
Catalytic oxidation and reduction reactions of hydrophilic carbon clusters with NADH and cytochrome C: features of an electron transport nanozyme†
 ab
Lizanne G.
Nilewski,
c
Kimberly
Mendoza,
c
Almaz
Jalilov,
d
James M.
Tour
*cfg
and
Thomas A.
Kent
*ach
ab
Lizanne G.
Nilewski,
c
Kimberly
Mendoza,
c
Almaz
Jalilov,
d
James M.
Tour
*cfg
and
Thomas A.
Kent
*ach
Abstract
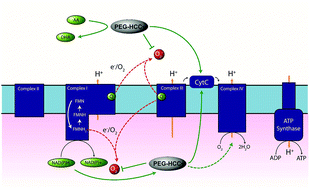
Previously, our group reported on the promising efficacy of poly(ethylene glycol)-hydrophilic carbon clusters (PEG-HCCs) to work as broadly active and high capacity antioxidants in brain ischemia and injury models including stroke and traumatic brain injury coupled with hemorrhagic shock. PEG-HCCs are a carbon nanomaterial derived from harsh oxidation of single wall carbon nanotubes and covalently modified with poly(ethylene glycol). They retain no tubular remnants and are composed of a highly oxidized carbon core functionalized with epoxy, peroxyl, quinone, ketone, carboxylate, and hydroxyl groups. HCCs are the redox active carbon core of PEG-HCCs, which have a broad reduction potential range starting at +200 mV and extending to −2 V. Here we describe a new property of these materials: the ability to catalytically transfer electrons between key surrogates and proteins of the mitochondrial electron transport complex in a catalytic fashion consistent with the concept of a nanozyme. The estimated reduction potential of PEG-HCCs is similar to that of ubiquinone and they enabled the catalytic transfer of electrons from low reduction potential species to higher reduction electron transport complex constituents. PEG-HCCs accelerated the reduction of resazurin (a test indicator of mitochondrial viability) and cytochrome c by NADH and ascorbic acid in solution. Kinetic experiments suggested a transient tertiary complex. Electron paramagnetic resonance demonstrated NADH increased the magnitude of PEG-HCCs’ intrinsic radical, which then reduced upon subsequent addition of cytochrome c or resazurin. Deconvolution microscopy identified PEG-HCCs in close proximity to mitochondria after brief incubation with cultured SHSY-5Y human neuroblastoma cells. Compared to methylene blue (MB), considered a prototypical small molecule electron transport shuttle, PEG-HCCs were more protective against toxic effects of hydrogen peroxide in vitro and did not demonstrate impaired cell viability as did MB. PEG-HCCs were protective in vitro when cells were exposed to sodium cyanide, a mitochondrial complex IV poison. Because mitochondria are a major source of free radicals in pathology, we suggest that this newly described nanozyme action helps explain their in vivo efficacy in a range of injury models. These findings may also extend their use to mitochondrial disorders.
- This article is part of the themed collection: Celebrating our 2020 Prize and Award winners
 Please wait while we load your content...
Please wait while we load your content...

