
Amplified luminescence in organo-curium nanocrystal hybrids†
Peter
Agbo
a,
Alexander
Müller
b,
Leticia
Arnedo-Sanchez
a,
Peter
Ercius
b,
Andrew M.
Minor
bc and
Rebecca J.
Abergel
 *ad
*ad
aChemical Sciences Division Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA. E-mail: abergel@berkeley.edu
bNational Center for Electron Microscopy, Molecular Foundry, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
cDepartment of Materials Science and Engineering, University of California, Berkeley, CA 94720, USA
dDepartment of Nuclear Engineering, University of California, Berkeley, CA 94720, USA
First published on 10th April 2019
Abstract
We present the first report of ligand-sensitized, actinide luminescence in a lanthanide nanoparticle host. Amplified luminescence of 248Cm3+ doped in a NaGdF4 lattice is achieved through optical pumping of a surface-localized metal chelator, 3,4,3-LI(1,2-HOPO), capable of sensitizing Cm3+ excited states. The data suggest the possibility of using such materials in theranostic applications, with a ligand-sensitized actinide or radio-lanthanide serving the dual roles of a nuclear decay source for radiotherapeutics, and as a luminescent center or energy transfer conduit to another emissive metal ion, for biological imaging.
The exploration of lanthanide luminescence in nanoparticle structures over the last decade has found much of its motivation behind their potential use in lasing, spectral conversion and biomedical imaging applications.1–6 The result has been a broad body of literature produced in the areas of sensitized lanthanide luminescence, lanthanide spectral conversion, and actinide photospectroscopy in lanthanide host materials.3,6–13 Additionally, significant investigations have delved into creating f-element materials capable of serving the dual purpose of targeted radiotherapeutics and photoluminescent cell/tissue-imaging agents.1,14–17 Less work has considered the prospect of combining these research areas, through the synthesis of ligand-sensitized lanthanide nanoparticles with highly radioactive actinide co-dopants for radiotherapy applications. The dearth of research in this area motivated an investigation into the luminescent properties of curium-doped, NaGdF4 nanoparticles featuring a surface-bound chelator, 3,4,3-LI(1,2-HOPO),18 hereafter 3,4,3 (Fig. 1). This study marks the first of its kind, with an actinide successfully doped into the hexagonal NaGd(Y)F4-type lattices that have become common host crystals for solid-state lanthanide luminescence studies. Nanoparticles were synthesized from metal acetate precursors in a 1-oleic acid/1-octadecene mixture and were decorated with 3,4,3 by substitution of 1-oleate ligands, following methods adapted from the literature.19 While we use a low-activity 248Cm isotope for the sake of safety, these results should be applicable to the more radioactive 243Cm and 244Cm, if not other actinides and lanthanide isotopes as well.
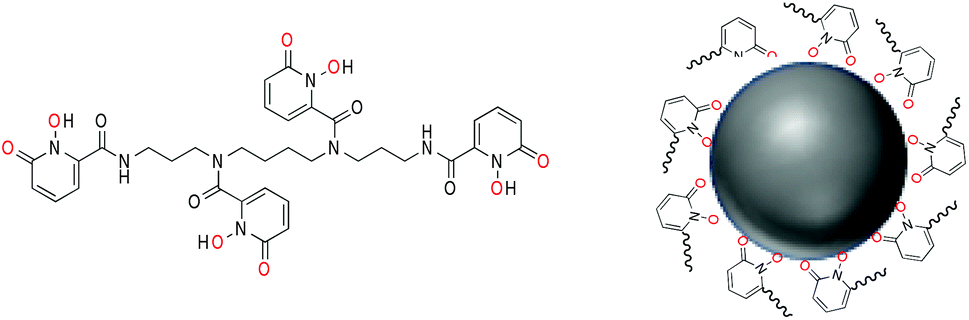 | ||
| Fig. 1 (left) Molecular structure of octadentate 3,4,3-LI(1,2-HOPO) with metal-binding oxygen atoms highlighted in red; (right) schematic depiction of nanoparticle surface binding upon deprotonation of the 1,2-HOPO functionalities. | ||
The Cm-doped, NaGdF4 formed ill-defined nanoparticles without distinct surface facets. Many nanoparticles were approximately spherical and up to 10 nm in size (Fig. 2a), but several larger nanoparticles seem to have formed by coalescence. The presence of distinct necking-like features supports our hypothesis (Fig. 2b). This is in accordance with reports of a low surface energy.20 Electron diffraction patterns over large areas confirmed the hexagonal β-NaGdF4 phase (Fig. 2c).21
 | ||
| Fig. 2 (a) High-angle annular dark field scanning TEM (HAADF-STEM) image of several nanoparticles; (b) high-resolution TEM (HRTEM) image of a few nanoparticles, (c) electron diffraction pattern of a large area. | ||
Using energy-dispersive X-ray spectroscopy (EDS), the Cm-content was quantified as being below 1% (Fig. 3a and b). Excitation at 357 nm into the ligand band of 3,4,3-modified, Cm-doped nanoparticles results in a broad, ligand-centered emission (453 nm) derived from 3,4,3 singlet/triplet relaxation, and a narrow emission peaking at 598 nm (6D7/2 → 8S7/2) arising from Cm3+ excited state decay (Fig. 4). Excitation spectra acquired by monitoring the 598 nm signal reveal that luminescence at this wavelength responds to excitation over the range 300–360 nm (ligand), while also demonstrating a sensitivity to f–f transitions originating from direct excitation of the curium center between ca. 390–500 nm, with a peak at 397 nm assigned to Cm3+ 8S7/2 → 6IJ (Fig. 4).9
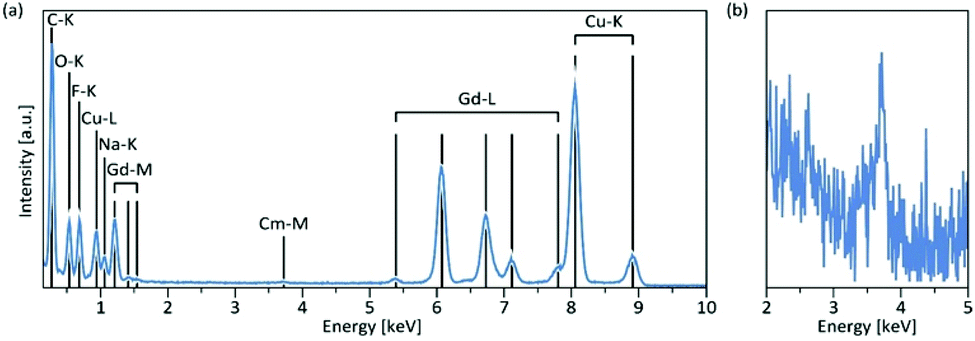 | ||
| Fig. 3 (a) Typical EDS spectrum of Cm-NaGdF4. Cu and C peaks are due to the grid onto which the nanoparticles were deposited, whereas oxygen is a common residual gas in the microscope column; (b) close-up of the weak Cm–M peak. The energy scale is the same as in (a), but the intensity scale has been stretched approximately 40-times. | ||
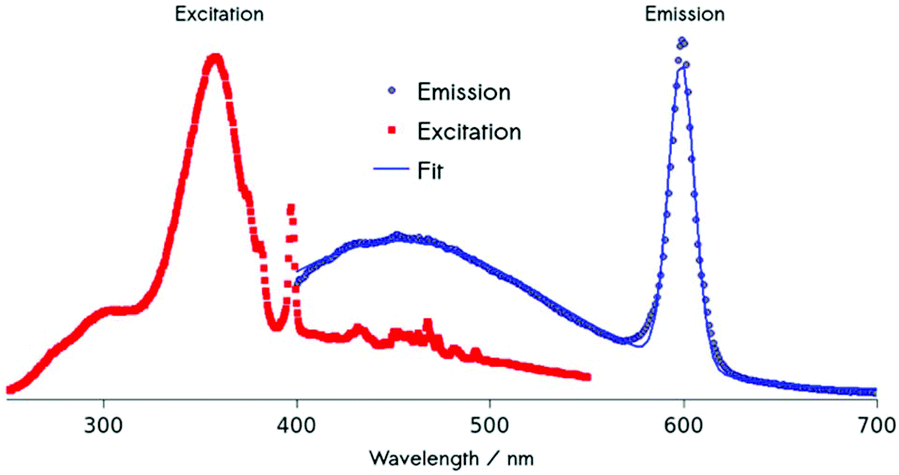 | ||
| Fig. 4 Steady-state photoluminescence (PL) of Cm-doped NaGdF4-3,4,3 nanoparticles. Red: Excitation spectrum measured at Cm3+ (6D7/2 → 8S7/2) emission at 598 nm. The peak at 350 nm is due to ligand absorption; the strong narrow excitation band at ca. 400 nm is the result of direct light absorption by Cm3+. Blue: PL spectrum representing a summation of ligand and Cm3+ ion emission. | ||
Further evidence of surface-bound ligand sensitization is seen through comparison of luminescence in the washes of the ligand-modification reactions with the nanoparticle suspension. Following a series of centrifugation/wash cycles to remove excess 3,4,3, a residual luminescence assigned to formation of the 3,4,3-Cm molecular complex is observed, with roughly 1/40th the emission intensity of the nanoparticle sample being present in the supernatant of the third wash. In addition, formation of the molecular complex in the washes is also evidenced by a significant (12 nm) shift in the Cm3+ emission band observed in the washes at wavelengths previously reported for Cm3+-3,4,3 (∼610 nm),22 which is notably distinct from the 598 nm emission wavelength of the sensitized nanocrystals (ESI†). Bathochromic Cm(III) fluorescence shifts, observed with the transition from the lowest energy crystal field level of the first excited 6D7/2 multiplet to the ground multiplet of the 8S7/2 state, have been qualitatively associated with trends in the nephelauxetic effect.23 The large ∼16 nm shift (∼450 cm−1) observed upon complexation of the free Cm(III) ion by 3,4,3 in aqueous solution is characteristic of strong ligand interactions that result in diminished electrostatic repulsion effects. Within the nanocrystals, the fluorescence shift is much less pronounced (∼4 nm or ∼120 cm−1) as the Cm ions interact with fluoride anions and the larger organic ligand is kept at the particle surface. While relatively small compared to several crystalline matrices, including Cs2NaYCl6,23,24 this shift is consistent with those observed in crystalline halides CmF3 and CmCl3 (∼90 and ∼82 cm−1, respectively),25 prior to corrections made for the crystal field splitting of the 6D7/2 multiplet. This is further confirmation of the constrained environment of the Cm ions within the nanoparticle host.
Co-substitution with Eu3+ reveals that Cm3+ → Eu3+ energy transfer is also possible, with direct photoexcitation of the curium ions at 400 nm resulting in the appearance of the europium 5D0 → 7F2 emission band at 612 nm. Population of europium's 5DJ manifold presumably arises from energetic exchange between the resonant 6D7/2 and 5D0 states in curium and europium centers, respectively (ESI†).
Measurements of quantum yields for the sensitization efficiency of the Cm-only system reveal a quantum yield of approximately 0.4% for the Cm3+ emission; factoring in ligand emission results in a total luminescence quantum yield of 1.2%. While these values are low, the curium doping levels employed here (0.075%) are far lower than those typically used in correspondent lanthanide luminescence studies. In past work, Eu3+ doping levels of 5% in NaGdF4 with this ligand set, were used.26 Despite the ∼70-fold dopant excess used in those previous studies relative to the curium investigations here, the sensitized-europium system was found to display a quantum efficiency only an order of magnitude (Φ = 3.3%), greater than what we observe for the curium system.26 It is worth noting that the 5% doping level used for the europium study did not place the system in a concentration-quenching regime, suggesting a significantly higher efficiency for ligand-curium energy transfer relative to the europium nanoparticle analog. This finding is consistent with earlier studies of luminescence sensitization in the 3,4,3-Cm and 3,4,3-Eu molecular complexes,22,27 for which respective quantum yield values of 45% and 7% had been reported in aqueous solutions buffered at physiological pH. There is, to our knowledge, no report of a better Cm sensitizing ligand than 3,4,3. However, higher brightness, larger quantum yield, and increased energy transfer efficiency could be obtained through confinement of the 3,4,3-Cm complex within a macromolecular cavity.28 Future work will explore variations in the parameters that influence energy transfer mechanisms, including different ligand architectures, both within protein matrices and in the nanoparticulate system discussed here, in order to optimize sensitization of the metal centers.
Time-dependent luminescence was investigated through pulsed excitation of the ligand band at 350 nm, with concurrent monitoring of sample emission at 598 nm (ESI†). We observed a triexponential luminescence decay, with rates of k1 = 647 s−1, k2 = 139 s−1, and k3 = 8.4 s−1, with their weighted-average yielding a mean decay time of 1.26 ms (normalized weights for the respective decay phases are c1 = 0.29, c2 = 0.55, c3 = 0.16). Notably, this average lifetime is much longer than those of the Cm aquo ion and the 3,4,3-Cm molecular complex (65 and 383 μs, respectively, in H2O)22 and more consistent with that of Cm embedded in a solid ThO2 crystalline host matrix (1380 μs).23 Incorporating past measurements of the 3,4,3 triplet state decay allowed determining the ligand-curium energy transfer efficiency as 0.22 (ESI†). The observed multiexponential emission is likely a result of the hypersensitive Cm3+ photoemission, which, unlike many f–f transitions, is sensitive to environment. In the case of the hexagonal NaGdF4 crystal system, the lanthanide/actinide ions occupy two, crystallographically-distinct sites. As a result, the lifetime of the Cm3+ excited state should be expected to be influenced by these distinct environments, leading to unique decay times for the 6D7/2 → 8S7/2 transition. In addition, any Cm ions residing on the solvent-exposed edges of a nanocrystal will be subject to solvent-coupled deactivation paths. These ions, particularly when exposed to a protic solvent such as ethanol, would display much shorter lifetimes than their solvent-insulated counterparts residing within the nanocrystal bulk. These considerations suggest multiple distinct Cm3+ ions within the crystals, each with a characteristic decay time for the Cm3+ excited state. In the case of peripheral Cm3+ ions subject to protic solvent quenching, the observed decay is likely dominated by solvent quenching processes, as a result of the known ability of OH oscillators to rapidly deplete electronic excited states. In this case, solvent-exposed ions at the nanocrystal edge would have similar rates of decay, despite the existence of the two distinct f-element sites in this crystal host. Superposition of these three unique forms of Cm3+ excited-state decays at 598 nm would then be expected to result in a composite triexponential decay function.
Our experiments confirm that interstitial doping of actinides in NaGdF4 hosts is indeed possible, opening up avenues to synthesize radioactive, luminescent nanocrystals for therapeutic and diagnostic medical applications. While there is no radioisotope of Cm that would display adequate properties for radiotherapeutic applications, isotopes such as the trivalent 177Lu and 225Ac are used for targeted immuno-therapy29–31 and are expected to exhibit coordination properties relatively similar to those of Cm within the nanoparticle hosts used in this study.32,33 One may envision designing systems that leverage the concerted insertion of luminescent f-block metals and medical f-block isotopes. Finally, the particular system examined in this study allows for the possibility of bio-imaging tissues at relatively low excitation powers, with the high extinction coefficient of 3,4,3 permitting relatively efficient photon absorption and Cm3+ luminescence relative to direct f–f actinide excitation.
Conflicts of interest
RJA and PA are listed as inventors on a patent application filed by the Lawrence Berkeley National Laboratory and describing inventions related to the research results presented here. The authors declare no other competing financial interests.Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division at the Lawrence Berkeley National Laboratory under Contract DE-AC02-05CH11231 (RJA). The microscopy work was performed at the Molecular Foundry, which is supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract DE-AC02-05CH11231.Notes and references
- J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC.
- X. Chen, L. Jin, W. Kong, T. Sun, W. Zhang, X. Liu, J. Fan, S. F. Yu and F. Wang, Nat. Commun., 2016, 7, 10304 CrossRef CAS PubMed.
- D. J. Garfield, N. J. Borys, S. M. Hamed, N. A. Torquato, C. A. Tajon, B. Tian, B. Shevitski, E. S. Barnard, Y. D. Suh, S. Aloni, J. B. Neaton, E. M. Chan, B. E. Cohen and P. J. Schuck, Nat. Photonics, 2018, 12, 402–407 CrossRef CAS.
- M. Schubert, A. Steude, P. Liehm, N. M. Kronenberg, M. Karl, E. C. Campbell, S. J. Powis and M. C. Gather, Nano Lett., 2015, 15, 5647–5652 CrossRef CAS PubMed.
- M. H. Werts, Sci. Prog., 2005, 88, 101–131 CrossRef CAS PubMed.
- W. Zou, C. Visser, J. A. Maduro, M. S. Pshenichnikov and J. C. Hummelen, Nat. Photonics, 2012, 6, 560 CrossRef CAS.
- L. J. Charbonnière, J.-L. Rehspringer, R. Ziessel and Y. Zimmermann, New J. Chem., 2008, 32, 1055–1059 RSC.
- Z. Huang and M. L. Tang, J. Am. Chem. Soc., 2017, 139, 9412–9418 CrossRef CAS PubMed.
- M. Illemassene, K. Murdoch, N. Edelstein and J. Krupa, J. Lumin., 1997, 75, 77–87 CrossRef CAS.
- M. Irfanullah, D. K. Sharma, R. Chulliyil and A. Chowdhury, Dalton Trans., 2015, 44, 3082–3091 RSC.
- S. Janssens, G. Williams and D. Clarke, J. Appl. Phys., 2011, 109, 023506 CrossRef.
- S. Li, X. Zhang, Z. Hou, Z. Cheng, P. Ma and J. Lin, Nanoscale, 2012, 4, 5619–5626 RSC.
- J. Zhang, C. M. Shade, D. A. Chengelis and S. Petoud, J. Am. Chem. Soc., 2007, 129, 14834–14835 CrossRef CAS PubMed.
- G. Chen, C. Yang and P. N. Prasad, Acc. Chem. Res., 2013, 46, 1474–1486 CrossRef CAS PubMed.
- X. Jin, F. Fang, J. Liu, C. Jiang, X. Han, Z. Song, J. Chen, G. Sun, H. Lei and L. Lu, Nanoscale, 2015, 7, 15680–15688 RSC.
- L. C. Mimun, G. Ajithkumar, M. Pokhrel, B. G. Yust, Z. G. Elliott, F. Pedraza, A. Dhanale, L. Tang, A.-L. Lin and V. P. Dravid, J. Mater. Chem. B, 2013, 1, 5702–5710 RSC.
- Z.-L. Wang, J. Hao and H. L. Chan, J. Mater. Chem., 2010, 20, 3178–3185 RSC.
- R. J. Abergel, P. W. Durbin, B. Kullgren, S. N. Ebbe, J. Xu, P. Y. Chang, D. I. Bunin, E. A. Blakely, K. A. Bjornstad, C. J. Rosen, D. K. Shuh and K. N. Raymond, Health Phys., 2010, 99, 401–407 CrossRef CAS PubMed.
- F. Wang, R. Deng and X. Liu, Nat. Protoc., 2014, 9, 1634 CrossRef CAS PubMed.
- X. Sun, B. Wang, I. Kempson, C. Liu, Y. Hou and M. Gao, Small, 2014, 10, 4711–4717 CrossRef CAS PubMed.
- J. H. Burns, Inorg. Chem., 1965, 4, 881–886 CrossRef CAS.
- M. Sturzbecher-Hoehne, B. Kullgren, E. E. Jarvis, D. D. An and R. J. Abergel, Chem. – Eur. J., 2014, 20, 9962–9968 CrossRef CAS PubMed.
- N. M. Edelstein, R. Klenze, T. Fanghänel and S. Hubert, Coord. Chem. Rev., 2006, 250, 948–973 CrossRef CAS.
- K. Murdoch, R. Cavellec, E. Simoni, M. Karbowiak, S. Hubert, M. Illemassene and N. Edelstein, J. Chem. Phys., 1998, 108, 6353–6361 CrossRef CAS.
- N. Stump, G. Murray, G. Del Cul, R. Haire and J. Peterson, Radiochim. Acta, 1993, 61, 129–136 CAS.
- P. Agbo, T. Xu, M. Sturzbecher-Hoehne and R. J. Abergel, ACS Photonics, 2016, 3, 547–552 CrossRef CAS.
- R. J. Abergel, A. D'Aleo, C. Ng Pak Leung, D. K. Shuh and K. N. Raymond, Inorg. Chem., 2009, 48, 10868–10870 CrossRef CAS PubMed.
- B. E. Allred, P. B. Rupert, S. S. Gauny, D. D. An, C. Y. Ralston, M. Sturzbecher-Hoehne, R. K. Strong and R. J. Abergel, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10342–10347 CrossRef CAS PubMed.
- J. G. Jurcic, M. Y. Levy, J. H. Park, F. Ravandi, A. E. Perl, J. M. Pagel, B. D. Smith, E. H. Estey, H. Kantarjian, D. Cicic and D. A. Scheinberg, Blood, 2016, 128, 4050 Search PubMed.
- C. Kratochwil, U. Haberkorn and F. L. Giesel, in Radionuclide Therapy of Metastatic Prostate Cancer, Seminars in Nuclear Medicine, Elsevier, 2019 Search PubMed.
- G. P. Nicolas, A. Morgenstern, M. Schottelius and M. Fani, J. Nucl. Med., 2019, 60, 167–171 CrossRef PubMed.
- M. G. Ferrier, E. R. Batista, J. M. Berg, E. R. Birnbaum, J. N. Cross, J. W. Engle, H. S. La Pierre, S. A. Kozimor, J. S. L. Pacheco and B. W. Stein, Nat. Commun., 2016, 7, 12312 CrossRef CAS PubMed.
- M. G. Ferrier, B. W. Stein, E. R. Batista, J. M. Berg, E. R. Birnbaum, J. W. Engle, K. D. John, S. A. Kozimor, J. S. Lezama Pacheco and L. N. Redman, ACS Cent. Sci., 2017, 3, 176–185 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, quantum yield data, time-resolved luminescence, energy transfer calculations, and Eu3+–Cm3+ excitation spectra. See DOI: 10.1039/c9nr01360a |
| This journal is © The Royal Society of Chemistry 2019 |