
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConformation switching of single native proteins revealed by nanomechanical probing without a pulling force†
Fabiola A.
Gutiérrez-Mejía
ab,
Christian P.
Moerland
ab,
Leo J.
van IJzendoorn
ab and
Menno W. J.
Prins
 *abc
*abc
aDepartment of Applied Physics, Eindhoven University of Technology (TU/e), Eindhoven, The Netherlands. E-mail: m.w.j.prins@tue.nl
bInstitute for Complex Molecular Systems (ICMS), TU/e, Eindhoven, The Netherlands
cDepartment of Biomedical Engineering, TU/e, Eindhoven, The Netherlands
First published on 24th September 2019
Abstract
Protein conformational changes are essential to biological function, and the heterogeneous nature of the corresponding protein states provokes an interest to measure conformational changes at the single molecule level. Here we demonstrate that conformational changes in single native proteins can be revealed by non-covalent antibody-targeting of specific domains within the protein, using nanomechanical probing without an applied pulling force. The protein of interest was captured between a particle and a substrate and three properties were quantified: the twist amplitude related to an applied torque, torsional compliance related to rotational Brownian motion, and translational Brownian displacement. Calcium-dependent conformation switching was studied in native human cardiac troponin, a heterotrimer protein complex that regulates the contraction and relaxation of heart muscle cells and is also a key biomarker for diagnosing myocardial infarction. The data reveal a change in mechanical properties upon conformation switching from the non-saturated to the calcium-saturated state, which in cardiomyocytes gives myosin motor proteins access to actin filaments. A clear increase was observed in the molecular stiffness for the calcium-saturated protein conformation. Using libraries of monoclonal antibodies, the nanomechanical probing of conformation by antibody targeting opens avenues for characterizing single native protein complexes for research as well as for diagnostic applications.
Introduction
Proteins are macromolecules of which subtle changes can have large consequences at the level of cells and tissues. Protein conformational changes modulate functions like molecular recognition, signal pathways, and mechanotransduction.1 Structural biological studies on protein conformations commonly employ X-ray crystallography, circular dichroism and nuclear magnetic resonance. These techniques measure average properties of large ensembles of molecules and are therefore not suited for heterogeneous systems, e.g. molecular systems with intrinsic disorder, with distributed molecular conformations, or with nonsynchronous molecular states. In contrast, single-molecule methods allow researchers to resolve heterogeneities in molecular structure and function. In the field of protein conformation, single molecule Förster resonant energy transfer (smFRET) has been developed for investigating changes of protein conformation.2–5 An important requirement of smFRET is that the protein under study needs to be labeled with a closely spaced pair of fluorescent moieties, which means that the protein needs to be chemically changed and cannot be studied in its native chemical form. Furthermore, single-molecule fluorescence techniques exhibit bleaching and blinking which limit the duration and stability of the molecular observations.In addition to the fluorescence based techniques, force spectroscopies have been developed in which single molecules are attached with a handle to a small mobile object, typically a particle or a tip.6–8 Important examples are optical tweezers (OT) and magnetic tweezers (MT) which have elucidated conformational changes of DNA9 and mechanisms of DNA replication by helicases.10 Optical tweezers have been successful in unravelling protein unfolding,11,12 protein misfolding13 and the kinetic properties of molecular motors such as kinesin14 while further studies on the mechanics of cell surfaces are in progress.15
In force spectroscopies, significant forces (pN to nN) are applied to the molecule of interest, passed on by the handles that are attached to the molecule. Such handles are typically introduced by covalent chemical modifications, which precludes studies of unmodified single native proteins. Furthermore, many native proteins are in fact protein complexes, consisting of multiple proteins stabilized by non-covalent protein–protein interactions. Such non-covalent protein bonds dissociate when a significant force is applied. Therefore methodologies with minimal forces are needed for revealing conformation switching in single unmodified native protein complexes.
Here we demonstrate the feasibility of a nanomechanical strategy to study conformational changes of single native protein complexes. We will abbreviate the methodology as nCAT: nanomechanical probing of conformation by antibody targeting. The sensing methodology is compatible with non-covalent protein–protein interactions because no pulling forces are applied, allowing studies on complexes of proteins with site-specific targeting using antibodies. We use the Ca2+ induced conformation change in the human cardiac troponin complex (cTn) as a molecular demonstrator system.
Experiments: the nCAT methodology
Fig. 1a sketches the nCAT experiment. Details on sample preparation and data analysis are given in the ESI section S1.† On the stage of a bright-field fluorescence microscope, a magnetic tweezers setup (Fig. 1b) is positioned with a flow cell (Fig. 1c). The protein of interest is captured between a particle and a substrate in the flow cell (Fig. 1d). The magnetic setup consists of four electromagnets with slanted pole tips, designed to apply a controlled torque on the particles without applying a force. The particle and the substrate are provided with a very low surface density of antibodies that bind to two epitopes of the protein. The methodology is applied to study calcium-induced changes of the native human cardiac troponin complex (cTn). This heterotrimer complex regulates cardiac muscle contraction16–20 and is the gold standard biomarker for the diagnosis of acute coronary syndrome,21,22 with approximately 200 million tests performed every year. Fig. 1e sketches how conformation switching occurs within the cTn complex. The complex has three functional subunits: the inhibitory subunit I (cTnI), the Ca2+ sensor subunit C (cTnC) and the tropomyosin-binding subunit T (cTnT). In the relaxed muscle state with low Ca2+ levels, cTnI blocks the myosin binding site on actin23 and thereby inhibits the actomyosin-ATPase activity.24 The sensor subunit cTnC has a dumbbell shape with globular lobes (N terminal and C terminal) that are connected through a flexible linker.25,26 Upon Ca2+ binding to the N lobe, a change in conformation occurs and the switch region of cTnI binds to a hydrophobic patch on the cTnC. This conformational change induces the displacement of the inhibitory region of cTnI from the myosin binding site and thereby leads to muscle contraction.27 Due to the sophistication of the molecular machinery and its physiological relevance, the troponin complex is a very interesting system for a study of conformation-dependent nanomechanical properties.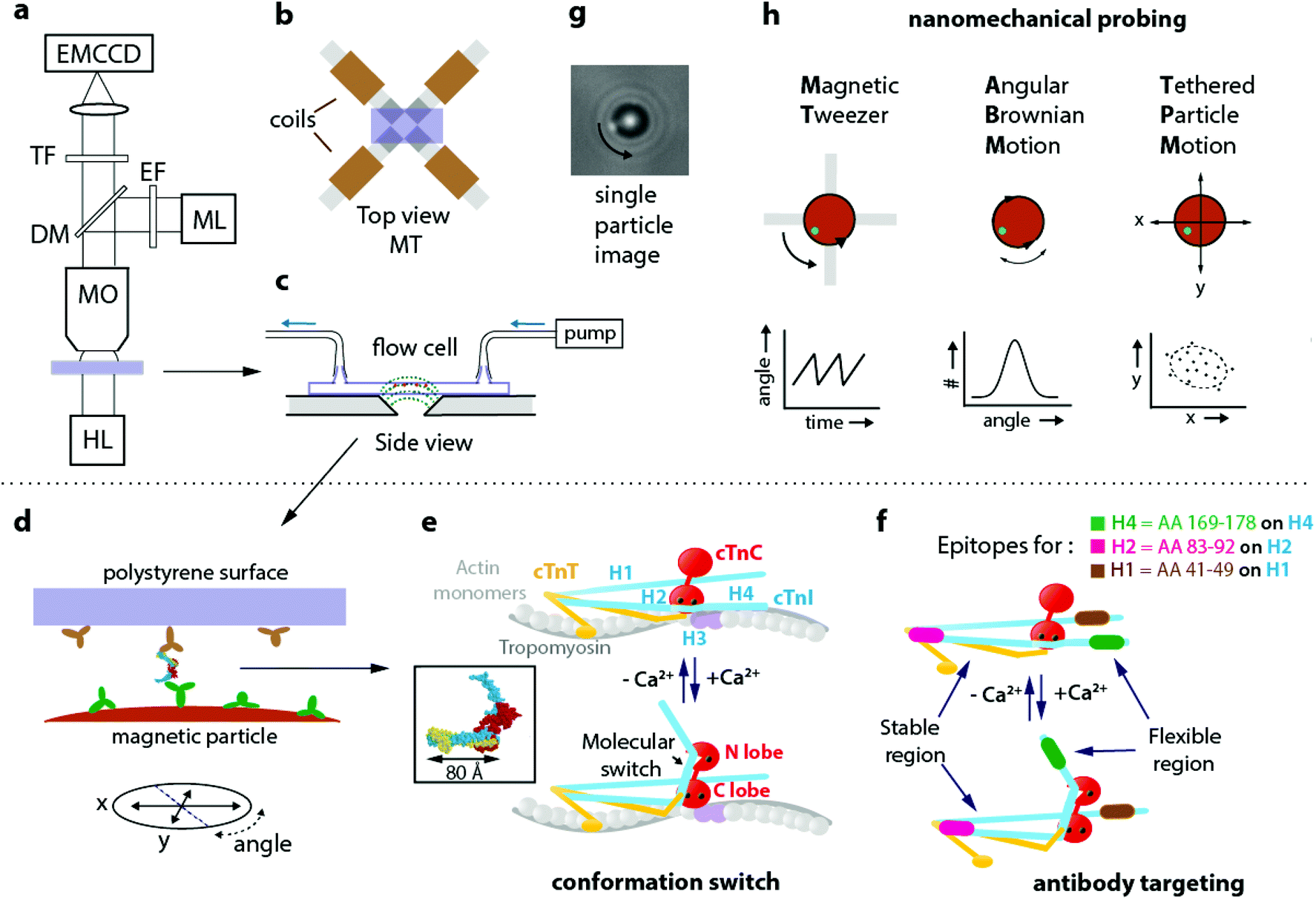 | ||
| Fig. 1 Nanomechanical probing of conformation by antibody targeting (nCAT) applied to a study of human cardiac troponin (cTn). (a) nCAT microscopy: the sample (purple) is illuminated by a halogen lamp (HL) and imaged in transmission mode with a water-immersion microscope objective (MO). A mercury lamp (ML) with excitation filter (EF) and dichroic mirror (DM) is used to excite fluorescent markers attached to magnetic particles in order to quantify angular orientation. The transmitted and reflected light are detected with an EMCCD positioned behind a transmission filter (TF). (b) Top view of the sample positioned on the magnetic tweezers (MT) system that consists of a quadrupole electromagnet with slanted pole tips. When currents are applied, the slanted tips create at the position of the particles a highly uniform magnetic field with an in-plane orientation. (c) Side view of the flow cell placed on the MT. The dotted lines (green) sketch the magnetic field lines. Magnetic particles (brown) are bound by a protein complex to the top polystyrene surface. (d) Monoclonal antibodies on the polystyrene surface of the flow cell and on the magnetic particles target epitopes on the protein complex, in this case cardiac troponin (cTn). (e) Schematic representation of the Ca2+ induced conformational change of the cTn complex according to Takeda et al.;18 the inset (reprinted by permission from Springer Nature, Nature, Structure of the core domain of human cardiac troponin in the Ca2+-saturated form, S. Takeda et al., Copyright 2003) depicts the molecular structure that was resolved by the authors. cTnI subunit in blue, cTnT in yellow, and cTnC in red. On cTnI the helical domains H1–H4 are indicated. (f) The cTnI epitopes targeted by the three monoclonal antibodies of this study: highlighted in green (H4 epitope, targeting aminoacids 169–178 on the H4 domain of cTnI), magenta (H2 epitope, targeting aminoacids 83–92 on the H2 domain of cTnI) and brown (H1 epitope, targeting aminoacids 41–49 on the H1 domain of cTnI). The flexible region of cTn and its Ca2+ induced conformational change are indicated. (g) Image of a magnetic particle as recorded by the EMCCD. A fluorescent fiducial marker is visible at the edge of the particle and allows to track particle rotation. (h) Nanomechanical probing modalities, one with magnetic field (MT) and two without applied magnetic field (ABM, TPM). A continuously rotating magnetic field of the MT induces protein twisting, recorded as the deformation angle vs time. Angular Brownian Motion (ABM) probes the rotational harmonic potential around the equilibrium angle of the molecular complex in the absence of a magnetic field. ABM is recorded as the probability vs angle. Tethered Particle Motion (TPM) probes the in-plane x,y positions in consecutive images and is recorded as a 2D scatter plot denoted as a motion pattern. The dashed line indicates the area of particle excursion. | ||
In this study, the cTn complex was specifically targeted using commercially available monoclonal antibodies that bind to the helical H4, H2 and H1 domains of cTnI within the native cTn I–C–T complex. Two targeting configurations are compared, for targeting a flexible and a stable region of cTn, see Fig. 1f. Antibody pair H4–H1 (depicted as green-brown) targets a flexible region that should switch under the influence of calcium. Antibody pair H2–H1 (depicted as magenta-brown) targets a region of cTnI that should be insensitive to calcium. In the experiment, the H1 antibodies were immobilized on the polystyrene coverslip by physisorption and the other antibodies (H4 or H2) were covalently coupled to 2.8 μm superparamagnetic particles using EDC-NHS chemistry. The preparation conditions were optimized to reach the single molecule regime by incubating a low concentration of the H1 antibody on the polystyrene coverslips, as explained in the ESI section S2.†
Both the angular orientation and position of a single particle are monitored by video microscopy (Fig. 1g) and serve to monitor the protein via three independent nanomechanical observables: the twist amplitude applying torque by the magnetic tweezer (MT), the torsional compliance measured by Angular Brownian Motion (ABM), and the translational compliance measured by the in-plane mobility of the particle by Tethered Particle Motion (TPM).
Fig. 1h sketches the principles of the nanomechanical measurements, one method with magnetic field (MT) and two methods without applied magnetic field (ABM and TPM). In the MT measurement, the particles are exposed to a rotating magnetic field that is generated by the magnetic tweezers. The field generates a magnetic torque in the particle and this torque twists the protein. As the field continues to rotate, a maximum twist angle is reached where the mechanical torque of the protein equals the maximum magnetic torque that can be delivered by the particle. Thereafter the particle flips back and the process starts anew. This repeated mechanism generates the sawtooth-like behaviour of the angle as a function of time. The maximum twist angle of the protein and the periodic angular excursions can be related to the torsional compliance of the protein, see ESI section S3.† The torsional molecular stiffness can also be measured by recording the angular thermal fluctuations of the particle, i.e. its Angular Brownian Motion (ABM). The measurement yields a histogram of angular positions and the width of the distribution is a direct measure for the torsional compliance of the protein in thermal equilibrium. Tethered Particle Motion (TPM) refers to the measurement of translational Brownian motion of a particle that is tethered to a substrate, yielding position histograms that are referred to as motion patterns. TPM has been used to study contour and persistence lengths of long macromolecules,28,29 but not yet to characterize changes in proteins. In our experiment the sandwich configuration of antibodies and cTn protein (mAb-cTn-mAb) acts as a stiff and short tether between particle and substrate. This short tether gives small yet clearly detectable motion patterns that relate to the flexibility of the proteins.
Results and discussion
Fig. 2a shows the MT measurements for the H4–H1 and the H2–H1 molecular configurations. The particle angle is shown as a function of time when the particles are exposed to a rotating magnetic field. A reproducible sawtooth-like behaviour is observed, in both clockwise and thereafter in anticlockwise direction, which relates to the torsional flexibility of the proteins captured between particle and substrate.30,31 The maximum excursion angle θmax is much larger for the H4–H1 antibody pair than for the H2–H1 antibody pair, indicating that the H4–H1 configuration is more flexible than the H2–H1 configuration. This is in agreement with the location of the epitopes, because the H4 helix is located in the intrinsically disordered region of cTnI5 while the H2 helix is stable. Fig. 2b shows the ABM histograms of the same particles and protein complexes of which the MT measurements were recorded in Fig. 2a. The H4–H1 experiment shows a larger torsional flexibility than the H2–H1 experiment, in agreement with the MT measurements. | ||
| Fig. 2 nCAT measurements targeting the flexible region (H4–H1, top row) and stable region (H2–H1, bottom row) of cTnI. The particles were bound to the substrate via an antibody sandwich with cTn I–C–T complex, as in Fig. 1d. The Ca2+ concentration was 1 nM. Top row: Particle bound through the H4–H1 antibody pair, targeting the mobile H4 helix of cTnI. Bottom row: Particle bound through the H2–H1 antibody pair, targeting the non-mobile H2 helix of cTnI. (a) Magnetic Tweezers (MT) measurements data in a rotating magnetic field. The angular orientation of the particle is shown for clockwise and anticlockwise rotations of the magnetic field. The static segment around t ≈ 6 s corresponds to a waiting time to allow reversal of the direction of field rotation. The field magnitude was 20 mT and the rotation frequency was 0.5 Hz. The maximum angular excursion is indicated with red arrows. (b) Angular Brownian Motion (ABM) measurements. Histograms of the angular fluctuations of the same particles as in panel a, but now without an applied magnetic field. The full width at half maximum (FWHM) of the distribution is indicated with red arrows. (c) Tethered Particle Motion (TPM) data, showing a scatter plot of recorded particle xy-positions, measured with an integration time of 50 ms. The ellipses enclose 95% of the data points. | ||
The torsional compliance has been quantified by calculating the torsion constant k of a linear Hookean torsional spring for both the MT and ABM measurements and are listed in Table 1. For the MT experiments, the spring constant of the troponin complex has been obtained by solving numerically the differential equation that describes the torque balance and finding the closest match to the experiment. The torque balance contains the applied magnetic torque and the counteracting torque of the molecular spring as well as hydrodynamic friction. The applied magnetic torque was calculated with a model of the magnetic particle that describes its internal structure with a distribution of coercivities and magnetization angles.32 Details of the calculation are given in the ESI section S3.† The values of the spring constants reported in Table 1 are averaged over the clockwise and anticlockwise directions. For the ABM experiments, the spring constants have been quantified by fitting by a Gaussian distribution and obtaining the spring constant k from the variance of the distribution corrected by motion blur. Details are given in the ESI sections S1 and S4.†
| MT | ABM | ||
|---|---|---|---|
| Configuration | [Ca2+] | k (Nm rad−1) | k (Nm rad−1) |
| H4–H1 (flexible region) | 1 nM | 1.0 ± 0.4 × 10−18 | 1.4 ± 0.8 × 10−18 |
| H4–H1 (flexible region) | 1 mM | 3.4 ± 1.0 × 10−18 | 3.2 ± 1.0 × 10−18 |
| H4–H1 (flexible region) | 1 nM | 1.8 ± 0.5 × 10−18 | 0.5 ± 0.3 × 10−18 |
| H2–H1 (stiff region) | 1 nM | 4.8 ± 1.0 × 10−18 | 5.3 ± 2.0 × 10−18 |
| H2–H1 (stiff region) | 1 mM | 4.8 ± 2.0 × 10−18 | 5.1 ± 2.0 × 10−18 |
| H2–H1 (stiff region) | 1 nM | 4.8 ± 2.0 × 10−18 | 4.9 ± 1.5 × 10−18 |
Remarkably, for both the H4–H1 and H2–H1 configurations, nearly all torsion constants quantified by MT and ABM agree within their uncertainties, which underlines the validity of the methods. The torsion constants in Table 1 show that at a low Ca2+ concentration of 1 nM the resulting torsion constants of the H4–H1 system are significantly lower than for the H2–H1 system, which is in agreement with the molecular picture sketched in Fig. 1e and f.
The TPM data of the same experiments are shown in Fig. 2c. The protein configuration acts as a stiff tether between particle and substrate, giving small particle excursions because of the short effective tether length (∼40 nm or less). The motion patterns are ellipsoidal rather than circular, in contrast to TPM data recorded with long dsDNA tethers.28,29 We observed a particle-to-particle variation of the ellipticity that is not correlated with the targeted region of the protein complex. We attribute the ellipsoidal shape to the fact that the protein complex is mechanically stiff, where the ellipsoidal pattern relates to its non-axially symmetric internal structure or to its inclination with respect to the substrate. Instead, we quantified the area of excursion of the particle A, as a measure for the bending compliance of the mAb-cTn-mAb protein complex. The data in Fig. 2c shows that the particle with the H4–H1 protein configuration explores a larger area than the H2–H1 configuration, which is in agreement with the larger torsional compliance of the H4–H1 configuration seen in the MT and ABM data.
These results provide solid ground for a study of calcium-induced changes of the cardiac troponin I–C–T complex. Calcium levels were modulated over time in a flow cell and single particles were monitored using MT, ABM, and TPM. Fig. 3 shows an example of a single-molecule cTn experiment with the H4–H1 antibody pair. CD spectroscopy and FRET experiments have shown that cTnC has two high-affinity Ca2+ binding sites in the C-lobe (KCa ∼ 3.2 ± 1.2 × 108 M−1) and one with lower affinity in the N-lobe that interacts with the motile H4 helix of cTnI (KCa ∼ 2.5 ± 2.0 × 106 M−1).33 Therefore the Ca2+ concentration was changed from 1 nM to 1 mM, and then back to 1 nM. The Ca2+ level was controlled using EGTA as a chelator agent and the concentration was monitored to ensure that the desired Ca2+ levels were reached, see ESI section S5.†
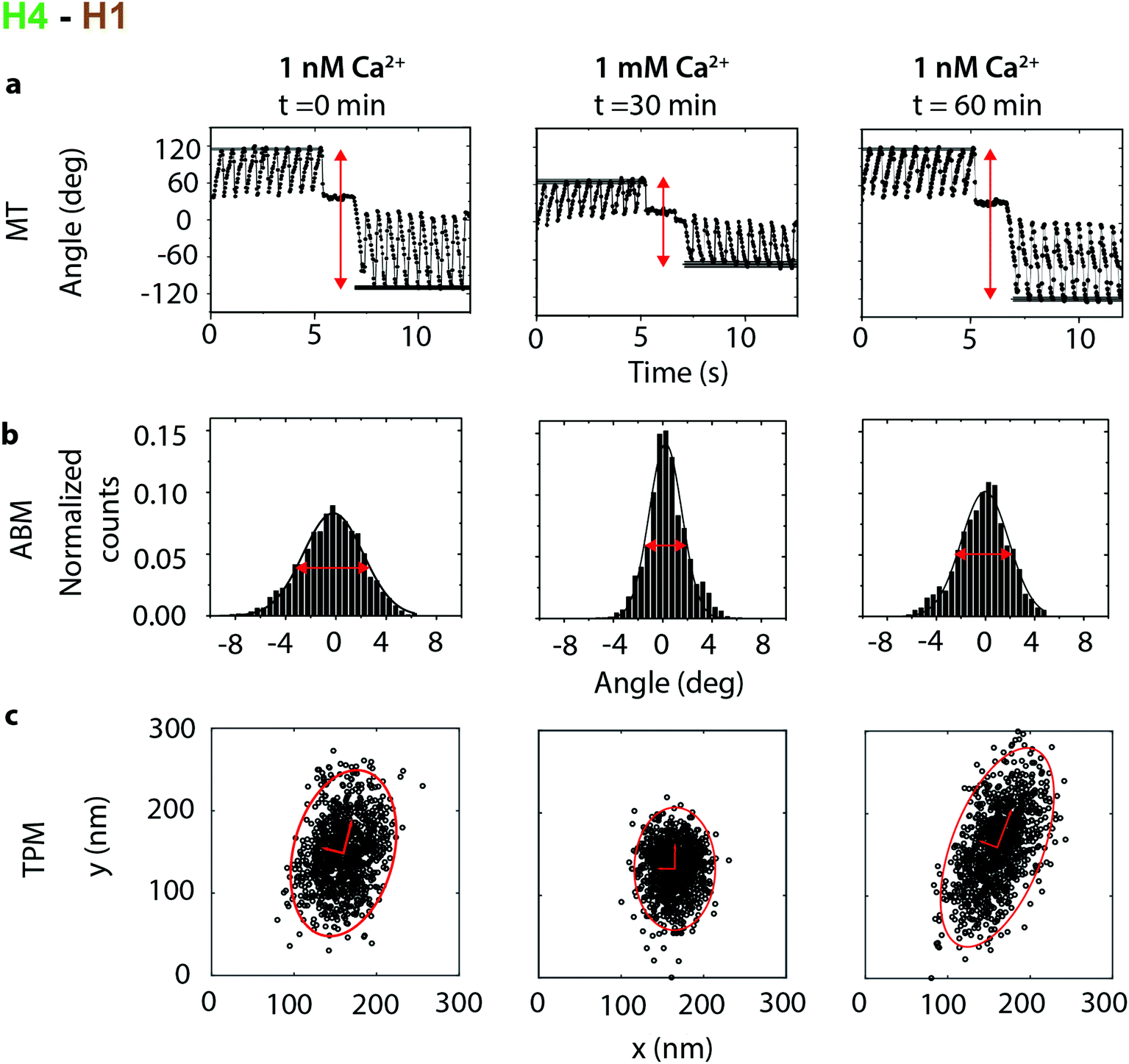 | ||
| Fig. 3 Calcium dependence recorded in an experiment with the H4–H1 antibody pair, targeting the motile H4 helix of cTnI. The initial Ca2+ concentration was 1 nM, thereafter the Ca2+ concentration was increased to 1 mM, and then the Ca2+ concentration was again reduced to 1 nM. At each concentration three data sets were recorded: (a) The angular deformation in a MT experiment, (b) angular Brownian motion and (c) tethered particle motion. Measurements at each Ca2+ concentration have been recorded with a time difference of ∼30 min in order to fully exchange the buffer in the flow cell (see ESI section S5†). | ||
The MT, ABM and TPM data all show a clear response to the calcium level. When the calcium concentration increased from 1 nM to 1 mM, the measurements revealed a decrease of θmax in the MT and a decrease of the full width at half maximum (FWHM) in the ABM measurements. The resulting torsion constants (Table 1) increase from an average 1.5 ± 0.6 × 10−18 Nm rad−1 at 1 nM Ca2+ to 3.4 ± 1 × 10−18 Nm rad−1 for a Ca2+ concentration of 1 mM, showing that the stiffness of the molecular complex increases upon Ca2+ binding. Additionally the area of excursion in the TPM experiments decreases from an average of 2.7 × 104 nm2 at 1 nM Ca2+ to 1.1 × 104 nm2 at 1 mM Ca2+, which also demonstrates the stiffening upon calcium binding.
This behaviour was reversible: when the Ca2+ concentration decreased from 1 mM to 1 nM, the absolute values of all parameters increased, although the original values were not exactly recovered. The ellipticity of the TPM motion patterns shows a significant particle-to-particle variation but does not show a correlation with the Ca2+ concentration.
As a control, calcium-modulated experiments were performed on the cTn complex targeted by the H2–H1 antibody pair (an example is shown in ESI section S6†), showing hardly any dependence on calcium levels. The corresponding torsion constants for the H2–H1 antibody pair in Table 1 confirm the absence of a Ca2+ dependence, although the errors are relatively large due to particle-to-particle variation.
Variability in protein response
In the experiments the particle and substrate were functionalized with antibodies using non-directional methods (physisorption on the substrate, covalent EDC chemistry on the particle). This induces a variety of antibody orientations,34–37 which is expected to generate variability in the nanomechanical parameters measured in different single-molecule experiments. The cTn I–C–T complex itself and surface roughness of particle and substrate can also be sources of variability. In order to establish the significance of the changes in the torsion constants for the H1–H4 versus the H1–H2 antibody pairs at different Ca2+ concentrations, we performed a statistical analysis (Fig. 4) and we analysed the calcium-modulated cTn experiments in terms of their types of response (Fig. 5).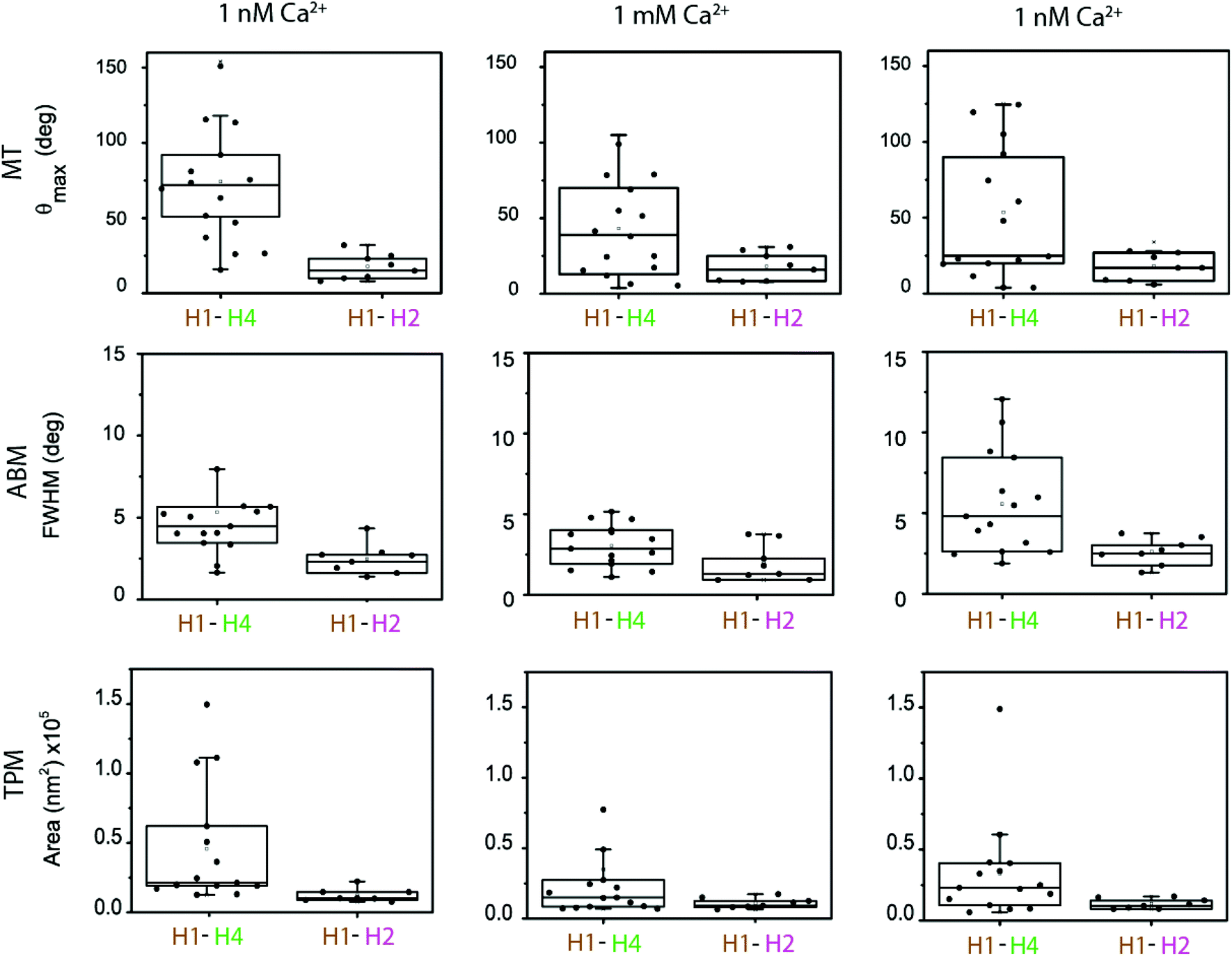 | ||
| Fig. 4 Data measured in multiple single-molecule experiments. In every experiment data were collected by the three measurement methods: MT, ABM, and TPM. Experiments were performed with the H4–H1 assay (n = 15) and the H2–H1 assay (n = 8). In every experiment, data was collected for the subsequent Ca2+ concentrations that are indicated at the top of the figure. | ||
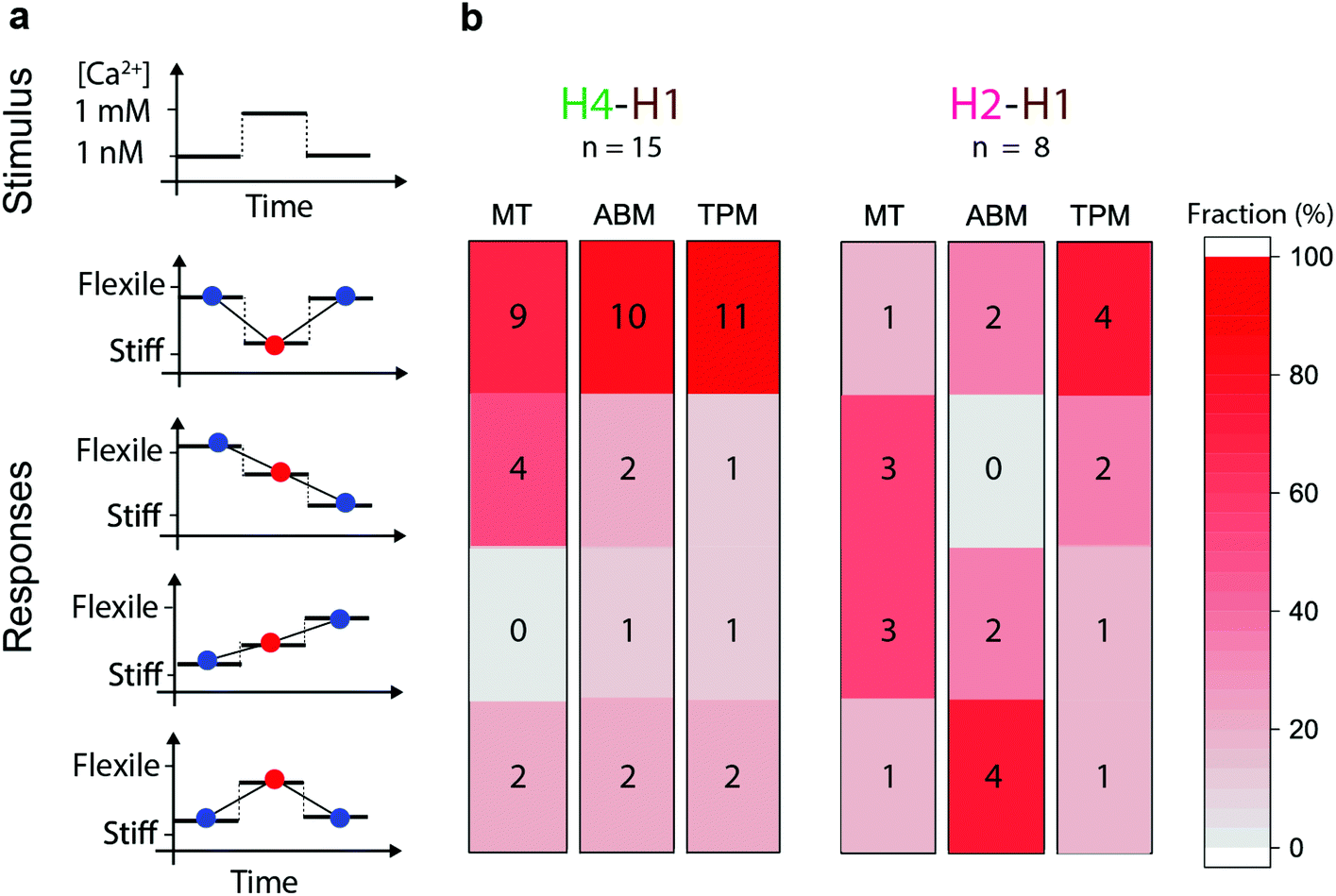 | ||
| Fig. 5 Relative changes of the three nanomechanical observables (MT, ABM, TPM) upon calcium modulation, for H4–H1 experiments (flexible region) and for H2–H1 experiments (stable region). (a) Sketch of possible responses to a calcium stimulus. The three connected data points refer to measurements on single particles at the following sequential Ca2+ levels: blue (initial 1 nM), red (1 mM), blue (final 1 nM). A downward step refers to stiffening; an upward step refers to increased flexibility. (b) Heat map of the measured response characteristics of the H4–H1 cTn complexes (n = 15) and the H2–H1 cTn complexes (n = 8), when exposed to a 1 nM–1 mM–1 nM Ca2+ concentration time profile. The numbers represent the number of particles exhibiting the indicated response. The red-grey color scale indicates the frequency, i.e. the fraction of particles with the indicated response. | ||
Fig. 4 shows the observables recorded with MTT, ABM and TPM, for both molecular configurations, and for the different subsequent calcium levels (1 nM, 1 mM, 1 nM). The figure shows the data as non-overlapping scatter points and boxes are included in the graph in order to show the distributional characteristics of the data. The lower and upper boundaries of the boxes indicate the first quartile Q1 and the third quartile Q3 of the data, respectively. The median is indicated as a line inside the box. The upper whisker is determined by Q3 + 1.5 × IQR and the lower whisker is Q1 − 1.5 × IQR, where IQR is the interquartile range. To asses significant differences, data was correlated with a T-test (p < 0.05).
The boxplots of the data indicate significant differences between the cTn targeted by the H4–H1 and by the H2–H1 antibodies. For all Ca2+ concentrations, the median values of the measured parameters are larger for the H4–H1 complex than for the H2–H1 complex. The differences are particularly clear for the data in the first column, which represents the start of the experiments, at a 1 nM Ca2+ concentration. All data show a large spread. At the start of the experiments, the MTT data points for the H4–H1 complex range between 20 and 150°, while the values for the H2–H1 cTn complex range between 10 and 20°.
The ABM and TPM data are in agreement with the MT measurements: larger median values and spread for the H4–H1 complex than for the H2–H1 complex. Our statistical analyses show that the difference between the two molecular configurations was significant for all three measurement methods at the start of the experiments (first column) and at the end of the experiment (third column).
Upon increase of Ca2+ concentration for the H4–H1 pair (second column in Fig. 4) the median of the distribution of MTT measurement decreases, together with a slight decrease of the width of the distribution. For the H2–H1 system, no noticeable change in the distribution is found. Consistently, the experimental observables from ABM and TPM also present an increase of stiffening upon calcium increase (lower medians) for the H4–H1 pair, and no evident effects for the H2–H1 complex. The T-tests show that the stiffening measured with MT and TPM upon increase of Ca2+ is significant, but it is not significant for the ABM data. The statistical analysis for the H2–H1 complex does not show significant differences for any of the three measurement techniques.
When subsequently the calcium concentration was decreased to 1 nM (third column in Fig. 4), the median of θmax for the H4–H1 complex did not fully recover. Hypotheses for the relative insensitivity of the molecular system to return to its flexible state might be a firm attachment of the complex as a result of the conformational change. Nevertheless, it is observed that the first quartile of the box and the upper whisker shift towards larger θmax, which does indicate that at least 50% of the population partially recovers after calcium removal. The ABM and TPM measurements did exhibit an increase of the median values, indicating more clearly the reversibility upon calcium exchange although not in all cases. The statistical analyses reflect this observation, in which only TPM measurements after recovery (second column) are significantly different to the measurements at 1 mM Ca2+ (third column). The H2–H1 system remained unchanged upon a decrease of the calcium concentration, since the medians and the quartile positions did not show significant differences in the statistical tests.
To further detail the quantification of the nCAT methodology, it will be interesting to study the role of surface functionalizations, antibody directionality, and to investigate other epitope binders such as Fab fragments, nanobodies and aptamers. See section S7 of the ESI† for a further discussion on possible sources of variabilities.
Fig. 5 shows the response types that have been observed for individual particles during modulation of the Ca2+ concentration in the flow cells. Fig. 5a shows the type of response and Fig. 5b the frequency of the response types in the three observables, for the H4–H1 and the H2–H1 configurations. A stiffening response corresponds to a decrease of the observed θmax in the MT measurement, a decrease of the FWHM in ABM, and a decrease of A in the TPM measurement; and the opposite for a loosening response. In spite of the variabilities, we see a remarkably clear difference between the H4–H1 and the H2–H1 experiments. The dominant response type in the H4–H1 experiments is, for each of the three observables, one of stiffening upon calcium increase and loosening upon subsequent calcium decrease (9–11 out of 15 experiments). In contrast, the H2–H1 data shows a disperse behaviour over all response types. Four H4–H1 experiments show reversible responses to calcium with a synchronous and consistent behaviour in all three observables, i.e. stiffening upon calcium increase and loosening upon subsequent calcium decrease.
The direction of calcium sensitivity observed in the H4–H1 experiment and the calcium insensitivity in the H2–H1 experiment are in agreement with the molecular picture sketched in Fig. 1. The H4–H1 antibody pair targets the highly flexible H4 helix (aminoacids 169–178 on cTnI).5,38 Takeda et al.18 determined that Ca2+ binding induces the displacement of residues 137–210. Recent FRET studies on the cTn complex39 show that the inhibitory region of cTnI (aminoacids 137–148) is flexible in the absence of Ca2+ and assumes a rigid confirmation in the Ca2+ saturated state due to binding of the H3 helix (aminoacids 148–163) to the N lobe of cTnC. In contrast, helix H2 and H1 are stabilized through multiple polar and van der Waals interactions with the other cTn subunits.19,40 Cordina et al.41 found with NMR that the H1 domain is tightly bound to the C lobe of cTnC, independent of the Ca2+ concentration. They did not find any evidence for calcium-induced changes in the H2 domain of cTnI, which is confirmed in our experiments.
The data in this paper show differences in calcium response between the H4–H1 and the H2–H1 experiments. In these experiments, the H1 antibodies were coupled to the glass substrate, and the H4 and H2 antibodies were coupled to the magnetic particle (see Fig. 1, and see Materials and methods). The H4 and H2 antibodies are both monoclonal mouse antibodies of the same IgG1 subclass, which means that their molecular similarity is very high, except for the antigen-binding regions, which bind respectively to the H4 and H2 helices on cTnI. All preparation protocols and experimental procedures were the same in the H4 and H2 experiments, which means that the physicochemical and molecular properties of the glass substrate and of the particles are exactly the same, with the exception of the antigen-binding regions of the antibodies. Therefore, we attribute the different calcium responses measured in the H4 and H2 experiments to the fact that the antibodies bind to different epitopes on the cTnI, and that the targeted regions of the cTn are differently affected by the calcium. This interpretation is in agreement with known mechanistic models of cTn, as discussed above.
However, it is important to also consider other possible explanations and evaluate if calcium might interact with other components in the system. Another explanation for the measured differences between the H4 and H2 experiments could be that the H4 antibody itself changes conformation upon calcium binding, while the H2 antibody does not. Another alternative might be that calcium causes bridging between the antigen-binding region of H4 and an alternative site on cTn, or between the antigen-binding region of H4 and the glass substrate, while calcium does not cause any bridging to the antigen-binding region of H2. These explanations seem unlikely, since the H4 and H2 antibodies were selected for differential epitope binding and not for differential calcium interactions, but the measurement data presented in this paper cannot exclude these possibilities. As a next step, to further study the role of conformation switching and exclude alternative interpretations, modulations could be studied in the driving pathway of conformation switching. For example, one could study a mutant cTn which does not change conformation upon calcium binding (e.g. due to a point mutation in the calcium binding site) or one could apply alternative cues that influence protein conformation (chemical, biochemical, physical).
Taken together, the nCAT measurement methodology indicates a sensitivity to conformation modulations within the native heterotrimer I–C–T cTn protein complex, revealed by targeting specific protein binding sites. To our knowledge this may be the first technique that can record conformation switching on a single native protein complex. The nCAT data is in agreement with molecular hypotheses of cTn and quantifies the conformation-dependent nanomechanical properties by a set of single-molecule observables.
Conclusions and outlook
This study shows how protein conformation switching in a single native protein complex can be quantified using nanomechanical probing in combination with antibody targeting. The non-covalent protein–protein system is stable over hours due to the fact that only minimal forces are applied. The methodology was demonstrated on the native cardiac troponin complex, a heterotrimer protein complex that regulates the contraction and relaxation of heart muscle cells by calcium-induced conformation changes. The technique enables studies on native protein complexes without the need for recombinant expression or chemical protein modification. This gives ample choice for studying specific functional regions of the protein, by using monoclonal antibodies from available libraries.The nCAT methodology can be applied for nanomechanical mapping of a wide range of native proteins and protein complexes, with studies on their conformations as a function of chemical, biochemical, and physical cues. Chemical effectors can for example be electrolytes, drugs, or enzymes. The single-molecule nature of the method enables studies of variabilities in proteins and protein function, which is particularly interesting for research on systems with large variability, e.g. heterogeneous complexes, and proteins with intrinsically disordered domains, as for example applies to long regions of the majority of regulatory proteins. The extent to which variabilities can be resolved scales with the number of particles and the duration of the experiment, which should be optimized for the question at hand.
While single-molecule techniques are applied to answer scientific questions, such methodologies also penetrate into bioanalytical sciences. Commercial analytical instruments are now available based on earlier academic single-molecule studies, in the fields of protein quantification42,43 and DNA sequencing,44,45 for example. The nanomechanical probing technique described in this paper can in the future be applied for bioanalysis because it is suited for measuring native proteins in biological samples, due to the use of the general concept of capturing a target molecule between two affinity binders, in a molecular sandwich arrangement. In this way the single-molecule technique may find application for biomarker characterization in near-patient testing46 and continuous monitoring47 technologies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Alexey Katrukha from HyTest, and Peter Zijlstra, Bedabrata Saha and Stefano Cappelli from Eindhoven University of Technology for helpful discussions; Melissa Bolleman for help in the experiments; and Maarten Merkx, Lorenzo Albertazzi, and Renko de Vries for critically reading the manuscript. This work was supported by the European Commission FP7 Marie-Curie project BIOMAX (no. 264737) and the FP7 NMP project NanoMag (no. 604448).References
- J. H. Ha and S. N. Loh, Protein conformational switches: From nature to design, Chem. – Eur. J., 2012, 18, 7984–7999 CrossRef CAS PubMed.
- P. R. Banerjee and A. A. Deniz, Shedding light on protein folding landscapes by single-molecule fluorescence, Chem. Soc. Rev., 2014, 43, 1172–1188 RSC.
- B. Schuler and W. A. Eaton, Protein folding studied by single-molecule FRET, Curr. Opin. Struct. Biol., 2008, 18, 16–26 CrossRef CAS PubMed.
- B. Schuler, A. Soranno, H. Hofmann and D. Nettels, Single-Molecule FRET Spectroscopy and the Polymer Physics of Unfolded and Intrinsically Disordered Proteins, Annu. Rev. Biophys., 2016, 45, 207–231 CrossRef CAS PubMed.
- L. A. Metskas and E. Rhoades, Conformation and Dynamics of the Troponin I C-Terminal Domain: Combining Single-Molecule and Computational Approaches for a Disordered Protein Region, J. Am. Chem. Soc., 2015, 137, 11962–11969 CrossRef CAS PubMed.
- K. K. C. Neuman and A. Nagy, Single-molecule force spectroscopy: optical tweezers, magnetic tweezers and atomic force spectroscopy, Nat. Methods, 2008, 5, 491–505 CrossRef CAS PubMed.
- C. Bustamente, W. Cheng and Y. X. Mejia, Revisting the central dogma one molecule at a time, Cell, 2011, 144, 480–497 CrossRef PubMed.
- Handbook of Single-Molecule Biophysics, ed. P. Hinterdorfer and A. van Oijen, Springer, 2009, e-ISBN: 978-0-387-76497-9 Search PubMed.
- F. Kriegel, E. Ermann and J. Lipfert, Probing the mechanical properties, conformational changes, and interactions of nucleic acids with magnetic tweezers, J. Struct. Biol., 2017, 197, 26–36 CrossRef CAS PubMed.
- S. Hodeib, R. Saurabh, M. Manosas, W. Zhang, D. Bagchi, B. Ducos, J.-F. Allemand, D. Bensimon and V. Croquette, Single molecule studies of helicases with magnetic tweezers, Methods, 2016, 105, 3–15 CrossRef CAS PubMed.
- J. Christof, M. Gebhardt, T. Bornschlögl and M. Rief, Full distance-resolved folding energy landscape of one single protein molecule, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2013–2018 CrossRef PubMed.
- J. Stigler and M. Rief, Calcium-dependent folding of single calmodulin molecules, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17814–17819 CrossRef CAS PubMed.
- P. O. Heidarsson, et al., Direct single-molecule observation of calcium-dependent misfolding in human neuronal calcium sensor-1, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 13069–13074 CrossRef CAS PubMed.
- B. Gutierrez-Medina, A. N. Fehr and S. M. Block, Direct measurements of kinesin torsional properties reveal flexible domains and occasional stalk reversals during stepping, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17007–17012 CrossRef CAS.
- A. Diz-Munoz, O. D. Weiner and D. A. Fletcher, In pursuit of the mechanics that shape cell surfaces, Nat. Phys., 2018, 14, 648–652 Search PubMed.
- W. Lehman, A. Galińska-Rakoczy, V. Hatch, L. S. Tobacman and R. Craig, Structural Basis for the Activation of Muscle Contraction by Troponin and Tropomyosin, J. Mol. Biol., 2009, 388, 673–681 CrossRef CAS PubMed.
- A. Narita, T. Yasunaga, T. Ishikawa, K. Mayanagi and T. Wakabayashi, Ca2+-induced switching of troponin and tropomyosin on actin filaments as revealed by electron cryo-microscopy, J. Mol. Biol., 2001, 308, 241–261 CrossRef CAS PubMed.
- S. Takeda, A. Yamashita, K. Maeda and Y. Maéda, Structure of the core domain of human cardiac troponin in the Ca(2+)-saturated form, Nature, 2003, 424, 35–41 CrossRef CAS PubMed.
- D. G. Vassylyev, S. Takeda, S. Wakatsuki, K. Maeda and Y. Maéda, Crystal structure of troponin C in complex with troponin I fragment at 2.3-A resolution, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4847–4852 CrossRef CAS PubMed.
- A. V. Gomes, J. D. Potter and D. Szczesna-Cordary, The role of troponins in muscle contraction, IUBMB Life, 2002, 54, 323–333 CrossRef CAS PubMed.
- G. Lippi, Biomarkers: Novel troponin immunoassay for early ACS rule-out, Nat. Rev. Cardiol., 2016, 13, 9–10 CrossRef CAS PubMed.
- T. Savukoski, et al., Troponin-specific autoantibody interference in different cardiac troponin I assay configurations, Clin. Chem., 2012, 58, 1040–1048 CrossRef CAS PubMed.
- H. M. Rarick, X. H. Tu, R. J. Solaro and A. F. Martin, The C terminus of cardiac troponin I is essential for full inhibitory activity and Ca2+sensitivity of rat myofibrils, J. Biol. Chem., 1997, 272, 26887–26892 CrossRef CAS PubMed.
- J. M. Chalovich and E. Eisenberg, Inhibition of actomyosin ATPase activity by troponin-tropomyosin without blocking the binding of myosin to actin, J. Biol. Chem., 1982, 257, 2432–2437 CAS.
- L. Spyracopoulos, et al., Calcium-induced structural transition in the regulatory domain of human cardiac troponin C, Biochemistry, 1997, 36, 12138–12146 CrossRef CAS PubMed.
- M. B. Abbott, et al., Regulatory domain conformational exchange and linker region flexibility in cardiac troponin C bound to cardiac troponin I, J. Biol. Chem., 2000, 275, 20610–20617 CrossRef CAS PubMed.
- M. V. Vinogradova, et al., Ca(2+)-regulated structural changes in troponin, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5038–5043 CrossRef CAS PubMed.
- P. C. Nelson, et al., Tethered particle motion as a diagnostic of DNA tether length, J. Phys. Chem. B, 2006, 110, 17260–17267 CrossRef CAS PubMed.
- E. W. A. Visser, L. J. van IJzendoorn and M. W. J. Prins, Particle Motion Analysis Reveals Nanoscale Bond Characteristics and Enhances Dynamic Range for Biosensing, ACS Nano, 2016, 10, 3093–3101 CrossRef CAS PubMed.
- X. J. A. Janssen, et al., Torsion stiffness of a protein pair determined by magnetic particles, Biophys. J., 2011, 100, 2262–2267 CrossRef CAS PubMed.
- A. Van Reenen, F. A. Gutiérrez-Mejía, L. J. van IJzendoorn and M. W. J. Prins, Torsion profiling of proteins using magnetic particles, Biophys. J., 2013, 104, 1073–1080 CrossRef CAS PubMed.
- F. A. Gutierrez Mejia, Proteins with a twist: torsion profiling of proteins at the single molecule level, PhD thesis, Eindhoven University of Technology, 2016, pp. 15–32 Search PubMed.
- M. J. Holroyde, S. P. Robertson, J. D. Johnson, R. J. Solaro and J. D. Potter, The calcium and magnesium binding sites on cardiac troponin and their role in the regulation of myofibrillar adenosine triphosphatase, J. Biol. Chem., 1980, 255, 11688–11693 CAS.
- B. Saha, T. H. Evers and M. W. J. Prins, How antibody surface coverage on nanoparticles determines the activity and kinetics of antigen capturing for biosensing, Anal. Chem., 2014, 86, 8158–8166 CrossRef CAS PubMed.
- Y. Liu and J. Yu, Oriented immobilization of proteins on solid supports for use in biosensors and biochips: a review, Microchim. Acta, 2016, 183, 1–19 CrossRef CAS.
- I. Vikholm and W. M. Albers, Oriented Immobilization of Antibodies for Immunosensing, Langmuir, 1998, 14, 3865–3872 CrossRef CAS.
- J. P. Gering, L. Quaroni and G. Chumanov, Immobilization of antibodies on glass surfaces through sugar residues, J. Colloid Interface Sci., 2002, 252, 50–56 CrossRef CAS PubMed.
- T. M. Blumenschein, D. B. Stone, R. J. Fletterick, R. A. Mendelson and B. D. Sykes, Dynamics of the C-terminal region of TnI in the troponin complex in solution, Biophys. J., 2006, 90, 2436–2444 CrossRef CAS PubMed.
- J. J. Jayasundar, J. Xing, J. M. Robinson, H. C. Cheung and W. J. Dong, Molecular dynamics simulations of the cardiac troponin complex performed with fret distances as restraints, PLoS One, 2014, 9, e87135 CrossRef PubMed.
- P. M. Hwang, F. Cai, S. E. Pineda-Sanabria, D. C. Corson and B. D. Sykes, The cardiac-specific N-terminal region of troponin I positions the regulatory domain of troponin C, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14412–14417 CrossRef CAS PubMed.
- N. M. Cordina, et al., Ca2+-induced PRE-NMR changes in the troponin complex reveal the possessive nature of the cardiac isoform for its regulatory switch, PLoS One, 2014, 9, e112976 CrossRef PubMed.
- J. Todd, B. Freese, A. Lu, D. Held, J. Morey, R. Livingston and P. Goix, Ultrasensitive Flow-based Immunoassays Using Single-Molecule Counting, Clin. Chem., 2007, 53, 1990–1995 CrossRef CAS PubMed.
- D. M. Rissin, C. W. Kan, T,G Campbell, S. C. Howes, D. R. Fournier, L. Song, T. Piech, P. P. Patel, L. Chang, A. J. Rivnak, E. P. Ferrell, J. D. Randall, G. K. Provuncher, D. R. Walt and D. C. Duffy, Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations, Nat. Biotechnol., 2010, 28, 595–599 CrossRef CAS PubMed.
- D. Branton, D. W. Deamer, A. Marziali, H. Bayley, S. A. Benner, T. Butler, M. Di Ventra, G. Slaven, A. Hibbs, X. Huang, S. B. Jovanovich, P. S. Krstic, S. Lindsay, X. S. Ling, C. H. Mastrangelo, A. Meller, J. S. Oliver, Y. V. Pershin, J. M. Ramsey, R. Riehn, G. V. Soni, V. Tabard-Cossa, M. Wanunu, M. Wiggin and J. A. Schloss, The potential and challenges of nanopore sequencing, Nat. Biotechnol., 2008, 26, 1146–1153 CrossRef CAS.
- J. Eid, et al., Real-Time DNA Sequencing from Single Polymerase Molecules, Science, 2009, 323, 133–138 CrossRef CAS PubMed.
- A. van Reenen, A. M. de Jong, J. M. J. den Toonder and M. W. J. Prins, Integrated lab-on-chip biosensing systems based on magnetic particle actuation – a comprehensive review, Lab Chip, 2014, 14, 1966–1986 RSC.
- E. W. A. Visser, J. Yan, L. J. van IJzendoorn and M. W. J. Prins, Continuous Biomarker Monitoring by Particle Mobility Sensing with Single Molecule Resolution, Nat. Commun., 2018, 9, 2541 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr01448a |
| This journal is © The Royal Society of Chemistry 2019 |