
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWet spinning and radial self-assembly of a carbohydrate low molecular weight gelator into well organized hydrogel filaments†
Anaïs
Chalard
ab,
Pierre
Joseph
b,
Sandrine
Souleille
b,
Barbara
Lonetti
 a,
Nathalie
Saffon-Merceron
c,
Isabelle
Loubinoux
d,
Laurence
Vaysse
d,
Laurent
Malaquin
b and
Juliette
Fitremann
*a
a,
Nathalie
Saffon-Merceron
c,
Isabelle
Loubinoux
d,
Laurence
Vaysse
d,
Laurent
Malaquin
b and
Juliette
Fitremann
*a
aIMRCP, Université de Toulouse, CNRS, Bat 2R1, 118 Route de Narbonne, 31062 Toulouse Cedex 9, France. E-mail: fitreman@chimie.ups-tlse.fr
bLAAS-CNRS, Université de Toulouse, CNRS, UPS, Toulouse, France
cUniversité de Toulouse, UPS and CNRS, ICT FR2599, 118 route de Narbonne, 31062 Toulouse, France
dTONIC, Toulouse NeuroImaging Center, Université de Toulouse, Inserm, UPS, France
First published on 10th June 2019
Abstract
In this work, we describe how a simple single low molecular weight gelator (LMWG) molecule – N-heptyl-D-galactonamide, which is easy to produce at the gram scale – is spun into gel filaments by a wet spinning process based on solvent exchange. A solution of the gelator in DMSO is injected into water and the solvent diffusion triggers the supramolecular self-assembly of the N-heptyl-D-galactonamide molecules into nanometric fibers. These fibers entrap around 97% of water, thus forming a highly hydrated hydrogel filament, deposited in a well organized coil and locally aligned. This self-assembly mechanism also leads to a very narrow distribution of the supramolecular fiber width, around 150 nm. In addition, the self-assembled fibers are oriented radially inside the wet-spun filaments and at a high flow rate, fibers are organized in spirals. As a result, this process gives rise to a high control of the gelator self-assembly compared with the usual thermal sol–gel transition. This method also opens the way to the controlled extrusion at room temperature of these very simple, soft, biocompatible but delicate hydrogels. The gelator concentration and the flow rates leading to the formation of the gel filaments have been screened. The filament diameter, its internal morphology, the solvent exchange and the velocity of the jet have been investigated by video image analysis and electron microscopy. The stability of these delicate hydrogel ropes has been studied, revealing a polymorphic transformation into macroscopic crystals with time under some storage conditions. The cell viability of a neuronal cell line on the filaments has also been estimated.
Introduction
In the context of tissue engineering, we are interested in the development of Low Molecular Weight Gelators (LMWGs or simply molecular gelators) based on saccharides as scaffolds for cell culture. They form a family of materials currently emerging for cell culture, opening the way to innovative approaches in this field. These soft materials are not based on polymers but on the self-assembly of small molecules. The self-assembly phenomenon provides fibrillary structures that entrap water and result in the formation of gels (Fig. 1). Since the self-assembly results from low energy interactions (hydrogen bonds, hydrophobic interactions, π–π stacking, etc.), the stability, the mechanical properties, the amount of molecules released and the clearance are different from those of polymers. Also, the self-assembly leads to a wide diversity of fibrillary organizations that are not observed with polymers. Yet it has been shown that the structure of these fibers at the micrometric/nanometric scale has an impact on the behavior of the cells seeded in these gels.1,2 Several LMWGs are suitable for cell culture.3–8 Most of them are based on peptides, but other families of LMWGs are also promising for these applications, based on carbohydrates,2,9–16 nucleosides17–23 or other building blocks.24 Our objective was to explore some of the simplest members of this family, for cell culture applications and also for other applications in materials chemistry.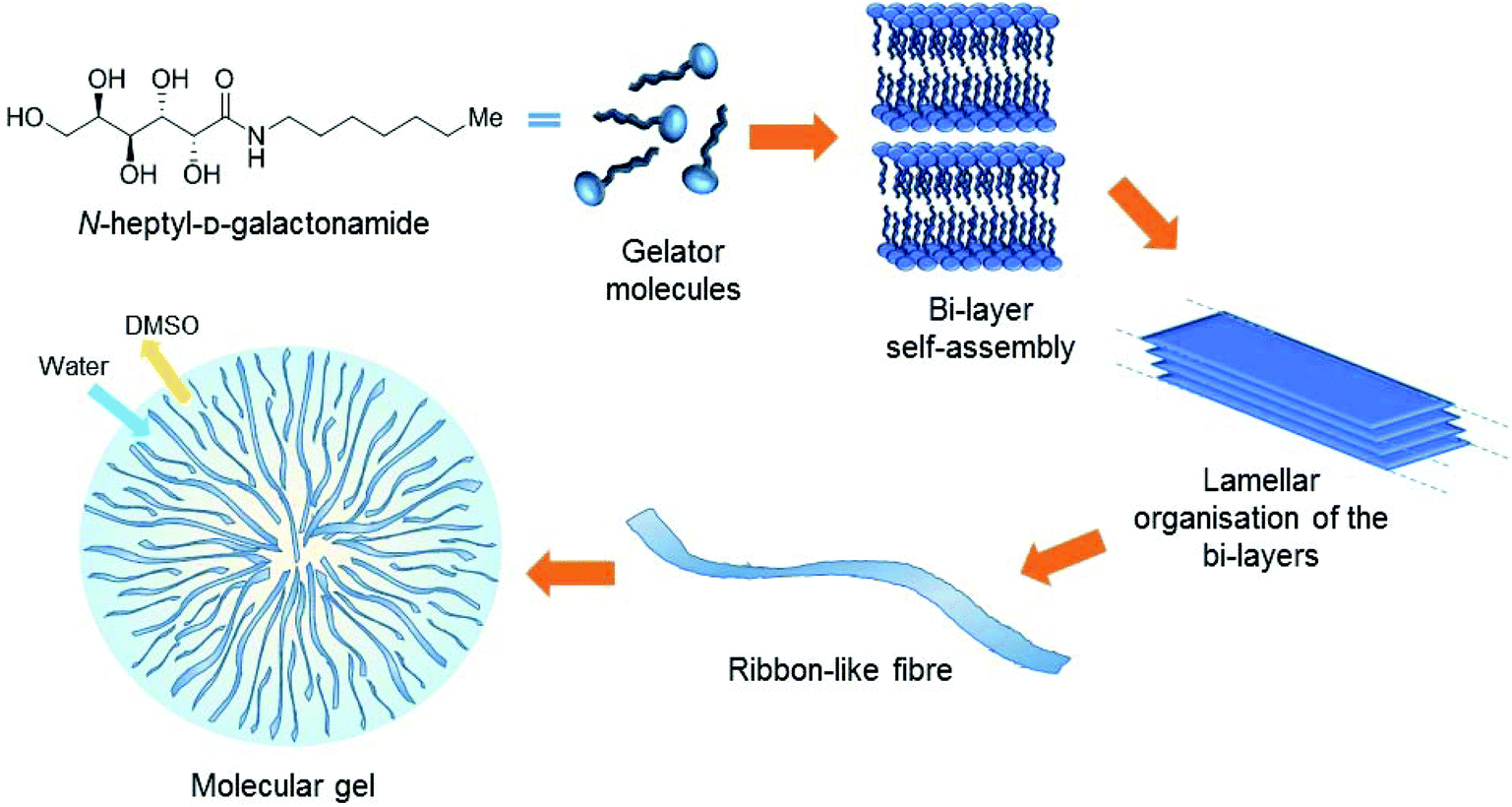 | ||
| Fig. 1 Structure of the Gal-C7 molecule and the self-assembly mechanism. | ||
In this context, we recently showed that a very simple synthetic molecule of a low molecular weight, the N-heptyl-D-galactonamide (Gal-C7, M.W. 293) – which is easy to produce at the gram scale – provided good results for the culture of a neuronal cell line (Neuro2A) and adult human neural stem cells (hNSCs).2 It has been related notably to the very low rigidity of these gels, which is known to be suitable for the growth and differentiation of neuronal cells.25 Still in the context of neuronal cell culture, it has been shown that oriented scaffolds are able to guide the development of neurites and axons in one preferential direction and can help in a suitable tissue reconstruction.26–28 Thus, for the scaffolding purpose, and also more generally for extending the scope of LMWG applications, our objective was to explore how fluidics (or microfluidics) can drive the self-assembly of the gelator into well-defined structures and shapes.
In the context of LMWGs, examples that tackle the control of self-assembly by using external fields still remain scarce. Applying well-controlled external triggers should bring interesting self-assembling effects and may result in new kinds of self-assembled materials with more controlled properties. Some studies described the use of shear stress,26,29–33 controlled reagent exchanges in microfluidic devices,34–38 electrical39 or magnetic fields,40,41 surface effects42–44 or ice growth45 to control self-assembly. While microfluidics is often used to control the self-assembly of micelles, particles, liquid crystals and some polymers,46,47 it is scarcely the case for molecular gels. Concerning the Gal-C7 hydrogel, the direct shearing of the gel to promote alignment was not possible because this kind of gel undergoes strong synaeresis when a mechanical stress is applied. For this reason, we explored a different way to work with this hydrogel.
Despite its galactonamide polar head and a quite short alkyl chain, the Gal-C7 molecule has very low solubility in water. It appeared that it might be possible to take advantage of this low solubility to trigger the self-assembly of the molecules by precipitation in water under controlled conditions. This principle is used for polymers in the technique of wet spinning. It consists in dissolving a polymer in a good solvent (this solution is called the “dope”) and extruding it into a non-solvent (called the “coagulation bath”) that triggers the precipitation of the polymer by counter diffusion. Wet spinning has regained interest recently for the production of fibers made out of biocompatible polymers such as collagen,48 poly(L-lactic acid)49 or polycaprolactone,50 alginate,51 chitosan,52–54 silkworm55 or spider silk,56,57 and notably for tissue engineering applications. Another advantage of wet spinning is that it can be scaled down to microfluidic systems in order to form micrometric or even nanometric yarns; and also to achieve different structures and organizations of the fibers: hollow, multi-component, grooved or even coded fibers can be fabricated by using the right types and layout of channels.58 Furthermore, wet spinning can be adapted to additive manufacturing and 3D printing techniques, through the precise deposition of the wet-spun fibers in three dimensions.59,60 Despite the use of the solvent exchange principle for preparing “bulk” molecular gels,61 very scarce examples describe the use of this physicochemical principle to spin fibers of supramolecular materials and non-polymeric “small” molecules while triggering their self-assembly. One study deals with the self-assembly by wet spinning of emulsion droplets charged with a copolymer.62 Otherwise few studies implemented electrospinning techniques for spinning small molecules without a polymer carrier (and without a coagulation bath), such as non-polymer liquid crystals,63 self-assembling peptides,64,65 tannic acid,66 cyclodextrins67,68 or lipids.69 In these cases, fibers made of the pure compound, thus, which are not gels, are obtained.
In this work we report the production of highly hydrated gel filaments, resulting from the self-assembly of a small molecule by solvent exchange, the N-heptyl-D-galactonamide under wet spinning conditions. The influence of the spinning parameters on gelation such as the flow rate, the dope concentration (i.e. the concentration of the gelator solution in DMSO) or the needle gauge is studied. The internal microscopic organization of the gel filaments was investigated by electron microscopy and scattering techniques, showing a well-defined organization of the self-assembled nanometric fibers. The diameters of the dope jet and of the resulting filament, and the velocity of the jet in the coagulation bath have also been investigated. Finally, the stability and the lifespan of the gel filaments were determined under different conditions, revealing polymorphism.
Since the gel itself is formed by the self-assembly of the molecule into nanometric fibers, for the sake of clarity, throughout the rest of the paper, the terms “filament” or “rope” will be used for the fibers of the gel resulting from the wet spinning process. The term “fiber” will stand exclusively for the nanometric fibers resulting from the self-assembly process of the Gal-C7 molecule, structuring the core of the gel filament.
Results and discussion
Application of wet spinning to N-heptyl-D-galactonamide
The N-heptyl-D-galactonamide (Gal-C7) hydrogels are usually prepared by cooling a 100 °C-heated solution of the gelator, at a typical concentration around 0.5 wt%. Upon cooling, the supramolecular fibers self-assemble (Fig. 1), resulting in a very wide size distribution (see Fig. 7g).2 Some of these fibers reach a millimetric length and several microns of width. The sol–gel transition occurs at high temperature, around 65 °C, precluding notably the introduction of living matter before setting. In addition, these gels are known to be mechanically very delicate. When a shear stress is applied, the gel undergoes synearesis, leading to water expulsion and the formation of a compact fiber mat. For this reason, this gel cannot be injected. To solve both problems, and also to study in a larger context different ways to self-assemble these molecules, we were looking for solutions to trigger the formation of the hydrogel at room temperature by fluidic methods.The Gal-C7 molecule is soluble in a few organic solvents, including DMSO (dimethylsulfoxide), hexafluoropropanol or methanol, and it is very poorly soluble in water at room temperature. DMSO was first selected because it is the most suited solvent for further biological applications: even though it can be detrimental for cells at high concentrations, it is non-toxic at lower ones and can be easily washed from the gel since it is miscible with water. By dissolving Gal-C7 in DMSO and injecting the solution in water, interestingly, we observed that a continuous filament of gel was produced (Fig. 2). This result was not really expected, since most of the time, the precipitation of a solute into a non-solvent bath leads to discontinuous aggregates. In the case of the Gal-C7 solution, both the properties of the solvent and the self-assembly mechanism promoted the formation of a continuous filament instead of discrete precipitation. As a matter of fact, DMSO is denser than water and it dives into the bath forming a continuous string instead of quickly mixing with water (Fig. 2d).
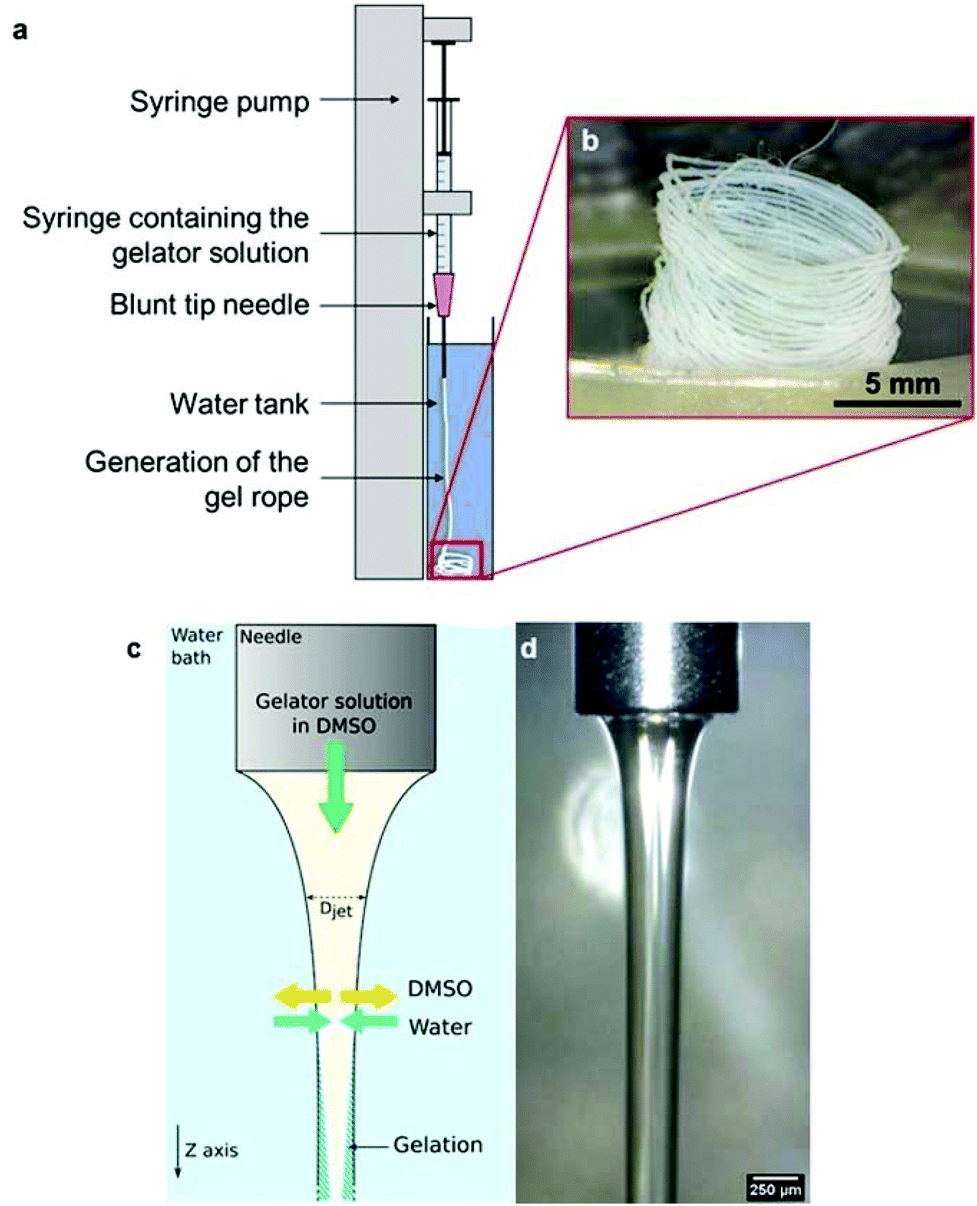 | ||
| Fig. 2 a. Experimental setup used for the generation of a continuous gel filament. b. Coiled filament of the gel obtained after the extrusion of a 4 wt% Gal-C7 solution in DMSO (50 μl min−1, 20G needle, ID 600 μm). c. Principle of the wet spinning technique: the gelator solution in DMSO is extruded through the syringe and a counter diffusion between DMSO and water occurs, resulting in the progressive gelation of the jet. d. Observation of the DMSO jet after the needle exit (30 μl min−1). | ||
When extruding the DMSO gelator solution in the water bath, coagulation occurs by solvent exchange by counter diffusion. When water meets the gelator molecules, it triggers their self-assembly into nanometric fibers that retain water and thus form the hydrogel. Due to a simple effect related to the mechanics of ropes,70 the gel falling in the solution often wraps in a coil, giving in those cases spontaneously locally aligned filaments.
Conditions for the formation of a continuous filament of the gel
In order to determine the most suited conditions to obtain Gal-C7 filaments, and also to check the robustness of the process, phase diagrams were established by setting the following parameters: the applied flow rate, the concentration of the Gal-C7 solution and the needle gauge. The bath temperature was kept constant at 22 °C (room temperature). The flow rates applied by the syringe pump varied from 0.5 to 200 μL min−1, the solution concentrations from 1.25 to 5 wt% and the needle inner diameters (ID) from 160 to 600 μm (30G to 20G). The phase diagrams were then based on simple visual observations and are presented in Fig. 3 (the different experiments at a given concentration have been slightly shifted from each other for clarity). Three regimes were identified (with a color code given on Fig. 3), separated by two transition regions: (i) the gel forms a clog directly after exiting the needle (red); (ii) a stable continuous filament is obtained (purple); and (iii) the gelation does not occur before reaching the bottom of the bath and no filament is formed (light blue). Between these well defined regimes, two transition regions were observed: (iv) a filament is formed but the needle is clogged after a few seconds (pink); and (v) the gelation occurs but to a rather lower extent in the bath (blue).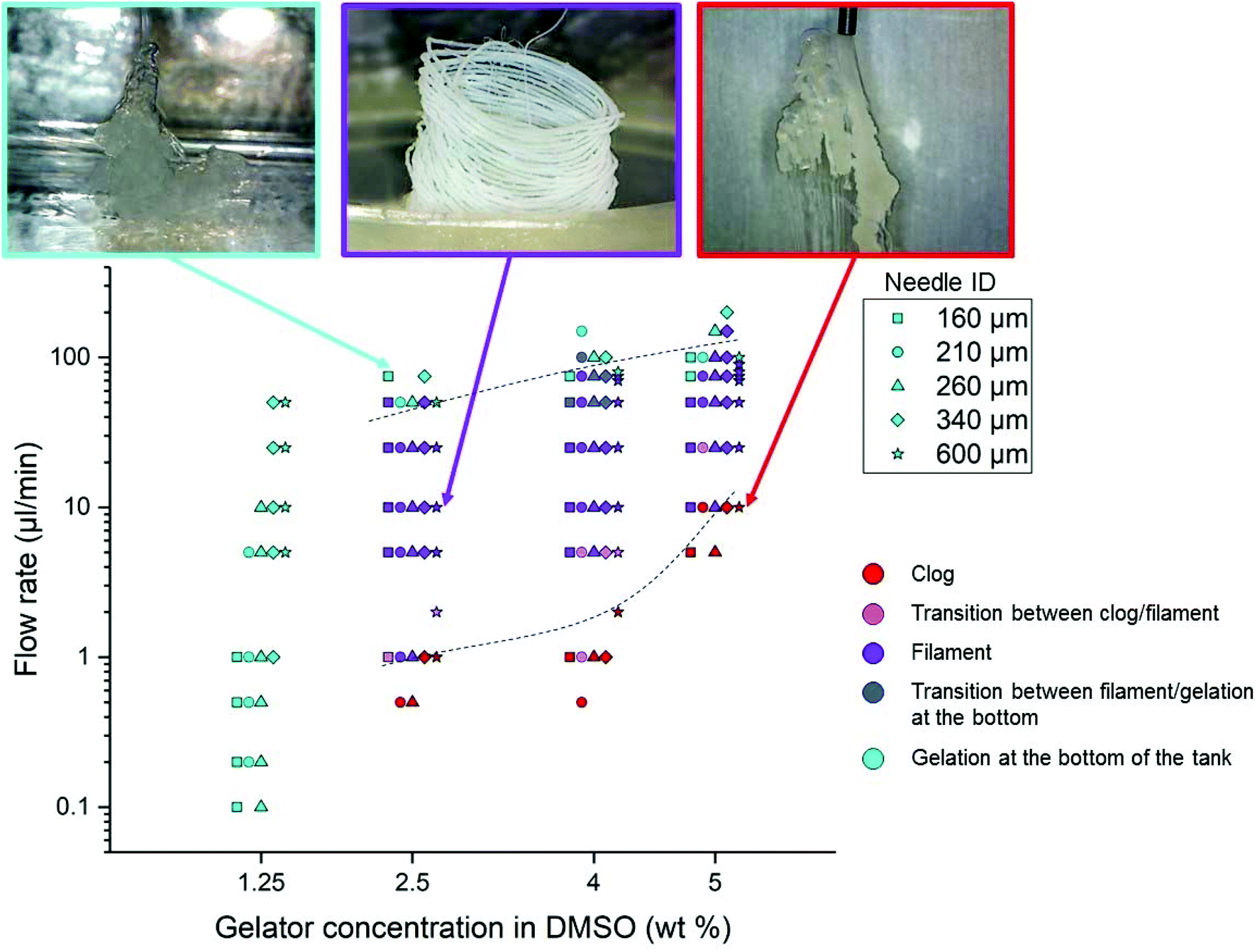 | ||
| Fig. 3 Flow rate/concentration phase diagrams for the Gal-C7 wet spinning. The different needle inner diameters (see inset for “Needle ID”) are represented by different marker shapes (squares, diamonds, stars, etc.). On the X-axis is represented the different concentrations of the gelator solution in DMSO. All the experiments were performed at precise concentrations (1.25/2.5/4 and 5 wt%), but for displaying the results of these experiments in the same diagram, the markers were slightly shifted from each other. Photographs of the different states of the gel are given on the top (from left to right): gelation at the bottom of the tank; filament of the gel; clog at the exit of the needle. | ||
The main information we extracted from this diagram is as follows. First, it provides the conditions of the concentration and flow rate to obtain the desired continuous gel filaments. They can thus be formed with Gal-C7 concentrations between 2.5 and 5 wt% and flow rates between 5 and 50 μL min−1. When the concentration was too low (1.25 wt%), we were not able to find any condition for which a continuous filament was formed. This result illustrates the fact that a continuous gel filament can be obtained only when the supramolecular fibers are dense enough to be more or less intertwined (see part 4). If the concentration of the molecule is too low, the supramolecular fiber network is too sparse to sustain a continuous string. Secondly, when the concentration increases, a higher flow rate must be applied to avoid clogging. More generally, at any concentration, if the flow rate is too low, a clog tends to form at the exit of the needle. Conversely, when the flow rate is too high, the solution does not have time to gel before reaching the bottom of the bath. These results are directly explained by the contact time of the DMSO/Gal-C7 solution with water. If the flow rate is too slow, water can diffuse early in the DMSO/GalC7 solution, resulting in clogging. If the flow rate is too high, the water does not have the time to diffuse inside the DMSO liquid rope before it reaches the bottom. It means that there is an optimal flow rate window within which the jet speed and the water diffusion match to trigger the gelation into a continuous filament. Concerning the needle diameter, surprisingly, this parameter does not seem to influence much the position of the limits in the diagram.
Characterization of the spinning process
The gelation mechanism and the solvent exchange occurring in the wet spinning process were investigated with different methods. | ||
| Fig. 4 a1–3: Characterization of the DMSO diffusing out of the forming gel filament ≈1 cm after the needle exit. White light (a1) and fluorescence (a2) observations of the jet and the superimposition of the two pictures (a3). b1–3: Characterization of the water diffusion throughout the fall of the DMSO solution. The pictures were taken at the following distances: 0 cm (b1), ≈3 cm (b2) and ≈6 cm (b3) after the needle exit. | ||
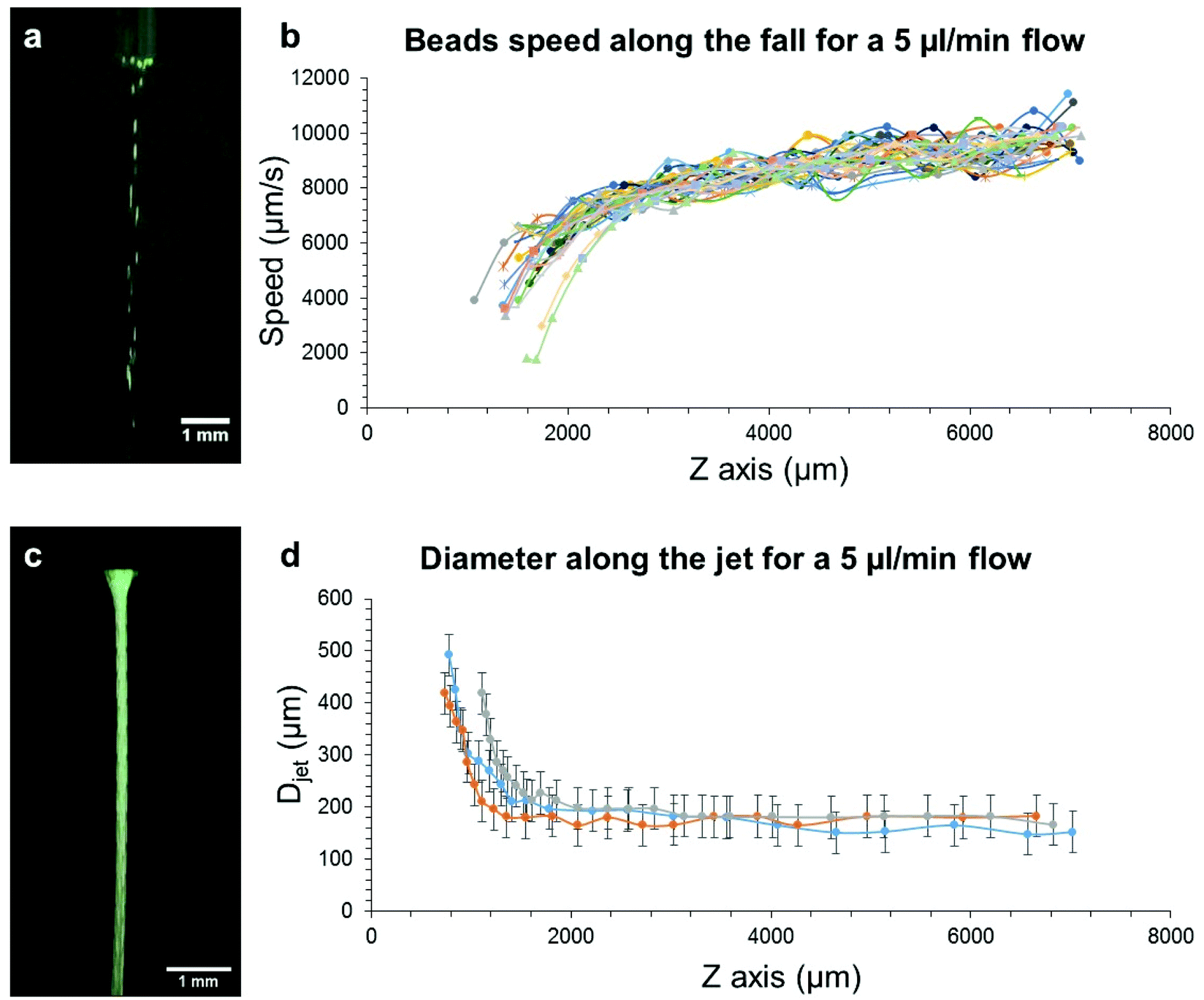 | ||
| Fig. 5 a. Snapshot of the video acquired to measure the jet speed. b. Speed of the fluorescent beads along their fall with a 5 μl min−1 flow rate and a needle ID of 600 μm (20G). c. Mean flow profile obtained from the flow of fluorescent beads in the DMSO/Gal-C7 solution at 5 μl min−1 (needle ID: 600 μm, 20G); different frames from the video were superimposed resulting in this image. d. Diameter along the jet measured on the same photograph as c.; three different experiments were performed. | ||
What can be seen in Fig. 5b and d is that the velocity of the beads is consistent with the shape of the DMSO jet since the velocity increases as the diameter of the jet decreases. This thinning of the jet is actually due to gravity that pulls the DMSO jet, making it take this hyperbolic shape at the exit of the needle. After a few millimeters, the jet velocity is quasi constant as well as the jet diameter, probably due to a balance between the gravitational force and viscous friction that establishes between the jet and the resting solution surrounding it. The mean jet velocity for a 5 μl min−1 flow rate is of 8.3 ± 0.5 mm s−1 (see Table SI-1†). The effective flow rate (Qeff) was then calculated from the measured velocities and diameters, thanks to eqn (1):
| Qeff = v × S | (1) |
 | ||
| Fig. 6 The jet (a) and the resulting filament diameters (b) were measured for different flow rates and for different needle internal diameters. Two needle gauges were used: 20G (orange) and 27G (blue). Two gelator concentrations in DMSO were also used: 2.5 wt% (circles) and 4 wt% (squares). | ||
The first thing to note is that, the higher the flow rate, the wider the diameters. Moreover, the resulting gel filament diameter is much smaller than that of the DMSO jet after the needle exit. The needle gauge (orange versus blue curves) has a significant influence on the jet diameter (Fig. 6a) but it does not affect the gel filament diameter (Fig. 6b). Actually, the jet diameter is measured very close to the needle exit (2.3 mm after, whereas the total fall height is 80 mm) and at this distance, it is mainly set by the needle size. Different phenomena occur between 2.3 mm and 80 mm: the jet thinning continues slightly after 2.3 mm (Fig. 5d) and the progressive solvent mixing and gel formation may change the volume of the jet. These phenomena may explain the difference in diameters between the jet measured early during the fall and the final gel filament.
What is also noticeable is that the solution concentration does not significantly influence the diameter of the jet since it is possible to build a continuous curve out of the two concentration conditions, 2.5 and 4 wt%. This result tends to show that, at these concentrations in DMSO, Gal-C7 does not induce viscoelastic effects. In the case of the gel filament, this one is slightly thinner with a lower concentration (2.5 wt%) when the smaller needle is used (27G). This difference is amplified when the large needle (20G) is used: a discontinuity between the curves at 2.5 wt% and the curves at 4 wt% is observed. In this case, the more the gelator, the larger the gel filament. However, the effect of the concentration is quite small, showing that the filament and jet diameters seem to be mainly set by the flow rate and by gravity.
Microscopic characterization and composition of the filaments
The microscopic structure of the gel filaments and their composition were studied by optical and electron microscopy and by thermogravimetric analysis.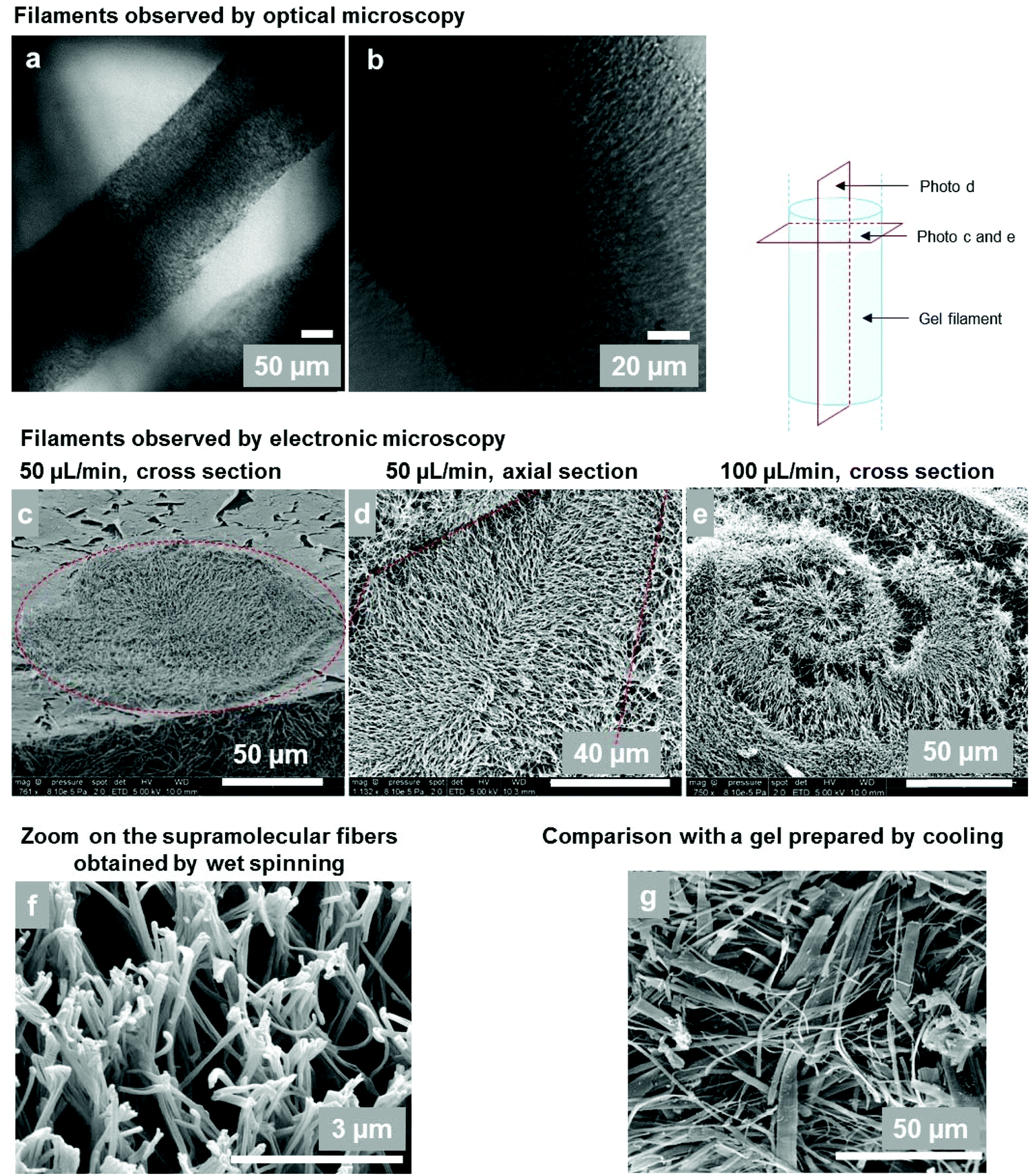 | ||
| Fig. 7 (a and b) Optical microscopy of gel filaments (4 wt%, 50 μl min−1, 20G needle ID = 600 μm). (c–e) Cryo-SEM observations of gel filament sections. (c) Cross section of a filament (4 wt%, 50 μl min−1, 20G needle). The dotted circle shows the section of the rope. The resulting diameter is around 120 μm. (d) Axial section of a gel filament (5 wt%, 50 μl min−1). The dotted lines show the edges of the filament. The radial arrangement of the supramolecular fibers can be seen. (e) At a higher flow rate (5 wt%, 100 μl min−1), fibers organized in spirals are observed; (f) Supramolecular gel fibers at a higher magnification (5 wt%, 100 μl min−1). (g) For comparison: cryo-SEM of a bulk Gal-C7 gel prepared by cooling down a hot gelator solution in water (0.45 wt%) and not by wet spinning (see ref. 2). Some fiber widths can reach 15 μm. | ||
The microscopic structure of the wet-spun filaments was further analyzed by cryo-SEM, highlighting the internal organization of the wet-spun gel filaments of Gal-C7 (Fig. 7c–e). They are sustained by a network of supramolecular fibers organized in radially oriented clusters. Interestingly, when the flow rate is high (100 μL min−1), fibers organized in spirals are observed, with a spiral size around 200 μm, matching the filament section (Fig. 7e). The gelator acts as a tracer of the diffusion mechanism during the spinning process. The gel network is also very different from the one obtained in “bulk gels” prepared by cooling down a hot gelator solution in water (Fig. 7g).2 First, the size of the constituting fibers is much reduced in the case of wet-spun gels compared to that of the bulk ones (Fig. 7g): the mean fiber width is around 140 nm for wet-spun hydrogels whereas the median width for bulk Gal-C7 hydrogels is 1.5 μm. Secondly, the width distribution of the supramolecular fibers: 136 ± 37 nm, is rather narrow in the wet-spun filaments, while in the “bulk gel” a very large distribution: 1.46 ± 2 μm is observed (see Fig. SI-2†). This result shows that the wet spinning enables a high control of the supramolecular fiber formation. Finally, the spatial organization of the fibers in the core of the filament is also striking. There seems to be a general radial organization of the fibers inside the filaments, which can be outlined on the cross section (Fig. 7c) or on the axial section (Fig. 7d), even if not all the fibers follow this pattern. Indeed, this organization results from the radial diffusion of water inside the liquid rope. In Fig. 7d, the edges of a filament can be outlined: the fibers mostly grew perpendicularly to the edge of the filament and go towards its center. As a consequence, the fibers are locally aligned at a microscopic level by this process. The wet spinning techniques thus enable obtaining radially organized supramolecular fibers with a narrow size distribution.
Cryo-SEM also confirmed the filament diameters on some pictures, such as that in Fig. 7a with a diameter around 120 μm, which matches the optical observations.
Lifetime of the wet-spun filaments
| Conditions | Exposed to air | Under a coverslip |
|---|---|---|
| Water (RT) | 2 | 3 |
| PBS (RT) | <1 | 2 |
| DMEM/FBS (RT) | >34 | >34 |
| DMEM/FBS (37 °C) | <1 | 4 |
Throughout the days, the filaments vanished progressively and the gelator recrystallized to form macroscopic translucent crystals (Fig. 8). This phenomenon occurred whatever the solution used, except for DMEM/FBS at room temperature where crystals were not observed even after more than a month. Interestingly, DMEM/FBS seems to stabilize the gel filaments for a long time, perhaps because of the numerous solutes of DMEM/FBS, but only at room temperature. Between these two states, spikes on the edges of the filaments can also be observed after some time, as seen in Fig. 8c.
 | ||
| Fig. 8 Evolution of the morphology of Gal-C7 filaments with time in water, without a coverslip: (a) just after extrusion, (b and c) 2 days after extrusion and (d) 3 days after extrusion. Scale bars: 100 μm (a and d) and 50 μm (b and c). | ||
The morphological transformation of low molecular weight gelators into crystals is a phenomenon already known.72–77 Notably, it has been described for similar molecules belonging to the family of N-alkyl-aldonamides.78 For example N-octyl-D-gluconamide, which has a different stereochemistry of the polar head, crystallizes readily. Interestingly, in the case of N-heptyl-D-galactonamide, the gels prepared by cooling the hot gelator solutions are stable for months, while the wet-spun gels offer a way to observe this transition.
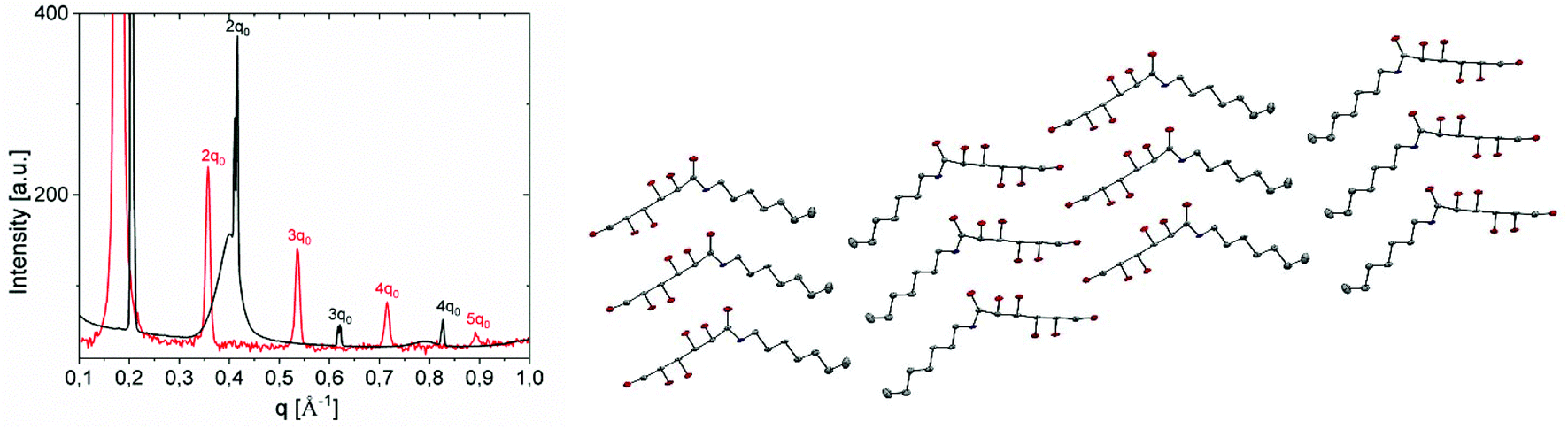 | ||
| Fig. 9 (a) SAXS spectra of a fresh Gal-C7 wet-spun gel (red curve) and Gal-C7 crystals (black curve). (b) Molecular packing of Gal-C7 in the crystals (H atoms have been omitted for clarity). | ||
The crystals obtained from the morphological transformation were also analyzed by SAXS, WAXS and X-ray diffraction. SAXS highlights a lamellar order with a smaller spacing of 30.5 Å (peak position, 0.204 Å−1) (Fig. 9a and SI-5b†). The crystallographic structure was determined from X-ray diffraction of bigger crystals (SI-7†). The N-heptyl-D-galactonamide (Gal-C7) crystallizes in a monoclinic system (space group P21). The molecular packing is a symmetric tail-to-tail bilayer sheet comparable to that of the gulonamide79 and talonamide80 analogues (Fig. 9b), but is in contrast to the head-to-tail packing observed in the gluconamide analogue.81 All fatty chains run parallel to each other but quite intriguingly an angle of ∼120° is observed between the packed chains at the junction of the tail-to-tail arrangement. An angle of 90 °C has also been described previously in the case of N-octyl-D-talonamide.80 The occurrence of an angle in the packing of the hydrophobic chains has also been been modeled in the case of Fmoc-derived gelators.82 In the crystal, intermolecular O–H⋯O and N–H⋯O hydrogen bonds link the molecules into a three-dimensional network (Fig. SI-7b and Table SI-7†). The stereochemistry of the galactonamide polar head enables the development of a strong hydrogen-bond pattern between hydroxyl groups of adjacent crystal sheets that drives the tail-to-tail molecular packing. In the case of gluconamide, the stereochemistry of the hydroxyl groups is different and leads to a head-to-tail packing. Still for crystals, the WAXS region looks like a powder diffraction spectrum with well defined peaks, while in the case of the gel filament, the same region of the spectrum shows much larger peaks with peak positions different compared with crystals (see Fig. SI-6†). This observation indicates a probable difference in the molecular conformation of the Gal-C7 molecule between the supramolecular fibers and the crystals. Together with the difference in the water content, it may explain the spacing difference between the two kinds of assembly.
Biocompatibility and filament lifetime under cell culture conditions
The suitability of Gal-C7 gels obtained by wet spinning for cell culture was assessed. For this purpose, the cell viability of a neuronal cell line (Neuro2A) on this material was evaluated with a live–dead cell assay. Overall, the majority of the cells found on the wet-spun gel filament were alive after 3 days. However in all experiments, some dead cells were gathered in some places of the gel (Fig. 10). It looks as if a toxic phenomenon was concentrated in some parts. It means that a lower cell viability is obtained on the wet-spun filaments compared with “bulk gels” on which a cell viability of 98% was obtained.2 From thermogravimetric measurements, it is found that there is no DMSO left in the wet-spun filaments and, in addition, extensive medium washing processes are performed before cell culture. Thus, it seems unlikely that DMSO induced the toxicity. Alternatively, perhaps other chemical, biochemical or physical factors (fiber size, surface morphology, rigidity, nutrient diffusion etc.) may explain this effect. Otherwise, despite the long lifetime observed for the filaments wet-spun in DMEM/FBS at room temperature (part 5), at 37 °C, this lifetime is much reduced. As a consequence, only a few fragments were left after 3 days of culture. The wet-spun gels obtained from Gal-C7 alone are thus too fragile to sustain cell culture and the process has to be improved to get filaments adapted for this application. | ||
| Fig. 10 (a) Neuro2A cells on a gel filament observed directly by bright-field microscopy and (b) Neuro2A cells on a gel filament after live/dead cell staining (3 days of culture). The gel filaments are visible under the cells. Scale bars: 50 μm. | ||
Conclusion
In this work, we have shown that wet spinning can be used to organize a low molecular weight gelator into continuous hydrogel filaments with controlled diameters. Moreover, the process led to a high degree of organization of the supramolecular fibers inside the filament: first, the fibers are very homogeneous in size. Secondly, as a result of the radial diffusion of the water inside the liquid rope, they are oriented radially in the filament and are locally aligned. It means that the wet spinning process, applied to this LMWG, enabled better control of its self-assembly as compared to standard procedures based on the cooling of a hot gelator solution. From the point of view of the material properties, the filament obtained is mainly composed of water and can be easily dissolved by simple washing processes. As a consequence, it could be useful as a sacrificial material. The controlled self-assembly of this gelator could alternatively be used in combination with other compounds to organize them in the space. Finally, wet spinning of Gal-C7 could allow the precise deposition of the gel on a surface, opening the way to its application to 3D-printing.Experimental section
N-Heptyl-D-galactonamide was synthesized according to the procedure described in the study in ref. 2. It can be purchased from Innov'Orga (Reims, France).Extrusion of the hydrogel filaments
The dope solutions of N-heptyl-D-galactonamide were prepared by dissolving the molecule at room temperature in dimethyl sulfoxide (purchased from Fisher, 99%, non-anhydrous) with the help of ultrasound and at different mass concentrations (2.5, 4 or 5 wt%). The solutions were extruded in a bath of ultrapure water (200 mL, bath height of approximately 8 cm) at room temperature (21–23 °C) with a syringe pump (CETONI neMESYS 290N), controlled by the neMESYS UserInterface software. Different gauges of blunt-tip needles, flow rates and concentrations of the solution were used. The videos acquired during these experiments were acquired with a DinoLite digital microscope (model AM4013MTL-FVW) under white light. Between each condition, the needle was cleaned and the bath was changed to fresh ultrapure water.Characterization of the solvent exchange
To assess the diffusion of DMSO out of the jet, 1 μm fluorescent beads (FluoSpheres, Invitrogen, diameter of 1 μm, 1.1010 beads per ml, ref F13080) were dissolved at 10% in volume in a 4 wt% gelator solution in DMSO. This concentration is sufficient so that the fluorescence is homogeneous in the jet. This resulting solution was then extruded at 50 μl min−1 according to the usual wet spinning protocol with a 23G needle (ID of 340 μm). Images were then acquired with two microscope cameras along the jet at different distances after the needle exit, one with white light (DinoLite model AM4013MTL-FVW) and the other with fluorescent light (DinoLite model AM4115T-GFBW, excitation at 480 nm and emission filter at 510 nm). Composite images were then created by overlaying fluorescence images on top of the white light ones with the ImageJ software.To assess the diffusion of water inside the DMSO jet, 5 mg of salicylaldehyde azine were dissolved into 1 ml of a 4 wt% Gal-C7 solution in DMSO and this solution was extruded at 50 μl min−1 with a 20G needle (ID of 600 μm) into a water bath. A video was then taken along the falling jet with the microscope camera suited for fluorescence observations at different distances after the needle exit (DinoLite model AM4115T-GFBW, excitation at 480 nm and emission filter at 510 nm).
Estimation of the jet velocity
For the speed analyses, 15 μm diameter fluorescent beads (purchased from Thermo Fisher Scientific) were used. The beads were suspended in DMSO to reach a concentration suitable for speed analysis (rather low to be able to distinguish the beads with the naked eye in the videos). The suspension was then mixed in the solution of Gal-C7 in DMSO. This last solution was then extruded at 5 or 10 μl min−1 and the DMSO jet was filmed with the DinoLite digital microscope suited for fluorescence acquisitions (model AM4115T-GFBW, excitation at 480 nm and emission filter at 510 nm). Three experiments were performed for each flow rate. The velocity was analyzed thanks to the ManualTracking plugin from ImageJ. For 10 μl min−1, a total of 23 beads were tracked throughout the three videos and 30 beads for 5 μl min−1. The measured jet velocity is the mean of the mean velocities of all the analyzed beads ± the associated standard deviation. To obtain the jet diameter under these conditions, several frames of the acquired videos were superimposed, while being careful of taking ones in which the jet was stable. Three different superimpositions at different times were done. The resulting pictures then allowed the measurement of diameters, which were then plotted along the Z axis. The error bars are an estimation of the error made when measuring the diameters (the value of two pixels in the picture, namely 40 μm). The error in the predicted velocity was calculated with the formula: Δv = v(ΔQ/Q + Δd/d), with v being the calculated velocity, Q the applied flow rate, ΔQ the error in the flow rate (= 0.00002 mm3 s−1 because the minimal flow that can be applied by the syringe pump is 1 nl min−1 with a 1 ml syringe), d the jet diameter and Δd the error in the measured diameter (40 μm).Measurements of the jet diameters
The jet diameters were measured from several snapshots from the videos of the extrusion. The videos were acquired with the DinoLite digital microscope (model AM4013MTL-FVW) under white light and with maximum magnification. The extrusions were recorded for approximately 2 min and snapshots of the videos were further taken around every 30 s. The jet diameters were then measured from these snapshots with ImageJ, at a distance of 2.3 mm after the extremity of the needle (maximum distance considering the picture size). The mean and standard deviation of the measured diameters were then calculated for each condition.Optical transmission microscopy of the filaments
For microscopic observations, the wet-spun gel filaments were retrieved thanks to a sieve in which a circular microscope slide was placed and plunged into the bath. The diameters of the wet-spun filaments were measured by transmission optical microscopy on an Olympus inverted microscope (bright field, ×10). At least three pictures at different points of the filament were taken for each condition and the mean and standard error of the diameters (measured with ImageJ) were calculated.Cryo-SEM observations
Gel filaments were prepared according to the wet spinning previously described and retrieved thanks to a sieve placed in the coagulation bath. The most robust filaments, corresponding to the conditions: 4 wt%; 50 μl min−1 (Fig. 4c) and 5 wt%; 50 μl min−1 (Fig. 4d), were studied. A part of the gel filament was then deposited on the cryo-SEM cane and frozen at −220 °C in liquid nitrogen. The frozen sample was fractured at −145 °C under vacuum in a cryo-transfer system chamber (Quorum PP3000 T). The sublimation was performed at −95 °C for 30 min. The sample was metalized with Pd for 60 s and introduced in the microscope. The temperature was kept at −145 °C. Images were recorded with an FEG FEI Quanta 250 microscope, at 5 kV for the acceleration voltage. Some of them with a suitable magnification and orientation (around 10 to 20 measures per gel) provided a rough estimation of the fiber thickness.Thermo-gravimetric analysis
Gal-C7 gel filaments were prepared by wet spinning: they were extruded at 50 μl min−1 with a 4 wt% solution of Gal-C7 in DMSO and a 20G blunt-tip needle. A sieve was placed at the bottom of the water tank to retrieve the formed filaments. The filaments were kept in the sieve in a very small volume of water for one hour before measurement. About ten milligrams of filaments were then placed in a crucible and the water surrounding the filaments was wiped off before analysis. The temperature program was as follows: heating from 30.00 °C to 300.00 °C at 1.00 °C min−1, then heating from 300.00 °C to 600.00 °C at 10.00 °C min−1 and holding for 10.0 min at 600.00 °C (under nitrogen, 100.0 ml min−1). The composition of the gel was then calculated as follows: the mass of each component was measured between the plateaus on the curve and the percentage of the total initial mass was then calculated. TGA was also performed with the Gal-C7 solid alone (Fig. SI-2b†) and with a solution of 1 wt% DMSO in pure water (Fig. SI-2c†) as references.Study of the lifetime of the filaments in solutions
30 μl of Gal-C7 in DMSO (4 wt%) were extruded in approximately 200 ml of ultrapure water (≈8 cm of fall), with a 20G needle at 50 μL min−1, and the resulting filaments were retrieved thanks to a glass slide placed in a sieve at the bottom of the water tank. After spinning, the glass slide was rapidly put in a well of a 12-well cell culture plate and 1 ml of the different solutions (ultrapure water, PBS (Phosphate Buffer Saline) 10×, or DMEM (Dulbecco's Modified Eagle's medium) supplemented with 10% FBS (Fetal Bovine Serum) and 1% penicillin–streptomycin) was added inside the well. In some cases, another glass slide was put on top of the liquid while being careful to not let it sink in the liquid (an air bubble keeps the slide from sinking). The plate was then kept either at room temperature (21–23 °C) or at 37 °C in a stove in the case of trials with DMEM. The filaments were then observed every day by optical microscopy (upright Olympus microscope, bright field, ×5 or ×10). At least five different pictures were taken for each well. The diameters of the resulting fibers were then manually measured with the ImageJ software and the mean and standard deviation were calculated for each day.Small angle X-ray scattering (SAXS)
Scattering measurements were performed on freshly wet-spun gels of Gal-C7 obtained from a 5 wt% DMSO solution extruded at 25 μl min−1. Crystals were obtained from wet-spun filaments obtained from a 2.5 wt% DMSO solution extruded at 25 μl min−1 and aged for two weeks. A XEUSS 2.0 SAXS/WAXS laboratory beamline equipped with a Cu source (E = 8 keV) and a pixel detector PILATUS3 1 M from Detrics was used. The sample-to-detector distance was 0.387 m (beam size: 0.8 mm × 0.8 mm) allowing a q range between 0.022 Å−1 and 1.6 Å−1. Samples were mounted on a sample holder for gels with a kapton window. Measurements were carried out at 25 °C.Crystallographic data collection and structure determination for Gal-C7
The data were collected at low temperature (193 K) on a Bruker-AXS APEX II QUAZAR diffractometer equipped with a 30 W air-cooled microfocus source, using MoKα radiation (λ = 0.71073 Å). Phi- and omega-scans were used. The data were integrated with SAINT, and an empirical absorption correction with SADABS was applied.83 The structure was solved by direct methods (ShelXT)84 and refined using the least-squares method on F2 (ShelXL).85 All non-H atoms were refined with anisotropic displacement parameters. The H atoms were refined isotropically at calculated positions using a riding model, except on oxygen and nitrogen atoms. These H atoms were located by a difference Fourier map. Selected data for Gal-C7: C13H27NO6, M = 293.35, monoclinic, space group P21, a = 4.957(3) Å, b = 5.007(4) Å, c = 30.60(2) Å, β = 92.980(8)°, V = 758.4(9) Å3, Z = 2, crystal size: 0.40 × 0.20 × 0.01 mm3, 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 967 reflections collected (3063 independent, Rint = 0.1024), 204 parameters, 1 restraint, R1 [I > 2σ(I)] = 0.0784, wR2 [all data] = 0.1680, largest diff. peak and hole: 0.345 and −0.294 e Å−3.
967 reflections collected (3063 independent, Rint = 0.1024), 204 parameters, 1 restraint, R1 [I > 2σ(I)] = 0.0784, wR2 [all data] = 0.1680, largest diff. peak and hole: 0.345 and −0.294 e Å−3.
Cell culture assays
100 μl of a 4 wt% Gal-C7 solution in DMSO were extruded according to the previously described protocol in a bath of sterile water and with a sterile syringe and needle. The gel filaments were transferred into the wells of a 12-well cell culture plate. The filaments were then rinsed 3 times (for 2 hours, overnight and 4 hours) with 2 ml of DMEM (Dulbecco's modified Eagle's medium) supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin at room temperature. 50000 cells of a neuronal cell line (Neuro2A, ATCC) were then seeded on the scaffolds with 2 mL of cell culture medium and incubated for 3 days at 37 °C under a humidified atmosphere containing 5% CO2. Fluorescence live–dead staining assay (Molecular probe) was used according to the manufacturer's instructions to visualize the proportion of viable cells in green (calcein AM) and nonviable cells in red (ethidium homodimer-1). The samples were observed using a Zeiss Axioskop microscope equipped with a Cool SNAPfx camera (Photometrics).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Christophe Coudret for the kind gift of salicylaldehyde azine and helpful discussion about molecules with aggregation induced emission; Pierre Roblin (IR, CNRS, LGC Toulouse) for support in SAXS measurements and FR FERMAT, Université de Toulouse, France-CPER IMATECBIO and for providing access to SAXS instrument XEUSS 2.0 XENOCS; and Bruno Payré, Isabelle Fourquaux, Dominique Goudounèche from CMEAB “Centre de Microscopie Electronique Appliquée à la Biologie” for TEM and cryo-TEM. This work was supported by the French National Research Agency (A. C.'s grant and financial support, ANR “Neuraxe”, grant N°ANR-15-CE07-0007-01) and as part of the MultiFAB project which has received funding from FEDER European Regional Funds and Région Occitanie – France (Grant Number: 16007407/MP0011594). The European Union is also acknowledged for its financial support for equipment (FEDER-35477: “Nano-objets pour la biotechnologie”).References
- G.-F. Liu, D. Zhang and C.-L. Feng, Angew. Chem., Int. Ed., 2014, 53, 7789–7793 CrossRef CAS PubMed.
- A. Chalard, L. Vaysse, P. Joseph, L. Malaquin, S. Souleille, B. Lonetti, J.-C. Sol, I. Loubinoux and J. Fitremann, ACS Appl. Mater. Interfaces, 2018, 10, 17004–17017 CrossRef CAS PubMed.
- J. Zhou, J. Li, X. Du and B. Xu, Biomaterials, 2017, 129, 1–27 CrossRef CAS PubMed.
- K. J. Skilling, F. Citossi, T. D. Bradshaw, M. Ashford, B. Kellam and M. Marlow, Soft Matter, 2014, 10, 237–256 RSC.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- X.-Q. Dou and C.-L. Feng, Adv. Mater., 2017, 1604062 CrossRef PubMed.
- M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2015, 15, 13–26 CrossRef PubMed.
- S. Koutsopoulos, J. Biomed. Mater. Res., Part A, 2016, 104, 1002–1016 CrossRef CAS PubMed.
- V. M. P. Vieira, A. C. Lima, M. de Jong and D. K. Smith, Chem. – Eur. J., 2018, 24, 15112–15118 CrossRef CAS PubMed.
- T. Tsuzuki, M. Kabumoto, H. Arakawa and M. Ikeda, Org. Biomol. Chem., 2017, 15, 4595–4600 RSC.
- I. S. Okafor and G. Wang, Carbohydr. Res., 2017, 451, 81–94 CrossRef CAS PubMed.
- Y. Ohsedo, M. Oono, K. Saruhashi and H. Watanabe, RSC Adv., 2014, 4, 48554–48558 RSC.
- S. Datta and S. Bhattacharya, Chem. Soc. Rev., 2015, 44, 5596–5637 RSC.
- X. Xiao, J. Hu, X. Wang, L. Huang, Y. Chen, W. Wang, J. Li and Y. Zhang, Chem. Commun., 2016, 52, 12517–12520 RSC.
- N. Baccile, L. V. Renterghem, P. L. Griel, G. Ducouret, M. Brennich, V. Cristiglio, S. L. K. W. Roelants and W. Soetaert, Soft Matter, 2018, 14, 7859–7872 RSC.
- M. Kamada, C. Pierlot, V. Molinier, J.-M. Aubry and K. Aramaki, Colloids Surf., A, 2018, 536, 82–87 CrossRef CAS.
- L. Latxague, M. A. Ramin, A. Appavoo, P. Berto, M. Maisani, C. Ehret, O. Chassande and P. Barthélémy, Angew. Chem., Int. Ed., 2015, 54, 4517–4521 CrossRef CAS PubMed.
- M. G. F. Angelerou, P. W. J. M. Frederix, M. Wallace, B. Yang, A. Rodger, D. J. Adams, M. Marlow and M. Zelzer, Langmuir, 2018, 34, 6912–6921 CrossRef CAS PubMed.
- K. J. Skilling, B. Kellam, M. Ashford, T. D. Bradshaw and M. Marlow, Soft Matter, 2016, 12, 8950–8957 RSC.
- J. Baillet, V. Desvergnes, A. Hamoud, L. Latxague and P. Barthélémy, Adv. Mater., 2018, 30, 1705078 CrossRef PubMed.
- M. A. Ramin, L. Latxague, K. R. Sindhu, O. Chassande and P. Barthélémy, Biomaterials, 2017, 145, 72–80 CrossRef CAS PubMed.
- L. Latxague, A. Gaubert and P. Barthélémy, Molecules, 2018, 23, 89 CrossRef PubMed.
- D. Yuan, X. Du, J. Shi, N. Zhou, A. A. Baoum, K. O. A. Footy, K. O. Badahdah and B. Xu, Beilstein J. Org. Chem., 2015, 11, 1352–1359 CrossRef CAS PubMed.
- T. Das, M. Häring, D. Haldar and D. Díaz Díaz, Biomater. Sci., 2018, 6, 38–59 RSC.
- D. Tarus, L. Hamard, F. Caraguel, D. Wion, A. Szarpak-Jankowska, B. van der Sanden and R. Auzély-Velty, ACS Appl. Mater. Interfaces, 2016, 8, 25051–25059 CrossRef CAS PubMed.
- A. Li, A. Hokugo, A. Yalom, E. J. Berns, N. Stephanopoulos, M. T. McClendon, L. A. Segovia, I. Spigelman, S. I. Stupp and R. Jarrahy, Biomaterials, 2014, 35, 8780–8790 CrossRef CAS PubMed.
- C. Davoust, B. Plas, A. Béduer, B. Demain, A.-S. Salabert, J. C. Sol, C. Vieu, L. Vaysse and I. Loubinoux, Stem Cell Res. Ther., 2017, 8, 253 CrossRef PubMed.
- L. Vaysse, F. Conchou, B. Demain, C. Davoust, B. Plas, C. Ruggieri, M. Benkaddour, M. Simonetta-Moreau and I. Loubinoux, Behav. Neurosci., 2015, 129, 423–434 Search PubMed.
- J. Fitremann, B. Lonetti, E. Fratini, I. Fabing, B. Payré, C. Boulé, I. Loubinoux, L. Vaysse and L. Oriol, J. Colloid Interface Sci., 2017, 504, 721–730 CrossRef CAS PubMed.
- E. R. Draper, O. O. Mykhaylyk and D. J. Adams, Chem. Commun., 2016, 52, 6934–6937 RSC.
- B. D. Wall, S. R. Diegelmann, S. Zhang, T. J. Dawidczyk, W. L. Wilson, H. E. Katz, H.-Q. Mao and J. D. Tovar, Adv. Mater., 2011, 23, 5009–5014 CrossRef CAS PubMed.
- S. Zhang, M. A. Greenfield, A. Mata, L. C. Palmer, R. Bitton, J. R. Mantei, C. Aparicio, M. O. de la Cruz and S. I. Stupp, Nat. Mater., 2010, 9, 594–601 CrossRef CAS PubMed.
- Q. Tong, L. Zhang, Y. Li, B. Li and Y. Yang, Soft Matter, 2018, 14, 6353–6359 RSC.
- I. Ziemecka, G. J. M. Koper, A. G. L. Olive and J. H. van Esch, Soft Matter, 2013, 9, 1556–1561 RSC.
- M. Numata, Chem. – Asian J., 2015, 10, 2574–2588 CrossRef CAS PubMed.
- D. Hermida-Merino, M. Trebbin, S. Foerster, F. Rodriguez-Llansola and G. Portale, Macromol. Symp., 2015, 358, 59–66 CrossRef CAS.
- D. Kiriya, M. Ikeda, H. Onoe, M. Takinoue, H. Komatsu, Y. Shimoyama, I. Hamachi and S. Takeuchi, Angew. Chem., Int. Ed., 2012, 51, 1553–1557 CrossRef CAS PubMed.
- M. Numata, R. Nogami and A. Kitamura, ChemNanoMat, 2018, 4, 175–182 CrossRef CAS.
- S. Tamaru, M. Ikeda, Y. Shimidzu, S. Matsumoto, S. Takeuchi and I. Hamachi, Nat. Commun., 2010, 1, 20 CrossRef PubMed.
- R. Contreras-Montoya, A. B. Bonhome-Espinosa, A. Orte, D. Miguel, J. M. Delgado-López, J. D. G. Duran, J. M. Cuerva, M. T. Lopez-Lopez and L. Á. de Cienfuegos, Mater. Chem. Front., 2018, 2, 686–699 RSC.
- P. van der Asdonk, M. Keshavarz, P. C. M. Christianen and P. H. J. Kouwer, Soft Matter, 2016, 12, 6518–6525 RSC.
- C. Vigier-Carrière, F. Boulmedais, P. Schaaf and L. Jierry, Angew. Chem., Int. Ed., 2018, 57, 1448–1456 CrossRef PubMed.
- J. Rodon Fores, M. L. Martinez Mendez, X. Mao, D. Wagner, M. Schmutz, M. Rabineau, P. Lavalle, P. Schaaf, F. Boulmedais and L. Jierry, Angew. Chem., Int. Ed., 2017, 56, 15984–15988 CrossRef CAS PubMed.
- M. G. F. Angelerou, B. Yang, T. Arnold, J. Rawle, M. Marlow and M. Zelzer, Soft Matter, 2018, 14, 9851–9855 RSC.
- L. Fan, J.-L. Li, Z. Cai and X. Wang, ACS Nano, 2018, 12, 5780–5790 CrossRef CAS PubMed.
- Y. Dou, B. Wang, M. Jin, Y. Yu, G. Zhou and L. Shui, J. Micromech. Microeng., 2017, 27, 113002 CrossRef.
- M. Numata, Y. Takigami, M. Takayama, T. Kozawa and N. Hirose, Chem. – Eur. J., 2012, 18, 13008–13017 CrossRef CAS PubMed.
- A. Yaari, Y. Schilt, C. Tamburu, U. Raviv and O. Shoseyov, ACS Biomater. Sci. Eng., 2016, 2, 349–360 CrossRef CAS.
- K. D. Nelson, A. Romero, P. Waggoner, B. Crow, A. Borneman and G. M. Smith, Tissue Eng., 2003, 9, 1323–1330 CrossRef CAS PubMed.
- K. Tuzlakoglu, I. Pashkuleva, M. T. Rodrigues, M. E. Gomes, G. H. van Lenthe, R. Müller and R. L. Reis, J. Biomed. Mater. Res., Part A, 2010, 92, 369–377 CrossRef CAS PubMed.
- Y. Yang, X. Liu, D. Wei, M. Zhong, J. Sun, L. Guo, H. Fan and X. Zhang, Biofabrication, 2017, 9, 045009 CrossRef PubMed.
- S. Hirano, Macromol. Symp., 2001, 168, 21–30 CrossRef CAS.
- M. Desorme, A. Montembault, T. Tamet, P. Maleysson, T. Bouet and L. David, J. Appl. Polym. Sci., 2019, 136, 47130 CrossRef.
- B. R. Lee, K. H. Lee, E. Kang, D.-S. Kim and S.-H. Lee, Biomicrofluidics, 2011, 5, 022208 CrossRef PubMed.
- R. Madurga, A. M. Gañán-Calvo, G. R. Plaza, G. V. Guinea, M. Elices and J. Pérez-Rigueiro, Green Chem., 2017, 19, 3380–3389 RSC.
- N. Weatherbee-Martin, L. Xu, A. Hupe, L. Kreplak, D. S. Fudge, X.-Q. Liu and J. K. Rainey, Biomacromolecules, 2016, 17, 2737–2746 CrossRef CAS PubMed.
- Y. Hsia, E. Gnesa, R. Pacheco, K. Kohler, F. Jeffery and C. Vierra, J. Visualized Exp., 2012, 65, e4191 Search PubMed.
- J. Cheng, Y. Jun, J. Qin and S.-H. Lee, Biomaterials, 2017, 114, 121–143 CrossRef CAS PubMed.
- C. Mota, S.-Y. Wang, D. Puppi, M. Gazzarri, C. Migone, F. Chiellini, G.-Q. Chen and E. Chiellini, J. Tissue Eng. Regener. Med., 2017, 11, 175–186 CrossRef CAS PubMed.
- D. Puppi, C. Migone, A. Morelli, C. Bartoli, M. Gazzarri, D. Pasini and F. Chiellini, J. Bioact. Compat. Polym., 2016, 31, 531–549 CrossRef CAS.
- S. M. Hashemnejad and S. Kundu, Langmuir, 2017, 33, 7769–7779 CrossRef CAS PubMed.
- R. V. Bell, C. C. Parkins, R. A. Young, C. M. Preuss, M. M. Stevens and S. A. F. Bon, J. Mater. Chem. A, 2016, 4, 813–818 RSC.
- M. Hecht, B. Soberats, J. Zhu, V. Stepanenko, S. Agarwal, A. Greiner and F. Würthner, Nanoscale Horiz., 2018, 4, 169–174 RSC.
- M. Maleki, A. Natalello, R. Pugliese and F. Gelain, Acta Biomater., 2017, 51, 268–278 CrossRef CAS PubMed.
- G. Singh, A. M. Bittner, S. Loscher, N. Malinowski and K. Kern, Adv. Mater., 2008, 20, 2332–2336 CrossRef CAS.
- M. Allais, D. Mailley, P. Hébraud, D. Ihiawakrim, V. Ball, F. Meyer, A. Hébraud and G. Schlatter, Nanoscale, 2018, 10, 9164–9173 RSC.
- A. Celebioglu, F. Topuz, Z. I. Yildiz and T. Uyar, Carbohydr. Polym., 2019, 207, 471–479 CrossRef CAS PubMed.
- H. Yoshida, K. Kikuta and T. Kida, Beilstein J. Org. Chem., 2019, 15, 89–95 CrossRef CAS PubMed.
- M. G. McKee, J. M. Layman, M. P. Cashion and T. E. Long, Science, 2006, 311, 353–355 CrossRef CAS PubMed.
- M. Habibi, N. M. Ribe and D. Bonn, Phys. Rev. Lett., 2007, 99, 154302 CrossRef CAS PubMed.
- E. R. Draper, T. O. McDonald and D. J. Adams, Chem. Commun., 2015, 51, 6595–6597 RSC.
- W. Liyanage, W. W. Brennessel and B. L. Nilsson, Langmuir, 2015, 31, 9933–9942 CrossRef CAS PubMed.
- Y. Wang, L. Tang and J. Yu, Cryst. Growth Des., 2008, 8, 884–889 CrossRef CAS.
- B. P. Krishnan and K. M. Sureshan, ChemPhysChem, 2016, 17, 3062–3067 CrossRef CAS PubMed.
- J. Liu, F. Xu, Z. Sun, Y. Pan, J. Tian, H.-C. Lin and X. Li, Soft Matter, 2016, 12, 141–148 RSC.
- D. Krishna Kumar and J. W. Steed, Chem. Soc. Rev., 2014, 43, 2080–2088 RSC.
- K. A. Houton, K. L. Morris, L. Chen, M. Schmidtmann, J. T. A. Jones, L. C. Serpell, G. O. Lloyd and D. J. Adams, Langmuir, 2012, 28, 9797–9806 CrossRef CAS PubMed.
- J. H. Fuhrhop, P. Schnieder, E. Boekema and H. Wolfgang, J. Am. Chem. Soc., 1988, 110, 2861–2867 CrossRef CAS.
- C. André, P. Luger and S. Svenson, Carbohydr. Res., 1992, 230, 31–40 CrossRef.
- C. André, P. Luger, S. Svenson and J.-H. Fuhrhop, Carbohydr. Res., 1993, 240, 47–56 CrossRef.
- V. Zabel, A. Müller-Fahrnow, R. Hilgenfeld, W. Saenger, B. Pfannemüller, V. Enkelmann and W. Welte, Chem. Phys. Lipids, 1986, 39, 313–327 CrossRef CAS.
- S. M. Hashemnejad, M. M. Huda, N. Rai and S. Kundu, ACS Omega, 2017, 2, 1864–1874 CrossRef CAS.
- SADABS, Program for Data Correction, Bruker-AXS Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Jet velocity analysis and fiber width distribution histograms; thermogravimetric analysis; and SAXS, WAXS and RX. See DOI: 10.1039/c9nr02727k |
| This journal is © The Royal Society of Chemistry 2019 |