
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the membrane permeability of polymersome nanoreactors developed by aqueous emulsion polymerization-induced self-assembly†
Spyridon
Varlas
 a,
Jeffrey C.
Foster
a,
Panagiotis G.
Georgiou
ab,
Robert
Keogh
ab,
Jonathan T.
Husband
a,
David S.
Williams
ac and
Rachel K.
O'Reilly
*a
a,
Jeffrey C.
Foster
a,
Panagiotis G.
Georgiou
ab,
Robert
Keogh
ab,
Jonathan T.
Husband
a,
David S.
Williams
ac and
Rachel K.
O'Reilly
*a
aSchool of Chemistry, University of Birmingham, B15 2TT, Birmingham, UK. E-mail: r.oreilly@bham.ac.uk
bDepartment of Chemistry, University of Warwick, Gibbet Hill Road, CV4 7AL, Coventry, UK
cDepartment of Chemistry, College of Science, Swansea University, SA2 8PP, Swansea, UK
First published on 15th June 2019
Abstract
Polymeric vesicles (or polymersomes) are hollow bilayer structures consisting of an inner aqueous compartment enclosed by a hydrophobic membrane. Vesicular constructs are ubiquitous in nature and perform a variety of functions by compartmentalizing molecules into disparate environments. For polymer chemists, the synthesis of vesicles can be readily accomplished using polymerization-induced self-assembly (PISA), whereby pure vesicle morphologies can be easily accessed by tuning initial reaction parameters. Research into polymersomes is motivated primarily by the fact that hydrophilic cargo such as drug molecules, DNA, or enzymes can be encapsulated and protected from the often harsh conditions of the surrounding environment. A key factor governing the capability of vesicles to retain and protect their cargo is the permeability of their hydrophobic membrane. Herein, we demonstrate that membrane permeability of enzyme-loaded epoxy-functionalized polymersomes synthesized by aqueous emulsion PISA can be modulated via epoxide ring-opening with various diamine cross-linkers and hydrophobic primary amines. In general, membrane cross-linking or amine conjugation resulted in increased polymersome membrane thickness. Membrane modification was also found to decrease permeability in all cases, as measured by enzymatically-catalysed oxidation of an externally administered substrate. Functionalization with hydrophobic amines resulted in the largest reduction in enzyme activity, suggesting significant blocking of substrate diffusion into the central aqueous compartment. This procedurally facile strategy yields meaningful insight into how the chemical structure of the membrane influences permeability and thus could be generally applied to the formulation of polymeric vesicles for therapeutic applications.
Introduction
Structural organization is an essential feature of nature's toolbox for maintaining all forms of life. Evolution of compartmentalized environments on both cellular and subcellular level (i.e. organelles) allows for vital biological reactions to occur selectively in confined spaces that simultaneously separate and protect them from external detrimental agents.1,2 Communication and transport of energy, nutrients and other signaling molecules between such compartments is achieved via metabolic pathways that in most cases involve diffusion through semi-permeable or stimuli-responsive membranes.2–4Inspired by nature, researchers have developed methodologies to design minimal synthetic analogues that mimic these complex systems.5 Among them, self-assembled bilayer nanostructures such as liposomes and amphiphilic block copolymer vesicles (also referred to as polymersomes) have been studied extensively for their application as functional artificial organelles and catalytic nanoreactors due to their ability to incorporate both hydrophilic and hydrophobic molecules into their domains.6–12 Additionally, polymersomes have attracted significant research interest owing to their higher chemical versatility, physical stability and more facile functionalization in comparison to liposomes.13–15
Until recently, preparation of polymeric vesicles was achieved by multi-step conventional block copolymer self-assembly strategies in solution, such as solvent-switch or thin-film rehydration, at low polymer concentrations (≤1% w/w) that in the majority of cases require the use of organic solvents.16–20 Over recent years, aqueous polymerization-induced self-assembly (PISA) has been established as a powerful single-step approach for in situ fabrication of block copolymer nano-objects at high solids concentrations (typically 10–30% w/w) that allows for access to higher-order morphologies, such as worm-like micelles and polymersomes, in a reproducible manner.21–26
In particular, development of single-phase block copolymer vesicles via PISA in dispersed aqueous media has been primarily achieved using reversible addition–fragmentation chain-transfer (RAFT) polymerization,22,27,28 as well as non-radical methodologies such as ring-opening metathesis polymerization (ROMP).29,30 However, a limited number of reports centered upon polymersomes prepared under emulsion PISA conditions have been introduced in the literature thus far,31–34 while the factors that allow for higher-order morphologies other than kinetically trapped spheres to be accessed under emulsion polymerization conditions remain currently unclear.
Importantly, different methodologies to conduct visible-light initiated PISA (photo-PISA) for synthesis of nano-objects at ambient reaction temperatures either by using special photoinitiators and photoredox catalysts or via the “photoiniferter” mechanism of chain transfer agents (CTAs) have been recently reported.35–41 Aqueous photo-PISA has enabled the direct non-disruptive encapsulation of inorganic nanoparticles,36,42 as well as other sensitive (bio)molecules, such as fluorophores,36,43 and proteins/enzymes,43–48 into polymeric vesicles for the efficient construction of delivery vehicles, therapeutics, and catalytic nanoreactors with biologically relevant applications.49
To date, studies in the field of cell-mimicking enzyme-loaded polymersome nanoreactors have been pioneered and extensively investigated by the van Hest, Lecommandoux, Meier, Battaglia, and Voit groups.50–54 The membranes of such nanoconstructs were rendered permeable toward substrate molecules and catalysis products upon incorporation of channel-forming transmembrane proteins,52,55 DNA nanopores,56 or stimuli-responsive moieties51,54 into their hydrophobic domains. Furthermore, a few reports were based upon the inherent permeability of the polymersome membranes for passive diffusion of small molecules between their outer and inner aqueous compartments.57,58
More recently, our group has demonstrated the preparation of enzyme-loaded poly(ethylene glycol)-b-poly(2-hydroxypropyl methacrylate) (PEG-b-PHPMA) nanoreactors via one-pot aqueous photo-PISA that were able to communicate through their inherently permeable and relatively hydrated membranes, allowing for catalytic cascade reactions to occur inside separated compartments.43 Moreover, encapsulation of a therapeutic enzyme into the same system resulted in nanoreactors intended for leukemia treatment, whilst protection of the cargo from antibody binding and proteolytic degradation owing to the size-selective permeability of the PHPMA membrane was also presented.46 In a different study by our group, incorporation of a channel-forming porin protein into the membrane of PEG-b-PHPMA vesicles led to a significant permeability enhancement.47 Other attempts to enhance the membrane permeability of PISA polymersomes by incorporating pH-responsive units into their core-forming blocks for drug release applications have also been reported recently.59,60
Herein, enzyme-loaded epoxy-functionalized polymersome nanoreactors of well-defined characteristics and inherent permeability were developed via aqueous RAFT-mediated emulsion photo-PISA at mild temperature using a mixture of 2-hydroxypropyl methacrylate (HPMA) and glycidyl methacrylate (GlyMA) as the core-forming monomers. The pendant epoxide groups of PGlyMA units provided a reactive handle for straightforward post-PISA functionalization of the membrane through nucleophilic ring-opening reactions induced by a series of primary amines and cross-linking diamines.61–63 In all cases, modification of the chemical composition of the core-forming block resulted in a distinct increase of vesicular membrane thickness and as a consequence in less-hydrated nanoreactors with tunable permeability toward small molecule substrates, as determined by enzymatic assays. Enhanced blocking efficiency was evident upon increasing the hydrophobicity of the nucleophile employed, allowing for identification of valuable structure–property relationships. Overall, our findings expand the current knowledge on membrane characteristics of semi-permeable nanocompartments and could facilitate the design of biomembrane-mimicking nanostructures and artificial “nanofactories” with programmed size-selective permeability via one-pot PISA.
Experimental section
Materials and methods
Materials and characterization techniques used are included in the ESI.†Synthetic procedures
A typical synthetic procedure to achieve epoxy-functionalized PEG113-b-P(HPMA320-co-GlyMA80) diblock copolymer vesicles at [solids] = 10% w/w via aqueous RAFT-mediated emulsion photo-PISA is described (Fig. 1A).43,47 PEG113 macro-CTA (9.1 mg, 1.7 × 10−6 mol, 1 eq.), HPMA (79 mg, 5.5 × 10−4 mol, 320 eq.) (80 mol%) and GlyMA (19.9 mg, 1.4 × 10−4 mol, 80 eq.) (20 mol%) were dispersed in deionized (DI) water (0.9 mL) with vigorous agitation using a vortex mixer for 2 min in a sealed 15 mL scintillation vial bearing a magnetic stirrer bar. The resulting monomer-in-water emulsion solution was degassed by sparging with N2(g) for 15 min. The sealed vial was incubated at 37 °C with magnetic stirring under 405 nm light irradiation for 2 hours to ensure full monomer conversion. After this period, the reaction mixture was exposed to air and allowed to cool to room temperature. FT-IR, 1H-NMR in methanol-d4 and SEC analyses in DMF + 5 mM NH4BF4 of the pure diblock copolymers were performed after lyophilization of an aliquot of particles. DLS analysis and dry-state and cryo-TEM imaging were performed on samples after dilution to an appropriate analysis concentration. 1H-NMR (400 MHz, methanol-d4): δ (ppm) 4.78 (br s, OH), 4.40 (br s, CH2 of PGlyMA side chain), 4.03 and 3.87 (br s, CH and CH2 of PHPMA side chain), 3.66 (br s, CH2CH2O of PEG), 3.33 (br s, CH of PGlyMA epoxide ring), 2.91 and 2.73 (br s, CH2 of PGlyMA epoxide ring) 2.30–1.80 (br m, CH2 of PHPMA and PGlyMA backbone), 1.45–0.81 (br m, CH3 of PHPMA and PGlyMA backbone and CH3 of PHPMA side chain).
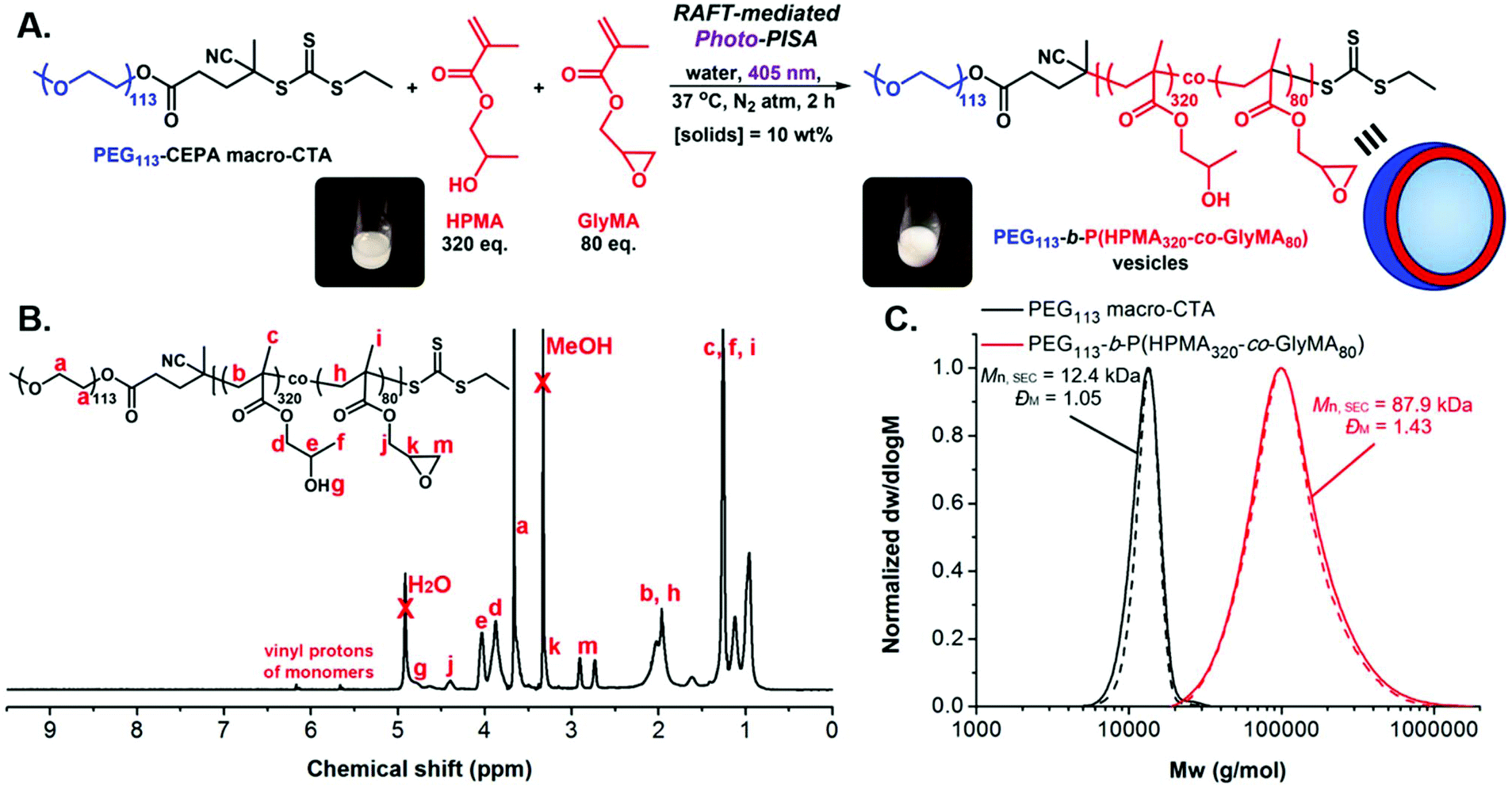 | ||
| Fig. 1 (A) Schematic illustration of the synthetic route employed for the preparation of PEG113-b-P(HPMA320-co-GlyMA80) diblock copolymer vesicles at [solids] = 10% w/w via aqueous RAFT-mediated emulsion photo-PISA (405 nm irradiation), using a PEG113 macro-CTA. Insets show images of the polymerization solution vial before (left) and after (right) photo-PISA. (B) Conversion 1H-NMR spectrum of crude PEG113-b-P(HPMA320-co-GlyMA80) diblock copolymer in methanol-d4, showing near quantitative monomer consumption (>98%). (C) Normalized SEC RI (solid lines) and SEC UV (dashed lines) molecular weight distributions for PEG113 macro-CTA (black traces) and PEG113-b-P(HPMA320-co-GlyMA80) diblock copolymer (red traces), along with their corresponding Mn and ĐM values calculated based on PMMA standards using DMF + 5 mM NH4BF4 as the eluent. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for the removal of unreacted monomer and free HRP enzyme.
000 rpm prior to kinetic colorimetric analysis. Successful ring-opening of PGlyMA units was confirmed by FT-IR spectroscopy of a lyophilized sample. The above protocol was also followed for 1-naphthylmethylamine (NMA) and 1-adamantanemethylamine (AMA). In the case of cross-linking diamines poly(ethylene glycol)n diamine (n = 23, 46) (PEGnDA), ethylenediamine (C2DA), 1,3-diaminopropane (C3DA), 1,4-diaminobutane (C4DA), hexamethylenediamine (C6DA), and p-xylylenediamine (PXDA), a [diamine]/[epoxide] molar ratio = 1.0 was maintained.
000 rpm for the removal of unreacted monomer and free HRP enzyme.
000 rpm prior to kinetic colorimetric analysis. Successful ring-opening of PGlyMA units was confirmed by FT-IR spectroscopy of a lyophilized sample. The above protocol was also followed for 1-naphthylmethylamine (NMA) and 1-adamantanemethylamine (AMA). In the case of cross-linking diamines poly(ethylene glycol)n diamine (n = 23, 46) (PEGnDA), ethylenediamine (C2DA), 1,3-diaminopropane (C3DA), 1,4-diaminobutane (C4DA), hexamethylenediamine (C6DA), and p-xylylenediamine (PXDA), a [diamine]/[epoxide] molar ratio = 1.0 was maintained.
For Michaelis–Menten kinetics determination, activity of purified HRP-loaded PEG113-b-P(HPMA320-co-GlyMA80) vesicles before and after post-PISA membrane functionalization using BA, NMA, and PXDA was evaluated at different [DMB] (0–6 mM) with H2O2 under saturating conditions. Vesicles at 20× dilution (0.5% w/w solids content) in 100 mM PB 5.5 (120 μL) were diluted with 100 mM PB 5.5 (20 μL) in a 96-well plate microwell. DMB of appropriate concentration (0–30 mM, 40 μL) was then added. Finally, a 35% w/v aqueous solution of hydrogen peroxide (20 μL) was added, and the change in absorbance was monitored in an identical manner. Absorbance values were corrected against particle absorbance at t = 0 min and reported as ΔAbs492 nm. In each case, the average initial slope of three repeat measurements (V0) for the first 10 min of the assay was used for construction of Michaelis–Menten kinetic plots and was normalized against Vmax (Table S3†). Calculated 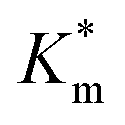 values are presented as mean ± standard error.
values are presented as mean ± standard error.
A similar process was followed for activity testing of the free enzyme after a series of control experiments at appropriate [HRP]. The free HRP solutions at final [HRP] = 1 U mL−1 in 100 mM PB 5.5 (20 μL) were diluted with 100 mM PB 5.5 (120 μL) in a 96-well plate microwell. DMB (2 mM, 40 μL) was then added. Finally, a 35% w/v aqueous solution of hydrogen peroxide (20 μL) was added, and the change in absorbance was monitored in an identical manner. In all cases, measurements were performed in at least triplicate and results are reported as their average values.
Results and discussion
Synthesis of epoxy-functionalized PEG113-b-P(HPMA320-co-GlyMA80) polymersomes via aqueous RAFT-mediated emulsion photo-PISA
Following related PISA studies previously reported by our group and others,36,40 a water-soluble poly(ethylene glycol) macromolecular chain transfer agent (PEG113 macro-CTA) was first synthesized through an esterification reaction between poly(ethylene glycol) monomethyl ether (PEG113-OH) and an acid-functionalized CTA (esterification efficiency = 93%, Mn, SEC = 12.4 kg mol−1, ĐM = 1.05). Aqueous RAFT-mediated photoinitiated PISA (photo-PISA) of a mixture of commercially available water-miscible 2-hydroxypropyl methacrylate (HPMA) and water-immiscible glycidyl methacrylate (GlyMA) as the core-forming monomers was achieved under emulsion polymerization conditions, using PEG113 macro-CTA as both the steric stabilizer block and the surfactant for stabilization of the heterogeneous monomer-in-water solution.64 Dispersed monomer droplets of varying size (5–30 μm) were observed upon optical microscopy imaging of the formed emulsion solution prior to polymerization (Fig. S2†).Single-phase epoxy-functionalized PEG113-b-P(HPMA-co-GlyMA) diblock copolymer vesicles were developed upon irradiation of the polymerization solution under 405 nm visible light in the absence of an externally added photoinitiator or catalyst (“photoiniferter” mechanism of trithiocarbonates)35,65,66 at 37 °C, targeting DPPHPMA = 320 (80 mol%) and DPPGlyMA = 80 (20 mol%) at 10% w/w solids content (Fig. 1A). Emulsion photo-PISA was carried out at mild temperature to ensure quantitative retention of epoxy functional groups, as epoxides can undergo partial hydrolysis in aqueous solution at elevated temperatures (60–100 °C).61 Importantly, 99% of PGlyMA pendant epoxy groups remained intact after photo-PISA, as calculated from 1H-NMR spectroscopy by comparing the integral ratio of the peaks corresponding to epoxy proton signals at 2.73 and 2.91 ppm (I2.73/2.91 ppm = 1.98) to the peak of the methacrylic backbone –CH2– protons at 1.50–2.30 ppm (I1.50–2.30 ppm = 10.00) (Fig. S3†). A kinetic study revealed that near complete monomer conversion (>98%) was achieved after 2 hours of irradiation time, as calculated by 1H-NMR spectroscopy, whilst the onset of particle micellization accompanied by an increase in polymerization rate was observed after ∼20 min of irradiation time (ca. 17% monomer conversion) (Fig. 1B and S4A†). The distribution of PGlyMA units along the growing polymer chain was also investigated during kinetics monitoring and the calculated HPMA/GlyMA molar ratio varied between 3 and 4 throughout the whole duration of photo-PISA, suggesting the formation of random copolymers (Fig. S4B†). The prepared PEG113-b-P(HPMA320-co-GlyMA80) diblock copolymer possessed monomodal molecular weight distribution with relatively low dispersity value, as determined by SEC analysis (Mn, SEC = 87.9 kg mol−1, ĐM = 1.43) (Fig. 1C).
Dynamic light scattering (DLS) analysis revealed the formation of nano-objects with a monomodal size distribution and mean hydrodynamic diameter (Dh) of 188.3 ± 5.6 nm, low polydispersity (PD = 0.18 ± 0.02) and a smooth, single exponential decay autocorrelation function with optimal signal-to-noise ratio (Y-intercept >0.9) (Fig. 2A and B). The absence of charges on the outer surface of the developed particles was confirmed by microelectrophoretic analysis at neutral pH (measured zeta-potential = −1.87 ± 0.10 mV). Dry-state transmission electron microscopy (TEM) imaging confirmed the successful development of well-defined PEG113-b-P(HPMA320-co-GlyMA80) unilamellar vesicles of spherical shape and uniform size (Ddry-state = 170.3 ± 23.0 nm) (Fig. 2C and S5A†). Additionally, the characteristics of PEG113-b-P(HPMA320-co-GlyMA80) vesicles in solution were studied by cryogenic TEM (cryo-TEM) imaging. Average vesicle size measured from particle counting analysis based on cryo-TEM was in good agreement with values determined by DLS and dry-state TEM analyses (Dcryo = 175.5 ± 24.1 nm), while calculated average membrane thickness was Mave = 28.0 ± 3.0 nm (Fig. 2D–F and S5B†).
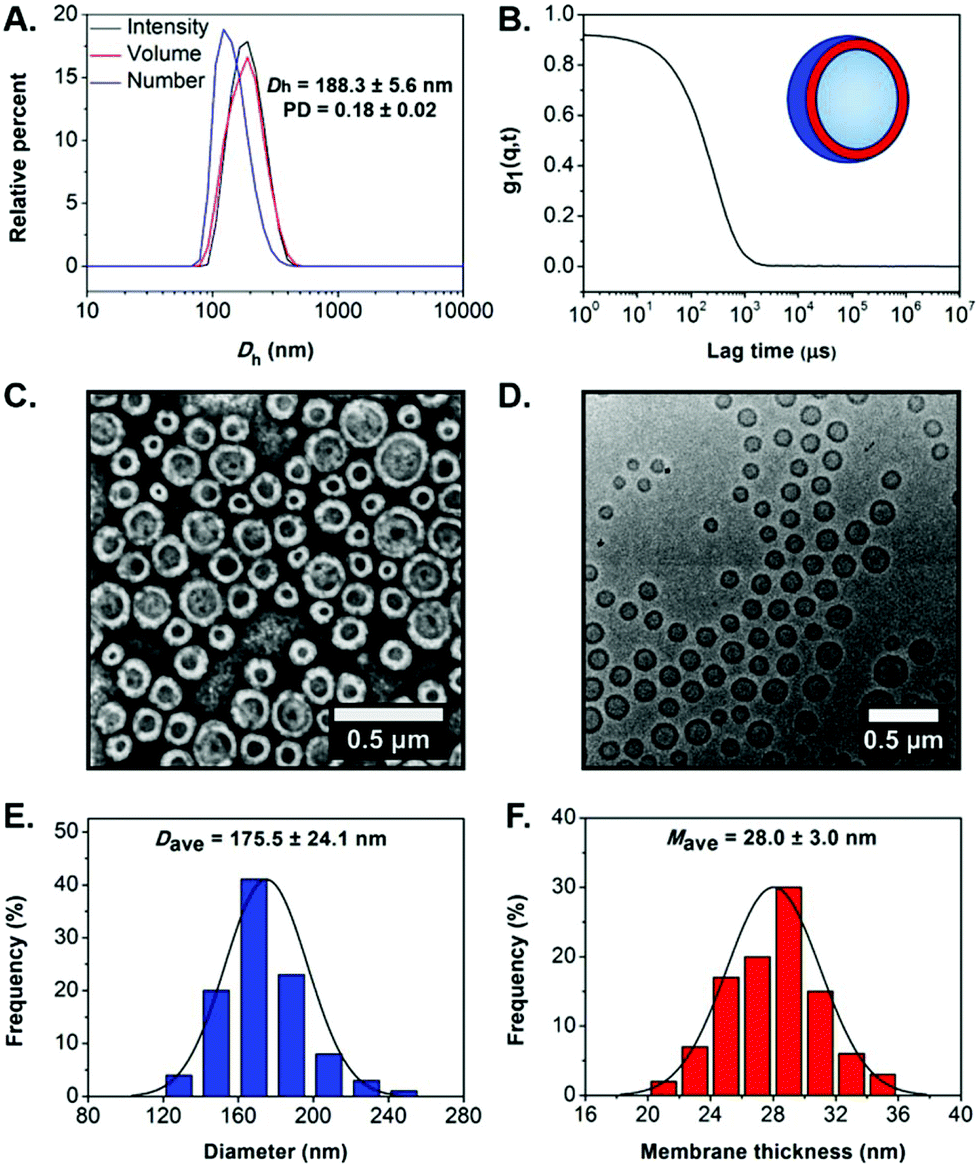 | ||
| Fig. 2 (A) Intensity-weighted size distributions along with average Dh and PD values (the error shows the standard deviation from 5 repeat measurements), and (B) autocorrelation function obtained by DLS for empty PEG113-b-P(HPMA320-co-GlyMA80) vesicles. (C) Representative dry-state TEM image, stained with 1 wt% uranyl acetate (UA) solution, and (D) representative cryo-TEM image of empty PEG113-b-P(HPMA320-co-GlyMA80) vesicles. (E) Histogram of size distribution, and (F) histogram of membrane thickness distribution along with calculated average diameter and membrane thickness values, respectively, measured from particle analysis based on cryo-TEM images for empty PEG113-b-P(HPMA320-co-GlyMA80) vesicles. In each case, at least 100 particles were analyzed. | ||
Preparation of catalytic enzyme-loaded PEG113-b-P(HPMA320-co-GlyMA80) polymersome nanoreactors via one-pot aqueous emulsion photo-PISA
Next, PEG113-b-P(HPMA320-co-GlyMA80) polymersomes were used as a nanocarrier platform for loading of a model hydrophilic enzyme to prepare catalytically active epoxy-functionalized nanoreactors.43,47 Enzyme horseradish peroxidase (HRP) was encapsulated into the inner aqueous compartment of the vesicles by performing a one-pot emulsion photo-PISA process under the same mild reaction conditions described herein in the presence of an aqueous HRP solution for the development of enzyme-loaded PEG113-b-P(HPMA320-co-GlyMA80) polymersome nanoreactors. Purification of the vesicle sample for complete removal of free HRP was achieved by consecutive centrifugation/resuspension cycles in either deionized (DI) water or 100 mM phosphate buffer (pH = 5.5, PB 5.5).Importantly, the encapsulation process of HRP into PEG113-b-P(HPMA320-co-GlyMA80) polymersomes did not alter their overall characteristics as judged by DLS analysis and TEM imaging (Fig. S6†). Comparable Dh and PD values were measured for HRP-loaded vesicles with respect to non-HRP-loaded vesicles (Dh = 182.6 ± 2.5 nm, and PD = 0.14 ± 0.04), whilst average vesicle size and average membrane thickness measured from cryo-TEM images were Dcryo = 172.5 ± 25.5 nm and Mave = 27.9 ± 2.6 nm, respectively.
HRP is known to catalyze the oxidation reaction of colourless substrate 3,3′-dimethoxybenzidine (DMB) to a coloured dimer product (red-brown), which can be monitored by measuring the change in absorbance at λ = 492 nm over time via kinetic colorimetric analysis. This assay provides a read-out of enzyme activity and consequently of polymersome membrane permeability. Notably, PEG113-b-P(HPMA320-co-GlyMA80) vesicles were found to be inherently permeable toward small molecule DMB, although they presented reduced enzyme activity compared to their reported PEG113-b-PHPMA400 counterparts presumably due to increased membrane hydrophobicity in the former case.43 Although nanoreactors with near identical membrane thickness values were developed in both cases, the presence of an additional hydrophobic core-block component in the case of PEG113-b-P(HPMA320-co-GlyMA80) vesicles resulted in particles with less hydrated membranes and reduced permeability, making the exchange of substrates between their outer and inner compartment a more difficult process.47 Control experiments to assess the activity of free HRP after incubation at 37 °C for 2 hours in 10% w/w HPMA or HPMA/GlyMA (80:20 molar ratio) monomer solutions in DI water under 405 nm irradiation (photo-PISA conditions) revealed quantitative retention of activity in both cases compared to untreated enzyme, showing that the enzyme function was not affected by either monomer (Fig. S7†). In addition, high-performance liquid chromatography (HPLC) and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-ToF MS) were employed to determine potential modification of HRP by ring-opening of the epoxide groups via its lysine residues. Notably, incubation of free HRP with a water-soluble small molecule epoxide (glycidol) for 2 hours in DI water showed no evident modification upon comparison with the untreated enzyme (Fig. S8†).
Membrane functionalization of enzyme-loaded PEG113-b-P(HPMA320-co-GlyMA80) vesicles via ring-opening of epoxide groups using primary amines
Incorporation of PGlyMA units and thus the presence of pendant epoxy groups within the hydrophobic membrane of prepared HRP-loaded nanoreactors provides a reactive handle that allows for post-PISA functionalization of the polymersomes with nucleophilic compounds. A commonly utilized procedure involves the ring-opening of epoxide groups by primary amines under mild reaction conditions.61–63 To this extent, we envisioned that introducing hydrophobic or cross-linking moieties into the polymersome membrane would result in permeability reduction or nanoreactors completely impermeable toward small molecules.A series of commercially available primary amines and diamines (cross-linking agents) of varying hydrophobicity were chosen as nucleophiles for ring-opening of PGlyMA epoxy groups and subsequent vesicle membrane modification (Scheme 1). Post-polymerization functionalization was achieved by mixing purified HRP-loaded PEG113-b-P(HPMA320-co-GlyMA80) vesicle solutions in DI water at [solids] = 1% w/w with different amines at room temperature for 18 hours. In all cases, the [amine]/[epoxide] molar ratio was constantly maintained at 2.0 (i.e. [diamine]/[epoxide] = 1.0).61–63 Following this period, membrane-functionalized polymersomes were transferred into PB 5.5 (optimum pH value of HRP) by one centrifugation/resuspension cycle prior to kinetic colorimetric analysis for enzyme activity and nanoreactor permeability determination. FT-IR spectroscopy was used for the confirmation of successful ring-opening of PGlyMA units in each case by monitoring the characteristic asymmetric vibration peaks of epoxide groups at 849 and 909 cm−1 before and after ring-opening reactions (Fig. S11, S14 and S16†). It should also be noted that incubation of free HRP in DI water at room temperature for 18 hours as a control experiment resulted in no loss of enzyme activity (>99% retention of activity), compared to incubation of the enzyme in PB 5.5 for the same period of time (Fig. S9†). Additionally, a control experiment for comparison of the activity of non-epoxy-functionalized and purified HRP-loaded PEG113-b-PHPMA400 vesicles before and after incubation with the utilized primary amine and diamine molecules at room temperature for 18 hours in DI water, highlighted that the prolonged presence of amines in the particle dispersion solutions did not affect the catalytic activity of HRP (>95% retention of activity in all cases – control vesicle solutions after incubation with CnDA cross-linkers could not be resuspended in PB 5.5 and hence their activity was not assessed) (Fig. S10†).
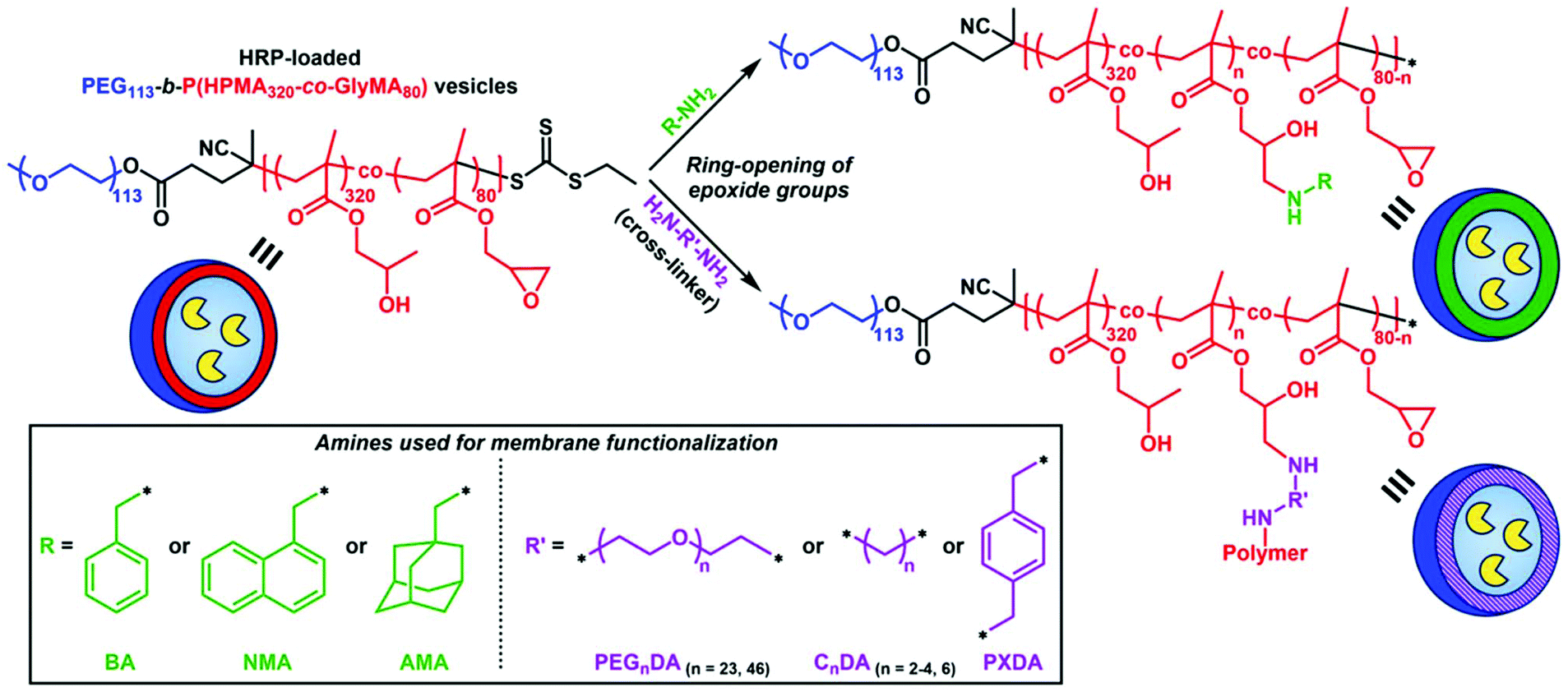 | ||
| Scheme 1 Schematic illustration of the membrane functionalization procedure of HRP-loaded PEG113-b-P(HPMA320-co-GlyMA80) polymersome nanoreactors via ring-opening of pendant PGlyMA epoxide groups using a series of primary amines as nucleophiles to yield polymersomes with controlled permeability. | ||
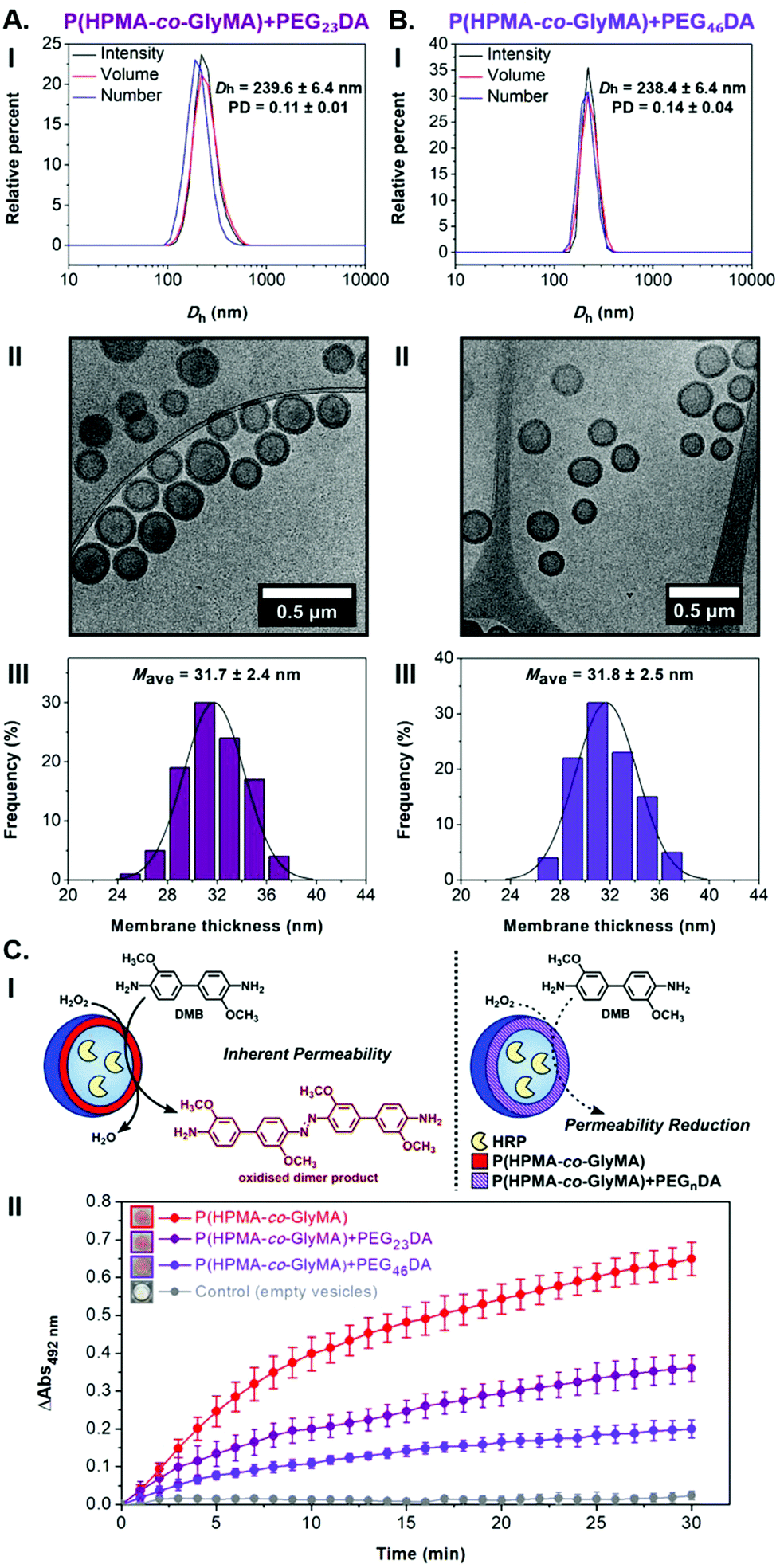 | ||
| Fig. 3 (A) For HRP-loaded PEG113-b-P(HPMA320-co-GlyMA80) + PEG23DA vesicles, and (B) for HRP-loaded PEG113-b-P(HPMA320-co-GlyMA80) + PEG46DA vesicles: (I) Intensity-weighted size distributions along with average Dh and PD values obtained by DLS (the error shows the standard deviation from 5 repeat measurements), (II) representative cryo-TEM images, and (III) histograms of membrane thickness distribution along with calculated average membrane thickness values measured from particle analysis based on cryo-TEM images. In each case, at least 100 particles were analyzed. (C) Schematic illustration of HRP-catalyzed oxidation reaction of DMB to a red-brown dimer product, detected by kinetic colorimetric assay, taking place inside inherently permeable HRP-loaded PEG113-b-P(HPMA320-co-GlyMA80) vesicles versus cross-linked HRP-loaded PEG113-b-P(HPMA320-co-GlyMA80) + PEGnDA vesicles (I), and enzymatic activity of the purified empty (grey line), HRP-loaded (red line), and HRP-loaded PEGnDA-functionalized (purple lines) PEG113-b-P(HPMA320-co-GlyMA80) vesicles (end point = 30 min, λ = 492 nm) (the insets show the end-point microwells in each case) (II). | ||
Surprisingly, activity comparison between encapsulated HRP into inherently permeable PEG113-b-P(HPMA320-co-GlyMA80) vesicles and PEGnDA cross-linked PEG113-b-P(HPMA320-co-GlyMA80) vesicles showed a significant enzyme activity decrease for the latter ones. More specifically, an absorbance decrease of 45 ± 5% was measured from kinetic colorimetric analysis after 30 min in the case of cross-linked PEG113-b-P(HPMA320-co-GlyMA80) + PEG23DA vesicles compared to their non-functionalized epoxide-containing counterparts, while a further absorbance decrease of 69 ± 4% was identified upon increasing the molecular weight of the PEGnDA cross-linker (n = 46) (Fig. 3C and S13†). The observed decrease in HRP activity demonstrates the permeability reduction of PEGnDA-functionalized nanoreactors toward DMB and hydrogen peroxide. Contrary to the observed particle swelling that would suggest a potential permeability enhancement, this can be understood in terms of the considerably thicker and more densely packed vesicular membranes after cross-linking that create an additional diffusive barrier which in turn hinders the passage of small molecule substrates to reach the active enzyme site.43
FT-IR spectroscopy was used to confirm the successful modification of the polymersome membrane (i.e. disappearance of the characteristic bands of epoxide groups at 849 and 909 cm−1) (Fig. S14†). Interestingly, DLS analysis of the CnDA-functionalized HRP-loaded polymersomes revealed a size decrease to 125–130 nm (PD = 0.12–0.13) for the shorter C2DA and C3DA cross-linkers compared to the original PEG113-b-P(HPMA320-co-GlyMA80) vesicles which was attributed to shrinkage of the particles upon cross-linking with hydrophobic compounds that results in exclusion of water molecules from the membrane. In the case of longer C4DA and C6DA cross-linkers, a vast Dh increase was measured to 1497 ± 266 nm and 1389 ± 149 nm, respectively, accompanied by a noticeable polydispersity increase (PD = 0.23–0.24) (Fig. 4A). These findings suggest the occurrence of particle agglomeration in the latter cases due to the development of inter-vesicular interactions resulting in the apparent population of particles with increased size. This was also evident by visual inspection of the PISA solutions upon functionalization after one week, where macroscopic precipitation was visible to some extent for PEG113-b-P(HPMA320-co-GlyMA80) + C4/6DA assemblies, implying the occurrence of time-dependent coagulation in aqueous media. The morphology retention of HRP-loaded PEG113-b-P(HPMA320-co-GlyMA80) + CnDA polymersomes was also verified by dry-state and cryo-TEM imaging after post-PISA functionalization (Dcryo = 116–120 nm), for which near identical vesicle formulations were observed in all cases. A negligible population of aggregated particles could only be observed in the case of C4DA and C6DA-functionalized polymersomes that supports the DLS findings described above. The average membrane thickness of the prepared nanoreactors was also calculated from particle counting analysis in each case. Increased membrane thickness values of Mave = 30–32 nm were measured in all cases, comparable to PEGnDA-functionalized vesicles (Fig. 4B and S15†).
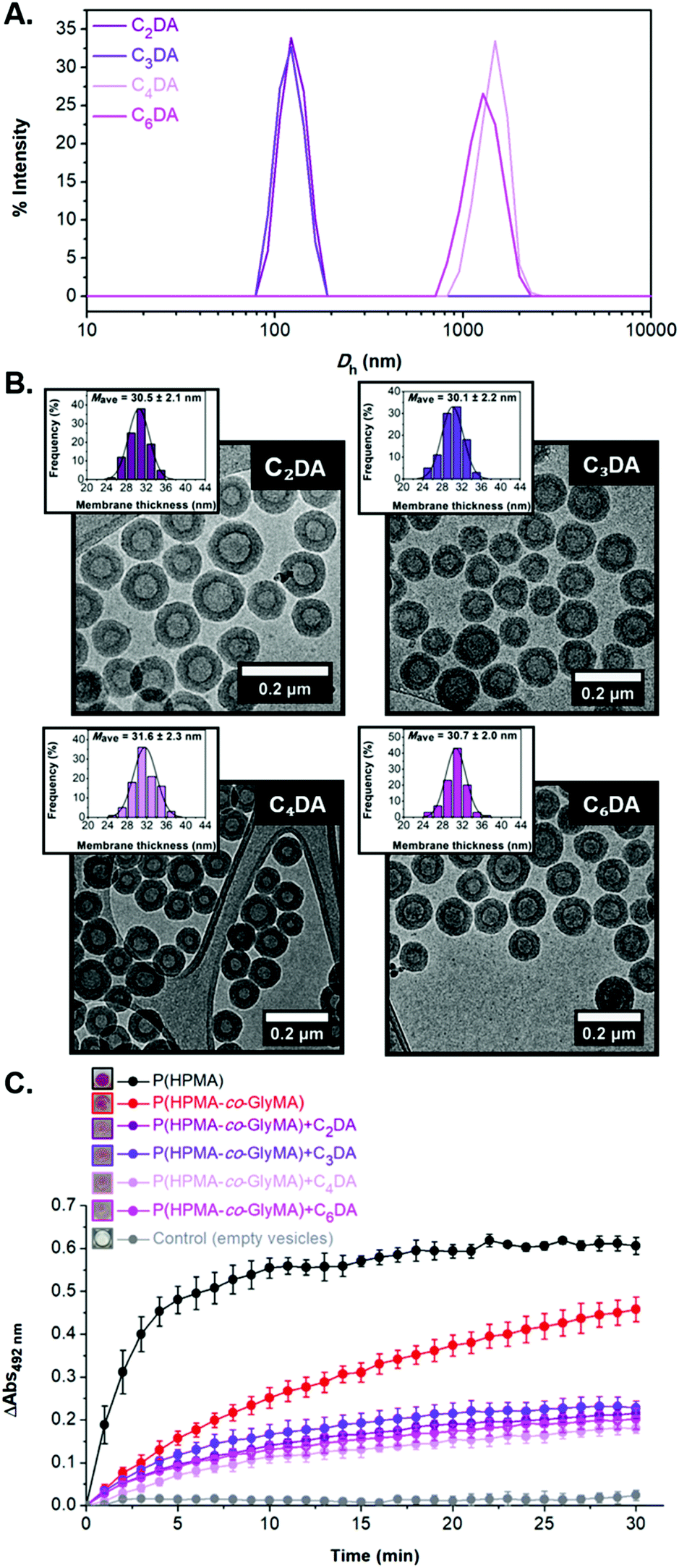 | ||
| Fig. 4 (A) Intensity-weighted size distributions along with average Dh and PD values obtained by DLS (the error shows the standard deviation from 5 repeat measurements) for HRP-loaded PEG113-b-P(HPMA320-co-GlyMA80) + CnDA (n = 2, 3, 4, 6) vesicles. (B) Representative cryo-TEM images of HRP-loaded PEG113-b-P(HPMA320-co-GlyMA80) + CnDA vesicles, and corresponding histograms of membrane thickness distribution along with calculated average membrane thickness values measured from particle analysis based on cryo-TEM images. In each case, at least 100 particles were analyzed. (C) Enzymatic activity of the purified empty (grey line), HRP-loaded (red line), and HRP-loaded CnDA-functionalized (purple/pink lines) PEG113-b-P(HPMA320-co-GlyMA80) vesicles, and HRP-loaded PEG113-b-P(HPMA)400 vesicles (black line) (end point = 30 min, λ = 492 nm) (the insets show the end-point microwells in each case). | ||
Similar to PEGnDA cross-linked vesicles, assessment of the enzyme activity of HRP-loaded PEG113-b-P(HPMA320-co-GlyMA80) + CnDA nanoreactors through kinetic colorimetric analysis revealed an absorbance decrease ranging from 50% to 61% depending on the cross-linker molecule used as compared to their PEG113-b-P(HPMA320-co-GlyMA80) counterparts (Fig. 4C). The absorbance decrease measured was approximately the same in all cases, whilst there was no apparent trend based on the length of the cross-linker used for ring-opening of PGlyMA units (i.e. increasing the CnDA length didn't result in an analogous HRP activity decrease). Overall, membrane cross-linking of the vesicles using CnDA molecules and subsequent introduction of additional hydrophobicity yielded particles with thicker and possibly less hydrated membranes with markedly reduced permeability.
In the case of BA-functionalized vesicles, Dh and PD values comparable with those of the original HRP-loaded vesicles were measured from DLS analysis (Dh = 203.4 ± 2.8 nm and PD = 0.08) (Fig. 5A-I and II). Dry-state TEM imaging was first used to confirm that post-PISA functionalization did not alter their morphology and uniform size (Fig. 5A-III and S17A-I†), while size and membrane thickness measurements were carried out from particle counting analysis based on cryo-TEM images (Fig. 5A-IV to VI and S17A-II†). Importantly, the average diameter of BA membrane-modified vesicles was in good agreement with DLS results (Dcryo, BA = 175.4 ± 27.7 nm), whereas a significant increase in average membrane thickness was also observed (Mave, BA = 33.2 ± 2.9 nm) compared to PEG113-b-P(HPMA320-co-GlyMA80) vesicles.
 | ||
| Fig. 5 (A) For HRP-loaded PEG113-b-P(HPMA320-co-GlyMA80) + BA vesicles, (B) for HRP-loaded PEG113-b-P(HPMA320-co-GlyMA80) + NMA vesicles, and (C) for HRP-loaded PEG113-b-P(HPMA320-co-GlyMA80) + PXDA vesicles: (I) Intensity-weighted size distributions along with average Dh and PD values (the error shows the standard deviation from 5 repeat measurements), and (II) autocorrelation functions obtained by DLS, (III) representative dry-state TEM images, stained with 1 wt% UA solution, and (IV) representative cryo-TEM images, (V) histograms of size distribution, and (VI) histograms of membrane thickness distribution along with calculated average diameter and membrane thickness values, respectively, measured from particle analysis based on cryo-TEM images. In each case, at least 100 particles were analyzed. | ||
Near identical well-defined vesicle formulations of low PD and similar Dh values were identified by DLS analysis and dry-state TEM imaging in the case of NMA and PXDA amines (Fig. 5B-I to III, C-I to III and S17†). Similar to HRP-loaded PEG113-b-P(HPMA320-co-GlyMA80) + BA nanoreactors, average Dcryo values were in good agreement with light scattering results, whilst an increased average membrane thickness was calculated for PXDA-functionalized vesicles (Mave, PXDA = 31.1 ± 2.7 nm). Additionally, a further membrane thickness increase to Mave, NMA = 37.6 ± 2.8 nm was calculated upon PGlyMA ring-opening using a more hydrophobic amine (i.e. NMA), showing an evident relationship between the structure/hydrophobicity of the nucleophile and the membrane thickness of the resulting vesicles (Fig. 5B-IV to VI, C-IV to VI and S17†).
The activity of HRP-loaded PEG113-b-P(HPMA320-co-GlyMA80) + BA/NMA/PXDA polymersomes was assessed by kinetic colorimetric analysis, monitoring the enzyme-catalyzed peroxidation of DMB in line with above described assays (Fig. 6A). The rate of DMB peroxidation was clearly affected by the physicochemical nature of the polymersome (particularly Mave), with reductions in substrate processing correlating to increasing thickness of the diffusional barriers provided by the functionalized membrane. At a fixed substrate concentration (0.4 mM DMB and 1.1 M H2O2), there was a small reduction in product formation for the PXDA cross-linked polymersomes of 31 ± 4% upon comparison with the original HRP-loaded PEG113-b-P(HPMA320-co-GlyMA80) vesicles that dropped to >80% in the case of BA and NMA-functionalized nanoreactors (Fig. 6B). Although these values correlate well with measured Mave values (i.e. NMA > BA > PXDA > PGlyMA) and showcase how post-PISA chemical modification affects the membrane character altering its relative permeability, it was important to expand this data to explore the effect of substrate concentration; measuring the observable permeability. No change to the actual Km of the intact enzyme was taken into consideration as such fundamental changes to activity are not generally associated with encapsulation.67 Normalized kinetic data was presented in terms of V0/Vmax due to the focus on comparisons between equilibrium substrate dissociation constant (Km) of the nanoreactor-loaded enzyme rather than absolute activity; an [enzyme]-dependent parameter.
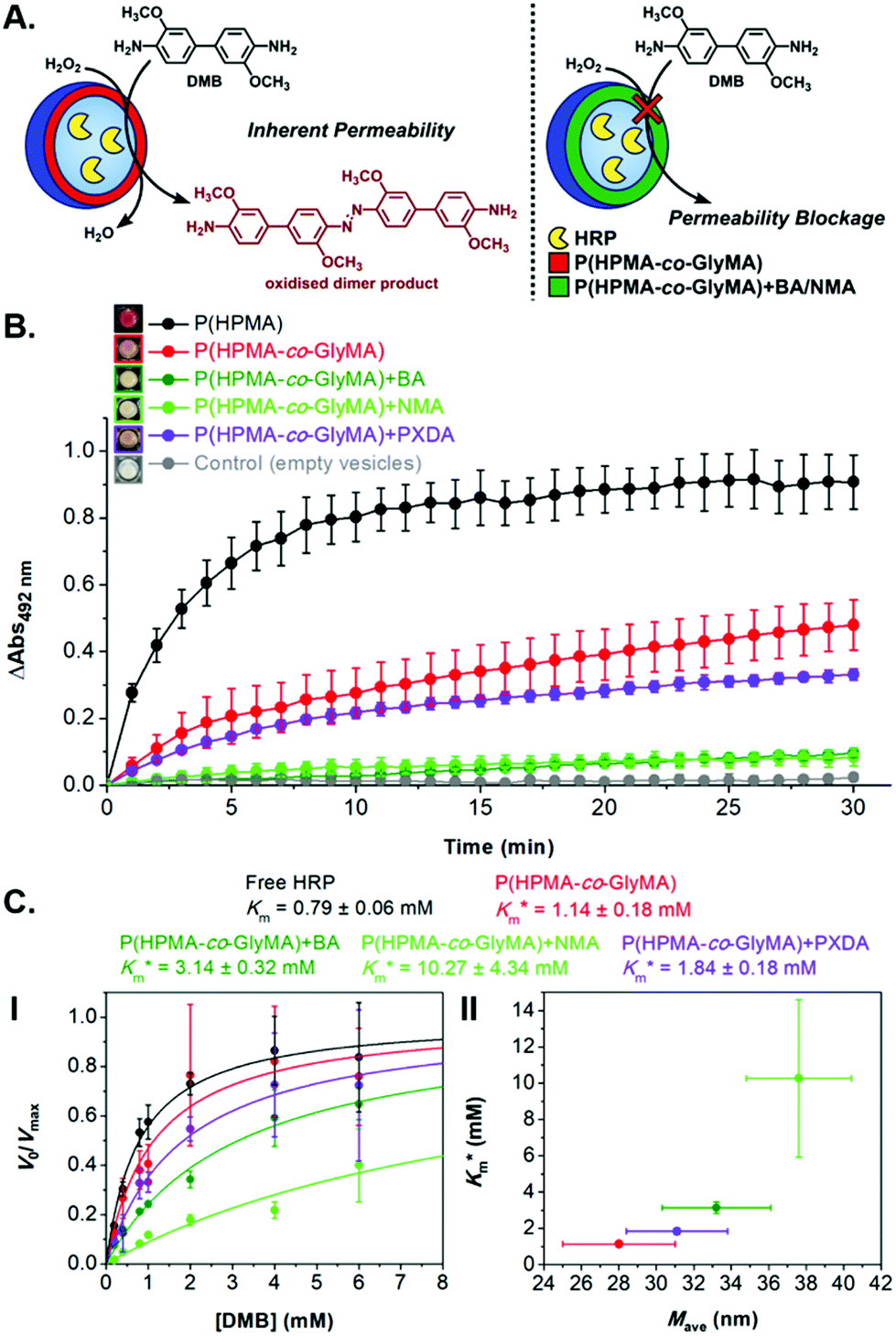 | ||
Fig. 6 (A) Schematic illustration of HRP-catalyzed oxidation reaction of DMB to a red-brown dimer product, detected by kinetic colorimetric assay, taking place inside inherently permeable HRP-loaded PEG113-b-P(HPMA320-co-GlyMA80) vesicles versus impermeable HRP-loaded PEG113-b-P(HPMA320-co-GlyMA80) + BA/NMA vesicles, and (B) enzymatic activity of the purified empty (grey line), HRP-loaded (red line), HRP-loaded PXDA-functionalized (purple line), and HRP-loaded BA/NMA-functionalized (green lines) PEG113-b-P(HPMA320-co-GlyMA80) vesicles, and HRP-loaded PEG113-b-P(HPMA)400 vesicles (black line) (end point = 30 min, λ = 492 nm) (the insets show the end-point microwells in each case). (C) Normalized Michaelis–Menten kinetic plots of free HRP and HRP-loaded PEG113-b-P(HPMA320-co-GlyMA80) + BA/NMA/PXDA vesicles (I), and calculated 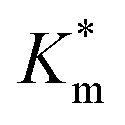 values (mean ± standard error) for each sample as a function of average nanoreactor membrane thickness (Mave) (II). values (mean ± standard error) for each sample as a function of average nanoreactor membrane thickness (Mave) (II). | ||
Systematically varying [DMB] (with [H2O2] under saturating conditions) gave a more detailed insight into the effect of membrane modification on the performance of nanoreactors, where the trend identified previously was confirmed and could be elucidated with greater detail (Fig. 6C-I and S18†). As compared to the Km value of the free enzyme at 0.79 ± 0.06 mM, the 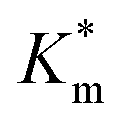 of HRP-loaded PEG113-b-P(HPMA320-co-GlyMA80) nanoreactors increased by ca. 30% (up to 1.14 ± 0.18 mM) as a result of enzyme compartmentalization within the semi-permeable P(HPMA320-co-GlyMA80) membrane. This effect could be tuned by introducing cross-linking or chemical modification. PXDA cross-linking of polymersomes further increased
of HRP-loaded PEG113-b-P(HPMA320-co-GlyMA80) nanoreactors increased by ca. 30% (up to 1.14 ± 0.18 mM) as a result of enzyme compartmentalization within the semi-permeable P(HPMA320-co-GlyMA80) membrane. This effect could be tuned by introducing cross-linking or chemical modification. PXDA cross-linking of polymersomes further increased 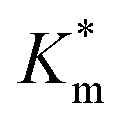 by ca. 30% (up to 1.84 ± 0.18 mM), in accordance with earlier data under fixed substrate concentration conditions. Interestingly, using such kinetic assays, we were able to resolve the effect of BA and NMA modification on the permeability of the polymersome membrane. Indeed, post-PISA membrane modification and insertion of benzylic (BA) groups gave rise to a 70% increase in
by ca. 30% (up to 1.84 ± 0.18 mM), in accordance with earlier data under fixed substrate concentration conditions. Interestingly, using such kinetic assays, we were able to resolve the effect of BA and NMA modification on the permeability of the polymersome membrane. Indeed, post-PISA membrane modification and insertion of benzylic (BA) groups gave rise to a 70% increase in  , which increased by a further 230% when using bulkier naphthyl (NMA) groups (up to 3.14 ± 0.32 & 10.27 ± 4.34 mM, respectively). Overall, the observed rate of substrate turnover of the unmodified polymersomes was ca. 90% higher than that of the NMA-functionalized nanoreactors. Relating these values for
, which increased by a further 230% when using bulkier naphthyl (NMA) groups (up to 3.14 ± 0.32 & 10.27 ± 4.34 mM, respectively). Overall, the observed rate of substrate turnover of the unmodified polymersomes was ca. 90% higher than that of the NMA-functionalized nanoreactors. Relating these values for 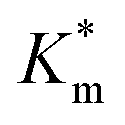 to the measured values of average membrane thickness (Mave), it is apparent that their relationship is non-linear, which means that the physicochemical origin of this tunability cannot be fully explicated by the influence of chemical modifier upon the membrane dimensions but is also related to the nature of the new membrane (Fig. 6C-II). In this instance, we can understand the increasing
to the measured values of average membrane thickness (Mave), it is apparent that their relationship is non-linear, which means that the physicochemical origin of this tunability cannot be fully explicated by the influence of chemical modifier upon the membrane dimensions but is also related to the nature of the new membrane (Fig. 6C-II). In this instance, we can understand the increasing 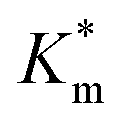 value (and decreasing permeability) between BA and NMA membranes arising due to the increasing non-covalent aromatic interactions between polymer chains (i.e. increasing the density and reducing the porosity of the bilayer).
value (and decreasing permeability) between BA and NMA membranes arising due to the increasing non-covalent aromatic interactions between polymer chains (i.e. increasing the density and reducing the porosity of the bilayer).
Overall, our findings provide a more in depth understanding of how relatively simple chemistries, used for the functionalization of polymersome membranes with increasingly more hydrophobic moieties, can tune the passive diffusion of small molecules into their aqueous inner lumen. This strategy has been utilized to achieve greater control over substrate processing by enzymes encapsulated within a polymersome nanoreactor.
Conclusions
In summary, we report a facile strategy to modulate the membrane permeability of polymeric vesicles. Epoxy-containing, enzyme-loaded vesicle nanoreactors obtained via aqueous emulsion PISA were functionalized with a series of diamine cross-linkers or hydrophobic primary amines using a simple procedure. Membrane modification resulted in increased thickness and reduced permeability relative to the non-functionalized particles. Of the compounds tested, the hydrophobic amines exhibited the most dramatic blocking effect on the vesicle membranes, reducing permeability by over 80% as determined by a colorimetric assay involving substrate oxidation by the encapsulated enzymes. This fundamental study reveals important insight into the relationship between membrane thickness, cross-linking density, or hydrophobicity and permeability. Such fine control over the diffusion of substrates across inherently permeable vesicular membranes has rarely been demonstrated and should inform particle design in future studies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the ERC (grant number 615142), EPSRC, and the University of Birmingham. D. S. W. thanks the Ser Cymru II programme for support; this project received funding from the European Union's Horizon 2020 research and innovation under the Marie Skłodowska-Curie grant agreement no. 663830. Advanced BioImaging Research Technology Platform, BBSRC ALERT14 award BB/M01228X/1, is thanked for supporting cryo-TEM characterization and Dr S. Bakker (University of Warwick) is thanked for cryo-TEM assistance. Dr M. C. Arno (University of Birmingham) is thanked for optical microscopy assistance.Notes and references
- J. W. Szostak, D. P. Bartel and P. L. Luisi, Nature, 2001, 409, 387–390 CrossRef CAS PubMed.
- W. Martin, Philos. Trans. R. Soc., B, 2010, 365, 847–855 CrossRef CAS PubMed.
- K. Sugano, M. Kansy, P. Artursson, A. Avdeef, S. Bendels, L. Di, G. F. Ecker, B. Faller, H. Fischer, G. Gerebtzoff, H. Lennernaes and F. Senner, Nat. Rev. Drug Discovery, 2010, 9, 597–614 CrossRef CAS PubMed.
- N. J. Yang and M. J. Hinner, in Site-Specific Protein Labeling: Methods and Protocols, ed. A. Gautier and M. J. Hinner, Springer New York, New York, NY, 2015, pp. 29–53, DOI:10.1007/978-1-4939-2272-7_3.
- M. Marguet, C. Bonduelle and S. Lecommandoux, Chem. Soc. Rev., 2013, 42, 512–529 RSC.
- R. Roodbeen and J. C. M. van Hest, BioEssays, 2009, 31, 1299–1308 CrossRef CAS PubMed.
- R. J. R. W. Peters, I. Louzao and J. C. M. van Hest, Chem. Sci., 2012, 3, 335–342 RSC.
- J. Xu, F. J. Sigworth and D. A. LaVan, Adv. Mater., 2010, 22, 120–127 CrossRef CAS PubMed.
- J. Gaitzsch, X. Huang and B. Voit, Chem. Rev., 2016, 116, 1053–1093 CrossRef CAS PubMed.
- B. C. Buddingh’ and J. C. M. van Hest, Acc. Chem. Res., 2017, 50, 769–777 CrossRef PubMed.
- P. A. Gale, J. T. Davis and R. Quesada, Chem. Soc. Rev., 2017, 46, 2497–2519 RSC.
- A. Belluati, I. Craciun, C. E. Meyer, S. Rigo and C. G. Palivan, Curr. Opin. Biotechnol., 2019, 60, 53–62 CrossRef CAS PubMed.
- J. F. Le Meins, O. Sandre and S. Lecommandoux, Eur. Phys. J. E, 2011, 34, 14 CrossRef PubMed.
- C. G. Palivan, R. Goers, A. Najer, X. Zhang, A. Car and W. Meier, Chem. Soc. Rev., 2016, 45, 377–411 RSC.
- E. Rideau, R. Dimova, P. Schwille, F. R. Wurm and K. Landfester, Chem. Soc. Rev., 2018, 47, 8572–8610 RSC.
- B. M. Discher, Y.-Y. Won, D. S. Ege, J. C. M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef CAS PubMed.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- J. Rodríguez-Hernández, F. Chécot, Y. Gnanou and S. Lecommandoux, Prog. Polym. Sci., 2005, 30, 691–724 CrossRef.
- S. Sugihara, A. Blanazs, S. P. Armes, A. J. Ryan and A. L. Lewis, J. Am. Chem. Soc., 2011, 133, 15707–15713 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- N. J. Warren, O. O. Mykhaylyk, D. Mahmood, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 1023–1033 CrossRef CAS PubMed.
- N. J. Warren, O. O. Mykhaylyk, A. J. Ryan, M. Williams, T. Doussineau, P. Dugourd, R. Antoine, G. Portale and S. P. Armes, J. Am. Chem. Soc., 2015, 137, 1929–1937 CrossRef CAS PubMed.
- W. Zhou, Q. Qu, Y. Xu and Z. An, ACS Macro Lett., 2015, 4, 495–499 CrossRef CAS.
- J. C. Foster, S. Varlas, B. Couturaud, Z. Coe and R. K. O'Reilly, J. Am. Chem. Soc., 2019, 141, 2742–2753 CrossRef CAS PubMed.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- X. Wang and Z. An, Macromol. Rapid Commun., 2019, 40, 1800325 CrossRef PubMed.
- D. B. Wright, M. A. Touve, M. P. Thompson and N. C. Gianneschi, ACS Macro Lett., 2018, 7, 401–405 CrossRef CAS.
- J. C. Foster, S. Varlas, L. D. Blackman, L. A. Arkinstall and R. K. O'Reilly, Angew. Chem., Int. Ed., 2018, 57, 10672–10676 CrossRef CAS PubMed.
- W. Zhang, F. D'Agosto, O. Boyron, J. Rieger and B. Charleux, Macromolecules, 2012, 45, 4075–4084 CrossRef CAS.
- W. Zhang, F. D'Agosto, P.-Y. Dugas, J. Rieger and B. Charleux, Polymer, 2013, 54, 2011–2019 CrossRef CAS.
- S. Y. Khor, N. P. Truong, J. F. Quinn, M. R. Whittaker and T. P. Davis, ACS Macro Lett., 2017, 6, 1013–1019 CrossRef CAS.
- J. Tan, X. Dai, Y. Zhang, L. Yu, H. Sun and L. Zhang, ACS Macro Lett., 2019, 8, 205–212 CrossRef CAS.
- J. Yeow and C. Boyer, Adv. Sci., 2017, 4, 1700137 CrossRef PubMed.
- J. Tan, H. Sun, M. Yu, B. S. Sumerlin and L. Zhang, ACS Macro Lett., 2015, 4, 1249–1253 CrossRef CAS.
- J. Tan, Y. Bai, X. Zhang, C. Huang, D. Liu and L. Zhang, Macromol. Rapid Commun., 2016, 37, 1434–1440 CrossRef CAS PubMed.
- J. Yeow, O. R. Sugita and C. Boyer, ACS Macro Lett., 2016, 5, 558–564 CrossRef CAS.
- J. Tan, D. Liu, Y. Bai, C. Huang, X. Li, J. He, Q. Xu, X. Zhang and L. Zhang, Polym. Chem., 2017, 8, 1315–1327 RSC.
- L. D. Blackman, K. E. B. Doncom, M. I. Gibson and R. K. O'Reilly, Polym. Chem., 2017, 8, 2860–2871 RSC.
- B. Couturaud, P. G. Georgiou, S. Varlas, J. R. Jones, M. C. Arno, J. C. Foster and R. K. O'Reilly, Macromol. Rapid Commun., 2019, 40, 1800460 CrossRef PubMed.
- J. Tan, D. Liu, X. Zhang, C. Huang, J. He, Q. Xu, X. Li and L. Zhang, RSC Adv., 2017, 7, 23114–23121 RSC.
- L. D. Blackman, S. Varlas, M. C. Arno, A. Fayter, M. I. Gibson and R. K. O'Reilly, ACS Macro Lett., 2017, 6, 1263–1267 CrossRef CAS PubMed.
- J. Tan, D. Liu, Y. Bai, C. Huang, X. Li, J. He, Q. Xu and L. Zhang, Macromolecules, 2017, 50, 5798–5806 CrossRef CAS.
- J. Tan, X. Zhang, D. Liu, Y. Bai, C. Huang, X. Li and L. Zhang, Macromol. Rapid Commun., 2017, 38, 1600508 CrossRef PubMed.
- L. D. Blackman, S. Varlas, M. C. Arno, Z. H. Houston, N. L. Fletcher, K. J. Thurecht, M. Hasan, M. I. Gibson and R. K. O'Reilly, ACS Cent. Sci., 2018, 4, 718–723 CrossRef CAS PubMed.
- S. Varlas, L. D. Blackman, H. E. Findlay, E. Reading, P. J. Booth, M. I. Gibson and R. K. O'Reilly, Macromolecules, 2018, 51, 6190–6201 CrossRef CAS.
- S. Varlas, P. G. Georgiou, P. Bilalis, J. R. Jones, N. Hadjichristidis and R. K. O'Reilly, Biomacromolecules, 2018, 19, 4453–4462 CrossRef CAS PubMed.
- G. Cheng and J. Pérez-Mercader, Macromol. Rapid Commun., 2019, 40, 1800513 CrossRef PubMed.
- R. J. R. W. Peters, M. Marguet, S. Marais, M. W. Fraaije, J. C. M. van Hest and S. Lecommandoux, Angew. Chem., Int. Ed., 2014, 53, 146–150 CrossRef CAS PubMed.
- H. Che, S. Cao and J. C. M. van Hest, J. Am. Chem. Soc., 2018, 140, 5356–5359 CrossRef CAS PubMed.
- C. Nardin, S. Thoeni, J. Widmer, M. Winterhalter and W. Meier, Chem. Commun., 2000, 1433–1434 RSC.
- J. Gaitzsch, D. Appelhans, L. Wang, G. Battaglia and B. Voit, Angew. Chem., Int. Ed., 2012, 51, 4448–4451 CrossRef CAS PubMed.
- D. Gräfe, J. Gaitzsch, D. Appelhans and B. Voit, Nanoscale, 2014, 6, 10752–10761 RSC.
- T. Einfalt, R. Goers, I. A. Dinu, A. Najer, M. Spulber, O. Onaca-Fischer and C. G. Palivan, Nano Lett., 2015, 15, 7596–7603 CrossRef CAS PubMed.
- L. Messager, J. R. Burns, J. Kim, D. Cecchin, J. Hindley, A. L. B. Pyne, J. Gaitzsch, G. Battaglia and S. Howorka, Angew. Chem., Int. Ed., 2016, 55, 11106–11109 CrossRef CAS PubMed.
- S. F. M. van Dongen, M. Nallani, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Chem. – Eur. J., 2009, 15, 1107–1114 CrossRef CAS PubMed.
- I. Louzao and J. C. M. van Hest, Biomacromolecules, 2013, 14, 2364–2372 CrossRef CAS PubMed.
- W.-J. Zhang, C.-Y. Hong and C.-Y. Pan, ACS Appl. Mater. Interfaces, 2017, 9, 15086–15095 CrossRef CAS PubMed.
- X.-F. Xu, C.-Y. Pan, W.-J. Zhang and C.-Y. Hong, Macromolecules, 2019, 52, 1965–1975 CrossRef CAS.
- P. Chambon, A. Blanazs, G. Battaglia and S. P. Armes, Langmuir, 2012, 28, 1196–1205 CrossRef CAS PubMed.
- J. R. Lovett, L. P. D. Ratcliffe, N. J. Warren, S. P. Armes, M. J. Smallridge, R. B. Cracknell and B. R. Saunders, Macromolecules, 2016, 49, 2928–2941 CrossRef CAS PubMed.
- F. L. Hatton, J. R. Lovett and S. P. Armes, Polym. Chem., 2017, 8, 4856–4868 RSC.
- S. Piogé, T. N. Tran, T. G. McKenzie, S. Pascual, M. Ashokkumar, L. Fontaine and G. Qiao, Macromolecules, 2018, 51, 8862–8869 CrossRef.
- S. Muthukrishnan, E. H. Pan, M. H. Stenzel, C. Barner-Kowollik, T. P. Davis, D. Lewis and L. Barner, Macromolecules, 2007, 40, 2978–2980 CrossRef CAS.
- M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS PubMed.
- L. M. P. E. van Oppen, L. K. E. A. Abdelmohsen, S. E. van Emst-de Vries, P. L. W. Welzen, D. A. Wilson, J. A. M. Smeitink, W. J. H. Koopman, R. Brock, P. H. G. M. Willems, D. S. Williams and J. C. M. van Hest, ACS Cent. Sci., 2018, 4, 917–928 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, supplementary NMR, FT-IR and DLS data, additional dry-state and cryo-TEM images, and HRP control experiment activity results. See DOI: 10.1039/c9nr02507c |
| This journal is © The Royal Society of Chemistry 2019 |