
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Six state molecular revolver mounted on a rigid platform
Jan
Homberg
a,
Marcin
Lindner
 a,
Lukas
Gerhard
*a,
Kevin
Edelmann
ab,
Timo
Frauhammer
ab,
Yasmine
Nahas†
a,
Michal
Valášek
a,
Marcel
Mayor
*acd and
Wulf
Wulfhekel
ab
a,
Lukas
Gerhard
*a,
Kevin
Edelmann
ab,
Timo
Frauhammer
ab,
Yasmine
Nahas†
a,
Michal
Valášek
a,
Marcel
Mayor
*acd and
Wulf
Wulfhekel
ab
aInstitute of Nanotechnology, Karlsruhe Institute of Technology, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: lukas.gerhard@kit.edu; marcel.mayor@unibas.ch
bPhysikalisches Institut, Karlsruhe Institute of Technology, 76131 Karlsruhe, Germany
cDepartment of Chemistry, University of Basel, St. Johannsring 19, CH-4056 Basel, Switzerland
dLehn Institute of Functional Materials (LIFM), Sun Yat-Sen University (SYSU), Xingang Rd. W., Guangzhou, China
First published on 25th April 2019
Abstract
The rotation of entire molecules or large moieties happens at 100 ps time scales and the transition process itself is experimentally inaccessible to scanning probe techniques. However, the reversible switching of a molecule between more than two metastable states allows to assign a rotational switching direction. Rotational switching is a phenomenon that is particularly interesting with regard to possible applications in molecular motors. In this work, single tetraphenylmethane molecules deposited on a Au(111) surface were studied in a low temperature scanning tunneling microscope (STM). These molecules comprise rotational axes mounted on a tripodal sulfur-anchored stand and with the STM tip, we were able to induce transitions between six rotational states of the molecular motif. We were able to identify critical parameters for the onset of rotational switching and to characterize the influence of the local environment. The subtle difference between fcc and hcp stacking and the rotational state of neighboring molecules clearly influence the population of the rotational states.
1. Introduction
Molecular machines that are driven by chemical or electric means have been identified in a number of biological systems. At the nanoscale, thermal energy leads to Brownian motion that is superimposed to any driving force, which makes the realization of artificial molecular machines a challenging task. The real-time observation of molecular dynamics that typically happens on the timescale of 100 ps is impossible with subnanometer resolution scanning probe microscopy. One solution is to follow molecular motion in a stepwise manner. This becomes possible when the motion is comprised of switching events between more than two metastable states which have residence times that are sufficiently long to be resolved by microscopy. In this way, multi-step rotational switching has been observed for caged 2-D coordination networks,1 chlorophyll molecules,2 proton-transfer in porphyrins,3 double-decker complexes4,5 and molecular units mounted on dangling bond dimer6 or Ru ball bearings.7Molecular mechanical devices that can be controlled by an external trigger are of particular interest. A number of different switching mechanisms and different stimuli have been found in single molecules adsorbed on surfaces: mechanical deformation,8,9 electric field,10–13 light,14–16 voltage controlled change of the charge state17,18 and current19 have been used to trigger the switching. Whereas most molecules that have been found to exhibit such a switching behavior are basically two-dimensional,20 here we present three-dimensional molecules that possess a higher number of possible conformations and even when adsorbed on a surface, internal conformation changes are allowed. In general, those non-planar molecular platforms allow to effectively decouple a molecular head group from the metallic substrate.21,22 Such molecular structures might be particularly promising for the implementation of functional units. Deposition of such complex structures and imaging them with scanning probe techniques has turned out to be difficult and only few successful studies have been published.23–27
2. Results and discussion
Here we present a low-temperature scanning tunneling microscopy (STM) study of tetraphenylmethane molecules deposited on a Au(111) surface. Adsorbed on the surface, six different metastable states were found and we managed to induce reversible transitions between all six states. An elaborate analysis allows us to give the critical parameters that lead to rotational switching and to identify the influence of the substrate and the neighboring molecules. Two different tetraphenylmethane molecules presented in Fig. 1a and b were dissolved in dichloromethane (DCM) and sprayed through a pulse valve onto a clean Au(111) placed in a vacuum chamber at about 10−3 mbar (see Fig. 1c). With this method any thermal stress or charging of the molecules can be avoided while the amount of solvent and contamination of the sample is minimized. Details of the procedure are published in ref. 26 and 28. Due to the different positions of the sulfur atoms at the phenyl groups, we refer to them as meta and para molecules respectively. After deposition, samples were transferred into ultra-high vacuum and were annealed at 150 °C for about 60 min in order to promote deprotection of the thiol anchoring groups and proper adsorption of the molecules. Additionally, the amount of possible contaminations and solvent remnants was reduced (see Fig. 2a). All STM experiments presented here were carried out at 5 K. For all deposition parameters, we found a coexistence of ordered islands and areas of well isolated monomers. While we discussed these ordered islands in our previous work,28,29 the focus of this work is on the isolated monomers (see Fig. 2a and b). An influence of the coverage on this ratio could not be identified. We explain this coexistence by an incomplete deprotection of the sulfur anchor groups of some molecules which then are mobile enough to form ordered structures while the fully deprotected molecules are strongly bound to the surface with all three sulfur groups. This explains the reduced diffusion and the different appearance of the monomers. This hypothesis is supported by the identification of remaining acetyl protection groups on the molecular islands (see ref. 29). The well-known influence of the Au(111) reconstruction on the local diffusion of adsorbates30 is only weakly pronounced (see Fig. 2a and b). In close-up STM images all isolated molecules show an identical triangular motif with one edge being elevated as can be seen in Fig. 2b–f. We find six orientations of the molecular motif which differ by a rotation of 60° (see Fig. 2g and h).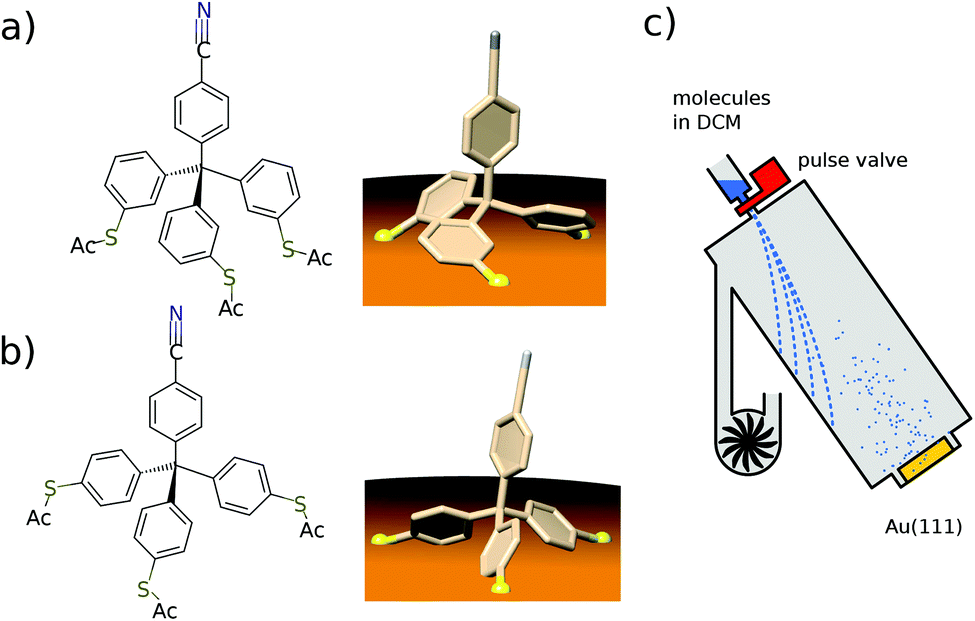 | ||
| Fig. 1 (a), (b) Chemical structure of the meta (a) and the para (b) tetraphenylmethane molecules and a 3D view of the expected adsorption configuration. (c) Scheme of the deposition setup used to spray the tetraphenylmethane molecules dissolved in dichloromethane (DCM) onto the Au(111) single crystal surface. | ||
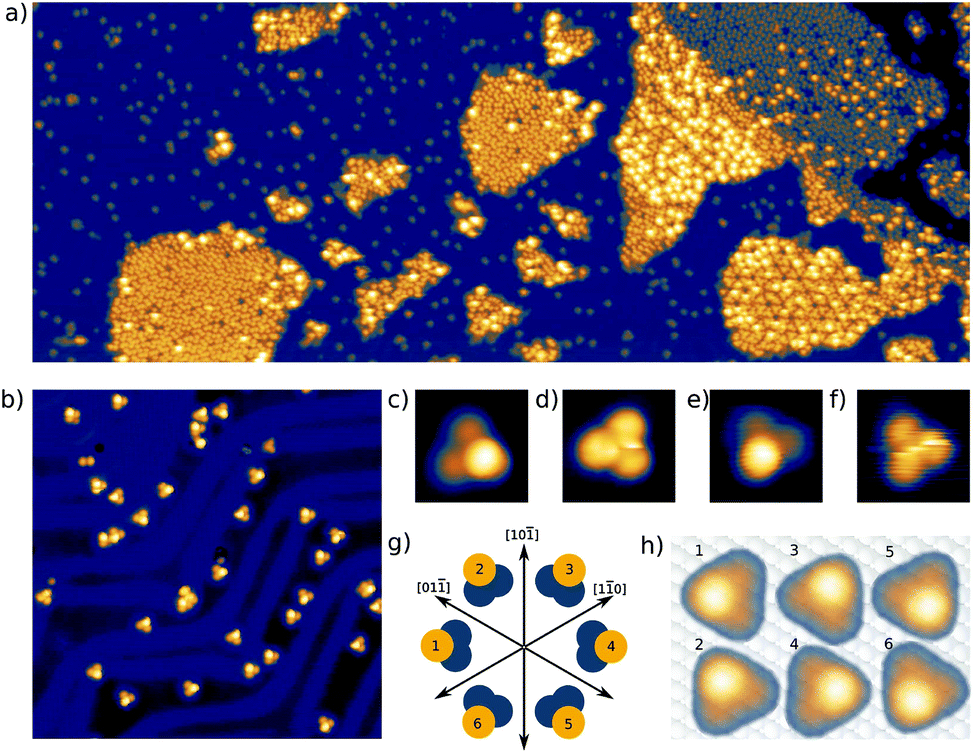 | ||
| Fig. 2 (a) Coexistence of isolated single molecules and ordered islands of para tetraphenylmethane molecules. The width of the image is 200 nm, recorded at a bias of 2.7 V and a setpoint of 5 pA. (b) Constant current STM image of isolated para tetraphenylmethane molecules adsorbed on a Au(111) surface. The width of the image is 30 nm. (c)/(d) Single para variant (0.8 V, 20 pA)/(0.15 V, 20 pA), (e)/(f) Single meta variant (0.7 V, 20 pA)/(0.04 V, 20 pA). The width of images (c)–(f) is 2 nm. (g) Schema showing the six existing different orientations with respect to the high symmetry directions of the Au(111) surface. (h) Schema showing the orientation of the molecular motif with respect to the lattice of the Au(111) surface, STM images to scale. | ||
At sufficiently high bias voltage and set point current, it is possible to induce transitions between the six different configurations. In order to image the rotational switching, we recorded images at alternating bias voltage: scans at 700 mV which are likely to induce a switching and read out scans at 120 mV which do not induce switching. An example of such a series of images of two para molecules is shown in Fig. 3a. The first image is a read out scan at 120 mV and a setpoint of 20 pA which does not induce any transitions. The second image is recorded at 700 mV (setpoint of 20 pA) and shows a transition of the lower molecule from one scan line to the other. The new configuration is then imaged at 120 mV, followed by the next scan at 700 mV that induces transitions in both molecules. We omitted the following scans at 700 mV and show the different orientations in the read out scans. The apparent rotation angle in successive read out scans at 120 mV is indicated at the arrows. Positive angles indicate clockwise rotation, negative angles indicate counter-clockwise rotation. We mostly observe rotations by 120°, rotations by 60° or 180° are observed only rarely. A similar behavior was found for the meta variant. A typical series of images showing a pair of molecules is depicted in Fig. 3b. The upper molecule in Fig. 3b does not show switching, most likely due to the nearby molecule above the scan frame. Bias and setpoint current in the experiments presented in Fig. 3a and b were chosen such that mainly single switching events occur during the high bias scans. Due to the statistical distribution of the time span between two switching events, this means that the tip often does not induce a switching during the high bias scan, that is we observe the molecule to be in the same state in two consecutive read out scans (see Fig. 3a). Intuitively, the bright edge of the triangle is identified with the protruding molecular head group that is oriented at an angle of approximately 80° with respect to the plane of the three sulfur atoms.28 Then, rotational steps of 120° can be related to the rotation of the inclined molecular head group, with the three metastable states corresponding to the threefold symmetric foot structure. Rarely, rotations of 60° or 180° can be observed, which correspond to a transition between “even” and “odd” orientations according to the numbering given in Fig. 2g and h. In other words, these rotations of 60° or 180° switch between two sets of three states with higher switching frequency.
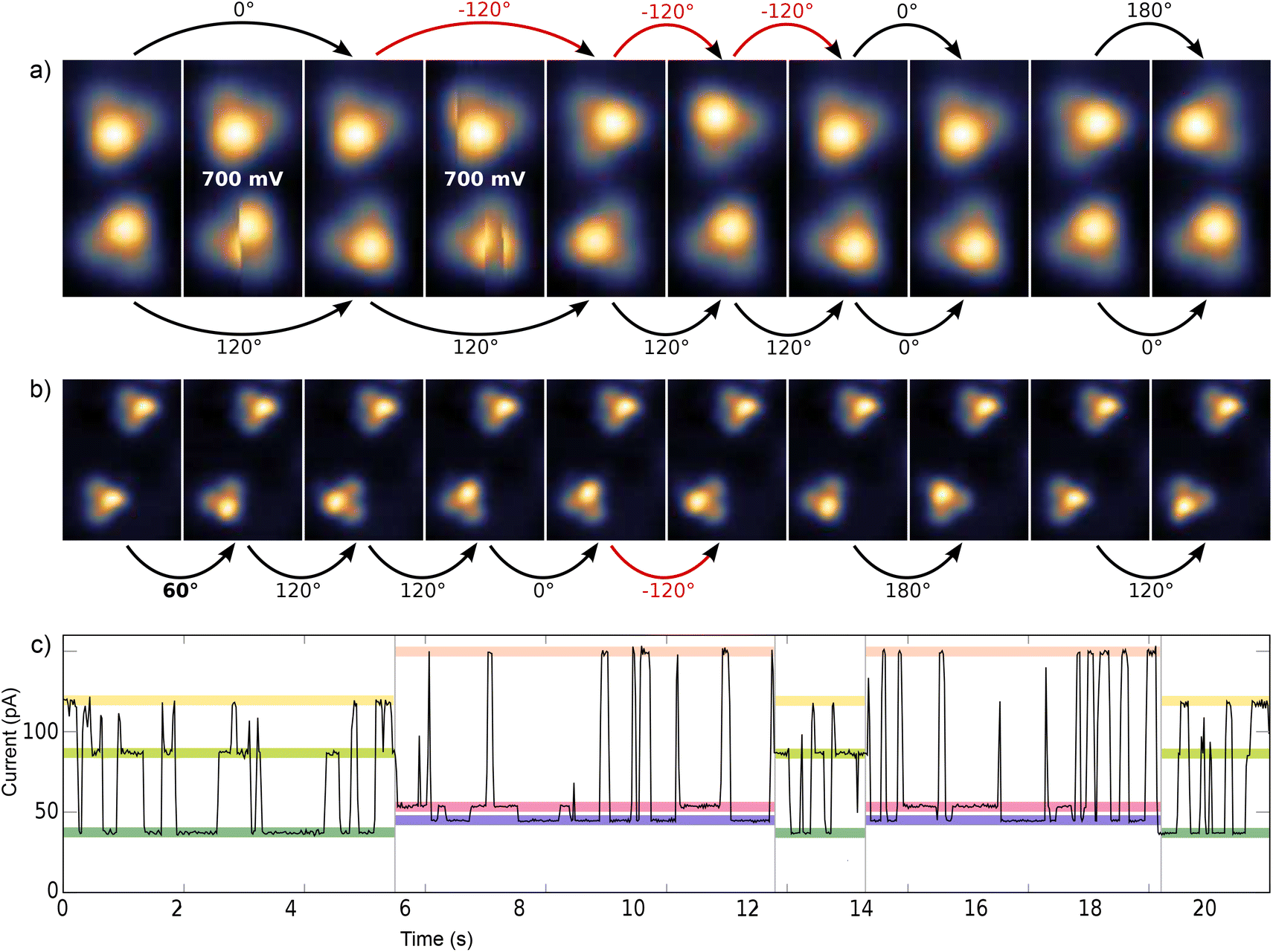 | ||
| Fig. 3 (a) Pair of para variant imaged at U = 120 mV and I = 20 pA, with a scan at 700 mV in between two images. Two of these scans at 700 mV are shown exemplarily. The width of the images is 2 nm. Slow scan in x direction. (b) Pair of meta variant imaged at U = 120 mV and I = 20 pA, with a scan at 700 mV in between two images. The width of the images is 3.37 nm. In (a) and (b) the rotation angle is indicated for consecutive read out scans. (c) Current trace at fixed tip position above a para variant with the two sets of three different current levels shown in color (recorded at a bias voltage of 800 mV). | ||
Typically, rotational motion of single molecules observed in STM is related to the relative rotation of the entire molecule with respect to the substrate. In stark contrast to most previous work on rotational switching, here, we discuss a rotatable axis mounted on a tripod which is in turn fixed on the substrate. A rotation around the center would require breaking of all three S–Au bonds followed by formation of new bonds in the rotated state. If this breaking of S–Au bonds is possible, however, also linear diffusion is expected. For both variants, the observed rotations never go along with a displacement of the molecule with respect to the surface in the sense of classical diffusion. Thus, we can rule out a rotation of the entire molecule around its center. A rotation around one of the S–Au bonds would require breaking of only two S–Au bonds. This lower barrier mechanism is thus expected to happen more often. However, such a rotation around one of the S–Au bonds would also involve a displacement of the molecular triangle for each rotation, which has never been observed in our STM experiments (see e.g.Fig. 3a and b). Thus, mechanisms that are based on a relative rotation of the entire molecule with respect to the substrate can safely be excluded. Instead, the observed transitions between the two sets of three states (rotation by 60° or 180°) must be based on internal reconfigurations of the molecular foot structure.
We suggest that the tetraphenylmethane scaffold adsorbed on the surface takes a propeller-like conformation (see Fig. 1a and b) which can have two different chiralities, which may explain two different internal configurations. As the protruding head group fully dominates the local density of states probed by the STM tip, the precise position of the sulfur atoms of the foot structure cannot be inferred from the images.
The striking similarity of the para and the meta variant lead us to perform various test experiments. We deposited a number of similar tetraphenylmethane-based molecules with slightly different head groups and we routinely deposited solvent without any molecules.28,29 However, the here described molecular triangles were only and always found when the molecules depicted in Fig. 1a and b were deposited. Thus, contamination of the deposition apparatus or the solvent can be excluded as the origin of the here described phenomena. Finally, we cannot exclude that the catalytic effect of the Au(111) surface induces a chemical modification of the molecules. Even though we lack a full explanation of the switching mechanism, we were able to characterize the switching behavior in more detail and thus to draw conclusions regarding intrinsic properties of the adsorbed molecules.
This switching can be induced by scanning above the molecule and also with the STM tip held at a fixed position. A typical time trace of the tunneling current, as shown in Fig. 3c above a para molecule, exhibits six different plateaus which represent the six different configurations of the molecule. It can be clearly seen that the threefold switching due to the rotation of the molecular head group occurs more frequently than the switching between the two sets of three states due to the internal reconfiguration of the molecule.
The probability to find a para molecule in one of the six different states depends on the domain it is adsorbed on. On the fcc areas of the reconstructed Au(111) surface31,32 the even orientations 2,4,6 (see Fig. 2g and h) are more likely than the odd orientations 1,3,5 (see Fig. 2g and h): (N(2,4,6)/N(1,3,5) = 172/42 = 4.1). On the hcp domains the odd configurations are found more often: N(2,4,6)/N(1,3,5) = 30/68 = 0.44. In this case, we can assume that the population of the different states that have different energies is determined by the Boltzmann distribution corresponding to the temperature at which the switching stops after cooling down from room temperature. Boltzmann statistics can then relate the observed population of the different states to their energy levels (kBT = 0.42 meV at 5 K):
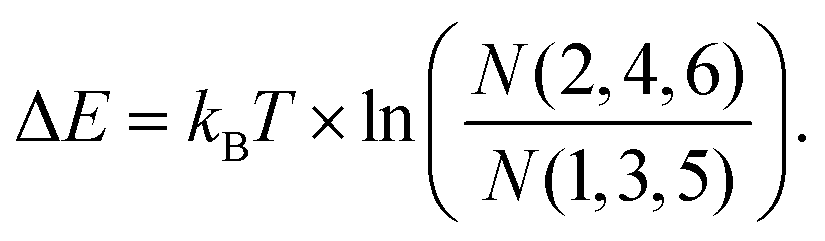
With this, we get a lower bound for the energy differences of ΔEfcc = E1,3,5fcc − E2,4,6fcc = 0.11 to 0.68 meV and ΔEhcp = E1,3,5hcp − E2,4,6hcp = −0.22 to −0.48 meV. In this way, the switching statistics of the molecules is a sensitive indicator of the stacking order of the underlying substrate. Also the switching statistics of individual para molecules, in particular the population of the odd and even states, are clearly different between the molecules adsorbed on fcc domains and those adsorbed on hcp domains as is depicted in Fig. 4a (Please note that the image in Fig. 4a is rotated by 60° compared to Fig. 2). Besides, it can be seen that molecules adsorbed on elbow sites of the herringbone reconstruction do not show any transitions (see red circles in Fig. 4a). This is in agreement with the statistics of the initial adsorption configuration mentioned above and indicates that the STM tip does not significantly influence the population of the different states. Therefore, we assume that the STM tip only enables the transitions by either lowering the energy barriers or by exciting the molecules over the barrier without a preferred direction. The latter would be in agreement with microscopic reversibility. Interestingly, the population of even (2,4,6 in Fig. 2g and h) and odd states (1,3,5 in Fig. 2g and h) of a para molecule also depends on the state of the neighboring molecule. After analyzing a series of about 2000 scans of the two para molecules presented in Fig. 3a, we found a negative correlation. The one molecule is more often in an even state when the other molecule is in an odd state: we find a population ratio of the second molecule M2even![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) : M2odd = 86:14 when the first molecule M1 is in an odd state. A ratio of M2even:M2odd = 80:20 is found when the first molecule is in an even state. We verified this effect for two different ensembles of five molecules adsorbed on fcc domains and analyzed the correlations of odd and even states of neighboring molecules. Correlations are significant for the first ensemble that stretches over about 7 nm (distance between the leftmost molecule M1 and the rightmost molecule M5 in Fig. 4b) The table below the image gives the corresponding Pearson correlation coefficients33 which are defined as covariance of the two variables (here: molecule A being in an odd/even state, molecule B being in an odd/even state) normalized by the standard deviations of the two variables. A positive (negative) correlation is indicated by a positive (negative) number (0 ≤ |r| ≤ 1) and no correlation is indicated by a value close to zero. We found both positive and negative correlations. For example, M1 and M2 show a negative correlation: M1even: M1odd = 20:80 when M2 is in an even state and M1even: M1odd = 78:22 when M2 is in an odd state. A positive correlation is found for M4 and M5: M4even: M4odd = 99:1 when M5 is in an even state, and M4even: M4odd = 13:86 when M5 is in an odd state. This intermolecular coupling also affects the population of the possible states of the whole ensemble. Fig. 4b shows the most likely configuration of the molecular states (M13;M26;M31;M42;M56) that is observed in 25/1370 scans (an equal population of all 65 = 7776 possible configurations would lead to an average of 0.18/1370). When only the difference between even and odd states is considered, the most probable configuration (M1odd;M2even;M3odd;M4even;M5even) is found in 572/1370 scans and its inverse (M1even;M2odd;M3even;M4odd;M5odd), which equally follows the preferred intermolecular coupling in another 132 scans. This means that in more than 50% of the scans, the configuration of the foot structure of all five molecules agrees with the sign of the correlations given in the table. In the case of M1 and M3, odd states are even more likely than even states, which is in contrast to the population statistics of all isolated para molecules adsorbed on fcc areas. Apparently, here the intermolecular coupling is stronger than the influence of the surface stacking. The presence of both positive and negative correlations excludes a simple dipolar interaction as the driving force. Furthermore, the surprisingly large change is unlikely to be carried by steric effects as the molecular centers are separated by more than 1.5 nm and the para molecules radially extend less than 0.5 nm. A coupling with oscillatory behavior induced by the surface state of noble metals has been observed for adatoms and small molecules.34,35 Also the π-systems of larger molecules were found to interact with the surface state electrons.36 In our STM images, the switching between even and odd states of the molecules appears as a rotation of the triangular foot structure by 60°. Therefore, it is fair to assume that this switching also affects the surface state that then mediates the interaction between neighboring molecules. While we cannot infer the chirality of a specific molecule from our STM images, we suppose that the intramolecular transition from odd to even states goes along with a change in chirality. This is particularly interesting against the background of the presence of a spin–orbit split surface state on the Au(111) surface37 that we propose as a potential mediator of chiral interaction, in contrast to short range forces that drive chiral recognition at close distance.38 A surface-state mediated interaction would oscillate with a periodicity of half the Fermi wavelength (λF = 3.76/3.27 nm)39 and would decay with the distance as 1/r2 (see ref. 34). Indeed, correlations are much smaller in the second ensemble that stretches over about 15 nm (see Fig. 4c). A quantitative analysis of this interaction would require a simulation of the scattering patterns of even and odd states and a much larger number of molecular ensembles, which is beyond the scope of this work.
: M2odd = 86:14 when the first molecule M1 is in an odd state. A ratio of M2even:M2odd = 80:20 is found when the first molecule is in an even state. We verified this effect for two different ensembles of five molecules adsorbed on fcc domains and analyzed the correlations of odd and even states of neighboring molecules. Correlations are significant for the first ensemble that stretches over about 7 nm (distance between the leftmost molecule M1 and the rightmost molecule M5 in Fig. 4b) The table below the image gives the corresponding Pearson correlation coefficients33 which are defined as covariance of the two variables (here: molecule A being in an odd/even state, molecule B being in an odd/even state) normalized by the standard deviations of the two variables. A positive (negative) correlation is indicated by a positive (negative) number (0 ≤ |r| ≤ 1) and no correlation is indicated by a value close to zero. We found both positive and negative correlations. For example, M1 and M2 show a negative correlation: M1even: M1odd = 20:80 when M2 is in an even state and M1even: M1odd = 78:22 when M2 is in an odd state. A positive correlation is found for M4 and M5: M4even: M4odd = 99:1 when M5 is in an even state, and M4even: M4odd = 13:86 when M5 is in an odd state. This intermolecular coupling also affects the population of the possible states of the whole ensemble. Fig. 4b shows the most likely configuration of the molecular states (M13;M26;M31;M42;M56) that is observed in 25/1370 scans (an equal population of all 65 = 7776 possible configurations would lead to an average of 0.18/1370). When only the difference between even and odd states is considered, the most probable configuration (M1odd;M2even;M3odd;M4even;M5even) is found in 572/1370 scans and its inverse (M1even;M2odd;M3even;M4odd;M5odd), which equally follows the preferred intermolecular coupling in another 132 scans. This means that in more than 50% of the scans, the configuration of the foot structure of all five molecules agrees with the sign of the correlations given in the table. In the case of M1 and M3, odd states are even more likely than even states, which is in contrast to the population statistics of all isolated para molecules adsorbed on fcc areas. Apparently, here the intermolecular coupling is stronger than the influence of the surface stacking. The presence of both positive and negative correlations excludes a simple dipolar interaction as the driving force. Furthermore, the surprisingly large change is unlikely to be carried by steric effects as the molecular centers are separated by more than 1.5 nm and the para molecules radially extend less than 0.5 nm. A coupling with oscillatory behavior induced by the surface state of noble metals has been observed for adatoms and small molecules.34,35 Also the π-systems of larger molecules were found to interact with the surface state electrons.36 In our STM images, the switching between even and odd states of the molecules appears as a rotation of the triangular foot structure by 60°. Therefore, it is fair to assume that this switching also affects the surface state that then mediates the interaction between neighboring molecules. While we cannot infer the chirality of a specific molecule from our STM images, we suppose that the intramolecular transition from odd to even states goes along with a change in chirality. This is particularly interesting against the background of the presence of a spin–orbit split surface state on the Au(111) surface37 that we propose as a potential mediator of chiral interaction, in contrast to short range forces that drive chiral recognition at close distance.38 A surface-state mediated interaction would oscillate with a periodicity of half the Fermi wavelength (λF = 3.76/3.27 nm)39 and would decay with the distance as 1/r2 (see ref. 34). Indeed, correlations are much smaller in the second ensemble that stretches over about 15 nm (see Fig. 4c). A quantitative analysis of this interaction would require a simulation of the scattering patterns of even and odd states and a much larger number of molecular ensembles, which is beyond the scope of this work.
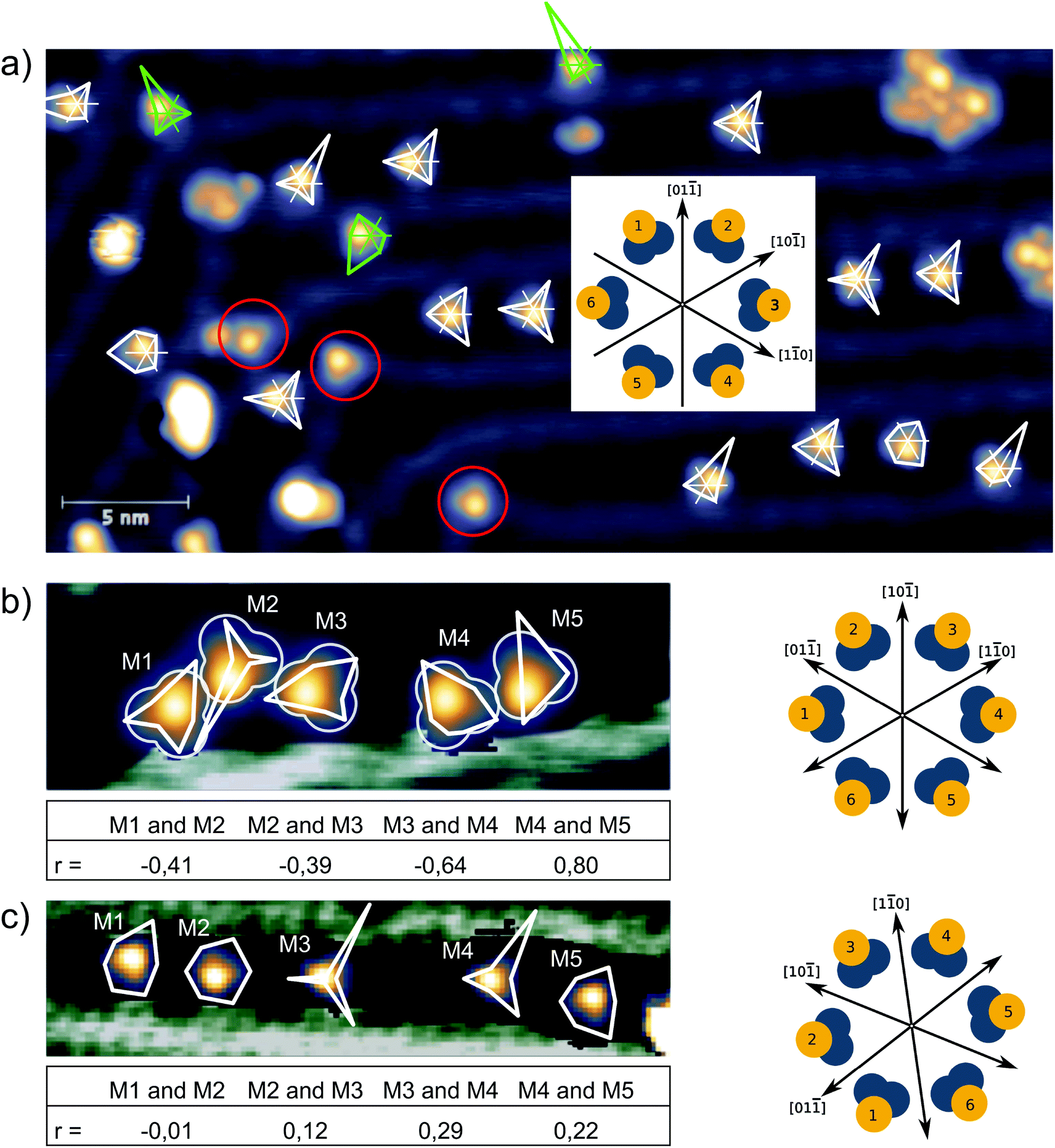 | ||
| Fig. 4 (a) The population of the six different states of 17 individual para molecules is analyzed in a series of 420 scans at I = 50 pA and U = 120 mV (imaging) alternating with 700 mV (switching), similar to Fig. 3a and b. The population of the six states is represented by polar graphs (white polygons on fcc areas and green polygons on hcp areas) right at the corresponding molecule with the definition of the different states defined in the inset. Three molecules located on the ridges of the reconstruction did not show any switching (red circles). The image is rotated by 60° compared to Fig. 2. (b), (c) Population in two different ensembles of five molecules with the corresponding Pearson correlation coefficient r given in the table below. (b) 1370 scans at U = 200 mV and I = 13 pA with scans at 800 mV in between two images. The width of the image is 12 nm. (c) 1000 scans at U = 200 mV and I = 50 pA, scans at 800 mV in between two images. The width of the image is 20 nm. | ||
These experiments show that the populations of rotational states of the foot structures of neighboring molecules are coupled and underline the influence of the local environment of a molecule on its switching behavior.40 Furthermore, these results strengthen our hypothesis of a thermal population of the different states, because a tip-induced switching direction would not lead to correlated behavior of neighboring molecules.
In the following, we further analyze the switching process. The residence times of the molecule in each of the states are plotted in Fig. 5a and show an exponential distribution which characterizes independent events that happen at a constant average rate. From the fit, the mean lifetime of a state and its inverse, the transition rate, can be determined. This transition rate was then determined in a new experiment as a function of the tunneling current with the bias voltage set at 0.4 V. This dependency is shown in Fig. 5b in a semi logarithmic plot. The transition rate increases with the increasing current. A fit to a power law would give a surprisingly low exponent of 0.25. An inelastic single electron process, however, would be characterized by an exponent close to 1 and higher order processes like excitation of higher vibrational modes (heating) would show an even higher exponent (see ref. 41 and 42). Therefore, we exclude an inelastic excitation as the mechanism. The increasing transition rate with the current, that is with decreasing distance between tip and sample, is instead in agreement with a mechanism related to the electric field in the junction. The voltage dependence of the transition rate is shown in Fig. 5c as a single current time trace. Obviously, at low voltages no switching takes place and higher voltages of both positive and negative polarity lead to higher transition rates. This voltage dependence of the switching rate is further studied at different currents (see Fig. 5d). We chose a logistic growth function multiplied by a linear term to reproduce the observed onset of the switching rate and the further increase for even higher voltages. In summary, although the exact mechanism remains elusive, our data speaks against a current induced switching process, but agrees better with an electric-field induced switching. In order to study a possible directionality in the switching direction, we recorded the sense of rotation of the switching by making use of a built-in function of the Nanonis scanning electronics named atom tracking. In doing so, the tip oscillates laterally above the molecule following the position of maximum apparent height, which allows to track the rotational-like switching of a single molecule over several days. Fig. 5e shows the smoothed position of the tip following the switching of a single molecule over several days. Obviously the tip lags behind the molecule. The obvious chirality of this curve is purely due to the tracking mechanism and is not related to the molecule itself. We believe that this oscillatory behavior of the tip in the atom tracking mode is responsible for the slightly preferred direction of rotation which we found in switching series of more than 10000 events, because the preferred direction was identical for different molecules and different polarities of the applied bias. As the here described molecules are not chiral before adsorption on the surface, both chiralities, which could impose a preferred sense of rotation for a specific molecule, must be equally present and thus both rotation directions must be found. If the preferred direction of rotation was related to the tunneling electrons, we would expect a reversed direction of the tunneling current to result in a reversed directionality of the rotational switching.
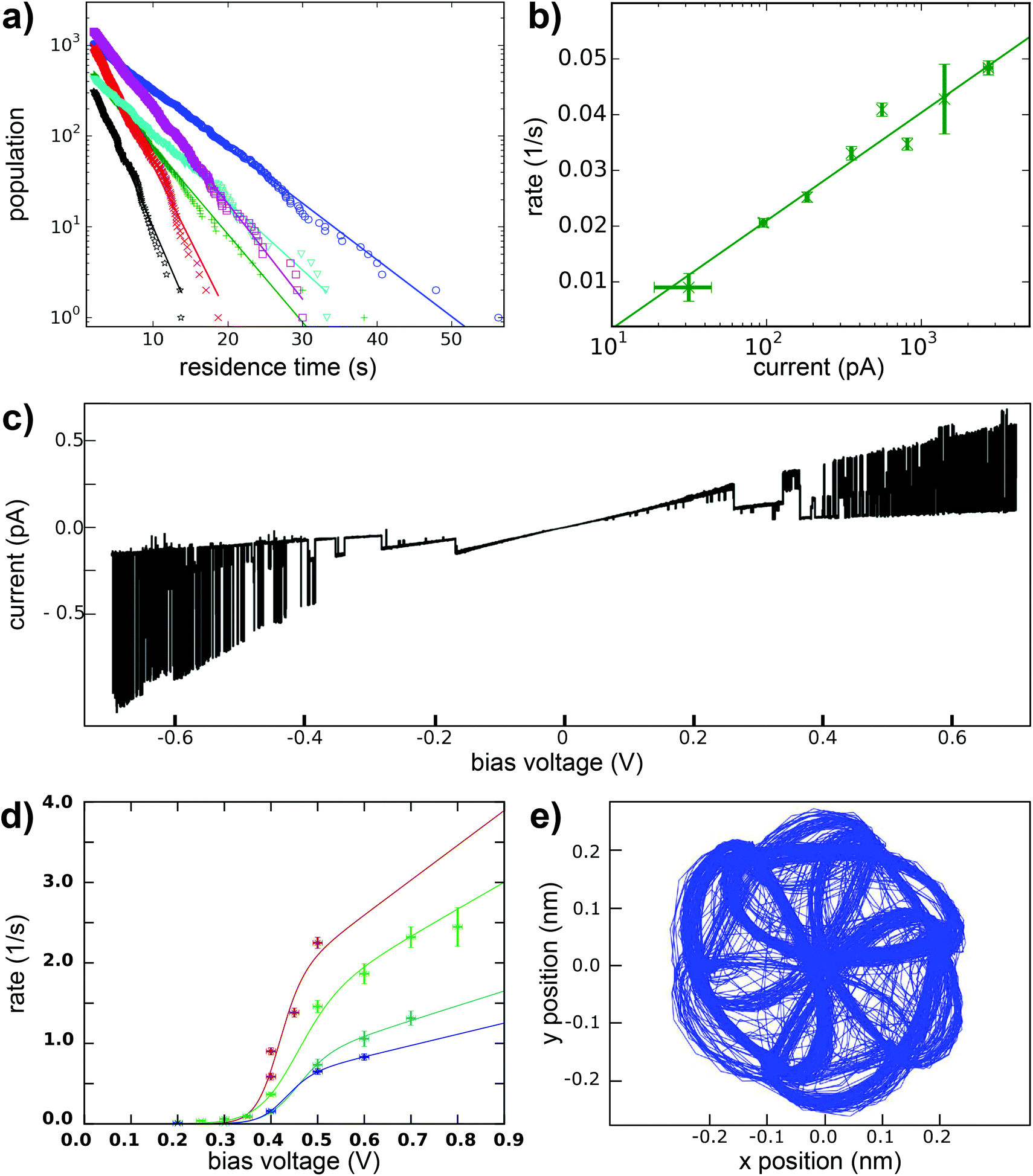 | ||
| Fig. 5 (a) Distribution of the measured residence times of the six states of a para molecule during the statistic switching at a voltage of 380 mV and a set point current of 40 pA. (b) Statistic switching as a function of the setpoint current at a voltage of 400 mV. (c) Current time trace as a function of the applied bias voltage at fixed z position. (d) Switching rate as a function of the applied voltage. (e) Tracking curve of the STM tip following the molecular head group. | ||
3. Conclusion
In summary, we have shown that tetraphenylmethane-based molecules are potential candidates for carrier platforms because they adsorb in a well-defined configuration with three sulfur groups that are strongly bonded to the Au(111) surface. We found that in the well-isolated single molecules six metastable conformational isomers exist in the adsorbed state. With the STM tip, we were able to induce a reversible switching between six rotational states, which allows to assign a rotation direction to the switching. These two characteristics of a molecular revolver are prerequisites for further functionalization as molecular motors. The population of the different states, which we relate to their energies via Boltzmann statistics, was found to depend on the atomic configuration of the substrate and the configuration of the neighboring molecules, even at distances clearly larger than the molecules diameter. This emphasizes the crucial role that is played by the local chemical and physical environment of a single molecule adsorbed on a surface. With this, our work shows that in order to fully exploit the advantages of the precision in chemical synthesis, the coupling to the contacting electrodes has to be defined with the same precision.4. Methods
STM measurements were carried out in a home-built STM in UHV at a temperature of 5.2 K. The STM tip was prepared by chemical etching of a tungsten wire and by repeated dipping into the gold surface. The Au(111) single-crystal surface was cleaned by several cycles of Ar+ sputtering and annealing to 700 K. The voltage is applied to the sample.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge financial support by the Helmholtz Research Program STN (“Science and Technology of Nanosystems”) and DFG Grants (MA 2605/6-1) and (GE 2989/2-1). T. F. acknowledges support from DFG Grant (WU349/13-1) and the Landesstiftung Baden-Württemberg. Y. N. acknowledges support from the Poitou-Charentes regional fellowship. M. M. acknowledges support by the 111 project (90002-18011002).References
- D. Kühne, F. Klappenberger, W. Krenner, S. Klyatskaya, M. Ruben and J. V. Barth, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21332 CrossRef PubMed.
- V. Iancu and S.-W. Hla, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13718 CrossRef CAS PubMed.
- W. Auwärter, K. Seufert, F. Bischoff, D. Ecija, S. Vijayaraghavan, S. Joshi, F. Klappenberger, N. Samudrala and J. V. Barth, Nat. Nanotechnol., 2012, 7, 41 CrossRef.
- J. Otsuki, Y. Komatsu, D. Kobayashi, M. Asakawa and K. Miyake, J. Am. Chem. Soc., 2010, 132, 6870 CrossRef CAS PubMed.
- Y. Zhang, H. Kersell, R. Stefak, J. Echeverria, V. Iancu, U. G. E. Perera, Y. Li, A. Deshpande, K.-F. Braun, C. Joachim, G. Rapenne and S.-W. Hla, Nat. Nanotechnol., 2016, 11, 706 CrossRef CAS PubMed.
- S. Godlewski, H. Kawai, M. Kolmer, R. Zuzak, A. M. Echavarren, C. Joachim, M. Szymonski and M. Saeys, ACS Nano, 2016, 10, 8499 CrossRef CAS PubMed.
- U. G. E. Perera, F. Ample, H. Kersell, Y. Zhang, G. Vives, J. Echeverria, M. Grisolia, G. Rapenne, C. Joachim and S.-W. Hla, Nat. Nanotechnol., 2013, 8, 46 CrossRef CAS PubMed.
- S. Y. Quek, M. Kamenetska, M. L. Steigerwald, H. J. Choi, S. G. Louie, M. S. Hybertsen, J. B. Neaton and L. Venkataraman, Nat. Nanotechnol., 2009, 4, 230 CrossRef CAS PubMed.
- M. Kiguchi, T. Ohto, S. Fujii, K. Sugiyasu, S. Nakajima, M. Takeuchi and H. Nakamura, J. Am. Chem. Soc., 2014, 136, 7327 CrossRef CAS PubMed.
- X. H. Qiu, G. V. Nazin and W. Ho, Phys. Rev. Lett., 2004, 93, 196806 CrossRef CAS PubMed.
- M. Alemani, M. V. Peters, S. Hecht, K.-H. Rieder, F. Moresco and L. Grill, J. Am. Chem. Soc., 2006, 128, 14446 CrossRef CAS PubMed.
- J. Wirth, N. Hatter, R. Drost, T. R. Umbach, S. Barja, M. Zastrow, K. Rück-Braun, J. I. Pascual, P. Saalfrank and K. J. Franke, J. Phys. Chem. C, 2015, 119, 4874 CrossRef CAS.
- L. Gerhard, K. Edelmann, J. Homberg, M. Valášek, S. G. Bahoosh, M. Lukas, F. Pauly, M. Mayor and W. Wulfhekel, Nat. Commun., 2017, 8, 14672 CrossRef PubMed.
- M. J. Comstock, N. Levy, A. Kirakosian, J. Cho, F. Lauterwasser, J. H. Harvey, D. A. Strubbe, J. M. J. Fréchet, D. Trauner, S. G. Louie and M. F. Crommie, Phys. Rev. Lett., 2007, 99, 038301 CrossRef PubMed.
- W. R. Browne and B. L. Feringa, Annu. Rev. Phys. Chem., 2009, 60, 407 CrossRef CAS.
- Y. Kim, T. J. Hellmuth, D. Sysoiev, F. Pauly, T. Pietsch, J. Wolf, A. Erbe, T. Huhn, U. Groth, U. E. Steiner and E. Scheer, Nano Lett., 2012, 12, 3736 CrossRef CAS.
- A. S. Blum, J. G. Kushmerick, D. P. Long, C. H. Patterson, J. C. Yang, J. C. Henderson, Y. Yao, J. M. Tour, R. Shashidhar and B. R. Ratna, Nat. Mater., 2005, 4, 167 CrossRef CAS PubMed.
- S. W. Wu, N. Ogawa, G. V. Nazin and W. Ho, J. Phys. Chem. C, 2008, 112, 5241 CrossRef CAS.
- P. Liljeroth, J. Repp and G. Meyer, Science, 2007, 317, 1203 CrossRef CAS PubMed.
- N. Henningsen, K. J. Franke, I. F. Torrente, G. Schulze, B. Priewisch, K. Rck-Braun, J. Doki, T. Klamroth, P. Saalfrank and J. I. Pascual, J. Phys. Chem. C, 2007, 111, 14843 CrossRef CAS.
- M. Valášek, M. Lindner and M. Mayor, Beilstein J. Nanotechnol., 2016, 7, 374 CrossRef PubMed.
- M. Valášek and M. Mayor, Chem. – Eur. J., 2017, 23, 13538 CrossRef PubMed.
- T. Kitagawa, Y. Idomoto, H. Matsubara, D. Hobara, T. Kakiuchi, T. Okazaki and K. Komatsu, J. Org. Chem., 2006, 71, 1362 CrossRef CAS PubMed.
- Y. Ie, T. Hirose, H. Nakamura, M. Kiguchi, N. Takagi, M. Kawai and Y. Aso, J. Am. Chem. Soc., 2011, 133, 3014 CrossRef CAS PubMed.
- S.-E. Zhu, Y.-M. Kuang, F. Geng, J.-Z. Zhu, C.-Z. Wang, Y.-J. Yu, Y. Luo, Y. Xiao, K.-Q. Liu, Q.-S. Meng, L. Zhang, S. Jiang, Y. Zhang, G.-W. Wang, Z.-C. Dong and J. G. Hou, J. Am. Chem. Soc., 2013, 135, 15794 CrossRef CAS PubMed.
- M. Valášek, K. Edelmann, L. Gerhard, O. Fuhr, M. Lukas and M. Mayor, J. Org. Chem., 2014, 79, 7342 CrossRef PubMed.
- N. Hauptmann, L. Groß, K. Buchmann, K. Scheil, C. Schütt, F. L. Otte, R. Herges, C. Herrmann and R. Berndt, New J. Phys., 2015, 17, 013012 CrossRef CAS.
- M. Lindner, M. Valášek, J. Homberg, K. Edelmann, L. Gerhard, W. Wulfhekel, O. Fuhr, T. Wächter, M. Zharnikov, V. Kolivoška, L. Pospíšil, G. Mészáros, M. Hromadová and M. Mayor, Chem. – Eur. J., 2016, 22, 13218 CrossRef CAS PubMed.
- M. Lindner, M. Valášek, M. Mayor, T. Frauhammer, W. Wulfhekel and L. Gerhard, Angew. Chem., Int. Ed., 2017, 56, 8290 CrossRef CAS PubMed.
- B. Fischer, H. Brune, J. V. Barth, A. Fricke and K. Kern, Phys. Rev. Lett., 1999, 82, 1732 CrossRef CAS.
- J. V. Barth, H. Brune, G. Ertl and R. J. Behm, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42, 9307 CrossRef CAS.
- V. Repain, J. M. Berroir, S. Rousset and J. Lecoeur, Appl. Surf. Sci., 2000, 162–163, 30 CrossRef CAS.
- K. Pearson, Proc. R. Soc. London, 1895, 58, 240 CrossRef.
- J. Repp, F. Moresco, G. Meyer, K.-H. Rieder, P. Hyldgaard and M. Persson, Phys. Rev. Lett., 2000, 85, 2981 CrossRef CAS PubMed.
- M. Kulawik, H.-P. Rust, M. Heyde, N. Nilius, B. A. Mantooth, P. S. Weiss and H.-J. Freund, Surf. Sci., 2005, 590, L253 CrossRef CAS.
- L. Gross, F. Moresco, L. Savio, A. Gourdon, C. Joachim and K.-H. Rieder, Phys. Rev. Lett., 2004, 93, 056103 CrossRef PubMed.
- S. LaShell, B. A. McDougall and E. Jensen, Phys. Rev. Lett., 1996, 77, 3419 CrossRef CAS PubMed.
- M. Lingenfelder, G. Tomba, G. Costantini, L. Colombi Ciacchi, A. De Vita and K. Kern, Angew. Chem., Int. Ed., 2007, 46, 4492 CrossRef CAS PubMed.
- F. Reinert, G. Nicolay, S. Schmidt, D. Ehm and S. Hüfner, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 115415 CrossRef.
- T. Kumagai, F. Hanke, S. Gawinkowski, J. Sharp, K. Kotsis, J. Waluk, M. Persson and L. Grill, Nat. Chem., 2014, 6, 41 CrossRef CAS PubMed.
- F. Elste, G. Weick, C. Timm and F. von Oppen, Appl. Phys. A, 2008, 93, 345 CrossRef CAS.
- T. Miyamachi, M. Gruber, V. Davesne, M. Bowen, S. Boukari, L. Joly, F. Scheurer, G. Rogez, T. K. Yamada, P. Ohresser, E. Beaurepaire and W. Wulfhekel, Nat. Commun., 2012, 3, 938 CrossRef PubMed.
Footnote |
| † Former affiliations: Laboratoire de Mécanique des Solides, UMR 7649 CNRS – Ecole polytechnique – Mines ParisTech, Route de Saclay, 91128 Palaiseau, France; Service de Physique de l'Etat Condensé UMR 3680 CNRS CEA, Bâtiment 772, Orme des merisiers, CEA Saclay 91191 Gif sur Yvette Cedex, France; Institut Pprime UPR 3346 CNRS – Université de Poitiers – ISAE ENSMA, Téléport 2, 11 boulevard Marie et Pierre Curie, 96962 Futuroscope Chasseneuil Cedex, France. |
| This journal is © The Royal Society of Chemistry 2019 |