
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-viral delivery of CRISPR/Cas9 complex using CRISPR-GPS nanocomplexes†
Piyush K.
Jain
 *abj,
Justin H.
Lo
bc,
Santosh
Rananaware
a,
Marco
Downing
a,
Apekshya
Panda
dg,
Michelle
Tai
e,
Srivatsan
Raghavan
fg,
Heather E.
Fleming
b and
Sangeeta N.
Bhatia
*bghi
*abj,
Justin H.
Lo
bc,
Santosh
Rananaware
a,
Marco
Downing
a,
Apekshya
Panda
dg,
Michelle
Tai
e,
Srivatsan
Raghavan
fg,
Heather E.
Fleming
b and
Sangeeta N.
Bhatia
*bghi
aDepartment of Chemical Engineering, University of Florida, Gainesville, FL 32611, USA
bMarble Center for Cancer Nanomedicine, Institute for Medical Engineering & Science, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
cDivision of Hematology and Oncology, Department of Internal Medicine, Vanderbilt University Medical Center, Nashville, TN 37232, USA
dDepartment of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
eDepartment of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
fDepartment of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA 02115, USA
gBroad Institute of Massachusetts Institute of Technology and Harvard, Cambridge, MA 02139, USA
hDepartment of Medicine, Brigham and Women's Hospital and Harvard Medical School, Boston, MA 02115, USA
iHoward Hughes Medical Institute, Cambridge, MA 02139, USA. E-mail: sbhatia@mit.edu
jUF Health Cancer Center, University of Florida, Gainesville, FL 32608. E-mail: jainp@ufl.edu
First published on 29th October 2019
Abstract
There is a critical need for the development of safe and efficient delivery technologies for CRISPR/Cas9 to advance translation of genome editing to the clinic. Non-viral methods that are simple, efficient, and completely based on biologically-derived materials could offer such potential. Here we report a simple and modular tandem peptide-based nanocomplex system with cell-targeting capacity that efficiently combines guide RNA (sgRNA) with Cas9 protein, and facilitates internalization of sgRNA/Cas9 ribonucleoprotein complexes to yield robust genome editing across multiple cell lines.
Introduction
The CRISPR (clustered regularly interspaced short palindromic repeats)/Cas9 (CRISPR associated protein 9) system has recently emerged as one of the most powerful genome editing tools available today, with a wide range of applications including gene disruption and correction, transcription and translation modulation, and imaging genomic elements.1–4 Originally adopted from the bacterial immune system, the CRISPR/Cas9 system consists of a single chimeric guide RNA (sgRNA) that directs the sgRNA/Cas9 complex to target DNA sequences inside cells, and a Cas9 nuclease that causes double-stranded cleavage of the bound DNA, which is subsequently repaired by various DNA repair mechanisms.5 Due to the ease of sgRNA design against any target DNA, CRISPR/Cas9 has been widely applied to create a range of animal models and treat genetic disorders in cells and animal models.6 Despite this major breakthrough, the clinical applications of this technology hinge on the safe and efficient delivery of CRISPR/Cas9 inside target cells.7 Therefore, in order to bring this field closer to using CRISPR/Cas in clinical applications as a treatment approach to genetic disorders, the development of a simple and efficient delivery system with targeting capabilities is much needed.A key challenge faced by the existing technology is the physical delivery of multiple components inside the nucleus of desired cell types while also maintaining their binding and catalytic activities.8 The CRISPR/Cas9 components are typically delivered in plasmid form using viral vectors such as lentivirus,9 adenovirus,10 baculovirus,11 or, most commonly, adeno-associated virus (AAV).12,13 Using AAVs is quite efficient. Serotype selection with tropism for different organs is possible. However, pre-existing immunity to AAVs and their relatively low packaging size limit AAV-mediated clinical applications.14 In addition, AAVs can result in long-term expression of Cas9 and sgRNA proteins, increasing the risk of off-target cleavage, which is a major concern in establishing the safety of CRISPR/Cas9-based therapeutics.7 Several non-viral methods that have been developed or repurposed to deliver CRISPR toolbox as plasmids encounter challenges. Their overall effects are delayed during the protein expression process. Therefore, transient non-viral delivery of Cas9 protein and sgRNA is an appealing goal. Yet again, this strategy faces challenges of combined protein and nucleic acid delivery to the target cell type and the desired intracellular compartment.
In order to address these challenges, transient delivery of sgRNA along with the Cas9 mRNA or Cas9 protein has been achieved using non-viral methods such as electroporation,15,16 hydrodynamic injection,17 microinjection,18 lipids,19–22 peptides,23–28 polyethylenimine in combination with other agents such as DNA nanoclew, graphene oxide, cholesterol,29–31 gold nanoparticles and other nanostructures,32–38 extracellular vesicles,39–42 virus-like particles,43 and biolistic delivery in plants.44,45 Apart from these methods, many hybrid frameworks have also been used to deliver CRISPR/Cas9.46–49 While some of these approaches have delivered CRISPR/Cas9 components in vivo and mediated gene correction, most are non-specific delivery methods with potential for toxicity. Accordingly, alternative particle-based strategies have also been developed that can target the delivery of CRISPR/Cas9 components to prostate cancer, osteosarcoma, and brain cancers in vivo.29,32,50,51 Although these promising methods offer targeted delivery, they require manipulation of sgRNA into a plasmid,29,50,51 or subcloning of sgRNA into minicircle DNA,32 and are compounded with complex particles that are difficult to tailor and require multistep synthesis.
Based on a tandem peptide nanocomplex system,52–58 previously developed by our lab to achieve targeted delivery of siRNA to tumors, we hypothesized a simple and modular delivery approach, CRISPR-GPS (Guiding Peptide Sequences). We discovered that it can co-deliver native or modified sgRNA and Cas9 protein to various cell types in vitro with comparable efficiency as lipofectamine® RNAiMAX, and may also offer the capacity to target specific cell types in vivo. Our engineered, modular tandem peptide nanocomplex system is composed of a C-terminal peptide-based targeting domain, a cell-penetrating peptide (CPP) domain for cellular internalization, and an N-terminal lipid tail for particle packaging and endosomal escape (Fig. 1).53 By optimizing both the ratios of individual nanocomplex components and the formulation strategy, we successfully delivered CRISPR/Cas9 in OVCAR8 cells (ovarian cancer), HeLa cells (cervical cancer), and 3TZ cells (a mouse fibroblast line derived from 3T6 fibroblasts) in vitro. Furthermore, we achieved robust CRISPR/Cas9-mediated disruption of a reporter gene in a functional assay.
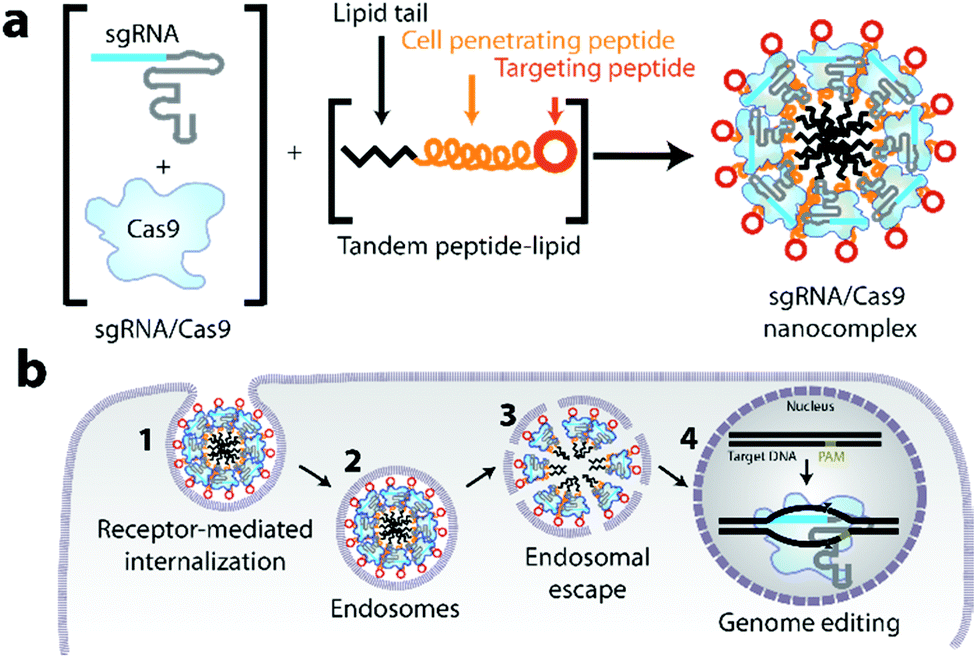 | ||
| Fig. 1 CRISPR-GPS (guiding peptide sequences) concept. (a) Schematic representation of the targeted tandem peptide constructs designed to deliver CRISPR/Cas9 components. A tandem peptide-lipid containing a cell penetrating peptide, a targeting peptide on its C-terminus, and a lipid tail on its N-terminus can be designed to package sgRNA and Cas9 in a nanocomplex. (b) The nanocomplex is taken up by target cells via receptor-mediated endocytosis, after which it escapes from endosomes so that the sgRNA/Cas9 complex can be internalized within the nucleus, due to the presence of an NLS sequence on Cas9, ultimately enabling genome editing. | ||
Results and discussion
Optimization of tandem peptides to form sgRNA, Cas9, and sgRNA/Cas9 nanocomplexes
Previously, our lab successfully developed a tandem peptide nanocomplex system for the delivery of siRNA to target tumors using a tandem peptide, mTP-LyP-1, where ‘m’ denotes a myristoyl group and acts as a lipid tail, ‘TP’ refers to transportan (sequence: GWTLNSAGYLLGKINLKALAALAKKIL, net charge +4), which is a cell penetrating peptide, and the LyP-1 (sequence: CGNKRTRGC, net charge +3) is a tumor targeting peptide that binds to the p32 receptor and contains a CendR motif.59,60 We sought to repurpose these tandem peptide nanocomplexes to deliver sgRNA/Cas9 protein as a ribonucleoprotein (RNP) complex inside cells. Although sgRNA/Cas9 is molecularly different and much larger than siRNA, we reasoned that similarly to siRNA (theoretical net charge −40), a Cas9 (theoretical net charge +22)/sgRNA (theoretical net charge −101) RNP complex should also be polyanionic (theoretical net charge −79) and therefore, has the potential to nanocomplex with tandem peptides that have successfully delivered siRNA in vivo.52,55,57,58 For targeting, we utilized a cyclic peptide, iRGD (sequence: CRGDKGPDC, net charge 0), which mediates tumor targeting and internalization by binding αvβ3/αvβ5 integrins and neuropilin-1.54,55 In addition, we designed and tested different lipid tails and the palmitoyl-TP-iRGD (pTP-iRGD) was found to be the most optimal candidate (data not shown).To test the ability of tandem peptides to complex RNP into nanocomplexes, we developed a gel retardation assay using a 5′-Cy5-labeled sgRNA (Genelink) and a Cy3-labeled Cas9 (PNA Bio) with unlabeled peptides. Using this assay, we observed a retardation in the mobility of the components as the peptide concentration was increased (Fig. 2a). Furthermore, the increase in the well-retention of sgRNA and Cas9 with higher concentrations of peptides suggests forming of nanocomplexes. Based on this finding, we sought to determine the minimal concentration of peptides required to assemble sgRNA and Cas9. We determined that pTP-iRGD can assemble Cas9 and sgRNA between a concentration of 15× to 60×, relative to sgRNA and Cas9 (Fig. 2a). Pre-complexation of sgRNA/Cas9 before the addition of tandem peptide was found to be the optimal strategy for incorporation of both, sgRNA and Cas9, relative to the addition of Cas9 after complexation of sgRNA with peptide, or the addition of sgRNA after complexing Cas9 with peptide (Fig. S1†). This order of formulation will also increase the likelihood that the sgRNA and Cas9 remain complexed upon their release from the endosomes. Interestingly, we found that tandem peptides can not only complex negatively-charged sgRNA but also complex overall positively-charged Cas9, suggesting non-electrostatic hydrophilic, hydrophobic, and van der Waals forces may be involved (Fig. 2a and S1†).
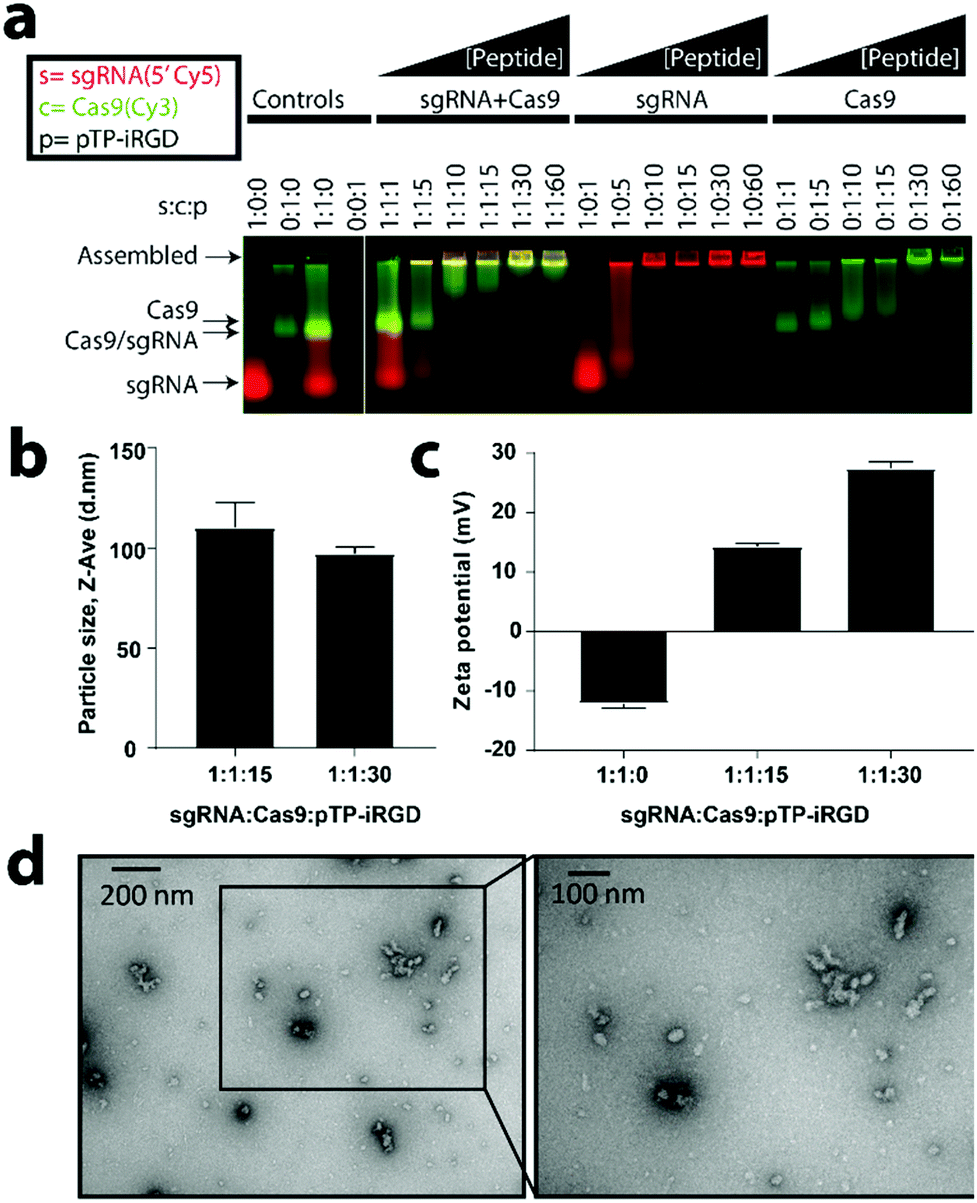 | ||
Fig. 2 Tandem peptides can assemble sgRNA/Cas9 RNP into nanocomplexes. (a) Effect of increasing the concentration of peptide-lipid (pTP-iRGD) on encapsulation of sgRNA/Cas9 RNP, sgRNA, or Cas9 (PNA Bio), as analyzed by an agarose-based gel retardation assay using equal concentrations of 5′-Cy5-labeled sgRNA and a Cy3-labeled Cas9 with increasing concentrations of unlabeled peptides. (b) Size analysis of particles using dynamic light scattering (DLS) for two different ratios of peptides relative to sgRNA and Cas9, as indicated above. Size analysis at other ratios can be found in Fig. S2.† Error bars indicate standard deviation (n = 3). (c) Zeta potential of nanocomplexes at the indicated ratios (n = 3). (d) Transmission electron microscopy (TEM) analysis of particles formed by combining 50 nM sgRNA and 50 nM of Cas9 with 1500 nM of pTP-iRGD (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:30) and imaged using 2% uranyl acetate as a counter stain. TEM analysis at other ratios can be found in Fig. S3.† :1:30) and imaged using 2% uranyl acetate as a counter stain. TEM analysis at other ratios can be found in Fig. S3.† | ||
CRISPR/Cas9 can be nanocomplexed with tandem peptides
To determine whether the sgRNA/Cas9/peptide complexes are actually nanocomplexes, we performed dynamic light scattering (DLS) and transmission electron microscopy (TEM) analyses. For different ratios of sgRNA/Cas9 and peptides, we found complexes of ranging sizes (mean ± SE) from 88.4 ± 1.9 nm to 115.3 ± 6.0 nm (Fig. 2b and S2†). In order to verify their nanocomplex nature and to determine shape of the particles, we performed TEM analysis at both 15× and 30× ratios after counter-staining with uranyl acetate (Fig. 2d and S3†). Interestingly, we observed that the particles showed smaller core sizes in the TEM than detected by DLS, possibly due to their hydrodynamic radii. While the liquid forms of Cas9 from various commercial sources (NEB, Clontech, and PNA Bio) formed nanocomplexes, large aggregates were observed when a lyophilized form of Cas9 (PNA Bio) was used instead (data not shown). In addition, we also observed particle aggregation when OptiMEM-diluted nanocomplexes were utilized and analysed under TEM 10 minutes post-dilution (Fig. S3†).When nanocomplexes were formed by combining equimolar amounts of sgRNA and Cas9, using different Cas9 sources mentioned above, we found that the zeta potential shifts become less negative, ranging from −16 to −7 mV at 0× peptide, −4 to 0 mV at 15× peptide, and +19 to +29 mV at 30× peptide relative to sgRNA/Cas9 (Fig. 2c and S4†). It was additionally found that Cas9 aggregates as the pH value nears its PI of 8 (Fig. S5†). As Cas9 loses its positive charge, its ability to complex with negatively charged sgRNA is reduced. We found that the loss of Cas9 interaction with the sgRNA leads to an increase in nanocomplex size (Fig. S6†). These findings are further indicative of the cationically-charged peptides interacting with the negatively-charged sgRNA/Cas9 to form nanocomplexes.
CRISPR-GPS achieves functional delivery of CRISPR/Cas9 components in a target-specific manner
Before testing the efficacy of CRISPR-GPS for genome editing in mammalian cells, we first validated expression of our chosen targeting peptide ligands, αvβ3 integrins on a GFP-expressing HeLa reporter cell line (Fig. S7†). Next, we tested different formulation strategies and ratios of peptides to co-deliver Cas9 and a guide RNA targeting GFP (sgGFP1) or a control scrambled guide (sgScr) previously validated by our lab and others.61 When we tested our integrin-targeting pTP-iRGD nanocomplexes to deliver sgGFP1, we observed significant knockdown of the GFP with sgGFP1 (∼28%) compared to the control sgScr (∼8%). Additionally, we did not observe significant knockdown of GFP when a control non-targeting peptide (mTP-ARAL) was utilized (Fig. 3a). Compared to RNAiMAX, we noticed a trend towards improved RNP delivery in these cells with CRISPR-GPS. However, these differences were not statistically significant. Using CRISPR-GPS, we observed no significant differences in the knockdown of GFP between the different formulation strategies (Fig. S7 and S9†) or different commercial sources of Cas9 (Fig. S9†). However, we observed significant toxicity with concentrations of peptides beyond 50×, with respect to the concentration of RNP (data not shown).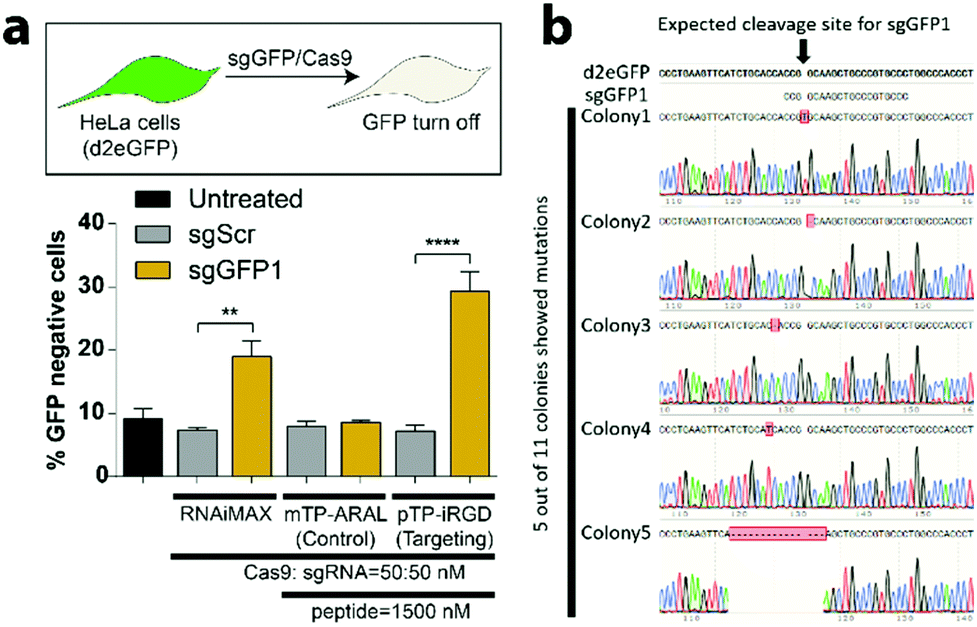 | ||
| Fig. 3 CRISPR-GPS delivers functional CRISPR/Cas9 components in a target-specific manner. (a) HeLa cells expressing destabilized GFP (d2eGFP) were plated in a 96-well plate and then treated with sgRNA (50 nM)/Cas9 (50 nM) RNP complex using either RNAiMAX (0.3 μL per well), 1500 nM of control non-targeting peptide (mTP-ARAL), or 1500 nM of integrin-targeting peptide (pTP-iRGD) in Opti-MEM media at the indicated ratios. After treatment for 5 h with the transfection mixture containing either a sgRNA against GFP (sgGFP1) or its scrambled sgGFP1 sequence (sgScr), cells were treated with the fresh cell culture media and GFP disruption was quantified using flow cytometry after 3 days. Untreated sample represents cells treated with OptiMEM without any transfection complex. Error bars represent standard deviation (n = 3) and the asterisks indicate **P < 0.01, ****P < 0.0001 analyzed by two-way ANOVA test. (b) The loss of GFP in HeLa/d2eGFP cells corresponds with the formation of indels near the sgRNA target cleavage site, as identified using an indel identification kit, followed by DNA sequencing (Clontech). 5 out of 11 colonies showed mutated DNA in the region of interest. | ||
Therefore, we selected 30× as the optimal ratio of sgRNA:Cas9:pTP-iRGD. This finding supports the hypothesis that the CRISPR-GPS system enables targeted delivery of CRISPR/Cas9 components inside cells. To confirm that the observed GFP disruption is due to insertions and/or deletions in genomic DNA mediated by CRISPR/Cas9, we performed indel identification using Clontech's indel identification kit. We detected indels at the intended cleavage site in 3 out of 11 colonies assayed (Fig. 3b). We also detected mutations in 2 other colonies that were present in close proximity to the cleavage site, and hence included them in our analysis. These results confirm that CRISPR-GPS can successfully deliver sgRNA/Cas9 RNP inside cells and mediate targeted genome editing in the nucleus.
CRISPR-GPS delivers functional CRISPR/Cas9 components in a GFP turn-on reporter system
To demonstrate that the system is generalizable to other guide RNAs and other cellular systems, we utilized CRISPR-GPS to turn on gene expression. For this purpose, we utilized a mouse fibroblast line, 3TZ, that harbors a STOP codon flanked by two FRT sequences, FRT-STOP-FRT-GFP, preventing the expression of GFP (Fig. 4a). By delivering a guide RNA targeting FRT sequences (sgFRT) along with Cas9, the expression of GFP can then be turned on. However, this system requires two different cuts instead of a single cut. Therefore, the efficiency of the turn on is expected to be lower than the GFP turn off. By transfecting these cells with CRISPR-GPS carrying sgFRT, Cas9, and pTP-iRGD, we observed significant increase in expression of GFP in over 12% of cells, similar to the result achieved by RNAiMAX-mediated delivery, at 10% (Fig. 4b). Using this platform, we also observed significant uptake of the nanocomplexes formed using control (ARAL) peptides when compared to iRGD. However, the GFP turn on efficiency was much lower (∼5%). These results confirm that CRISPR-GPS can deliver sgRNA/Cas9 RNP to different cell types in a target-specific manner and that the targeting peptide is important for higher delivery efficiency.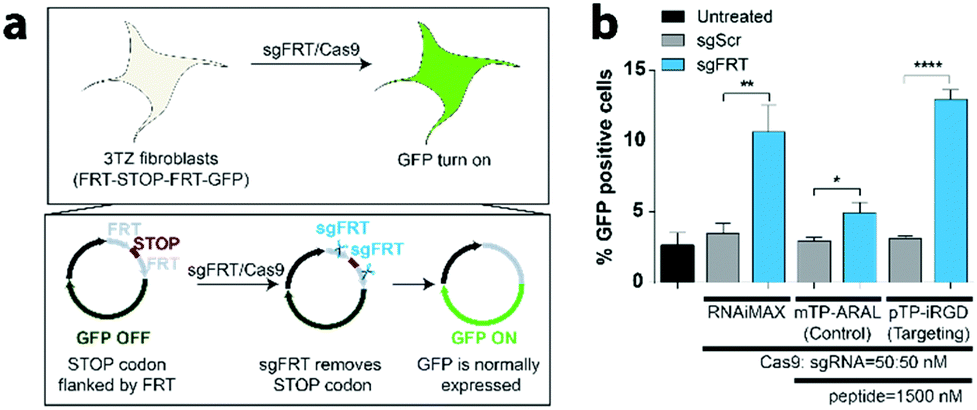 | ||
| Fig. 4 CRISPR-GPS delivers functional CRISPR/Cas9 components in a GFP turn-on reporter system. To test our CRISPR-GPS in a functional GFP turn-on reporter assay, we utilized mouse fibroblasts, 3TZ cells expressing a FRT-STOP-FRT cassette upstream of GFP (3TZ/FRT-STOP-FRT), where paired sgRNAs against FRT (sgFRT) can excise the stop sequence and turn on GFP expression. (a) Schematic representation of a sgFRT-based GFP turn-on system. (b) 3TZ/FRT-STOP-FRT cells were plated in a 96-well plate and then transfected with sgRNA (50 nM)/Cas9 (50 nM) RNP complexes using either RNAiMAX (0.3 μL per well), 1500 nM of control non-targeting peptide (mTP-ARAL), or 1500 nM of integrin-targeting peptide (pTP-iRGD) in OptiMEM media at the indicated ratios. After treatment for 3 h with the transfection mixture containing either sgRNA against FRT sequence (sgFRT) or a sgGFP1 scrambled sequence (sgScr), as described previously, cells were treated with the fresh cell culture media and GFP expression was quantified using flow cytometry after 3 days. Untreated sample represents cells treated with OptiMEM without any transfection complex. Error bars represent standard deviation (n = 3) and the asterisks indicate *P < 0.05, **P < 0.01, ****P < 0.0001 analyzed by two-way ANOVA test. | ||
CRISPR-GPS achieves CRISPR/Cas9 internalization and retains co-localization of sgRNA and Cas9 inside cells
To demonstrate that CRISPR-GPS is generalizable to other human cancer cell lines, we generated OVCAR8 cells expressing destabilized GFP (OVCAR8/d2eGFP) using lentiviral transduction, followed by antibiotic selection and cell sorting. To investigate the ability of integrin-targeted CRISPR-GPS to deliver RNP inside cells, we first tested the presence of αvb3 integrins in OVCAR8 cells by immunostaining and flow cytometry analysis, confirming expression of the desired target integrins by OVCAR8 cells (Fig. S7†). This finding makes it suitable for iRGD-mediated targeting. Similarly, we confirmed the expression of αvb3 integrins in 3TZ cells (Fig. S8†).Next, we used 5′-Cy5-labeled sgRNA (Cy5-sgRNA), unlabeled recombinant Cas9 carrying a nuclear localization signal (NLS) domain from different commercial sources, and unlabeled pTP-iRGD to prepare CRISPR-GPS nanocomplexes that were then applied to transfect OVCAR8/d2eGFP cells for either 1 h, 3 h, or 24 h. The cells were fixed at these time points and subsequently imaged using a fluorescent microscope. We detected a similar uptake of the CRISPR-GPS particles between different Cas9 sources when cells were transfected for 1 h or 3 h (data not shown). We also observed signs of toxicity based on the changes in morphology of the treated cells after 24 h of transfection in OptiMEM.
With these optimization parameters in mind, we tested for subcellular localization of the nanocomplex components using 5′-Cy5-labeled sgRNA (Cy5-sgRNA) and a Cy3-labeled Cas9 (Cy3-Cas9) formulated with unlabeled pTP-iRGD and transfected OVCAR8 cells co-expressing destabilized GFP for 3 h (Fig. 5a). Similarly, the cells were fixed at different time points and imaged using a wide-field microscope with 3D deconvolution capabilities. In addition, we compared the delivery efficiency of the CRISPR-GPS using RNAiMAX complexed with RNP at the same concentration (Fig. S10†). We observed that CRISPR-GPS successfully internalized the Cy5-sgRNA and Cy3-Cas9 inside cells after 3 h of transfection, as indicated by the yellow overlay (Fig. 5a). Furthermore, we noted that at least a subset of the complexes co-localized with the blue nuclear staining (appearing white, indicated with orange arrowheads), suggesting the particles escaped endosomes and reached the target organelle for genome editing. Interestingly, when we compared with RNAiMAX-mediated delivery, CRISPR-GPS achieved comparable particle delivery inside cells. Yet, at least qualitatively, the CRISPR-GPS-delivered particles appeared to co-localize Cy5-sgRNA with Cy3-Cas9 more efficiently than RNAiMAX. This effect was distinctly noticeable after 24 h (Fig. S11†).
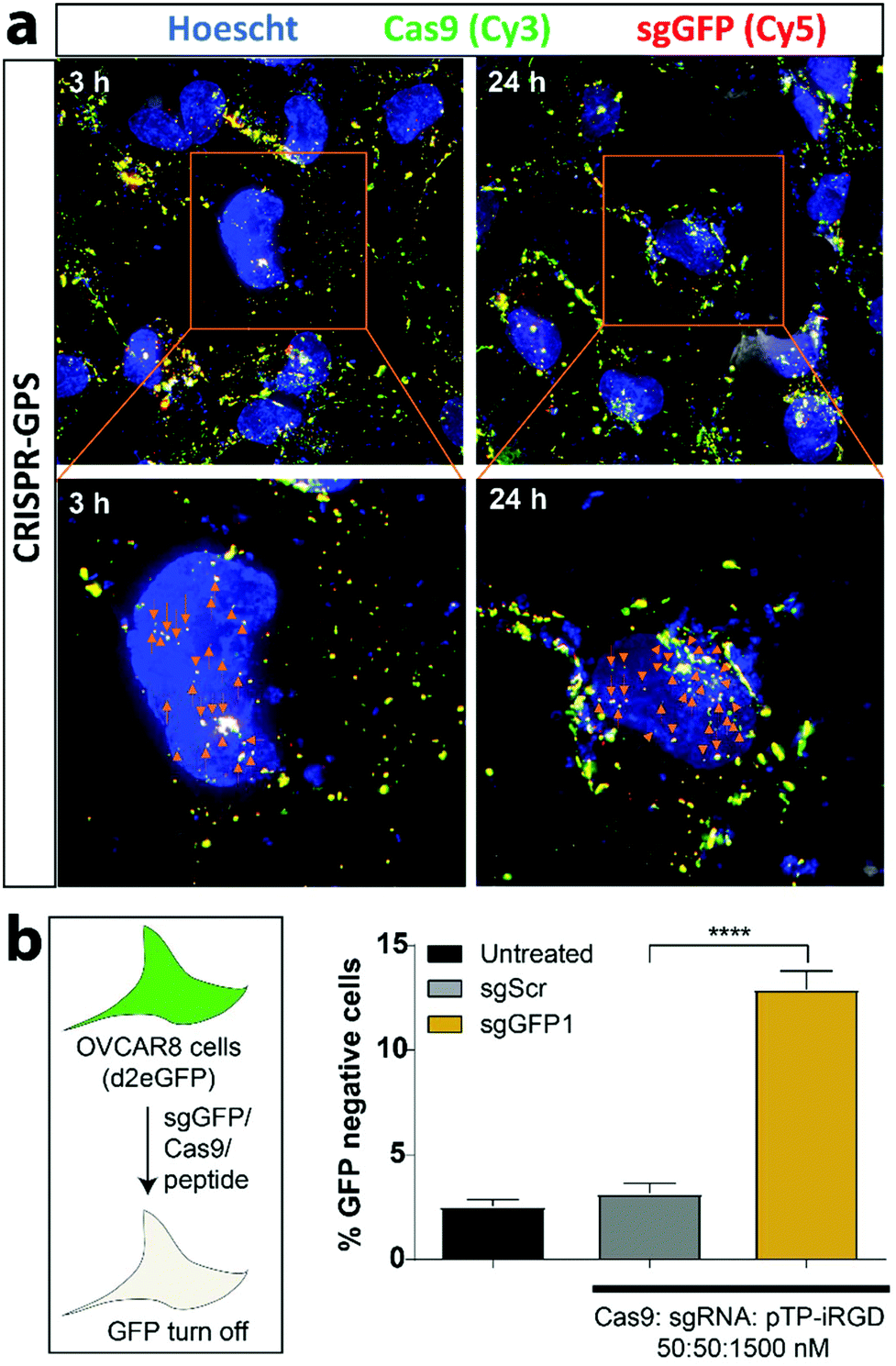 | ||
| Fig. 5 CRISPR-GPS delivers functional sgRNA/Cas9 cargo inside human tumor-derived OVCAR8 cells. Ovcar8/d2eGFP cells were treated with nanocomplexes containing 50 nM of 5′Cy5-labeled sgRNA and 50 nM of Cy3-labeled Cas9 and 1500 nM of pTP-iRGD in 500 uL of OptiMEM for 3 h and then the transfection mixture was replaced with fresh media. The cells were fixed using 4% paraformaldehyde at different time points (3 h or 24 h) and then imaged using an Olympus FV1200 Laser Scanning Confocal Microscope with an oil-immersed 60×/1.40, Plan Apo, IX70 objective. Images depict representative fields of cells (one out of three captured images) with blue nuclei (Hoescht) and examples of yellow, co-localized Cas9 (green) and sgRNA (red) complexes, which appear white when co-localized with the blue nuclear signal. Cas9/sgRNA complexes co-localized in the nucleus are highlighted with arrows. The experiment was repeated and showed similar data. (b) To assess CRISPR-GPS functionality in these cells, OVCAR8/d2eGFP cells were plated in a 96-well plate and transfected as in (a) for 6 h where the transfection mixture contained either a sgRNA against GFP (sgGFP1) or its scrambled sgGFP1 sequence (sgScr), followed by replacement of the media. GFP disruption was quantified using flow cytometry after 2 days. Untreated sample represents cells treated with OptiMEM without any transfection complex. Error bars represent standard deviation (n = 3) and the asterisks indicate ****P < 0.0001 analyzed by two-way ANOVA. | ||
Using unlabeled Cas9 and sgRNA in combination with 30× of pTP-iRGD in OVCAR8/d2eGFP cells, we confirmed that the delivered sgRNA/Cas9 complex was functional, based on the observed disruption of GFP expression in OVCAR8/d2eGFP cells (Fig. 5b). As expected, we also observed significant knockdown of GFP with sgGFP1/Cas9-transfected cells compared to the scrambled sequence guide (sgScr/Cas9)-transfected cells. Similarly, we also tested the delivery of sgRNA/Cas9 using our CRISPR-GPS system in OVCAR8 cells co-expressing eGFP and RFP. After 3 days, we noted that CRISPR-GPS mediated significant knockdown of eGFP when treated with sgGFP1 compared to sgScr (Fig. S9b†), with no signs of toxicity (Fig. S9c†). In order to further validate CRISPR-GPS for targeting endogenous genes, we transfected HEK293 cells expressing GFP under the control of an EF1a promoter with CRISPR-GPS carrying guide RNAs against CD71 (transferrin receptor)61 and analysed the levels of indels by Tracking of Indels by Decomposition (TIDE) analysis.62 We observed significant disruption of the CD71 gene with target sgRNA compared to scrambled sgRNA for both CRISPR-GPS and RNAiMAX (Fig. S12 and S13a–d†). These results not only validate that CRISPR-GPS delivers CRISPR/Cas9 complexes inside cells to the desired subcellular compartment, but also suggest that CRISPR-GPS can complex them for a durable period and still achieve functional delivery.
Conclusions
In summary, here we present CRISPR-GPS, a modular non-viral approach for the delivery of CRISPR/Cas9 RNPs. This method is easy to formulate into nanocomplexes and achieves effective genome editing across multiple cell lines without the need for transfection-mediated gene delivery. Although the CRISPR-GPS was found to be comparable to RNAiMAX in vitro, it offers a simple alternative approach for the delivery of CRISPR/Cas9 RNPs. This platform immediately enables targeted delivery of CRISPR/Cas9 in vitro for a wide range of genome engineering applications. Given the past success of tandem peptide nanocomplex-mediated delivery of siRNA, this platform may also offer the potential for application in vivo. The CRISPR-GPS system can be further extended to deliver other CRISPR/Cas system variants with immediate applications in the field of DNA and RNA editing.Conflicts of interest
S.N.B. is a director at Vertex, co-founder and consultant at Glympse Bio, consultant for Cristal, Maverick and Moderna, and receives sponsored research funding from Johnson & Johnson.Acknowledgements
We are grateful to Nisha Dalvie (Yale), Colin Buss (MIT), Dr Ester Kwon (UCSD), Mihir Doshi (U Mass), and Dr Jaideep Dudani (MIT) for helpful discussions. We are thankful to Prof. Tyler Jacks and his lab members (MIT) for providing us with 3TZ/FRT-STOP-FRT-GFP cells and guide RNA against FRT (sgFRT), and to Prof. Phillip A. Sharp and his lab members (MIT) for providing HeLa cells expressing destabilized GFP (HeLa-d2eGFP cells) and Prof. Wen Xue and his lab members (UMass Medical) for providing HEK293 cell line expressing GFP under EF1a promoter. We also thank the Koch Institute Swanson Biotechnology Core at MIT and the W. M. Keck Microscopy Facility at the Whitehead Institute, especially Nicki Watson. This study was supported in part by the Ludwig Center for Molecular Oncology, the Marie D. & Pierre Casimir-Lambert Fund, NCI CCNE Grant (U54-CA151884), and a Koch Institute Support Grant (5P30CA014051-48) from the National Cancer Institute (Swanson Biotechnology Center) and the start-up funds from the Herbert Wertheim College of Engineering at the University of Florida. S. N. B. is a Howard Hughes Medical Institute Investigator.Notes and references
- L. Cong, F. A. Ran, D. Cox, S. Lin, R. Barretto, N. Habib, P. D. Hsu, X. Wu, W. Jiang, L. A. Marraffini and F. Zhang, Science, 2013, 339, 819–823 CrossRef CAS PubMed.
- L. A. Gilbert, M. H. Larson, L. Morsut, Z. Liu, G. A. Brar, S. E. Torres, N. Stern-Ginossar, O. Brandman, E. H. Whitehead, J. A. Doudna, W. A. Lim, J. S. Weissman and L. S. Qi, Cell, 2013, 154, 442–451 CrossRef CAS.
- A. C. Komor, Y. B. Kim, M. S. Packer, J. A. Zuris and D. R. Liu, Nature, 2016, 533, 420–424 CrossRef CAS.
- J. D. Sander and J. K. Joung, Nat. Biotechnol., 2014, 32, 347–355 CrossRef CAS PubMed.
- M. Jinek, K. Chylinski, I. Fonfara, M. Hauer, J. A. Doudna and E. Charpentier, Science, 2012, 337, 816–821 CrossRef CAS PubMed.
- H. X. Wang, M. Li, C. M. Lee, S. Chakraborty, H. W. Kim, G. Bao and K. W. Leong, Chem. Rev., 2017, 117, 9874–9906 CrossRef CAS.
- A. C. Komor, A. H. Badran and D. R. Liu, Cell, 2017, 169, 559 CrossRef CAS.
- D. C. Luther, Y. W. Lee, H. Nagaraj, F. Scaletti and V. M. Rotello, Expert Opin. Drug Delivery, 2018, 15, 905–913 CrossRef CAS.
- A. M. Kabadi, D. G. Ousterout, I. B. Hilton and C. A. Gersbach, Nucleic Acids Res., 2014, 42, e147 CrossRef PubMed.
- I. Maggio, M. Holkers, J. Liu, J. M. Janssen, X. Chen and M. A. Goncalves, Sci. Rep., 2014, 4, 5105 CrossRef CAS PubMed.
- M. Mansouri, I. Bellon-Echeverria, A. Rizk, Z. Ehsaei, C. Cianciolo Cosentino, C. S. Silva, Y. Xie, F. M. Boyce, M. W. Davis, S. C. Neuhauss, V. Taylor, K. Ballmer-Hofer, I. Berger and P. Berger, Nat. Commun., 2016, 7, 11529 CrossRef CAS PubMed.
- P. Mali, L. Yang, K. M. Esvelt, J. Aach, M. Guell, J. E. DiCarlo, J. E. Norville and G. M. Church, Science, 2013, 339, 823–826 CrossRef CAS PubMed.
- R. J. Platt, S. D. Chen, Y. Zhou, M. J. Yim, L. Swiech, H. R. Kempton, J. E. Dahlman, O. Parnas, T. M. Eisenhaure, M. Jovanovic, D. B. Graham, S. Jhunjhunwala, M. Heidenreich, R. J. Xavier, R. Langer, D. G. Anderson, N. Hacohen, A. Regev, G. P. Feng, P. A. Sharp and F. Zhang, Cell, 2014, 159, 440–455 CrossRef CAS.
- P. Colella, G. Ronzitti and F. Mingozzi, Mol. Ther.–Methods Clin. Dev., 2018, 8, 87–104 CrossRef CAS.
- S. Kim, D. Kim, S. W. Cho, J. Kim and J. S. Kim, Genome Res., 2014, 24, 1012–1019 CrossRef CAS PubMed.
- M. Hashimoto and T. Takemoto, Sci. Rep., 2015, 5, 11315 CrossRef CAS PubMed.
- H. Yin, W. Xue, S. Chen, R. L. Bogorad, E. Benedetti, M. Grompe, V. Koteliansky, P. A. Sharp, T. Jacks and D. G. Anderson, Nat. Biotechnol., 2014, 32, 551–553 CrossRef CAS PubMed.
- D. Chaverra-Rodriguez, V. M. Macias, G. L. Hughes, S. Pujhari, Y. Suzuki, D. R. Peterson, D. Kim, S. McKeand and J. L. Rasgon, Nat. Commun., 2018, 9, 3008 CrossRef.
- J. A. Zuris, D. B. Thompson, Y. Shu, J. P. Guilinger, J. L. Bessen, J. H. Hu, M. L. Maeder, J. K. Joung, Z. Y. Chen and D. R. Liu, Nat. Biotechnol., 2015, 33, 73–80 CrossRef CAS.
- X. Yu, X. Liang, H. Xie, S. Kumar, N. Ravinder, J. Potter, X. de Mollerat du Jeu and J. D. Chesnut, Biotechnol. Lett., 2016, 38, 919–929 CrossRef CAS PubMed.
- M. Wang, J. A. Zuris, F. T. Meng, H. Rees, S. Sun, P. Deng, Y. Han, X. Gao, D. Pouli, Q. Wu, I. Georgakoudi, D. R. Liu and Q. B. Xu, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2868–2873 CrossRef CAS.
- Z. Y. He, Y. G. Zhang, Y. H. Yang, C. C. Ma, P. Wang, W. Du, L. Li, R. Xiang, X. R. Song, X. Zhao, S. H. Yao and Y. Q. Wei, Hum. Gene Ther., 2018, 29, 223–233 CrossRef CAS.
- S. Ramakrishna, A. B. Kwaku Dad, J. Beloor, R. Gopalappa, S. K. Lee and H. Kim, Genome Res., 2014, 24, 1020–1027 CrossRef CAS PubMed.
- I. Lostale-Seijo, I. Louzao, M. Juanes and J. Montenegro, Chem. Sci., 2017, 8, 7923–7931 RSC.
- B. Suresh, S. Ramakrishna and H. Kim, Methods Mol. Biol., 2017, 1507, 81–94 CrossRef CAS.
- T. DelGuidice, J. P. Lepetit-Stoffaes, L. J. Bordeleau, J. Roberge, V. Theberge, C. Lauvaux, X. Barbeau, J. Trottier, V. Dave, D. C. Roy, B. Gaillet, A. Garnier and D. Guay, PLoS One, 2018, 13, e0195558 CrossRef PubMed.
- X. Guan, Z. Luo and W. Sun, J. Biol. Chem., 2018, 293, 17306–17307 CrossRef CAS PubMed.
- H. X. Wang, Z. Song, Y. H. Lao, X. Xu, J. Gong, D. Cheng, S. Chakraborty, J. S. Park, M. Li, D. Huang, L. Yin, J. Cheng and K. W. Leong, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4903–4908 CrossRef CAS PubMed.
- C. Liang, F. Li, L. Wang, Z. K. Zhang, C. Wang, B. He, J. Li, Z. Chen, A. B. Shaikh, J. Liu, X. Wu, S. Peng, L. Dang, B. Guo, X. He, D. W. T. Au, C. Lu, H. Zhu, B. T. Zhang, A. Lu and G. Zhang, Biomaterials, 2017, 147, 68–85 CrossRef CAS PubMed.
- N. Ryu, M. A. Kim, D. Park, B. Lee, Y. R. Kim, K. H. Kim, J. I. Baek, W. J. Kim, K. Y. Lee and U. K. Kim, Nanomedicine, 2018, 14, 2095–2102 CrossRef CAS PubMed.
- H. Yue, X. Zhou, M. Cheng and D. Xing, Nanoscale, 2018, 10, 1063–1071 RSC.
- Z. Chen, F. Liu, Y. Chen, J. Liu, X. Wang, A. T. Chen, G. Deng, H. Zhang, J. Liu, Z. Hong and J. Zhou, Adv. Funct. Mater., 2017, 27, 1703036 CrossRef PubMed.
- J. S. Ha, J. S. Lee, J. Jeong, H. Kim, J. Byun, S. A. Kim, H. J. Lee, H. S. Chung, J. B. Lee and D. R. Ahn, J. Controlled Release, 2017, 250, 27–35 CrossRef CAS PubMed.
- J. D. Finn, A. R. Smith, M. C. Patel, L. Shaw, M. R. Youniss, J. van Heteren, T. Dirstine, C. Ciullo, R. Lescarbeau, J. Seitzer, R. R. Shah, A. Shah, D. Ling, J. Growe, M. Pink, E. Rohde, K. M. Wood, W. E. Salomon, W. F. Harrington, C. Dombrowski, W. R. Strapps, Y. Chang and D. V. Morrissey, Cell. Rep., 2018, 22, 2227–2235 CrossRef CAS PubMed.
- R. Mout, M. Ray, G. Yesilbag Tonga, Y. W. Lee, T. Tay, K. Sasaki and V. M. Rotello, ACS Nano, 2017, 11, 2452–2458 CrossRef CAS.
- K. Lee, M. Conboy, H. M. Park, F. Jiang, H. J. Kim, M. A. Dewitt, V. A. Mackley, K. Chang, A. Rao, C. Skinner, T. Shobha, M. Mehdipour, H. Liu, W. C. Huang, F. Lan, N. L. Bray, S. Li, J. E. Corn, K. Kataoka, J. A. Doudna, I. Conboy and N. Murthy, Nat. Biomed. Eng., 2017, 1, 889–901 CrossRef CAS PubMed.
- P. Wang, L. Zhang, W. Zheng, L. Cong, Z. Guo, Y. Xie, L. Wang, R. Tang, Q. Feng, Y. Hamada, K. Gonda, Z. Hu, X. Wu and X. Jiang, Angew. Chem., Int. Ed., 2018, 57, 1491–1496 CrossRef CAS PubMed.
- W. Zhou, H. Cui, L. Ying and X. F. Yu, Angew. Chem., Int. Ed., 2018, 57, 10268–10272 CrossRef CAS PubMed.
- L. A. Campbell, L. M. Coke, C. T. Richie, L. V. Fortuno, A. Y. Park and B. K. Harvey, Mol. Ther., 2019, 27(1), 151–163 CrossRef CAS PubMed.
- C. Montagna, G. Petris, A. Casini, G. Maule, G. M. Franceschini, I. Zanella, L. Conti, F. Arnoldi, O. R. Burrone, L. Zentilin, S. Zacchigna, M. Giacca and A. Cereseto, Mol. Ther.–Nucleic Acids, 2018, 12, 453–462 CrossRef CAS.
- Y. Lin, J. Wu, W. Gu, Y. Huang, Z. Tong, L. Huang and J. Tan, Adv. Sci., 2018, 5, 1700611 CrossRef.
- Y. Liu, G. Zhao, C. F. Xu, Y. L. Luo, Z. D. Lu and J. Wang, Biomater. Sci., 2018, 6, 1592–1603 RSC.
- J. Kong, Y. Wang, J. Zhang, W. Qi, R. Su and Z. He, Angew. Chem., Int. Ed., 2018, 57, 14032–14036 CrossRef CAS PubMed.
- S. Svitashev, C. Schwartz, B. Lenderts, J. K. Young and A. Mark Cigan, Nat. Commun., 2016, 7, 13274 CrossRef CAS.
- H. Hamada, Y. Liu, Y. Nagira, R. Miki, N. Taoka and R. Imai, Sci. Rep., 2018, 8, 14422 CrossRef PubMed.
- S. K. Alsaiari, S. Patil, M. Alyami, K. O. Alamoudi, F. A. Aleisa, J. S. Merzaban, M. Li and N. M. Khashab, J. Am. Chem. Soc., 2018, 140, 143–146 CrossRef CAS.
- W. Sun, W. Ji, J. M. Hall, Q. Hu, C. Wang, C. L. Beisel and Z. Gu, Angew. Chem., Int. Ed., 2015, 54, 12029–12033 CrossRef CAS PubMed.
- M. Wang, J. A. Zuris, F. Meng, H. Rees, S. Sun, P. Deng, Y. Han, X. Gao, D. Pouli, Q. Wu, I. Georgakoudi, D. R. Liu and Q. Xu, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2868–2873 CrossRef CAS PubMed.
- L. Zhang, P. Wang, Q. Feng, N. Wang, Z. Chen, Y. Huang, W. Zheng and X. Jiang, NPG Asia Mater., 2017, 9, e441–e441 CrossRef CAS.
- L. Li, L. Song, X. Liu, X. Yang, X. Li, T. He, N. Wang, S. Yang, C. Yu, T. Yin, Y. Wen, Z. He, X. Wei, W. Su, Q. Wu, S. Yao, C. Gong and Y. Wei, ACS Nano, 2017, 11, 95–111 CrossRef CAS PubMed.
- S. Zhen, Y. Takahashi, S. Narita, Y. C. Yang and X. Li, Oncotarget, 2017, 8, 9375–9387 Search PubMed.
- Y. Ren, H. W. Cheung, G. von Maltzhan, A. Agrawal, G. S. Cowley, B. A. Weir, J. S. Boehm, P. Tamayo, A. M. Karst, J. F. Liu, M. S. Hirsch, J. P. Mesirov, R. Drapkin, D. E. Root, J. Lo, V. Fogal, E. Ruoslahti, W. C. Hahn and S. N. Bhatia, Sci. Transl. Med., 2012, 4, 147ra112 Search PubMed.
- Y. Ren, S. Hauert, J. H. Lo and S. N. Bhatia, ACS Nano, 2012, 6, 8620–8631 CrossRef CAS.
- Y. Ren, J. E. Sagers, L. D. Landegger, S. N. Bhatia and K. M. Stankovic, Sci. Rep., 2017, 7, 12922 CrossRef.
- J. H. Lo, L. Hao, M. D. Muzumdar, S. Raghavan, E. J. Kwon, E. M. Pulver, F. Hsu, A. J. Aguirre, B. M. Wolpin, C. S. Fuchs, W. C. Hahn, T. Jacks and S. N. Bhatia, Mol. Cancer Ther., 2018, 17, 2377–2388 CrossRef CAS.
- J. H. Lo, E. J. Kwon, A. Q. Zhang, P. Singhal and S. N. Bhatia, Bioconjugate Chem., 2016, 27, 2323–2331 CrossRef CAS PubMed.
- E. J. Kwon, M. Skalak, R. Lo Bu and S. N. Bhatia, ACS Nano, 2016, 10, 7926–7933 CrossRef CAS.
- A. P. Mann, P. Scodeller, S. Hussain, J. Joo, E. Kwon, G. B. Braun, T. Molder, Z. G. She, V. R. Kotamraju, B. Ranscht, S. Krajewski, T. Teesalu, S. Bhatia, M. J. Sailor and E. Ruoslahti, Nat. Commun., 2016, 7, 11980 CrossRef CAS.
- K. N. Sugahara, T. Teesalu, P. P. Karmali, V. R. Kotamraju, L. Agemy, O. M. Girard, D. Hanahan, R. F. Mattrey and E. Ruoslahti, Cancer Cell, 2009, 16, 510–520 CrossRef CAS PubMed.
- L. Roth, L. Agemy, V. R. Kotamraju, G. Braun, T. Teesalu, K. N. Sugahara, J. Hamzah and E. Ruoslahti, Oncogene, 2012, 31, 3754–3763 CrossRef CAS PubMed.
- P. K. Jain, V. Ramanan, A. G. Schepers, N. S. Dalvie, A. Panda, H. E. Fleming and S. N. Bhatia, Angew. Chem., Int. Ed., 2016, 55, 12440–12444 CrossRef CAS PubMed.
- E. K. Brinkman, T. Chen, M. Amendola and B. van Steensel, Nucleic Acids Res., 2014, 42, e168 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr01786k |
| This journal is © The Royal Society of Chemistry 2019 |