. Brandenburg, L. Spree, V. Bon, S. Kaskel, A. Wolter, B. Buchner, S. Avdoshenko and A. A. Popov, Phys. Chem. Chem. Phys., 2018, 20, 11656 RSC; (c) D. S. Krylov, F. Liu, S. M. Avdoshenko, L. Spree, B. Weise, A. Waske, A. U. B. Wolter, B. Büchner and A. A. Popov, Chem. Commun., 2017, 53, 7901 RSC.
- R. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef.
- S. F. Yang, M. Kalbac, A. Popov and L. Dunsch, ChemPhysChem, 2006, 7, 1990 CrossRef CAS PubMed.
- T. Okazaki, K. Suenaga, Y. F. Lian, Z. N. Gu and H. Shinohara, J. Mol. Graphics, 2001, 19, 244 CrossRef CAS.
- M. Krause, X. J. Liu, J. Wong, T. Pichler, M. Knupfer and L. Dunsch, J. Phys. Chem. A, 2005, 109, 7088 CrossRef CAS PubMed.
- (a) A. A. Popov and L. Dunsch, J. Am. Chem. Soc., 2008, 130, 17726 CrossRef CAS PubMed; (b) R. Valencia, A. Rodriguez-Fortea, A. Clotet, C. de Graaf, M. N. Chaur, L. Echegoyen and J. M. Poblet, Chem. – Eur. J., 2009, 15, 10997 CrossRef CAS PubMed.
- (a) K. M. Sparta, M. Krug, U. Heinemann, U. Mueller and M. S. Weiss, J. Appl. Crystallogr., 2016, 49, 1085 CrossRef CAS; (b) U. Mueller, R. Förster, M. Hellmig, F. U. Huschmann, A. Kastner, P. Malecki, S. Pühringer, M. Röwer, K. Sparta, M. Steffien, M. Ühlein and P. Wilk, et al. , Eur. Phys. J. Plus, 2015, 130, 141 CrossRef.
- V. Dubrovin, L.-H. Gan, B. Büchner, A. A. Popov and S. M. Avdoshenko, Phys. Chem. Chem. Phys., 2019, 21, 8197 RSC.
- (a) H. Yang, C. M. Beavers, Z. Wang, A. Jiang, Z. Liu, H. Jin, B. Q. Mercado, M. M. Olmstead and A. L. Balch, Angew. Chem., Int. Ed., 2010, 49, 886 CrossRef CAS PubMed; (b) H. Yang, Z. Wang, H. Jin, B. Hong, Z. Liu, C. M. Beavers, M. M. Olmstead and A. L. Balch, Inorg. Chem., 2013, 52, 1275 CrossRef CAS PubMed; (c) H. Yang, H. Jin, X. Wang, Z. Liu, M. Yu, F. Zhao, B. Q. Mercado, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2012, 134, 14127 CrossRef CAS PubMed.
- M. M. Olmstead, A. de Bettencourt-Dias, J. C. Duchamp, S. Stevenson, H. C. Dorn and A. L. Balch, J. Am. Chem. Soc., 2000, 122, 12220 CrossRef CAS.
- S. F. Yang, S. I. Troyanov, A. A. Popov, M. Krause and L. Dunsch, J. Am. Chem. Soc., 2006, 128, 16733 CrossRef CAS PubMed.
- (a) J. Zhang, W. Yi, Z. Chen and X. Zhou, Dalton Trans., 2013, 42, 5826 RSC; (b) S. Anfang, K. Harms, F. Weller, O. Borgmeier, H. Lueken, H. Schilder and K. Dehnicke, Z. Anorg. Allg. Chem., 1998, 624, 159 CrossRef CAS; (c) K. L. M. Harriman, J. L. Brosmer, L. Ungur, P. L. Diaconescu and M. Murugesu, J. Am. Chem. Soc., 2017, 139, 1420 CrossRef CAS PubMed; (d) L. Zhao, S. Xue and J. Tang, Inorg. Chem., 2012, 51, 5994 CrossRef CAS PubMed; (e) P. Zhang, L. Zhang, C. Wang, S. Xue, S.-Y. Lin and J. Tang, J. Am. Chem. Soc., 2014, 136, 4484 CrossRef CAS PubMed.
- A. G. Orpen, L. Brammer, F. H. Allen, O. Kennard, D. G. Watson and R. Taylor, J. Chem. Soc., Dalton Trans., 1989, S81–S83 Search PubMed.
- S. Stevenson, H. M. Lee, M. M. Olmstead, C. Kozikowski, P. Stevenson and A. L. Balch, Chem. – Eur. J., 2002, 8, 4528 CrossRef CAS.
- A. Voevodin, L. Abella, E. Castro, D. W. Paley, L. M. Campos, A. Rodríguez-Fortea, J. M. Poblet, L. Echegoyen and X. Roy, Chem. – Eur. J., 2017, 23, 13305 CrossRef CAS PubMed.
- (a) J. Hafner, J. Comput. Chem., 2008, 29, 2044 CrossRef CAS PubMed; (b) G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS PubMed; (c) G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS; (d) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed; (e) S. Grimme, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 211 CAS.
- X. Aparicio-Anglès, N. Alegret, A. Clotet, A. Rodríguez-Fortea and J. M. Poblet, J. Phys. Chem. C, 2013, 117, 12916 CrossRef.
- L. Echegoyen, C. J. Chancellor, C. M. Cardona, B. Elliott, J. Rivera, M. M. Olmstead and A. L. Balch, Chem. Commun., 2006, 2653 RSC.
- A. A. Popov, J. Comput. Theor. Nanosci., 2009, 6, 292 CrossRef CAS.
- M. Krause, H. Kuzmany, P. Georgi, L. Dunsch, K. Vietze and G. Seifert, J. Chem. Phys., 2001, 115, 6596 CrossRef CAS.
- (a) Y. Morino, K. Kuchitsu and T. Shimanouchi, J. Chem. Phys., 1952, 20, 726 CrossRef CAS; (b) K. Kimura and M. Kimura, J. Chem. Phys., 1956, 25, 362 CrossRef CAS.
- (a) W. S. Benedict and E. K. Plyler, Can. J. Phys., 1957, 35, 1235 CrossRef CAS; (b) J. D. Swalen and J. A. Ibers, J. Chem. Phys., 1962, 36, 1914 CrossRef; (c) A. Rauk, L. C. Allen and K. Mislow, Angew. Chem., Int. Ed. Engl., 1970, 9, 400 CrossRef CAS; (d) J. M. Lehn, Nitrogen inversion, in Dynamic Stereochemistry, Fortschritte der Chemischen Forschung, Springer, Berlin, Heidelberg, 1970, vol. 15/3, pp. 311–377 Search PubMed.
- (a) C. C. Wilson, Crystallogr. Rev., 2009, 15, 3 CrossRef CAS; (b) H. B. Bürgi and S. C. Capelli, Acta Crystallogr., Sect. A: Found. Crystallogr., 2000, 56, 403 CrossRef PubMed.
- (a) J. P. Gordon, H. J. Zeiger and C. H. Townes, Phys. Rev., 1955, 99, 1264 CrossRef CAS; (b) J. P. Gordon, H. J. Zeiger and C. H. Townes, Phys. Rev., 1954, 95, 282 CrossRef CAS.
- M. Oxborrow, J. D. Breeze and N. M. Alford, Nature, 2012, 488, 353 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional details of HPLC separation mass-spectra and X-ray diffraction analysis and optimized coordinates. See DOI: 10.1039/c9nr03593a |
| This journal is © The Royal Society of Chemistry 2019 |