
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanoscale J-aggregates of poly(3-hexylthiophene): key to electronic interface interactions with graphene oxide as revealed by KPFM†
Elisa
Palacios-Lidón
 *a,
Emin
Istif‡
b,
Ana M.
Benito
b,
Wolfgang K.
Maser
*b and
Jaime
Colchero
*a
*a,
Emin
Istif‡
b,
Ana M.
Benito
b,
Wolfgang K.
Maser
*b and
Jaime
Colchero
*a
aDepartamento Física, Edificio CIOyN (Campus Espinardo), Universidad de Murcia, E-30100 Murcia, Spain. E-mail: elisapl@um.es; colchero@um.es
bInstituto de Carboquímica (ICB-CSIC), E-50018 Zaragoza, Spain. E-mail: wmaser@icb.csic.es
First published on 16th May 2019
Abstract
The nanoscale aggregate structure of conjugated polymers critically determines the performance of organic thin film optoelectronic devices. Their impact on electronic interface interactions with adjacent layers of graphene, widely reported to improve the device characteristics, yet remains an open issue, which needs to be addressed by an appropriate benchmark system. Here, we prepared discrete ensembles of poly(3-hexylthiophene) nanoparticles and graphene oxide sheets (P3HTNPs–GO) with well-defined aggregate structures of either J- or H-type and imaged their photogenerated charge transfer dynamics across their interface by Kelvin probe force microscopy (KPFM). A distinctive inversion of the sign of the surface potential and surface photovoltage (SPV) demonstrates that J-aggregates are decisive for establishing charge transfer interactions with GO. These enable efficient injection of photogenerated holes from P3HTNPs into GO sheets over a range of tens of nanometers, causing a slow SPV relaxation dynamics, and define their operation as an efficient hole-transport layer (HTL). Conversely, H-type aggregates do not facilitate specific interactions and entrust GO sheets the role of charge-blocking layers (CBLs). The direct effect of the aggregate structure of P3HT on the functional operation of GO as a HTL or CBL thus establishes clear criteria towards the rational design of improved organic optoelectronic devices.
Introduction
Poly(3-hexylthiophene) (P3HT) is a prominent member of the family of π-conjugated polymers commonly investigated as a photoactive layer in organic thin film opto-electronic devices owing to its ease of processing and its impressive photophysical performance.1 It is widely recognized that the conditions of film fabrication from solutions directly impact on their nanoscale morphology.2,3 The resulting packing of the P3HT polymer chains into crystalline domains exhibiting either dominant interchain (H-aggregates) or intrachain (J-aggregates) interactions critically determines the transport properties of the photogenerated charge carriers.1,4–9 In this context, J- and H-aggregates can be defined through the sign of the Coulomb interaction between adjacent chromophores, determined by the dipole–dipole coupling of the effective dipoles induced by optical excitation of a polymer chain. For J-aggregates, the Coulomb interaction is negative, while for H-aggregates, this interaction is positive.10 For P3HT-polymer chains, crystalline regions forming J-aggregates imply predominantly a “head to tail” interaction of chromophores (on the same chain) as compared to a “face to face” interaction in H-aggregates (on different chains). The HOMO level has been reported to be the highest for amorphous P3HT, intermediate for J-aggregates, and the lowest (the highest work function) for H-aggregates.11 The precise molecular arrangement and crystallization critically determine the optoelectronic properties of P3HT-based structures, in particular, the band-gap, the HOMO/LUMO levels, their energetic alignment, the charge-transfer behaviour and the optical properties. A detailed understanding and precise control of these properties are fundamental for their application in optoelectronic devices.Over the last few years, it has been found that the aggregate structure can be established prior to thin film deposition in a controllable manner by liquid phase self-assembly approaches rendering P3HT in the form of nanofibers and spherical nanoparticles, thus contributing to overcome the dependency on extrinsic processing conditions.11–15 In this context, we have recently shown that the crystalline chain packing of P3HT nanoparticles (P3HTNPs) can be controlled by sheets of graphene oxide (GO).16 Their presence during the liquid phase self-assembly process of P3HTNPs affords a significant change in their aggregate structure from H-type to J-type. Concomitantly, this enables the creation of P3HTNPs–GO charge transfer complexes exhibiting superior optoelectronic properties. The study of individual P3HTNPs–GO ensembles is fundamental to unambiguously determine the influence of the aggregate structure for establishing interface interactions between P3HT and GO sheets and its effect on the charge-transport properties of the photogenerated charge carriers. Such a study would clarify the yet somewhat arbitrarily operational functionality of GO sheets in layered P3HT–GO assemblies, which are reported to act as a hole-transport layer (HTL), electron-blocking layer, electron-transport layer (ETL) or hole-blocking layer.17,18
Kelvin Probe Force Microscopy (KPFM) is a highly versatile technique to elucidate the nanoscale electronic properties.19 Combined with controlled illumination, KPFM allows the study of local photophysical processes, providing important feedback on the design of improved materials to achieve enhanced performance of thin film optoelectronic devices.20 Many types of conjugated polymers have been investigated by KPFM in the dark and under illuminated conditions, revealing the direct impact of blend morphology and nanoscale donor–acceptor interface structures on the photoinduced charge transport processes and conduction pathways.21–25
In this work, we study the optoelectronic properties of individual P3HTNPs–GO nanoscale ensembles deposited on ultra-flat ITO surface. To elucidate the influence of the polymer aggregate structure on the interactions with the GO sheets and the photoinduced charge transport processes between the two components, the surface potential (SP) and surface photovoltage (SPV) have been monitored by KPFM with and without illumination. To gain insight into the relevance of the aggregate structure, we probed P3HTNPs–GO nanoscale ensembles prepared either by an ex situ mixing process (P3HTNPs are first synthesized in solution and then mixed with a GO solution) or by an in situ re-precipitation approach (P3HTNPs are directly synthesized in a GO solution), favouring aggregate structures in the P3HTNPs of either H-type or J-type aggregate, respectively.16 SP values clearly enable to distinguish between H- and J-type aggregates. In addition, an important sign inversion of the SPV indicates the presence of interface interactions between P3HTNPs and GO sheets and the formation of charge transfer complexes established exclusively for J-type aggregates formed in the in situ sample. Here, charge injection over a range of several tens of nanometers is observed causing a slow relaxation process. This endows GO the role of a hole-transport layer upon interaction with P3HT interfaces exhibiting J-type aggregates. In contrast, H-type P3HTNPs do not enable interactions and assign GO the role of a hole-blocking layer. This study underlines the important role of the type of the aggregate structure for defining interactions, photocharge transport, and operational functionality of GO as either a hole-transport layer or hole-blocking layer.
Experimental
Synthesis
P3HTNPs and P3HTNPs–GO ensembles were synthesized based on previously described protocols.16 Detailed information is provided in the ESI.†Preparation of discontinuous films
1 mL stock solutions of in situ P3HTNPs–GO, ex situ P3HTNPs–GO and P3HTNPs were diluted to 5 mL with deionized water in order to avoid the agglomeration of the individual nanostructures in a thin film. The bare GO dispersion was also diluted to a final concentration of 0.01 mg mL−1. The diluted samples were spray coated on ultra-flat ITO substrate (previously cleaned with isopropanol several times), which were placed on a heating plate (65 °C). The coated surface area was 0.5 cm × 0.5 cm. The spray gun passed a maximum of 5 times over the substrate, thus enabling the formation of discontinuous films consisting of samples in the form of isolated individual objects (Scheme 1).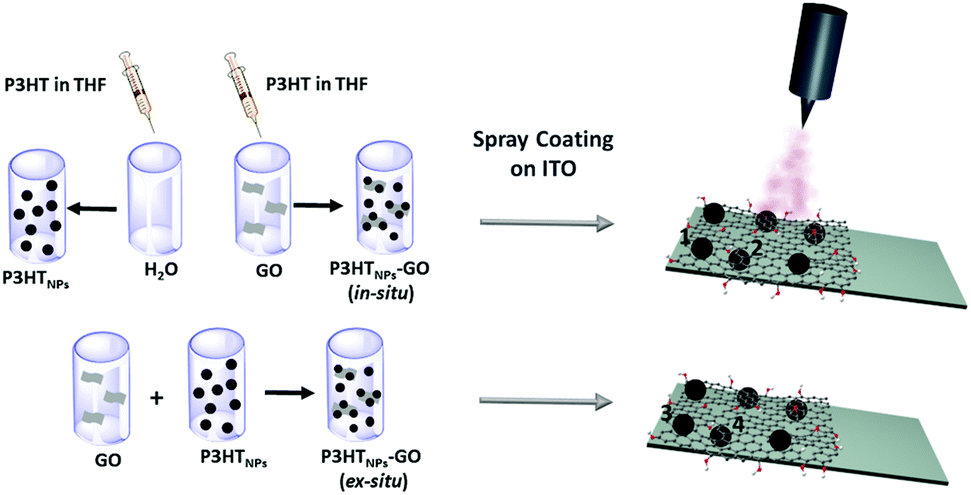 | ||
| Scheme 1 Left: Synthesis of P3HTNPs–GO ensembles by in situ and ex situ self-assembly processes. Right: Preparation of discontinuous films of in situ and ex situ P3HTNPs–GO ensembles on ultra-flat ITO substrates exposing individual isolated objects in different configurations: (1 and 3) P3HTNPS above, and (2 and 4) P3HTNPs below GO sheets for in situ and ex situ films, respectively. | ||
Experimental techniques
Scanning Force Microscopy (SFM) and KPFM measurements were carried out using a home-made system. Set-up and operation conditions are based on previously described protocols.26 Essentially, the SFM data was acquired using the oscillation amplitude as a signal for topography feedback (the so-called AM-SFM) at the very low reduction of the oscillation amplitude. The SP is measured using the force gradient of the electrostatic interaction induced by a harmonic tip–sample voltage. Our measurement protocol results in a non-perturbative low interaction mode and high resolution, low noise and quantitative SP values.27 Detailed information including illustration (Fig. S1†) is provided in the ESI.†Results and discussion
P3HTNPs and GO samples
First, the GO and the P3HTNPs samples are studied separately on ITO substrates. The GO topography (Fig. 1a) shows a discontinuous film of staked GO sheets with a lateral size of several micrometers and a sheet height of about 1.7 nm, in good agreement with previous values reported in the literature.28 The GO sheets are clearly distinguished in the corresponding KPFM image (Fig. 1b) appearing as large brighter regions compared to the ITO reference. In fact, it is possible to resolve SP differences (ΔSP) relative to the ITO substrate depending on the fact that if one (ΔSP1GO = +80 mV), two (ΔSP2GO = +60 mV) or more GO (ΔSP>2GO = +20 mV) sheets are stacked (Fig. S2†). This means that on regions with GO the work function of the surface is reduced.29 As explained in the ESI,† the sign convention for the SP is chosen in order to describe the position of the Fermi/HOMO level of the surface structures as compared to the Fermi level of the ITO substrate. A higher (more positive, brighter) SP, therefore, corresponds to a level that is higher than that of the ITO substrate and thus has a lower work function, a lower ionization energy and a lower electronegativity.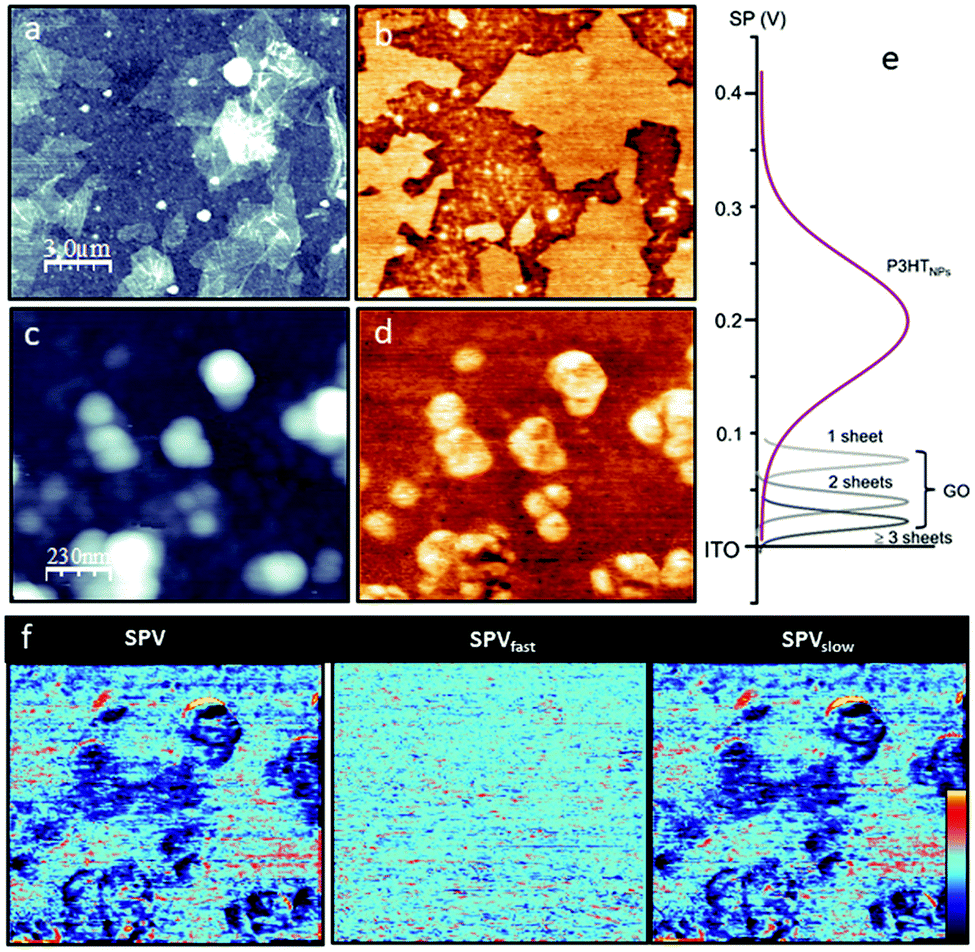 | ||
| Fig. 1 Comparison of GO and the P3HTNPs deposited on ITO as a substrate. (a) Topography (z = 20 nm) and (b) SPdark (−50 to +100 mV) images of the GO. (c) Topography (z = 200 nm) and (d) SPdark (−130 to +370 mV) images of the P3HTNPs. (e) SP histograms obtained from (b) and (d) for different sample regions. (f) SPV performed on P3HTNPs in the same region as that of (c) and (d) (z = ±100 mV). | ||
The topography of the P3HTNPs (Fig. 1c) shows spherical nanoparticles ranging from 50 to 100 nm that tend to agglomerate, probably during the deposition drying process (low magnification images in Fig. S3†). The P3HTNPs present mostly a bright SP contrast (Fig. 1d) with respect to the ITO substrate. The SP of the P3HTNPs is not homogeneous: at high resolution, it is possible to resolve a SP nanostructure within the nanoparticle not correlated with topographic features. The SP difference between high and low SP regions is as high as +250 mV, consistent with the existence of different types of polymer chain aggregates. The high SP regions correspond to more disordered or non-aggregated entangled polymer chains, while the low SP regions can be ascribed to molecularly ordered polymer chain aggregates of H-type characteristic, in agreement with recent findings.11 In these aggregates, the P3HT chains adopt a face-to-face orientation enabling effective π–π inter-chain stacking interactions, which leads to a lowering of the highest occupied molecular energy level, and a corresponding increase of the work function. Results from UV-Vis spectroscopy confirm the co-existence of ordered domains of H-aggregates, exhibiting weakly coupled excitonic interchain interactions and regions of non-aggregated polymer chains in the P3HTNPs.16
To access the optoelectronic properties on a nanometer scale and, in particular, the SPV, KPFM experiments are performed without and with illumination at λ = 535 nm. This wavelength ensures the excitation of the P3HTNPs systems and the involvement of their fundamental vibronic transitions relevant for the transport properties, as outlined in Fig. S4.† As discussed in more detail in the ESI,† in addition to the acquisition of whole SP images in darkness (SPdark), a “two pass” illumination method is applied, where each image line is scanned with and without illumination.26 From this “two pass” method, a pair of SP images (SPon, SPoff) is obtained. These two images allow the classification of the total SPV = SPon − SPdark into a fast contribution SPVfast = SPon − SPoff and a slow contribution SPVslow = SPoff − SPdark. SPV experiments carried out on the pristine materials show that both the GO and the ITO substrate present a null SPV (SPdark = SPon), as expected due to the negligible absorption of these materials at λ = 535 nm (results not shown). In contrast, under green illumination, the SPon of the P3HTNPs is shifted by about −50 mV (Fig. 1f), which is entirely due to slow optoelectronic processes. This well-known behavior is characteristic of the P3HT/ITO interface: holes are transferred to the ITO substrate and trapped at the interface, while electrons are pushed to the external surface.30,31 This explains the slow characteristic of the relaxation process (SPV ≈ SPVslow).
P3HTNPs–GO ensembles
To shed light on how the P3HTNPs polymer chain aggregation and the P3HTNPs–GO interface properties are modified by the intimate contact between GO and the P3HT chains during the in situ P3HTNPs formation, we compare the topography, the SPdark and the SPV images of ex situ and in situ P3HTNPs–GO samples (Fig. 2). Since our main purpose is to clarify the effect of the GO sheets in the P3HTNPs–GO ensembles, as a function of the preparation method, we will focus our study on P3HTNPs, which are located on GO layers in two different configurations, i.e. either “on top” or “sandwiched” between GO sheets, thus avoiding any direct contact to the ITO substrate.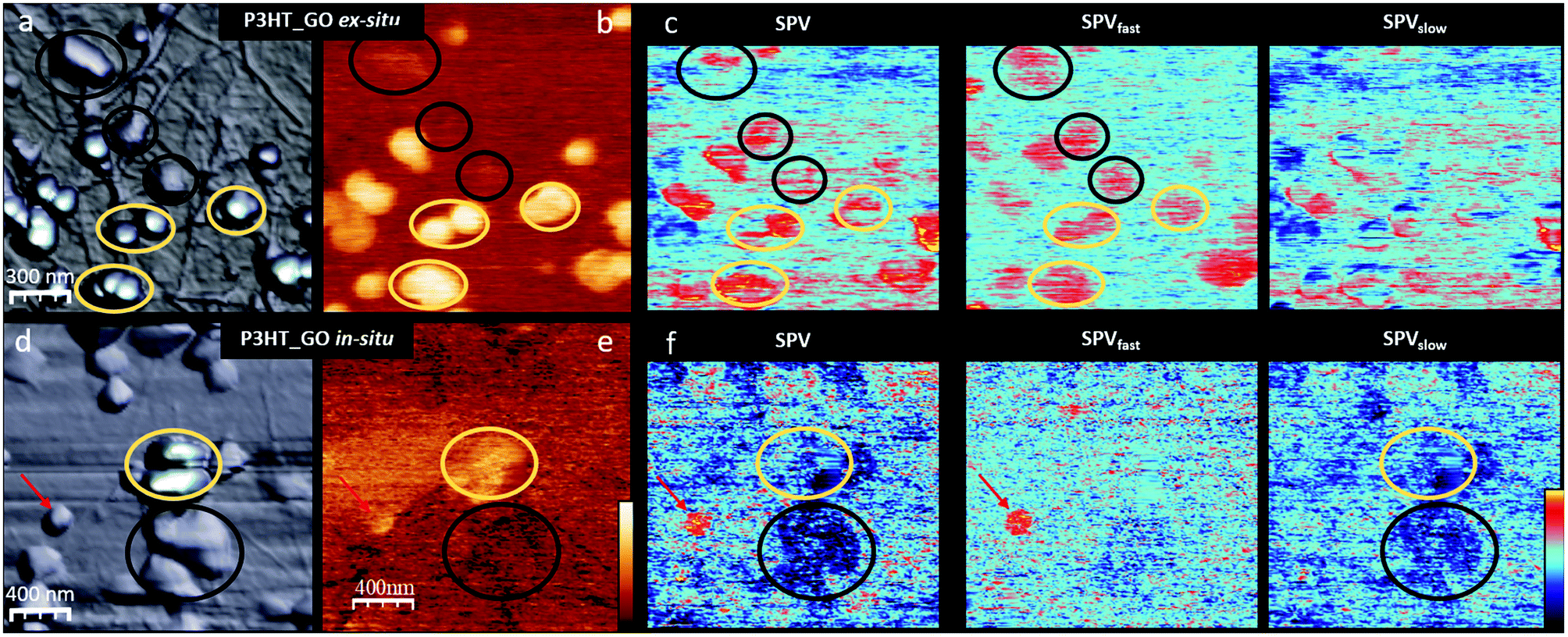 | ||
| Fig. 2 (a) and (d) 3D topography images (z scale 300 nm), (b) and (e) SPdark (colour scale (−50 to +100 mV) and (c) and (f) SPV images (z scale ± 200 mV) of the ex situ and in situ P3HTNPs–GO ensembles, respectively. | ||
Low magnification images show that in the ex situ sample, the P3HTNPs preserve their original size, while in the in situ sample, they tend to form larger aggregates. In both samples, from the topography (Fig. 2a and d) and the SPdark (Fig. 2b and e) images, we distinguish between two types of P3HTNPs exhibiting a very different SPdark value: those situated above and those localized between the GO sheets (marked with yellow and black circles, respectively, in Fig. 2). For both ex situ and in situ P3HTNPs, those placed on top of the GO sheet always present a large bright SPdark contrast. For the ex situ P3HTNPs (Fig. 2b), the ΔSP value with respect to the GO sheet is about +220 ± 50 mV. This value is similar to the one observed in the P3HTNPs directly deposited on ITO, indicating that the polymer chain aggregates within the P3HTNPs are unaltered during the ex situ mixing and confirming the previous spectroscopic results.16 We thus conclude that the properties of the ex situ P3HTNPs–GO ensembles can be completely understood knowing the individual properties of the P3HTNPs on the one hand and the GO on the other, as deduced from the data shown in Fig. 1.
The in situ P3HTNPs, in contrast, behave very differently. The SPdark value is reduced to +100 ± 50 mV (Fig. 2e). Taking into account the fact that the SPdark value is mainly related to the chain aggregates within the P3HTNPs that determine its local work function, the lower SPdark value suggests the existence of a higher number of J-type aggregates (crystalline regions formed by lamellae of folded P3HT chains in a head-to-tail conformation) in the in situ nanoparticles.12,14 Furthermore, the interpretation of lower SPdark values in terms of a higher number of J-type aggregates is fully consistent with the spectroscopic UV-Vis data of in situ P3HTNPs–GO samples.16 These reveal significantly reduced exciton-coupling constants, characteristic of J-aggregates with their highly planar P3HT chains, enabled through π–π interface interactions with GO during the in situ re-precipitation self-assembly process.
The SPdark difference between ex situ and in situ samples is even more drastic in the P3HTNPs covered by GO sheets. The ex situ nanoparticles show a slightly bright SPdark contrast due to the screening effect of the high permittivity GO sheet that reduces the apparent SP of the nanoparticle.32 However, the in situ P3HTNPs present an apparent dark contrast with respect to the GO. As will be discussed below, this SPdark contrast inversion cannot be explained assuming that the GO plays a passive role, acting just as a screening layer, but we should assume that the polymer chain aggregation is modified and that an electron transfer to the GO sheet is taking place. This fully agrees with the results from Raman, FTIR and XPS spectroscopy,16 thus confirming the formation of a ground-state charge-transfer complex on the scale of individual P3HTNPs.
SPV experiments (Fig. 2c and f) also reveal remarkable differences between the in situ and ex situ photoresponses of the P3HTNPs. In the ex situ samples, independently of the nanoparticle position (above or below GO sheets), the P3HTNPs present a positive SPV of about +100 mV. This behaviour is completely opposed to the one of in situ samples, where the P3HTNPs show a negative SPV of about −150 mV that is even more pronounced for the particles covered by GO sheets. Moreover, comparing the SPV image together with its corresponding SPVfast and SPVslow components, we find that the ex situ P3HTNPs almost recover their initial SPdark value instantly (in our experimental time scale <2 s), therefore the SPV ≈ SPVfast. In contrast, for the in situ P3HTNPs, we find SPV ≈ SPVslow, i.e. the photogenerated charges do not have enough time to relax during the “off” part of the two-pass method.33 It should be mentioned that in the in situ sample, a few particles present an ex situ-like behaviour (marked with a red arrow in Fig. 2(d), (f) and (g)). We attribute this performance to P3HTNPs that during the in situ self-assembly process could not establish interactions with GO and thus co-exist as a clearly discernable minor fraction in the in situ P3HTNPs–GO sample.
Taking into account that the SPV is sensitive not only to the polymer chain aggregation determining the photoexciton generation and charge separation within the particle but also to the nanoparticle/GO interface controlling the charge-injection from the P3HTNPs to the GO,34 we can extract several important conclusions: in the ex situ sample, the SPV sign and the time scale is opposite to the SPV of the P3HTNPs directly deposited on ITO (Fig. 1f). Thus, the SPV differences should be due to the presence of low conducting GO sheets in between the ITO substrate and the nanoparticle. In this situation, the GO acts as a blocking-charge layer, whereby the photogenerated charges do not leave the nanoparticle but are redistributed within it. In this situation, as schematically represented in Fig. 3a, the SPV possibly is related to the formation of an effective positive dipole. However, its origin is not easy to elucidate and it may arise from a number of possible processes.34 The photogenerated charges remain in proximity to the nanoparticle and can quickly recombine during the dark periods of the “two pass” method leaving only a small SPslow component. In contrast, in the in situ sample, the SPV behaviour is qualitatively similar to the one observed in the pristine P3HTNPs sample; however, a larger SPV effect is observed since the SPV value changes from −40 mV to −150 mV. This means that holes are more effectively transferred to the GO sheet, acting as hole-acceptor, while the particle remains negatively charged (Fig. 3b).17
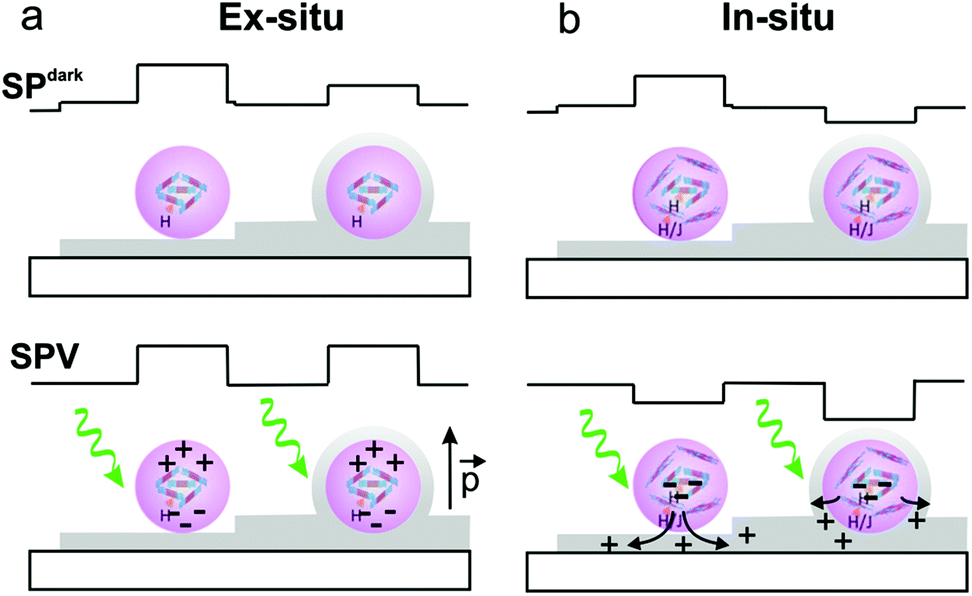 | ||
| Fig. 3 SPdark and SPV illustrations for ex situ (a) and (b) in situ P3HTNPs–GO ensembles probed in two different configurations, respectively: P3HTNPs (purple spheres indicating the aggregate type) on top of GO sheets (grey) and below (sandwiched between) GO sheets, under dark conditions (top) and under illumination (bottom). | ||
In this situation, the photogenerated charges are far apart and the equilibrium is slowly restored once the sample is brought again into darkness. The resulting charge-separation of the photogenerated charges in the P3HTNPs–GO hybrid system should thus be responsible for the enhanced photocurrents observed in thin film devices.
Ex situ samples are characterized by P3HTNPs not interacting with GO sheets, leading to a shielding effect under SPdark conditions and to quickly relaxing effective dipoles under illuminated conditions. In situ samples are characterized by donor–acceptor interactions leading to a P3HT–GO charge-transfer complex with reduced SP dark values and transfer of photogenerated holes to GO under illuminated conditions.
The fact that the SPV sign and the time scale of the relaxation processes differ depending on the ex situ or in situ P3HTNPs–GO prepared samples provides clear evidence that during the in situ preparation process P3HTNPs–GO charge-transfer complexes are formed. These are based on favourable donor–acceptor interface interactions involving J-type aggregates with a more planar chain conformation as evidenced by the SPdark. This results in significantly reduced exciton-coupling constants of the more planar P3HT chains interacting with GO enabling the transfer of photoexcited holes to GO sheets and thus the effective separation of the photogenerated charges (see also sections S1.1 and S4 in the ESI†).
To further understand the in situ P3HTNPs–GO nanohybrids formation, and how it determines the photogenerated charge transport through the P3HTNPs–GO interface, we will focus on the P3HTNPs situated below the GO sheet, where the interface is directly exposed to the SFM tip, compared to the nanoparticles located above the GO sheet, where the interface is hidden by the particle itself. A detailed inspection of the in situ P3HTNPs–GO interface by means of high-resolution images (Fig. 4) reveals that the dark SPdark of the P3HTNPs below the GO sheet has a SP sub-structure that clearly differs from the surrounding GO. Here, large low SP domains comprising most of the P3HTNPs coexist with smaller high SP regions (Fig. 4b). The existence of the low SP domains, related to higher local electronegativity, indicates that at these regions, a negative charge-transfer from the P3HTNPs to the GO is taking place, modifying the local GO work function, confirming the formation of donor–acceptor P3HTNPs–GO charge-transfer complexes, as discussed above.
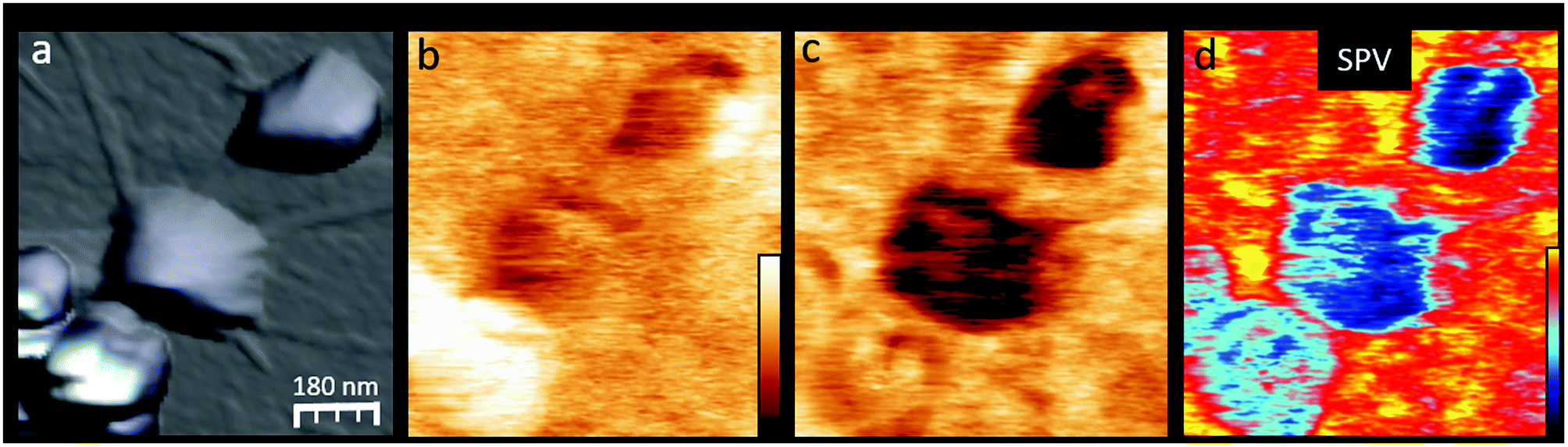 | ||
| Fig. 4 High magnification images of an in situ P3HTNPs–GO sample region. (a) Topography (z = 200 nm), (b) SPdark (z = 300 mV), (c) SPon (z = 300 mV), and (d) SPV = SPon − SPdark (z = −230–70 mV) images. | ||
Under illumination (Fig. 4c), the entire P3HTNP becomes more electronegative (SPon is downshifted). Although a certain SPon particle sub-structure is also observed, there is no direct correlation with the original SPdark one, as further evidenced by the SPV image (Fig. 4d). Moreover, small positively charged patches appear at the surrounding GO sheet confirming that the photogenerated holes are effectively injected from the P3HTNPs to the GO sheets over distances of several tens of nanometers, demonstrating the role of the interacting GO sheets as a HTL. The successful charge-separation of the photogenerated charges disclosed in this KPFM study explains well the findings of enhanced photocurrents in films of P3HTNPs–GO donor–acceptor ensembles and thus further highlights their value as a photoactive layer.16
Conclusions
In summary, we present a comprehensive KPFM study on individual P3HTNPs–GO ensembles. Probing for the first time their nanoscale photogenerated charge-transfer dynamics, we demonstrate that the type of aggregate structure acquired by the conjugated polymer chains critically defines the operational functionality of GO sheets and thus the overall photophysical performance of the P3HTNPs–GO ensemble. SPdark images clearly visualize the existence of crystalline domains in individual P3HTNPs and uncover an important change from H-type to J-type aggregates for the in situ P3HTNPs–GO ensembles, discernable by lowering of SPdark by up to 120 meV. Moreover, both SPdark and SPV images disclose a distinctive positive or negative voltage sign for P3HTNPs of ex situ or in situ ensembles, respectively. This unique contrast inversion undoubtedly proves the creation of donor–acceptor interactions between P3HTNPs and GO and the formation of P3HTNPs–GO charge-transfer complexes, which involve J-type aggregates being established exclusively during the in situ liquid phase self-assembly process. Additionally, the two-pass SPV method allows for discerning between quick relaxation processes associated with the creation of effective dipoles within the P3HTNPs of ex situ P3HTNPs–GO ensembles and a slow relaxation dynamics originated by the dissociation of photogenerated charges in the in situ P3HTNPs–GO donor–acceptor complex. Although in the ex situ ensembles GO thus acts as a charge-blocking layer, in the in situ charge-transfer complex GO adapts the role of a HTL, facilitating the efficient injection of photogenerated holes from the interacting P3HTNPs over distances of several tens of nanometers. Our findings on the critical role of the aggregate structure for defining the role of GO sheets and establishing favourable phototransport properties in the P3HTNPs–GO benchmark ensembles are of general character and provide pathways to a rational design of conjugated polymer and graphene oxide ensembles offering improved optoelectronic performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has received funding from the Spanish MINECO (project grants ENE 2016-79282-C5-1-R and ENE 2016-79282-C5-4-R) and the associated EU Regional Development Funds. E. P. L. and J. C. acknowledge funding from Fundación Séneca, Comunidad Autónoma de la Región de Murcia (Plan de Actuación 2018, Ref. 20860/PI/18). A. M. B., E. I., and W. K. M. acknowledge funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowksa-Curie grant agreement no. 642742. They also acknowledge the Gobierno de Aragón (Grupo Reconocido DGA T03_17R) and the associated EU Regional Development Funds.Notes and references
- K. Tremel and S. Ludwigs, Adv. Polym. Sci., 2014, 265, 39–82 CrossRef CAS.
- D. M. DeLongchamp, B. M. Vogel, Y. Jung, M. C. Gurau, C. A. Richter, O. A. Kirillov, J. Obrzut, D. A. Fischer, S. Sambasivan, L. J. Richter and E. K. Lin, Chem. Mater., 2005, 17, 5610–5612 CrossRef CAS.
- M. T. Dang, L. Hirsch, G. Wantz and J. D. Wuest, Chem. Rev., 2013, 113, 3734–3765 CrossRef CAS PubMed.
- F. C. Spano, Chem. Phys., 2006, 325, 22–35 CrossRef CAS.
- J. Clark, J.-F. Chang, F. C. Spano, R. H. Friend and C. Silva, Appl. Phys. Lett., 2009, 94, 163306 CrossRef.
- S. T. Turner, P. Pingel, R. Steyrleuthner, E. J. Crossland, S. Ludwigs and D. Neher, Adv. Funct. Mater., 2011, 21, 4640–4652 CrossRef CAS.
- F. C. Spano and C. Silva, Annu. Rev. Phys. Chem., 2014, 65, 477–500 CrossRef CAS PubMed.
- N. Kleinhenz, N. Persson, Z. Xue, P. H. Chu, G. Wang, Z. Yuan, M. A. McBride, D. Choi, M. A. Grover and E. Reichmanis, Chem. Mater., 2016, 28, 3905–3913 CrossRef CAS.
- S. Y. Son, Y. Kim, J. Lee, G.-Y. Lee, W.-T. Park, Y.-Y. Noh, C. E. Park and T. Park, J. Am. Chem. Soc., 2016, 138, 8096–8103 CrossRef CAS PubMed.
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS PubMed.
- M. Baghgar and M. D. Barnes, ACS Nano, 2015, 9, 7105–7112 CrossRef CAS PubMed.
- G. Nagarjuna, M. Baghgar, J. A. Labastide, D. D. Algaier, M. D. Barnes and D. Venkataraman, ACS Nano, 2012, 6, 10750–10758 CrossRef CAS PubMed.
- E. T. Niles, J. D. Roehling, H. Yamagata, A. J. Wise, F. C. Spano, A. J. Moule and J. K. Grey, J. Phys. Chem. Lett., 2012, 3, 259–263 CrossRef CAS.
- M. Baghgar, J. A. Labastide, F. Bokel, R. C. Hayward and M. D. Barnes, J. Phys. Chem. C, 2014, 118, 2229–2235 CrossRef CAS.
- N. E. Persson, P.-H. Chu, M. McBride, M. Grover and E. Reichmanis, Acc. Chem. Res., 2017, 50, 932–942 CrossRef CAS PubMed.
- E. Istif, J. Hernandez-Ferrer, E. P. Urriolabeitia, A. Stergiou, N. Tagmatarchis, G. Fratta, M. J. Large, A. B. Dalton, A. M. Benito and W. K. Maser, Adv. Funct. Mater., 2018, 1707548 CrossRef.
- S.-S. Li, K.-H. Tu, C.-C. Lin, C.-W. Chen and M. Chhowalla, ACS Nano, 2010, 4, 3169–3174 CrossRef CAS PubMed.
- J. Liu, M. Durstock and L. Dai, Energy Environ. Sci., 2014, 7, 1297 RSC.
- Kelvin Probe Force Microscopy: From Single Charge Detection to Device Characterization, ed. S. Sadewasser and T. Glatzel, Springer, Cham, Switzerland, 2018 Search PubMed.
- C. Groves, O. G. Reid and D. S. Ginger, Acc. Chem. Res., 2010, 43, 612–620 CrossRef CAS PubMed.
- H. Hoppe, T. Glatzel, M. Niggemann, A. Hinsch, M. C. Lux-Steiner and N. S. Sariciftci, Nano Lett., 2005, 5, 269–274 CrossRef CAS PubMed.
- A. Liscio, G. P. Veronese, E. Treossi, F. Suriano, F. Rossella, V. Bellani, R. Rizzoli, P. Samori and V. Palermo, J. Mater. Chem., 2011, 21, 2924–2931 RSC.
- E. J. Spadafora, R. Demadrille, B. Ratier and B. Grévin, Nano Lett., 2010, 10, 3337–3342 CrossRef CAS PubMed.
- G. Shao, M. S. Glaz, F. Ma, H. Ju and D. S. Ginger, ACS Nano, 2014, 8, 10799–10807 CrossRef CAS PubMed.
- D. J. Ellison, J. Y. Kim, D. M. Stevens and C. D. Frisbie, J. Am. Chem. Soc., 2011, 133, 13802–13805 CrossRef CAS PubMed.
- E. Escasain, E. Lopez-Elvira, A. M. Baro, J. Colchero and E. Palacios-Lidon, Nanotechnology, 2011, 22, 375704 CrossRef CAS PubMed.
- E. Palacios-Lidon, J. Abellan, J. Colchero, C. Munuera and C. Ocal, Appl. Phys. Lett., 2005, 87, 154106–154108 CrossRef.
- K. A. Mkhoyan, A. W. Contryman, J. Silcox, D. A. Stewart, G. Eda, C. Mattevi, S. Miller and M. Chhowalla, Nano Lett., 2009, 9, 1058–1063 CrossRef CAS PubMed.
- J. Li, X. Qi, G. Hao, K. Huang and J. Zhong, Fullerenes, Nanotubes, Carbon Nanostruct., 2015, 23, 777–781 CrossRef CAS.
- N. Rujisamphan, T. Supasai and T. Dittrich, Appl. Phys. A: Mater. Sci. Process., 2016, 122, 77 CrossRef.
- F. E. Osterloh, M. A. Holmes, L. Chang, A. J. Moulé and J. Zhao, J. Phys. Chem. C, 2013, 117, 26905–26913 CrossRef CAS.
- K. S. Kumar, S. Pittala, S. Sanyadanam and P. Paik, RSC Adv., 2015, 5, 14768–14779 RSC.
- C. Rogero, D. F. Pickup, J. Colchero, E. Azaceta, R. Tena-Zaera and E. Palacios-Lidón, ACS Appl. Mater. Interfaces, 2016, 8, 16783–16790 CrossRef CAS PubMed.
- J. Zhao and F. E. Osterloh, J. Phys. Chem. Lett., 2014, 5, 782–786 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr01491h |
| ‡ Present address: Univ. Bordeaux, CNRS, Bordeaux-INP, LCPO, UMR 5629, F-33600 Pessac, France. |
| This journal is © The Royal Society of Chemistry 2019 |