
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanoscale distribution of TLR4 on primary human macrophages stimulated with LPS and ATI†
Jan
Neumann
 ab,
Kira
Ziegler
a,
Márton
Gelléri
b,
Janine
Fröhlich-Nowoisky
a,
Fobang
Liu
a,
Iris
Bellinghausen
c,
Detlef
Schuppan
de,
Udo
Birk‡
b,
Ulrich
Pöschl
a,
Christoph
Cremer
ba and
Kurt
Lucas
*a
ab,
Kira
Ziegler
a,
Márton
Gelléri
b,
Janine
Fröhlich-Nowoisky
a,
Fobang
Liu
a,
Iris
Bellinghausen
c,
Detlef
Schuppan
de,
Udo
Birk‡
b,
Ulrich
Pöschl
a,
Christoph
Cremer
ba and
Kurt
Lucas
*a
aMultiphase Chemistry Department, Max Planck Institute for Chemistry, Hahn-Meitner-Weg 1, 55128 Mainz, Germany. E-mail: k.lucas@mpic.de
bInstitute of Molecular Biology, Ackermannweg 4, 55128 Mainz, Germany
cDepartment of Dermatology, University Medical Center of the Johannes Gutenberg University, 55131 Mainz, Germany
dInstitute of Translational Immunology, University Medical Center of the Johannes Gutenberg University, Langenbeckstraße 1, 55131 Mainz, Germany
eDivision of Gastroenterology, Beth Israel Deaconess Medical Center, Harvard Medical School, 330 Brookline Ave, Boston, MA 02215, USA
First published on 30th April 2019
Abstract
Toll-like receptor 4 (TLR4) plays a crucial role in the recognition of invading pathogens. Upon activation by lipopolysaccharides (LPS), TLR4 is recruited into specific membrane domains and dimerizes. In addition to LPS, TLR4 can be stimulated by wheat amylase-trypsin inhibitors (ATI). ATI are proteins associated with gluten containing grains, whose ingestion promotes intestinal and extraintestinal inflammation. However, the effect of ATI vs. LPS on the membrane distribution of TLR4 at the nanoscale has not been analyzed. In this study, we investigated the effect of LPS and ATI stimulation on the membrane distribution of TLR4 in primary human macrophages using single molecule localization microscopy (SMLM). We found that in unstimulated macrophages the majority of TLR4 molecules are located in clusters, but with donor-dependent variations from ∼51% to ∼75%. Depending on pre-clustering, we found pronounced variations in the fraction of clustered molecules and density of clusters on the membrane upon LPS and ATI stimulation. Although clustering differed greatly among the human donors, we found an almost constant cluster diameter of ∼44 nm for all donors, independent of treatment. Together, our results show donor-dependent but comparable effects between ATI and LPS stimulation on the membrane distribution of TLR4. This may indicate a general mechanism of TLR4 activation in primary human macrophages. Furthermore, our methodology visualizes TLR4 receptor clustering and underlines its functional role as a signaling platform.
Introduction
Toll-like receptors (TLR) represent a family of pattern recognition receptors (PRRs) that are part of the innate immune system.1,2 TLRs detect damage and pathogen associated molecular patterns (DAMPs, PAMPs), which subsequently triggers the production and release of inflammatory mediators. One of the best studied TLRs is TLR4, which is sensitive to lipopolysaccharide (LPS), a cell wall component of Gram-negative bacteria.3,4 Moreover, TLR4 detects a broad spectrum of different molecules that are summarized as DAMPs, such as high-mobility group protein B1 (HMGB1) or heat-shock protein 60 (HSP60). TLR4 plays a central role in many acute and chronic inflammatory diseases such as chronic obstructive pulmonary disease (COPD), allergic asthma or non-celiac wheat sensitivity (NCWS).5–8Stimulation of TLR4 through LPS is facilitated by a complex interaction between multiple molecules. First, LPS is bound by the soluble lipopolysaccharide binding protein (LBP), which transfers LPS to the co-receptor CD14. The LPS-CD14 complex in turn interacts with the TLR4-myeloid differentiation protein-2 (MD-2) heterodimer.9–11 Binding of LPS leads to the dimerization of the TLR4/MD-2 complex followed by a change in receptor conformation and finally to the translocation of the transcription factor nuclear factor ‘kappa-light-chain-enhancer’ of activated B-cells (NF-κB) to the nucleus.2
Stimulation and subsequent downstream signaling of TLR4 is accompanied by a re-localization of TLR4 and its accessory molecules into specific domains of the cell membrane (reviewed in Płóciennikowska et al.,12 Ruysschaert and Lonez13). Thereby, TLR4 is recruited into CD14 cholesterol-rich membrane microdomains after treatment with LPS.14 Since these domains are smaller than 200 nm,15,16 they are inaccessible by conventional microscopy. With the advent of super-resolution microscopy techniques,17–26 various studies have investigated the membrane distribution of TLR4 at the single molecule level. It has been reported that in human glioblastoma cells TLR4 clusters are present with a cluster size of ∼50 nm.27 In mouse macrophages, another study reported an increased TLR4 cluster size from ∼380 nm to ∼520 nm after simulation with LPS.28 Moreover, it was shown that the oligomeric state of TLR4 in HEK 293 cells is affected, depending on the presence of the co-receptors MD2 and CD14 as well as on the LPS chemotype used.29
In general, it is assumed that the existence of receptor clusters and the recruitment of receptors into specific domains creates a local environment that facilitates downstream signaling and thus represents a functional or regulatory mechanism.30,31 Thus, an increase in the cluster size or density of TLR4 on the membrane upon stimulation apparently promotes immune responses. Of note, in the course of stimulation the entire TLR4/MD-2/CD14 complex is endocytosed,32 which might cause a depletion of TLR4 on the cell surface.
Our prior studies have shown that TLR4 can be stimulated by amylase trypsin inhibitors (ATI) from wheat and thus act as natural activators of innate immunity in monocytes, macrophages and dendritic cells.7,33–39 Moreover, ATI are major allergens in Baker's asthma.40 ATI are a family of non-gluten proteins found in different cereals containing gluten such as wheat, rye or barley.35 With wheat as a major staple in modern societies, the daily intake of ATI is estimated at 0.5 g to 1.5 g per person.35 Nutritional ATI are resistant to intestinal proteolysis and are major causes of non-celiac wheat sensitivity,6,34,41 driving intestinal and extraintestinal inflammation.33,35,37,38
During inflammation, endogenous reactive oxygen and nitrogen species are formed that are capable of inducing protein nitration. Moreover, during highly polluted environmental conditions, proteins can be nitrated exogenously.42–44 In a previous study, we have demonstrated that nitrated ATI induced enhanced TLR4 stimulation, pro-inflammatory cytokine secretion and T-cell proliferation.39
To investigate if the elevated immune reactions are caused by a change in TLR4 surface representation and distribution, we tested native and nitrated ATI as well as the natural TLR4 agonist LPS. We used single molecule localization microscopy (SMLM) to observe their effect on the spatial distribution of TLR4 in primary human macrophages.
Experimental
Cell culture
Primary human macrophages were derived from peripheral blood mononuclear cells (PBMC) isolated from leucocyte-enriched buffy coats from healthy donors (Transfusion Center, University Medical Center of the Johannes Gutenberg University, Mainz, Germany; for all donors: IgE < 20 kU l−1, food- and aero-allergy negative, (CAP 0)) using Ficoll-Paque (1.077 g ml−1, Biochrom) density centrifugation. After two washing steps with phosphate-buffered saline (PBS, Thermo Fisher Scientific), PBMC were diluted in Isocove's Modified Dulbecco's medium (IMDM, Lonza) containing 1% antibiotic antimycotic solution (Sigma Aldrich) and 3% heat-inactivated (30 min at 56 °C) autologous plasma. To enrich CD14+ monocytes, 2.5 × 106 PBMC were seeded per endotoxin-free glass coverslip (R. Langenbrinck GmbH, thickness of 0.17 mm ± 0.005 mm), which had been placed in a 6-well plate (Greiner Bio-One). After 30 min incubation at 37 °C and 5% CO2, cells were washed three times with pre-warmed PBS and the remaining monocytes were incubated for 6 days in IMDM supplemented with granulocyte macrophage colony stimulating factor (GM-CSF, 200 Units ml−1, Immunex) and 2% heat-inactivated autologous plasma.Treatment with LPS, ATI or nitrated ATI
Primary human macrophages grown on glass coverslips were washed once with pre-warmed PBS. Next, cells were incubated with either 100 ng ml−1 lipopolysaccharide (LPS-EB; from E. coli O111:B4, Invivogen), 12.5 μg ml−1 ATI (α-amylase inhibitor from Triticum aestivum, Type I, Sigma Aldrich) or 12.5 μg ml−1 tetranitromethane (TNM)-nitrated ATI. All stimuli were diluted in complete cell culture medium and incubation was done at 37 °C and 5% CO2 for 15 min and 30 min. Afterwards, cells were washed once in PBS followed by a two-step fixation process.45,46 First, cells were fixed in cold PBS containing 4% formaldehyde (Thermo Fisher Scientific) and 0.2% glutaraldehyde (Sigma Aldrich). After 15 min at 4 °C, a second fixation step was performed with the same fixation buffer for 30 min at room temperature. Afterwards, cells were washed three times in PBS and stored at 4 °C in PBS until further use for immunostaining.Immunostaining
Fixed cells were quenched for 7 min in 0.1% (w/v) sodium borohydride (NaBH4, Sigma Aldrich). After three washing steps in PBS, cells were blocked in 2% bovine serum albumin (BSA, Cell Signaling Technology) dissolved in PBS for 60 min. Next, cells were incubated for 60 min at room temperature with 10 μg ml−1 mouse monoclonal anti-TLR4-antibody (ab22048, Abcam) diluted in blocking buffer. The performance of this TLR4 antibody has been validated in a previous study by Zeuner et al.,27 which showed similar results in TLR4 imaging and clustering. Then, samples were washed three times with PBS and subsequently incubated with 20 μg ml−1 F(ab′)2 goat anti-mouse Alexa Fluor 647 secondary antibody (A-21237, Thermo Fisher Scientific) diluted in blocking buffer for 60 min at room temperature. After incubation, the sample was washed again three times in PBS and stained cells were post-fixed with 4% formaldehyde in PBS for 10 min at room temperature. After three additional washing steps in PBS, the sample was embedded in Vectashield H-1000 (Vector Laboratories) and sealed using picodent twinsil (picodent Dental Produktions- und Vertriebs GmbH).Localization microscopy
Experiments were conducted on a custom-built localization microscope (for details see Fig. S1†). Alexa Fluor 647 was excited using a 647 nm laser (Obis, Coherent). For SMLM imaging, the laser beam was expanded 3.125-fold, resulting in an illuminated area of ∼640 μm2 in the sample plane (area of 1/e2 width of Gaussian profile). The collimated laser beam was coupled into the back port of the microscope (DM RBE, Leica) and focused onto the back focal plane of the objective lens (100×/NA 1.49, Olympus). After passing an emission filter (bandpass 700/75 nm, Chroma Technology), emitted fluorescent light was collected on an electron multiplying charge-coupled device (EMCCD) camera (iXon 897 Ultra, Andor Technology) with an effective pixel size of 95 nm.Imaging and data processing
A region of interest (ROI) was placed on a single cell selected for SMLM imaging. The ROI typically covered an area of ∼50 μm2 to ∼400 μm2 of the plasma membrane. From each ROI, 5000 frames were acquired with a laser intensity of 6.5 kW cm−2 (measured in the sample plane) and an exposure time of 40 ms.To obtain the list of localizations, processing of the raw data stacks was done using Matlab (MathWorks) and the custom written software package Integrated Localization Environment (ILE; https://gitlab.com/microscopy/ILE). The localization algorithm of ILE, which accomplishes the extraction and position determination of single molecule signals from the raw data stacks, is based on fastSPDM.47 In brief, an initial background image was created from the first eight frames of the raw data stack. The background image was subtracted from the frames of the raw data stack and while looping through the frames of the raw data stack the background image was continuously adjusted to account for changes in background intensities during imaging. Next, signal peaks were extracted from the resulting background corrected images. A 7 × 7 px ROI was centered around each signal peak exceeding a standard deviation of 2 times the noise. The noise was considered to follow a Poisson noise model. Extracted signal peaks were passed for subsequent position determination. Positions with subpixel accuracy were determined by calculating the center of mass of each input ROI, which corresponds in the absence of background (due to background subtraction) to a maximum likelihood estimation (MLE). Close-by signals were detected by searching each ROI from its center towards its boundaries for local intensity minima. In case of local minima, the ROI was clipped and the signal was recalculated or discarded when the clipping resulted in a loss of more than 30% of the ROI intensity. An initial list of localizations was returned by the algorithm, listing x- and y-coordinates of each detected single molecule as well as information about its photon count and localization precision. Next, the list of localization was post-processed using ILE by first joining localizations occurring in consecutive frames and within 2.5 times of the mean localization precision. Drift correction was done by splitting the list of localization in 5 subsets and correlating the reconstructions of the subsets.48,49 The shift among the subsets was interpolated and used to correct the positions of the detected signals in the list of localizations. No further filtering of the list of localizations was applied. For reconstruction of SMLM images, the single molecule positions were blurred by a Gaussian with a standard deviation corresponding to the localization precisions of the respective signals.
Spatial point pattern analysis of SMLM data
A custom-written software package in Matlab (Localization Analyzer for Nanoscale Distributions (LAND), https://github.com/Jan-NM/LAND) was used to analyze spatial distribution of the detected single molecule signals. The list of localizations was pre-processed in a first step by reconstructing the images (via histogram binning) and cropping a rectangular ROI for each cell that showed a homogenous distribution on large scales; i.e. no visible gaps. For each cell, a new list of localizations was generated containing only the molecule signals within the rectangular ROI. The new list of localizations was used as experimental input data in the subsequent analysis, which was performed on the basis of the single molecule coordinates. Random data following complete spatial randomness (CSR) were generated using the density of signals of the experimental input data. In total, three different algorithms were used to characterize the point pattern: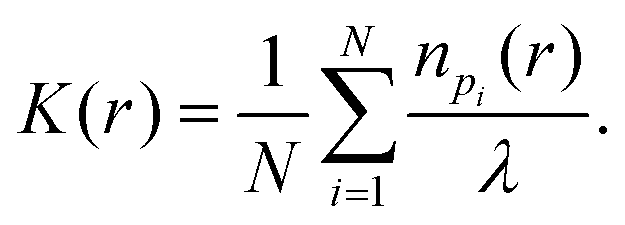
Here N denotes the total number of molecule signals that are investigated, n is the number of molecule signals within a distance r of a point pi and λ the density of molecule signals. The K-function was later transformed to obtain Ripley's H-function:

Subsequently, the Ripley's H-functions obtained from different donors from the same treatment were averaged and the maximum of Ripley's H-function was used as an estimate of the characteristic cluster size. Only maxima within a distance of 200 nm were considered.
Wide-field fluorescence microscopy of TLR4
Primary human macrophages were derived and isolated from PBMC as described before. Instead of glass coverslips, 2.5 × 106 cells per well were seeded in a glass bottom 12-well plate (Cellvis). Fixation and immunostaining of TLR4 was done in the same way as for the localization microscopy experiments. Nuclei were counterstained using 4′,6-diamidino-2-phenylindole (DAPI; Thermo Fisher Scientific) at a concentration of 300 nM in PBS for 3 min. After incubation, cells were washed three times in PBS. Instead of Vectashield H-1000, cells were imaged in PBS on the Opera Phenix High-Content Screening system (PerkinElmer). TLR4 stained cells (excitation 640 nm, emission 650 nm–760 nm) and DAPI stained nuclei (excitation 405 nm, emission 435 nm–480 nm) were imaged in non-confocal mode using a 20×/1.0 water immersion objective.Nitration of ATI with tetranitromethane (TNM) and endotoxin quantification
ATI were diluted in ultra-pure water to a final concentration of 1 mg ml−1. 4.55 μl TNM/methanol (4% v/v) were added to 500 μl of diluted ATI. The mixture was stirred for 3 h at room temperature. Residual TNM was removed using a PD-25 size exclusion column (GE Healthcare). The endotoxin content was quantified using a limulus amebocyte lysate (LAL) assay (Thermo Fisher Scientific). Nitrated ATI solutions with a final endotoxin content of less than 20 endotoxin units per ml (1.3 EU μg−1 protein) were used for experiments.Sample size
Experiments were conducted on primary human macrophages obtained from three different healthy donors. A total of at least 25 cells were evaluated per treatment. All values refer to the median. Number of cells used for Ripley's H-function and DBSCAN analysis can be found in Tables S1–S3.†Ethics statement
Buffy coats from healthy donors were obtained anonymously from the local blood bank (Transfusion Center, University Medical Center of the Johannes Gutenberg University, Mainz, Germany). The local ethical committee (Landesärztekammer Rheinland-Pfalz, Mainz, Germany) approved all experiments according to the guidelines of the 1964 Declaration of Helsinki (9th revision, 2013). Consent of participants was given in written form.Results and discussion
TLR4 is pre-clustered in unstimulated primary human macrophages
To investigate the large and small-scale distribution of TLR4 on the cell membrane, unstimulated macrophages stained for TLR4 were imaged by conventional fluorescence and localization microscopy (Fig. 1A + B). Conventional fluorescence images show that macrophages express TLR4. In line with other studies,53,54 we observed differences in TLR4 surface representation between cells and variations on individual cells (Fig. 1A, Fig. S4†). Using SMLM, finer structural details become visible compared to conventional fluorescence microscopy (Fig. 1B, Fig. S5†). An accumulation of TLR4 molecules in domains at the nanometer range was detected (Fig. 1C, ROI). To quantitatively evaluate the observed pattern, a grid-based density analysis was performed (Fig. 1D, ROI). It reveals that in unstimulated cells TLR4 clusters are present that are embedded in regions reduced in TLR4 surface representation. These regions contain small isolated clustered regions that can be associated with fluorophores belonging to a single TLR4 receptor. Normally, a single fluorophore may blink several times during SMLM imaging. This results in multiple detected signals per fluorophore, whose positions are distributed within the localization precision. Therefore, several fluorophores coupled to a single antibody generate small pseudo-clusters (Fig. S6, ESI Note 1†). Overall, our findings show that TLR4 molecules are present in a pre-clustered state on unstimulated macrophages, which confirms previous investigations on receptor clustering.55–57 | ||
| Fig. 1 TLR4 clusters on the cell membrane of primary human macrophages. (A) Conventional fluorescence microscopy image of unstimulated macrophages stained for TLR4. Intensity-coded visualization reveals differences in TLR4 expression levels between cells and variations in its surface representation on individual cells (white arrows). Scale bar = 50 μm. (B) Combined SMLM and wide-field image of an untreated macrophage stained for TLR4. Scale bar = 2 μm. (C) TLR4 SMLM image sections of macrophages treated with LPS-EB (100 ng ml−1), ATI (12.5 μg ml−1) or nitrated ATI (12.5 μg ml−1) for 15 min compared to medium control. The image section of the medium control was taken from the region of interest (ROI) indicated in panel 1B. SMLM images are intensity-coded, i.e. high intensity areas correspond to an accumulation of single molecule signals. Scale bar = 500 nm. (D) Grid-based density analysis of SMLM images from panel 1C. Blue areas indicate a dispersed distribution, whereas yellow areas show clustering of TLR4. | ||
Next, we investigated whether ATI and tetranitromethane (TNM)-nitrated ATI have an effect on the membrane distribution of TLR4. Therefore, macrophages were stimulated with ATI or nitrated ATI for 15 min and 30 min, respectively (ESI Note 2 + 3, Fig. S7†). LPS served as a positive control. Visual inspection and grid-based density analysis of SMLM images from the stimulated cells revealed a clustered distribution of TLR4 as observed in the unstimulated samples (Fig. 1C + D, Fig. S8†).
Donor-dependent TLR4 cluster formation upon stimulation with LPS and ATI
To analyze the effect of LPS and ATI further, the density of molecules on the membrane was calculated for the different donors (Fig. 2, Fig. S9A†). Already in the unstimulated state, the donors exhibit different levels of density of molecules on the membrane (Fig. 2A). With ∼80 signals per μm2, the lowest density of molecules was observed for donor 2, while the other donors showed densities 4 times higher in the unstimulated state. Differences among donors were also detected in response to LPS, ATI and nitrated ATI (Fig. 2B–D). Donor 2 in particular showed an increase in the density of molecules by about 390% and 250% compared to unstimulated cells after treatment with LPS and ATI for 15 min (Fig. 2C). In contrast, no change in molecule densities was found after treatment of macrophages from donor 2 with nitrated ATI. The substantial changes in the density of molecules seen for donor 2 after stimulation with LPS and ATI was not observed for the other donors (Fig. 2B + D). Apparently, these donors already had an elevated level in the density of molecules in the unstimulated state, suggesting that the ground state may influence later response to external stimuli.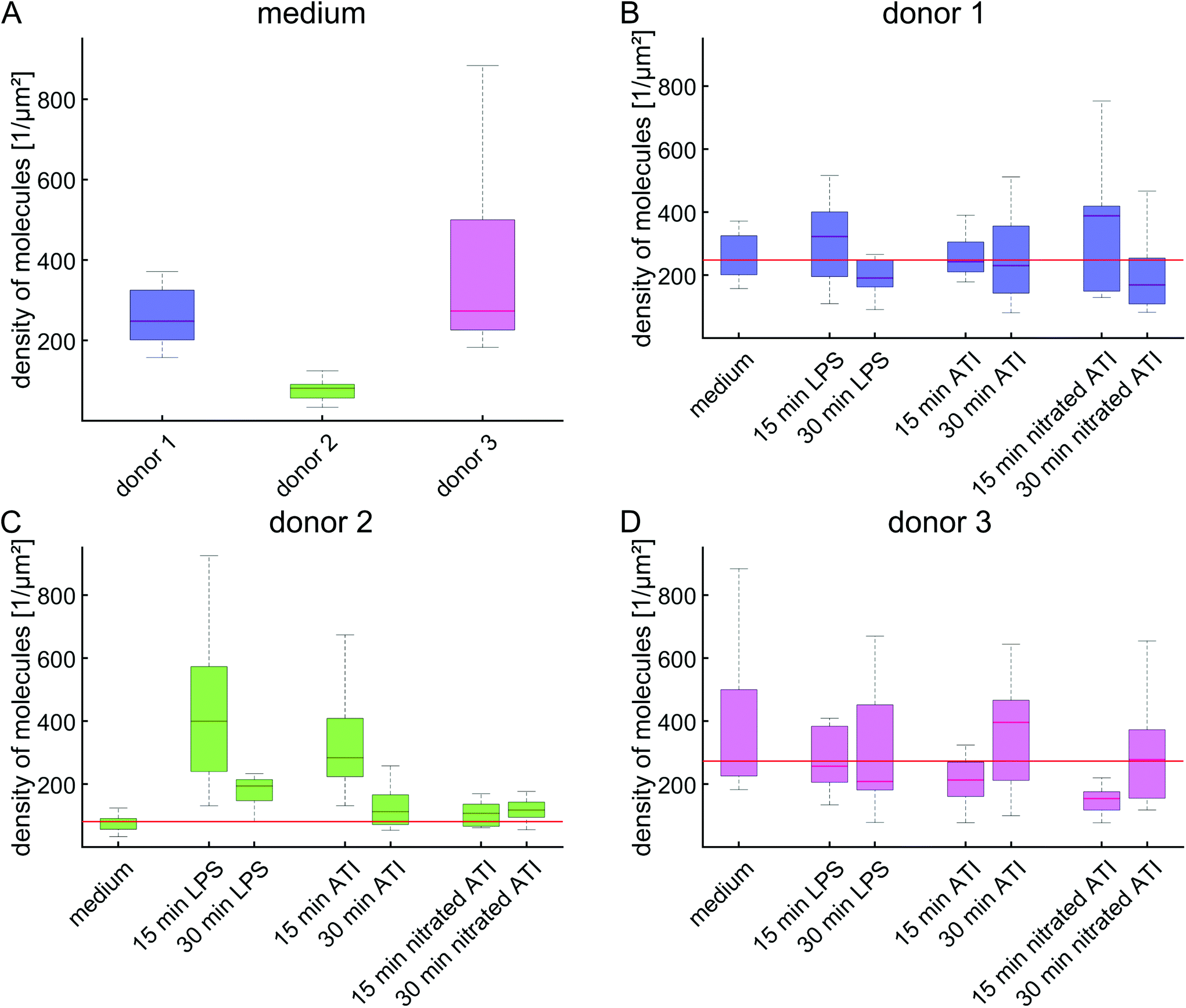 | ||
| Fig. 2 Molecule densities on the cell membrane are specific to the individual donors. (A) Density of molecules on unstimulated macrophages. The colors of the boxes represent the three different donors. (B–D) Density of molecules after stimulation with LPS, ATI or nitrated ATI, categorized according to the various donors. The boxes represent the median and the 25th to 75th percentile of the donors, whereas the whiskers cover 99.3% of the data. The horizontal red line indicates the median value of the medium control. | ||
To analyze the clustering behavior of TLR4, the fraction of clustered molecules and the density of clusters was calculated for each donor separately (Fig. 3, Fig. S9B + C†). Similar to the density of the molecules on the cell membrane, strong differences were present in the fraction of clustered molecules among the donors in the unstimulated state (Fig. 3A). These range from ∼51% for donor 2 to ∼75% for donor 1. For donor 2, a pronounced increase in the fraction of clustered molecules was found after stimulation, with LPS and ATI showing the highest increase of about 40% and nitrated ATI the lowest with ∼10%. After 15 min ATI stimulation, donor 1 showed an increase of about 7%, whereas donor 3 showed the strongest increase after 30 min stimulation with ATI and nitrated ATI. Furthermore, the cluster analysis revealed that on unstimulated cells, approximately 2 clusters per μm2 to 4.4 clusters per μm2 were present (Fig. 3B). This can be translated to a distance of neighboring clusters between 200 nm and 320 nm, respectively. Comparing the density of clusters vs. the density of molecules on the membrane, a positive correlation between both variables can be found (Fig. S10†). As a result, an increase in the density of clusters on the membrane was most pronounced for donor 2, forming about 8 clusters per μm2 after 15 min of stimulation with LPS and ATI. For donor 3, a broadening of the cluster density distribution was recognizable after 30 min stimulation, which may suggest a heterogeneous response to the stimuli. The observed densities of clusters on the membrane are in agreement with previous investigations on LPS stimulated glioblastoma cells.27
 | ||
| Fig. 3 Donor dependent clustering of TLR4 as analyzed by DBSCAN. The fraction of clustered molecules (A) and the density of clusters on the membrane (B) for the three different donors is shown. The color-coded boxes indicate the median and the 25th to 75th percentile for the individual donors. The dashed lines represent the median values of the medium control from the respective donors. The whiskers cover 99.3% of the data. In total 264 cells were evaluated. | ||
TLR4 cluster sizes remain unchanged after LPS or ATI stimulation
The clusters were further analyzed by extracting their diameter, the absolute number of molecules per cluster and the density of molecules within the clusters (Fig. 4). A median cluster diameter of about 44 nm was found (Fig. 4A). No significant changes in the median cluster diameter could be observed, as the variations for each condition stayed close to the median of unstimulated cells, within the interquartile range. To verify the results, Ripley's H-function was calculated (Fig. S11†) and the maximum of the function was used to derive the characteristic cluster size (Fig. 4B). Moreover, simulations were carried out to verify that the characteristic cluster size corresponds to the cluster diameter (Fig. S12†). As already found in the previous analysis, cluster size remained unchanged among the samples. Similar findings were made by Zeuner et al.,27 where the authors observed a cluster size of ∼50 nm on glioblastoma cells independent of the LPS stimuli applied. In contrast, Aaron et al.28 reported a TLR4 cluster size of ∼380 nm in unstimulated mouse macrophages, which increased to ∼520 nm after stimulation with LPS. Discrepancies between the studies could be attributed to the use of different cell lines, especially a different behavior of TLR4 in mouse cells compared to human cells.27 Alternatively, these discrepancies may be the result of inefficient photoswitching during localization imaging,27 which can lead to an apparently larger cluster size. It must be also noted that our measured cluster diameter is larger than actual diameters due to the indirect labeling system. This system consists of primary antibody and fluorescently labeled secondary F(ab′)2 fragments, which increases the distance between protein and detection molecule. Therefore, the measured cluster diameter represents an upper limit.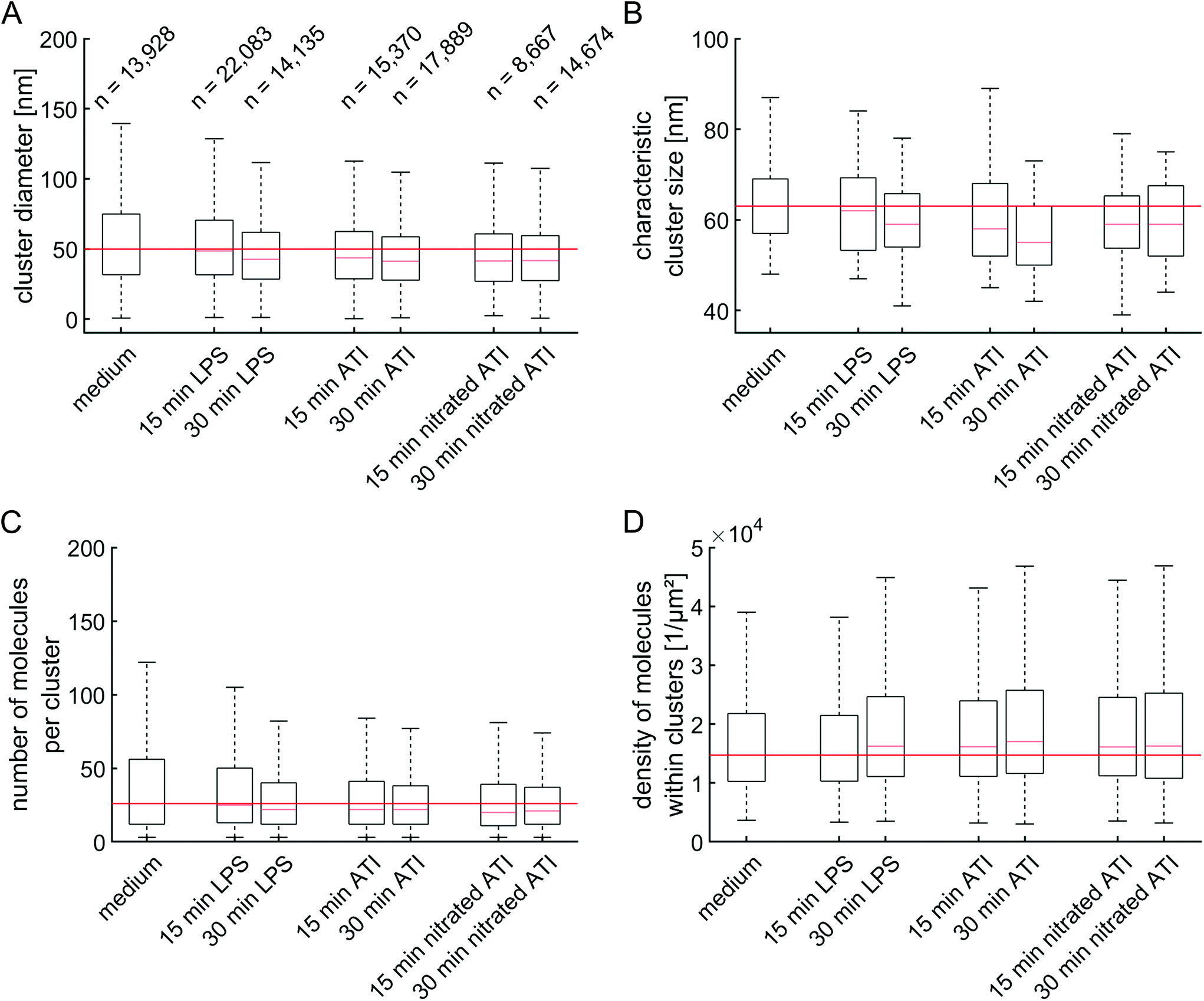 | ||
| Fig. 4 Unchanged cluster characteristics of TLR4 revealed by DBSCAN and Ripley's H-function. (A) Cluster diameter as obtained from DBSCAN analysis. The numbers above the boxes denote the number of identified clusters for panels A, C and D. (B) Average cluster size of TLR4 obtained from the maximum of Ripley's H-function of individual cells (Fig. S11†). (C) Number of molecules per cluster and (D) density of molecules within the clusters as obtained from DBSCAN analysis. All data are presented as box plots, showing the median and the 25th to 75th percentile as boxes. The whiskers cover 99.3% of the data. | ||
Regarding the absolute signals per cluster, on average 22 molecule signals were detected (Fig. 4C), resulting in an average density within the clusters of 1.6 × 104 molecules per μm2 (Fig. 4D). As already observed for the cluster diameter, no significant changes were observed.
Model of TLR4 receptor clustering
In our study, we have demonstrated that the majority of TLR4 molecules are already located in clusters in unstimulated primary human macrophages. Upon stimulation, donor-dependent variations in the fraction of clustered molecules accompanied by changes in the density of receptors and clusters on the membrane were observed. These changes may result from the superposition of TLR4 recruitment and endocytosis processes (Fig. 5). On the one hand, TLR4 receptors can be recruited into or around pre-existing clusters. On the other hand, new clusters can be generated by recruitment of receptors around monomers or pre-existing lower order oligomers. Moreover, any recruitment of receptors increases the local density of detected single molecule signals during SMLM imaging, thus exceeding the threshold beyond which DBSCAN detects a cluster. For both processes, replenishment of TLR4 receptors can happen from the Golgi apparatus58,59 or by translocation of receptors present on the membrane following stimulation, i.e. recruitment into cholesterol-rich microdomains, also referred as lipid rafts.14 These recruitment processes can also be assumed to take place continuously in unstimulated cells, but the rate might differ after stimulation. | ||
| Fig. 5 Schematic diagram of recruitment, clustering and depletion of membranous TLR4. TLR4 is glycosylated in the Golgi apparatus and subsequently recruited on the cell membrane.58,59 Upon stimulation, TLR4 molecules may be recruited into pre-existing clusters or form new clusters by re-arrangement of TLR4 monomers. Subsequently, TLR4 receptors are endocytosed,60,61 thus leading to a depletion of TLR4 from the membrane. | ||
Moreover, the TLR4 receptor complex can be endocytosed from the membrane after stimulation.60,61 Altogether, these processes determine the density of clusters on the membrane and the fraction of clustered molecules.
A recent study elucidated the ligand-specific receptor dimerization of TLR4 using stoichiometric labeling of TLR4 together with quantitative single molecule localization microscopy.29 In that study, the authors were able to determine the proportion of monomeric and dimeric TLR4 complexes depending on the LPS chemotype used. An extension of these experiments to ATI might help to understand whether the formation of clusters is accompanied by a change in the ratio of monomers to dimers upon ATI stimulation. Such effects would indicate stronger and focused signaling events, which would underline together with our results the importance of receptor clusters as signaling platforms.
Conclusions
In our study, we investigated the membrane distribution of TLR4 on primary human macrophages using SMLM. We focused on ATI and nitrated ATI, which have been identified as stimulating the TLR4/NF-κB pathway in macrophages and dendritic cells.33,39 Using different quantitative analysis methods, we found that the majority of TLR4 molecules are already located in clusters, whereby pronounced differences existed among the donors already in the unstimulated state. Upon stimulation with LPS or ATI, the overall fraction of clustered TLR4 molecules increased. Moreover, we observed an overall increase in the density of clusters up to ∼8 clusters per μm2 compared to ∼2 clusters per μm2 in the unstimulated state. Furthermore, we found that the clusters have a median diameter of about 44 nm, which did not change significantly after treatment. The donor-dependent differences underline the individuality of human samples and the need for future experiments on a larger cohort of donors to get a better understanding and comprehensive picture of TLR4 clustering and activation, as well as to extract possible patterns of different response types.Altogether, our findings suggest a donor-dependent formation of clusters before and after stimulation with LPS and ATI. This structural change could further affect functional or regulatory processes of the TLR4 downstream cascade inducing finally enhanced inflammation. Regardless of the TLR4 agonist used, we found similar structural changes suggesting a general mechanism of TLR4 cluster formation upon stimulation.
Author contributions
UP, CC, KL, UB and JN designed the research. JN and KZ performed the experiments. JN, MG, KL and JFN analyzed and interpreted the data. FL, IB and DS contributed materials and methods. JN, KZ, MG and KL wrote the paper. All authors contributed to the discussion, editing and proofreading of the manuscript.Funding disclosure
This work was supported by the Max Planck Graduate Center with the Johannes Gutenberg University Mainz (MPGC). DS was supported for ATI-related work by grants from the German Research Foundation (DFG) (DFG-Schu 646/17-1, DFG-Schu-646/20-1, the Collaborative Research Center TR128 “Multiple Sclerosis” project A08), and the Leibniz Foundation (project Wheatscan). IB was supported by DFG grant BE 4504/3-1.Conflicts of interest
The authors have no conflicting financial interests to disclose. UB and CC are also affiliated with LuciaOptics Forschungszentrum, a non-profit company based in Karlsruhe, Germany. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript or in the decision to publish the results.Acknowledgements
We thank Dr Sandra Ritz, Dr Maria Hanulova, Dr Jonas Schwirz, Dr Aleksander Szczurek, Florian Schock and Amine Gourram for continuous support and helpful discussions. Support by the Max Planck Graduate Center, the Mainz Program for Chemical Allergology and the joint Laboratory of Inflammation and Microscopy is gratefully acknowledged. We also acknowledge the IMB Microscopy and Histology Core Facility for support and the Deutsche Forschungsgemeinschaft (DFG) for funding the Opera Phenix High Content Spinning Disk Microscope (Project 402386039). We thank Dr Frank Helleis, Dr Thomas Klimach and the workshop of the MPI for Chemistry as well as the E-lab of IMB for help and support in the construction of the SMLM setup. Open Access funding provided by the Max Planck Society.References
- E. M. Y. Moresco, D. LaVine and B. Beutler, Curr. Biol., 2011, 21, R488–R493 CrossRef CAS PubMed.
- T. Kawasaki and T. Kawai, Front. Immunol., 2014, 5, 1–8 CAS.
- J. C. Chow, D. W. Young, D. T. Golenbock, W. J. Christ and F. Gusovsky, J. Biol. Chem., 1999, 274, 10689–10692 CrossRef CAS PubMed.
- B. Beutler, X. Du and A. Poltorak, J. Endotoxin Res., 2001, 7, 277–280 CrossRef CAS PubMed.
- L. Zuo, K. Lucas, C. A. Fortuna, C.-C. Chuang and T. M. Best, Front. Physiol., 2015, 6 DOI:10.3389/fphys.2015.00312.
- A. Fasano, A. Sapone, V. Zevallos and D. Schuppan, Gastroenterology, 2015, 148, 1195–1204 CrossRef CAS PubMed.
- D. Schuppan and V. Zevallos, Dig. Dis., 2015, 33, 260–263 CrossRef PubMed.
- S. Zundler and M. F. Neurath, Immunol. Rev., 2017, 278, 263–276 CrossRef CAS PubMed.
- J. da Silva Correia, K. Soldau, U. Christen, P. S. Tobias and R. J. Ulevitch, J. Biol. Chem., 2001, 276, 21129–21135 CrossRef CAS PubMed.
- T. L. Gioannini, A. Teghanemt, D. Zhang, N. P. Coussens, W. Dockstader, S. Ramaswamy and J. P. Weiss, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4186–4191 CrossRef CAS PubMed.
- B. S. Park, D. H. Song, H. M. Kim, B.-S. Choi, H. Lee and J.-O. Lee, Nature, 2009, 458, 1191–1195 CrossRef CAS PubMed.
- A. Płóciennikowska, A. Hromada-Judycka, K. Borzęcka and K. Kwiatkowska, Cell. Mol. Life Sci., 2014, 72, 557–581 CrossRef PubMed.
- J.-M. Ruysschaert and C. Lonez, Biochim. Biophys. Acta, Biomembr., 2015, 1848, 1860–1867 CrossRef CAS PubMed.
- M. Triantafilou, K. Miyake, D. T. Golenbock and K. Triantafilou, J. Cell Sci., 2002, 115, 2603–2611 CAS.
- L. J. Pike, J. Lipid Res., 2006, 47, 1597–1598 CrossRef CAS PubMed.
- M. Schürmann, N. Frese, A. Beyer, P. Heimann, D. Widera, V. Mönkemöller, T. Huser, B. Kaltschmidt, C. Kaltschmidt and A. Gölzhäuser, Small, 2015, 11, 5781–5789 CrossRef PubMed.
- S. W. Hell and J. Wichmann, Opt. Lett., 1994, 19, 780–782 CrossRef CAS PubMed.
- R. Heintzmann and C. Cremer, Proc. SPIE, 1999, 3568, 185–196 CrossRef.
- M. G. Gustafsson, J. Microsc., 2000, 198, 82–87 CrossRef CAS PubMed.
- K. A. Lidke, B. Rieger, T. M. Jovin and R. Heintzmann, Opt. Express, 2005, 13, 7052–7062 CrossRef PubMed.
- E. Betzig, G. H. Patterson, R. Sougrat, O. W. Lindwasser, S. Olenych, J. S. Bonifacino, M. W. Davidson, J. Lippincott-Schwartz and H. F. Hess, Science, 2006, 313, 1642–1645 CrossRef CAS PubMed.
- S. T. Hess, T. P. K. Girirajan and M. D. Mason, Biophys. J., 2006, 91, 4258–4272 CrossRef CAS PubMed.
- M. J. Rust, M. Bates and X. Zhuang, Nat. Methods, 2006, 3, 793–795 CrossRef CAS PubMed.
- A. Sharonov and R. M. Hochstrasser, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 18911–18916 CrossRef CAS PubMed.
- M. Heilemann, S. Van De Linde, M. Schüttpelz, R. Kasper, B. Seefeldt, A. Mukherjee, P. Tinnefeld and M. Sauer, Angew. Chem., Int. Ed., 2008, 47, 6172–6176 CrossRef CAS PubMed.
- P. Lemmer, M. Gunkel, D. Baddeley, R. Kaufmann, A. Urich, Y. Weiland, J. Reymann, P. Müller, M. Hausmann and C. Cremer, Appl. Phys. B, 2008, 93, 1–12 CrossRef CAS.
- M.-T. Zeuner, C. L. Krüger, K. Volk, K. Bieback, G. S. Cottrell, M. Heilemann and D. Widera, Biochim. Biophys. Acta, Mol. Cell Res., 2016, 1863, 3084–3095 CrossRef CAS PubMed.
- J. S. Aaron, B. D. Carson and J. A. Timlin, Small, 2012, 8, 3041–3049 CrossRef CAS PubMed.
- C. L. Krüger, M. T. Zeuner, G. S. Cottrell, D. Widera and M. Heilemann, Sci. Signaling, 2017, 10(503) DOI:10.1126/scisignal.aan1308.
- T. Lang and S. O. Rizzoli, Physiology, 2010, 25, 116–124 CrossRef CAS PubMed.
- M. F. Garcia-Parajo, A. Cambi, J. A. Torreno-Pina, N. Thompson and K. Jacobson, J. Cell Sci., 2014, 127, 4995–5005 CrossRef PubMed.
- N. Tanimura, S. Saitoh, F. Matsumoto, S. Akashi-Takamura and K. Miyake, Biochem. Biophys. Res. Commun., 2008, 368, 94–99 CrossRef CAS PubMed.
- Y. Junker, S. Zeissig, S.-J. Kim, D. Barisani, H. Wieser, D. A. Leffler, V. Zevallos, T. A. Libermann, S. Dillon, T. L. Freitag, C. P. Kelly and D. Schuppan, J. Exp. Med., 2012, 209, 2395–2408 CrossRef CAS PubMed.
- D. Schuppan, G. Pickert, M. Ashfaq-Khan and V. Zevallos, Best Pract. Res., Clin. Gastroenterol., 2015, 29, 469–476 CrossRef CAS PubMed.
- V. F. Zevallos, V. Raker, S. Tenzer, C. Jimenez-Calvente, M. Ashfaq-Khan, N. Rüssel, G. Pickert, H. Schild, K. Steinbrink and D. Schuppan, Gastroenterology, 2017, 152, 1100–1113.e12 CrossRef CAS PubMed.
- M. Cuccioloni, M. Mozzicafreddo, L. Bonfili, V. Cecarini, M. Giangrossi, M. Falconi, S. I. Saitoh, A. Maria Eleuteri and M. Angeletti, Sci. Rep., 2017, 7, 4–6 CrossRef PubMed.
- V. Zevallos, V. K. Raker, J. Maxeiner, P. Scholtes, K. Steinbrink and D. Schuppan, Eur. J. Nutr., 2018 DOI:10.1007/s00394-018-1681-6.
- I. Bellinghausen, B. Weigmann, V. Zevallos, J. Maxeiner, S. Reißig, A. Waisman, D. Schuppan and J. Saloga, J. Allergy Clin. Immunol., 2019, 143, 201–212.e4 CrossRef CAS PubMed.
- K. Ziegler, J. Neumann, F. Liu, J. Fröhlich-Nowoisky, C. Cremer, J. Saloga, K. Reinmuth-Selzle, U. Pöschl, D. Schuppan, I. Bellinghausen and K. Lucas, Front. Immunol., 2019, 9 DOI:10.3389/fimmu.2018.03174.
- A. S. Tatham and P. R. Shewry, Clin. Exp. Allergy, 2008, 38, 1712–1726 CAS.
- V. Leccioli, M. Oliveri, M. Romeo, M. Berretta and P. Rossi, Nutrients, 2017, 9, 1203 CrossRef PubMed.
- T. Franze, M. G. Weller, R. Niessner and U. Pöschl, Environ. Sci. Technol., 2005, 39, 1673–1678 CrossRef CAS PubMed.
- Y. K. Gruijthuijsen, I. Grieshuber, A. Stöcklinger, U. Tischlera, T. Fehrenbach, M. G. Weller, L. Vogel, S. Vieths, U. Pöschl and A. Duschl, Int. Arch. Allergy Immunol., 2006, 141, 265–275 CrossRef CAS PubMed.
- K. Reinmuth-Selzle, C. J. Kampf, K. Lucas, N. Lang-Yona, J. Fröhlich-Nowoisky, M. Shiraiwa, P. S. J. Lakey, S. Lai, F. Liu, A. T. Kunert, K. Ziegler, F. Shen, R. Sgarbanti, B. Weber, I. Bellinghausen, J. Saloga, M. G. Weller, A. Duschl, D. Schuppan and U. Pöschl, Environ. Sci. Technol., 2017, 51, 4119–4141 CrossRef CAS PubMed.
- K. A. K. Tanaka, K. G. N. Suzuki, Y. M. Shirai, S. T. Shibutani, M. S. H. Miyahara, H. Tsuboi, M. Yahara, A. Yoshimura, S. Mayor, T. K. Fujiwara and A. Kusumi, Nat. Methods, 2010, 7, 865–866 CrossRef CAS PubMed.
- P. Sengupta, T. Jovanovic-Talisman, D. Skoko, M. Renz, S. L. Veatch and J. Lippincott-Schwartz, Nat. Methods, 2011, 8, 969–975 CrossRef CAS PubMed.
- F. Grüll, M. Kirchgessner, R. Kaufmann, M. Hausmann and U. Kebschull, in Proceedings – 21st International Conference on Field Programmable Logic and Applications, IEEE, 2011, pp. 1–5.
- M. J. Mlodzianoski, J. M. Schreiner, S. P. Callahan, K. Smolková, A. Dlasková, J. Šantorová, P. Ježek and J. Bewersdorf, Opt. Express, 2011, 19, 15009–15019 CrossRef PubMed.
- Y. Wang, J. Schnitzbauer, Z. Hu, X. Li, Y. Cheng, Z.-L. Huang and B. Huang, Opt. Express, 2014, 22, 15982–15991 CrossRef PubMed.
- B. D. Ripley, J. R. Stat. Soc. Ser. B, 1977, 39, 172–212 Search PubMed.
- M. A. Kiskowski, J. F. Hancock and A. K. Kenworthy, Biophys. J., 2009, 97, 1095–1103 CrossRef CAS PubMed.
- M. Ester, H.-P. Kriegel, J. Sander and X. Xu, Proc. Second Int. Conf. Knowl. Discov. Data Min., 1996, pp. 226–231.
- K.-H. Tse, K. B. S. Chow, W. K. Leung, Y. H. Wong and H. Wise, Neuroscience, 2014, 279, 10–22 CrossRef CAS PubMed.
- N. Rashidi, M. Mirahmadian, M. Jeddi-Tehrani, S. Rezania, J. Ghasemi, S. Kazemnejad, E. Mirzadegan, S. Vafaei, M. Kashanian, Z. Rasoulzadeh and A.-H. Zarnani, J. Reprod. Infertil., 2015, 16, 72–81 Search PubMed.
- R. Kaufmann, P. Müller, G. Hildenbrand, M. Hausmann and C. Cremer, J. Microsc., 2011, 242, 46–54 CrossRef CAS PubMed.
- Y. S. Hu, H. Cang and B. F. Lillemeier, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7201–7206 CrossRef CAS PubMed.
- P. S. Boyd, N. Struve, M. Bach, J. P. Eberle, M. Gote, F. Schock, C. Cremer, M. Kriegs and M. Hausmann, Nanoscale, 2016, 8, 20037–20047 RSC.
- E. Latz, A. Visintin, E. Lien, K. A. Fitzgerald, B. G. Monks, E. A. Kurt-Jones, D. T. Golenbock and T. Espevik, J. Biol. Chem., 2002, 277, 47834–47843 CrossRef CAS PubMed.
- T. Ohnishi, M. Muroi and K. Tanamoto, Clin. Diagn. Lab. Immunol., 2003, 10, 405–410 CAS.
- H. Husebye, Ø. Halaas, H. Stenmark, G. Tunheim, Ø. Sandanger, B. Bogen, A. Brech, E. Latz and T. Espevik, EMBO J., 2006, 25, 683–692 CrossRef CAS PubMed.
- J. C. Kagan, T. Su, T. Horng, A. Chow, S. Akira and R. Medzhitov, Nat. Immunol., 2008, 9, 361–368 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr00943d |
| ‡ Current address: Hochschule für Technik und Wirtschaft Chur, Pulvermühlestrasse 57, 7000 Chur, Switzerland. |
| This journal is © The Royal Society of Chemistry 2019 |