
Improved oxygen evolution activity of IrO2 by in situ engineering of an ultra-small Ir sphere shell utilizing a pulsed laser†
Wenwu
Zhong‡
a,
Zhiping
Lin‡
*a,
Shangshen
Feng
a,
Da
Wang
b,
Shijie
Shen
 a,
Qinghua
Zhang
b,
Lin
Gu
b,
Zongpeng
Wang
*a and
Baizeng
Fang
*c
a,
Qinghua
Zhang
b,
Lin
Gu
b,
Zongpeng
Wang
*a and
Baizeng
Fang
*c
aSchool of Pharmaceutical and Materials Engineering, School of Advanced Study, Taizhou University, Taizhou, Zhejiang 318000, China. E-mail: zplin@tzc.edu.cn; wangzp@tzc.edu.cn
bInstitution of Physics, Chinese Academy of Sciences, Beijing, 100190, China
cDepartment of Chemical & Biological Engineering, University of British Columbia, 2360 East Mall, Vancouver, B.C. V6T 1Z3, Canada. E-mail: bfang@chbe.ubc.ca
First published on 11th February 2019
Abstract
Noble metal-based catalysts are vital electrocatalysts for the oxygen evolution reaction (OER), which is a half reaction among multiple renewable energy-related reactions. To fully exploit their potential as efficient OER catalysts, we developed a fast one-step strategy to engineer a unique nanostructure for the benchmark catalyst IrO2 utilizing an ultra-fast pulse laser, through which a shell of ultra-small Ir spheres with a diameter of ca. 2 nm is in situ engineered around the IrO2 core. The creation of the Ir sphere shell not only increases the electrochemical surface area, but also improves the electrical conductivity of the electrocatalyst. The as-engineered IrO2@Ir architecture exhibits extremely high electrocatalytic activity towards the OER, revealing an overpotential of 255 mV at 10 mA cm−2 and Tafel slope of 45 mV dec−1. These values are much lower than those observed for the unmodified structure. Furthermore, the catalytic performance is the best among all the noble metal-based OER catalysts. This work may open a new avenue to efficiently improve the catalytic activity of noble metal-based catalysts and significantly advance the development in the energy industry.
1. Introduction
The rapid depletion of fossil fuels and severe environmental problems resulting from the green house effect are pressing issues faced by human society.1 To upgrade the energy sectors and mitigate global warming, sustainable energy sources are being explored and developed, such as solar light, wind and wave power. Electrochemistry, which can reversibly convert electrical energy into chemicals, plays a central role in energy conversion, storage and transportation after electrical energy is harvested from these renewable energy sources.2,3 The oxygen evolution reaction (OER) is a fundamental process among several vital electrochemical reactions in the energy industry, including water splitting,3,4 CO2 reduction5 and metal–air batteries.6 Therefore, considerable efforts have been dedicated to elucidating the mechanism and details of the OER. The other half reaction of water splitting is the hydrogen evolution reaction (HER), which involves only two electron transfer steps;7 in contrast, the OER is a much more complex process involving four electron transfer steps with the breaking of strong O–H bonds and generation of O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds.8 As a result, the sluggish reaction kinetics and high overpotential of the OER have greatly hindered the technological progress of OER-related electrochemical industries.
O bonds.8 As a result, the sluggish reaction kinetics and high overpotential of the OER have greatly hindered the technological progress of OER-related electrochemical industries.
To reduce the overpotential and improve the practical effectiveness, efficient electrochemical catalysts for the OER have flourished over the past decades. Transition metal oxides,9 phosphides,10 sulphides and selenides11 have been proposed as cost-effective earth-abundant catalysts for the OER. Hydroxides are also efficient OER catalysts.12 More recently, bimetal alloys,13 bimetal phosphides14 and sulphides15 were developed with considerably lower overpotentials. Additionally, the influences of catalyst support and orientations on the OER performance have been explored.16–18
Other than the search for new active species, a general method to reduce the overpotential is to increase the content of the active sites. Hence, various morphologies of catalysts have been prepared and characterized.19 Besides the above mentioned earth-abundant catalysts, a more expensive but more stable and efficient class of catalysts for the OER is the noble metal based catalysts.20 Commercially used Ir and Ru-based catalysts provide state-of-the-art comprehensive performance for the OER process and are considered to be benchmarks for the evaluation of others.21 Several studies have focused on improving the activity of noble metal catalysts.16,18,22 However, the potential of noble metal-based OER catalysts has not been fully exploited.
In this study, we report a considerable improvement in the electrochemical activity of IrO2 for the OER in an alkaline environment. The ultra-fast pulse laser technology is introduced to modify the IrO2 precursor. A shell composed of ultra-small Ir spheres with a diameter of ca. 2.5 nm is engineered in situ on the surface of IrO2 particles due to the extremely rapid nanosecond-scale heating and cooling. Benefiting from the unique IrO2 core-Ir shell nanostructure, the IrO2@Ir architecture reveals a considerably low overpotential (i.e., 255 mV at 10 mA cm−2) and Tafel slope (i.e., 45 mV dec−1), which are much lower than the state-of-the-art IrO2 catalyst. The improvement in the catalytic activity can be mainly attributed to two aspects: first, being an efficient OER catalyst itself, the generation of ultra-small Ir spheres largely increases the active specific surface area by creating additional active sites. Second, the electrons in the Ir shell exhibit higher mobility than that in the IrO2 core, providing the core–shell catalyst with better electrical conductivity after the modification. Hence, this fast one-step approach for engineering the unique core–shell nanostructure to improve the electrocatalytic activity of IrO2 for the OER is believed to have a great potential to advance the technological developments in the energy industry.
2. Experimental
2.1 Pulse laser treatment
The as-purchased IrO2 powder (Alfa, Rutile structure, 99%) was adopted after ball-milling. A commercial Nd:YAG pulsed laser (Continuum PRII-8000, pulse duration = 8 ns, frequency = 10 Hz) was then used for the one-step treatment of IrO2 powder to generate the IrO2-Ir core–shell nanostructure. Typically, IrO2 powder was spread uniformly on a substrate as a square with a side length of 1 cm. Then the square sample was irradiated perpendicularly by the pulsed laser in a scanning manner. The experimental setup is illustrated in Fig. S1 (ESI†). The spot diameter of the laser is 6 mm, about half the side length of the square sample; thus, it involves three movements of the mobile stage in a single scanning progression. Five scans overall were performed for the preparation of the sample, with a redistribution of the IrO2 powder between each scan to homogenize the radiation on all the powder. The total duration of laser irradiation was 60 seconds with an optical power of 1 W. The pulsed laser treatment was performed under an ambient atmosphere and at room temperature.2.2 Characterizations
Scanning electron microscopy (SEM) and Energy Dispersive X-ray (EDX) spectra were measured by Hitachi S-4800. Transmission electron microscopy (TEM) and selected area electron diffraction (SAED) were performed with a TESCAN VEGA3 (TESCAN, Czech). High resolution transmission electron microscopy (HRTEM) was conducted on a scanning transmission electron microscope (STEM) coupled with a spherical aberration corrector, JEOL JEM-ARF2100F. ICP elemental analysis was conducted on Optima 8300DV. Powder X-ray diffraction (PXRD) data were collected on Bruker-D8 Advance with Cu Kα radiation and a graphite monochromator. X-ray photoelectron spectroscopy (XPS) analysis was conducted on a Thermo Fisher Scientific ESCALAB 250 Xi using an Al Kα radiation source (hν = 1486.6 eV). Raman spectra measurements were carried out on a Thermo DXR Raman Microscope using a 532 nm laser.2.3 Electrochemical measurements
All electrochemical measurements were conducted using an electrochemical workstation (CHI 660E, Chenhua) under a conventional three-electrode framework at a temperature of 25 ± 0.5 °C. The sample was loaded onto a glassy carbon electrode (GCE, 3 mm in diameter) as the working electrode. An Ag/AgCl electrode was chosen as the reference electrode and a graphite rod as the counter electrode. The catalyst solution was prepared by mixing 5 mg of catalyst in a solution containing 750 μL of deionized water, 250 μL of isopropyl alcohol, and 50 μL of 5 wt% Nafion solution, followed by sonication for 0.5 h. Then, 5 μL of the well-dispersed catalyst solution was dropped on the surface of the GCE and dried naturally in air. The OER performance was measured in O2-saturated 1 M KOH solution with a scan rate of 5 mV s−1. All potentials were corrected with iR to that of a reversible hydrogen electrode (RHE). The Tafel slopes were calculated from the corresponding linear polarization curves. Cyclic voltammetry (CV) measurements were recorded from 0.85 to 0.95 V versus RHE at scan rates ranging from 20 to 200 mV s−1 for the determination of the double-layer capacitances of the samples. Electrochemical impedance spectroscopy (EIS) measurements were carried out from 100 kHz to 0.1 Hz under a static potential of 1.5 V. The time dependence of potential was measured at a constant current density of 10 mA cm−2 for 10 h.2.4 SAED simulation method
SAED simulations were performed using a commercial software package, Single Crystal.3. Results and discussion
3.1 Engineering of IrO2@Ir core–shell structure
The formation process of the ultra-small Ir sphere shell outside the IrO2 core by pulsed laser treatment can be divided into two steps, as illustrated in Fig. 1. First, when illuminated by a pulsed laser (the setup is shown in Fig. S1, ESI†), IrO2 powder absorbs the energy emitted by the laser and is heated. The powder then melts and decomposes into Ir and O atoms. Note that the pulse is so transient that the melting and decomposing only occur on the very shallow surface of IrO2. Although IrO2 is a metal oxide, chemically it is a conductor.23 Therefore, the heating effect of the pulsed laser on IrO2 involves three processes: light energy harvested by free electrons, energy transportation through electron collisions, and heat transfer by electron–phonon interactions.24 Second, after the illumination is removed, the molten surface cools down rapidly due to the precipitous temperature gradient between the surface and its vicinity. During the cooling down step, active –O– atoms gather together to form OO bonds and then escape as O2. On the contrary, because the cooling is extremely rapid, the Ir atoms aggregate and deposit as tremendously ultra-small spheres on the surface of IrO2. The Ir spheres are so dense that they stack and form a shell outside the IrO2 core. In general, the pulsed laser treatment causes a thin layer of IrO2 to decompose near the surface of the IrO2 sample and re-deposit as Ir spheres within a time scale of several nanoseconds. This presumption is based on physical logics and evidenced by the surface characterizations of the as-prepared product, as discussed below.
 | ||
| Fig. 1 Schematic illustration of the in situ engineering of the Ir shell outside the IrO2 core. | ||
3.2 Formation mechanism of IrO2@Ir core–shell structure
To explore the formation mechanism of the IrO2 core-Ir shell nanostructure, IrO2 powder without laser treatment was also characterized. Henceforth, IrO2 samples before and after the laser treatment are denoted as b-IrO2 and a-IrO2, respectively. To examine the morphology of the samples, SEM analyses were performed, and representative SEM images of b-IrO2 and a-IrO2 are shown in Fig. S2 (ESI†). Both of the samples show a spherical morphology with a typical size of ∼50 nm. As expected, the SEM images do not exhibit a significant difference, considering the Ir shell is only several nanometers thick. However, the fragment or crump-like features are founded as traces of the melting process in the a-IrO2. Additionally, the O atom drain is evidenced by the EDX analysis of the atomic ratio of Ir to O (Table S1 and Fig. S2C, ESI†), which shows an increased atomic ratio of Ir to O after the pulsed laser treatment because some O atoms were lost as O2. We further confirmed the content of Ir in the b-IrO2 and a-IrO2 samples by ICP analysis. The results are summarized in Table S2† and are in good accordance with the results acquired from EDX. The content of Ir in the b-IrO2 is significantly less than that in the a-IrO2 sample.The b-IrO2 and a-IrO2 samples were further characterized by TEM and STEM analyses. From Fig. 2A, one can clearly see the core–shell structures. The thickness of the shell is ca. 2.4 nm, which is in good agreement with the diameter statistics of the ultra-small Ir spheres shown in Fig. S3 (ESI†). Fig. 2B was acquired on a STEM, from which a specific scenario can be seen: a cluster of small spheres (with diameters lower than 5 nm) are distributed uniformly on the surface of a much larger sphere (with a diameter over 20 nm). Fig. 2A and B confirm the core–shell structure from different profiles. A comparison of the TEM images of b-IrO2 and a-IrO2 is also shown in Fig. S4 and S5 (ESI†). Clearly, no shell was observed in the b-IrO2.
 | ||
Fig. 2 (A, B) Morphology of a-IrO2. The image shown in A was acquired on a traditional TEM, while that in B was acquired on a STEM with an aberration corrector; (C, D) HRSTEM images of the a-IrO2 and Ir spheres, respectively. The green cross markers in C partly indicate the positions where the Ir spheres are distributed. The inset in C illustrates the crystal structure of IrO2 viewed from the [010] direction, where the larger cyan spheres represent Ir atoms while the smaller pink spheres represent O atoms. The d spacing of 2.493 Å in C corresponds to the spacing of the (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) face of IrO2. The green dashed circles in D mark the Ir spheres. The green and red dots in the circles are Ir crystals viewed from the [01] and [01] directions, respectively. The d spacing of 2.197 Å in D corresponds to the spacing of the (111) plane of Ir; (E) SAED image of the a-IrO2. The red circles are the simulation results with electron beam incident from the [11] zone axis; (F) Fast Fourier Transform (FFT) of the HRTEM of Ir sphere, where the red circles are the simulated results with electron beam incident from the [01] zone axis. ) face of IrO2. The green dashed circles in D mark the Ir spheres. The green and red dots in the circles are Ir crystals viewed from the [01] and [01] directions, respectively. The d spacing of 2.197 Å in D corresponds to the spacing of the (111) plane of Ir; (E) SAED image of the a-IrO2. The red circles are the simulation results with electron beam incident from the [11] zone axis; (F) Fast Fourier Transform (FFT) of the HRTEM of Ir sphere, where the red circles are the simulated results with electron beam incident from the [01] zone axis. | ||
Fig. 2C shows an HRSTEM image taken near the edge of a large a-IrO2 sphere. The lattice arrangement coincides well with the IrO2 lattice viewed from the [010] direction, as shown in the inset. The d spacing of 2.493 Å marked in Fig. 2C agrees with the spacing of the (10) plane of IrO2 (2.586 Å). Therefore, the core is composed of IrO2. Fig. 2C also exhibits circular shadows, some of which are marked by green crosses at the center. An enlarged image is shown in Fig. S6 (ESI†) with reduced brightness to make the shadows more visible. We believe that the shadows were formed by the distribution of small Ir spheres. In Fig. 2D, a HRSTEM image of some smaller spheres scattered at the edge of a larger a-IrO2 sphere is displayed. The smaller spheres are surrounded by green circle markers. The green and red dots in circle “1” are the Ir lattice viewed from the [01] and [01] directions, respectively. We can that the image matches the HRSTEM image perfectly. Additionally, the d spacing of 2.197 Å in Fig. 2D agrees well with the spacing of the (11) plane of Ir (i.e., 2.217 Å). Therefore, the smaller spheres that constitute the shell are proven to be Ir. Further, a SAED image of a-IrO2 is shown in Fig. 2E, in which the spots perfectly match the simulation results (red circles) from the electron beam incidence from zone axis [11] of a-IrO2. The SAED image of Ir, however, is extremely hard to obtain due to the ultra-small size. Instead, Fast Fourier Transform (FFT) of the HRTEM of the Ir spheres (i.e., Fig. 2D) was performed, and the profile is shown in Fig. 2F, also in good agreement with the simulation results from the electron beam incidence from zone axis [01] of Ir. Other indexed planes of IrO2 and Ir are also shown in Fig. 2E and F, respectively. The crystal structure parameters of the a-IrO2 and Ir are listed in Table S3 (ESI†). It is also worthy to note that twin crystals are observed for the Ir spheres, as shown in circles “1” and “2” in Fig. 2D. The ultra-small size and the presence of the twin crystal boundaries reflect that the crystallization of Ir was a non-equilibrium process, implying that the cooling down step was extremely fast after the removal of laser.
In addition to electron microscopy, various spectral analyses were used for characterizing the samples. PXRD analyses were performed to investigate the chemical compositions of b-IrO2 and a-IrO2, and the XRD patterns are shown in Fig. 3A. For b-IrO2, the XRD pattern shows characteristic peaks of pure IrO2 (JCPDS #15-0870). For a-IrO2, besides the set of diffraction peaks from IrO2, additional diffraction peaks of Ir (JCPDS #46-1044) were also clearly observed. This result further proves that Ir was produced after the pulsed laser treatment. The Raman spectra of b-IrO2 and a-IrO2 (Fig. S7†) are exactly the same because pure metals do not exhibit well-defined Raman signals. This phenomenon is due to the weak interaction between the photons of the laser and the phonons in the metal lattices, which is mainly caused by the Coulomb screening effect and large wave vector mismatch. Fig. 3B shows the high-resolution XPS spectra of Ir 4f and O 1s in the IrO2 samples, and the XPS survey spectra for the IrO2 samples are shown in Fig. S8 (ESI†). The binding energy scale is referenced to the C 1s signal at 284.8 eV. The high-resolution XPS spectrum of Ir4+ 4f can be fitted with two doublets, as reported previously,18,25 which likely correspond to the screened and unscreened components of an Ir4+ state. Two dominant peaks located at 62.2 and 65.1 eV are ascribed to Ir4+ 4f7/2 and Ir4+ 4f5/2, respectively. For the a-IrO2, two additional peaks at a lower binding energy compared to b-IrO2 are clearly observed, suggesting a higher electron density at the Ir site. Additionally, the metallic Ir exhibits binding energies of 60.8 eV and 63.7 eV, corresponding to Ir0 4f7/2 and Ir0 4f5/2, respectively. Both of the binding energies of the metallic Ir reveal a 1.4 eV core-level shift compared to that of Ir4+.26 Similar results are observed for the O 1s XPS line of the IrO2 before and after the laser treatment. The position of the main peak of the doublet is located at 530.5 eV, corresponding to the O 1s signal. The additional broader feature with higher binding energy is considered to be the result of oxygen adsorbed on the surface.
 | ||
| Fig. 3 (A) XRD patterns of the b-IrO2 and a-IrO2, and (B) High resolution Ir 4f and O 1s XPS spectra of b-IrO2 and a-IrO2. | ||
3.3 Catalytic activity of the as-engineered IrO2@Ir
To evaluate the catalytic activity of the as-prepared a-IrO2 for the OER, we performed electrochemical measurements with the commercially used b-IrO2 as the baseline. In the measurements, the samples were drop-casted on GCE with a constant catalyst loading of ca. 0.35 ± 0.035 mg cm−2. The electrochemical OER tests were conducted in 1 M aqueous KOH solution. From the iR-corrected polarization curves (Fig. 4A), it is evident that b-IrO2 exhibits limited performance towards the OER, requiring an overpotential (η) of 353 mV (vs. RHE) to achieve a geometric current density of 10 mA cm−2 (j10). The η value (i.e., 353 mV) is very close to that (i.e., 320 mV) reported previously by other researchers.27 In contrast, a-IrO2 can successfully achieve j10 at an η of 255 mV (vs. RHE). The extrapolation of the linear region of η vs. log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) j (Fig. 4B) reveals Tafel slopes of 79 and 45 mV dec−1 for b-IrO2 and a-IrO2, respectively. The Tafel slope of 79 mV dec−1 observed for b-IrO2 is also in good agreement with that (i.e., 76 mV dec−1) reported previously for commercial IrO2.27 The key electrochemical performances of IrO2 before and after the laser treatment are also summarized in Fig. S9 (ESI†). One can easily conclude that the overpotential and Tafel slope are considerably improved after the pulsed laser modification. A comparison of the OER performance of the a-IrO2 and b-IrO2 electrocatalysts in alkaline solution with other representative OER catalysts reported in the literature is given in Table S4.† It is evident that the catalytic performance of a-IrO2 is state-of-the-art among noble metal-based OER catalysts and shows first-rate performance among all of the OER catalysts reported so far.28
j (Fig. 4B) reveals Tafel slopes of 79 and 45 mV dec−1 for b-IrO2 and a-IrO2, respectively. The Tafel slope of 79 mV dec−1 observed for b-IrO2 is also in good agreement with that (i.e., 76 mV dec−1) reported previously for commercial IrO2.27 The key electrochemical performances of IrO2 before and after the laser treatment are also summarized in Fig. S9 (ESI†). One can easily conclude that the overpotential and Tafel slope are considerably improved after the pulsed laser modification. A comparison of the OER performance of the a-IrO2 and b-IrO2 electrocatalysts in alkaline solution with other representative OER catalysts reported in the literature is given in Table S4.† It is evident that the catalytic performance of a-IrO2 is state-of-the-art among noble metal-based OER catalysts and shows first-rate performance among all of the OER catalysts reported so far.28
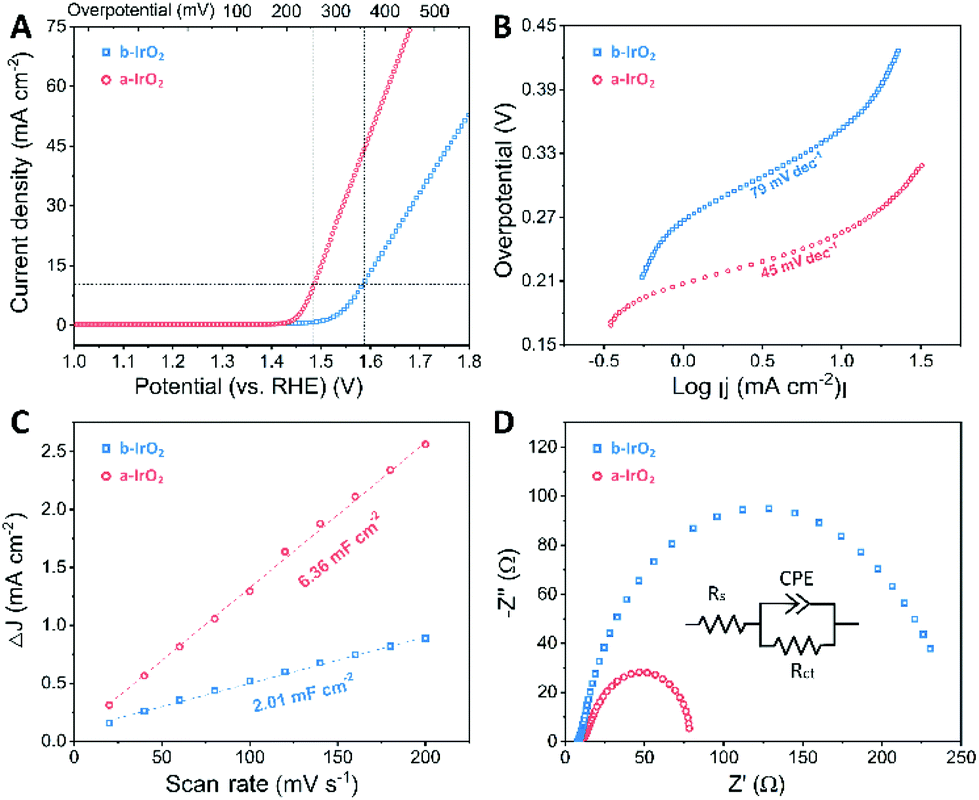 | ||
| Fig. 4 Electrochemical measurements of b-IrO2 and a-IrO2: (A) iR-corrected linear sweep polarization curves measured in 1 M KOH, (B) the corresponding Tafel slopes, (C) the derived double layer capacitance (Cdl), and (D) electrochemical impedance diagrams. | ||
To explore the underlying fundamentals of the improved electrochemical activity, the electrochemical surface area (ECSA) was estimated by double layer capacitance (Cdl), which was extracted from cyclic voltammetry cycles in the non-faradaic region between 0.85 and 0.95 V (vs. RHE) (Fig. S10, ESI†). Fig. 4C shows that the Cdl is 2.01 and 6.36 mF cm−2 for b-IrO2 and a-IrO2, respectively, indicating a much larger ECSA after exposure to the pulsed laser. The 3-fold larger ECSA can be ascribed to the generation of the ultra-small Ir spheres. Note that Ir is also an efficient OER catalyst; thus, the nanocrystallization of Ir considerably increases the physical specific area and the content of the active sites, resulting in a greatly enhanced ECSA. We further analysed the TOF (turnover frequency) and normalized the LSV curves by the ECSA (the details about the derivation are shown in the ESI†). The TOF of a-IrO2 is calculated to be 0.026 s−1 per active site at an overpotential of 270 mV, whereas the TOF value of b-IrO2 is only 0.0018 s−1 per active site. The normalized LSV curves (Fig. S11†) show that even after normalization by the ECSA, a-IrO2 displays a comparable intrinsic activity to b-IrO2. To better understand the electrode kinetics during the OER, electrochemical impedance spectroscopy (EIS) was measured (Fig. 4D). Both samples exhibit one capacitive semicircle, indicating that the OER process can be characterized by an equivalent circuit with a time constant. The diameter of the semicircle of a-IrO2 is much smaller than that of b-IrO2, suggesting a considerably smaller charge transfer resistance (Rct). This difference stems from the formation of the Ir shell with significantly increased electrical conductivity. The Nyquist plots were then fitted using an equivalent circuit, as shown in the inset of Fig. 4D. Rct was extracted from the fitting, with values of 70.08 and 238.8 Ω for b-IrO2 and a-IrO2, respectively. The Rct of a-IrO2 is less than one third of that of b-IrO2, suggesting an ultrafast faradaic process, which results in the superior OER kinetics. Some of the key electrochemical data obtained for IrO2 before and after the laser modification are summarized in Table S5 (ESI†).
Electrochemical stability is another important characteristic of OER catalysts, which was evaluated by chronopotentiometry measurements conducted at a constant current density of 10 mA cm−2. The time dependence of the overpotential observed for the as-engineered IrO2@Ir catalyst is shown in Fig. S12 (ESI†), exhibiting a comparable durability for a-IrO2 and commercial IrO2 (i.e., b-IrO2). In addition, according to the XRD pattern (Fig. S13†), XPS spectrum (Fig. S14†) and HRTEM images (Fig. S15†) of a-IrO2 after the stability test, it was found that the Ir shell still existed with minimal damage, implying a good stability of the as-developed electrocatalyst.
4. Conclusions
In summary, an innovative synthesis strategy, pulsed laser surface modification, was developed to create novel OER catalysts with a unique nanostructure and excellent electrocatalytic activity. This strategy involves only one-step pulsed laser treatment to in situ engineer a shell of ultra-small Ir spheres around the IrO2 core. When examined as an electrocatalyst for the OER, the as-engineered IrO2 core-Ir shell catalyst demonstrates an overpotential of 255 mV at 10 mA cm−2 and Tafel slope of 45 mV dec−1, both considerably lower than those of the commercially used IrO2. The introduction of the ultra-fine Ir spheres contributes to the improved OER activity in two main ways: the increased ECSA due to the enlarged physical specific area, and the reduced charge transfer resistance resulting from the enhanced electrical conductivity. This study provides a new avenue to improve the catalytic activity of noble metal-based catalysts and favors the technological progress in the energy industry.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the National Natural Science Foundation of China (No. 51572183) and the Natural Science Foundation of Zhejiang Province, China (No. LY15E010002).References
- J. Goldemberg, Science, 2007, 315, 808 CrossRef CAS PubMed; S. Chu and A. Majumdar, Nature, 2012, 488, 294 CrossRef PubMed; V. R. Stamenkovic, D. Strmcnik, P. P. Lopes and N. M. Markovic, Nat. Mater., 2016, 16, 57 CrossRef PubMed.
- I. Katsounaros, S. Cherevko, A. R. Zeradjanin and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2014, 53, 102 CrossRef CAS PubMed; D. Larcher and J.-M. Tarascon, Nat. Chem., 2015, 7, 19 CrossRef PubMed; B. E. Logan and K. Rabaey, Science, 2012, 337, 686 CrossRef PubMed; D. Kundu, E. Talaie, V. Duffort and L. F. Nazar, Angew. Chem., Int. Ed., 2015, 54, 3431 CrossRef PubMed; Y. Wang, N. Zhou, B. Fang, H. Li, X. Bi and H. Wang, Chem. Rev., 2015, 115, 3433 CrossRef PubMed; B. Fang, J. Kim, M. Kim and J. Yu, Acc. Chem. Res., 2013, 46, 1397 CrossRef PubMed; J. Wang, W. Cui, Q. Liu, Z. Xing, A. M. Asiri and X. Sun, Adv. Mater., 2016, 28, 215 CrossRef PubMed; I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef; A. Eftekhari and B. Fang, Int. J. Hydrogen Energy, 2017, 42, 25143 CrossRef; Z. Lin, C. Wang, Z. Wang, Q. Liu, C. Le, B. Lin and S. Chen, Electrochim. Acta, 2019, 294, 142 CrossRef.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. B. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, 4998 CrossRef PubMed.
- P. Tian, Y. Yu, X. Yin and X. Wang, Nanoscale, 2018, 10, 5054 RSC; Z. Liu, H. Yu, B. Dong, X. Yu and L. Feng, Nanoscale, 2018, 10, 16911 RSC; Y. Wang, B. Fang, X. Wang, A. Ignaszak, Y. Liu, A. Li, L. Zhang and J. Zhang, Prog. Mater. Sci., 2018, 98, 108 CrossRef CAS; R. Dangol, Z. Dai, A. Chaturvedi, Y. Zheng, Y. Zhang, K. Dinh, B. Li, Y. Zong and Q. Yan, Nanoscale, 2018, 10, 4890 RSC; T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520 RSC; M. Gong and H. Dai, Nano Res., 2015, 8, 23 CrossRef; C. Zhu, Z. Yin, W. Lai, Y. Sun, L. Liu, X. Zhang, Y. Chen and S. Chou, Adv. Energy Mater., 2018, 8, 1802327 CrossRef; Y. Wang, Y. Sun, F. Yan, C. Zhu, P. Gao, X. Zhang and Y. Chen, J. Mater. Chem. A, 2018, 6, 8479 RSC.
- F. Lyu, Y. Bai, Z. Li, W. Xu, Q. Wang, J. Mao, L. Wang, X. Zhang and Y. Yin, Adv. Funct. Mater., 2017, 27, 1702324 CrossRef CAS; J. P. Jones, G. K. S. Prakash and G. A. Olah, Isr. J. Chem., 2014, 54, 1451 CrossRef.
- E. McCalla, A. M. Abakumov, M. Saubanère, D. Foix, E. J. Berg, G. Rousse, M.-L. Doublet, D. Gonbeau, P. Novák and G. Van Tendeloo, Science, 2015, 350, 1516 CrossRef CAS PubMed; M. Sathiya, G. Rousse, K. Ramesha, C. P. Laisa, H. Vezin, M. T. Sougrati, M.-L. Doublet, D. Foix, D. Gonbeau and W. Walker, Nat. Mater., 2013, 12, 827 CrossRef PubMed; K. Yao, M. Risch, S. Sayed, Y. Lee, J. Harding, A. Grimaud, N. Pour, Z. Xu, J. Zhou, A. Mansour, F. Barde and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 2417 RSC; E. H. Yu, X. Wang, U. Krewer, L. Li and K. Scott, Energy Environ. Sci., 2012, 5, 5668 RSC.
- D. Wang, D. Zhang, C. Tang, P. Zhou, Z. Wu and B. Fang, Catal. Sci. Technol., 2016, 6, 1952 RSC; Z. Wu, C. Tang, P. Zhou, Z. Liu, Y. Xu, D. Wang and B. Fang, J. Mater. Chem. A, 2015, 3, 13050 RSC; Z. Wu, B. Fang, Z. Wang, C. Wang, Z. Liu, F. Liu, W. Wang, D. Wang and D. Wilkinson, ACS Catal., 2013, 3, 2101 CrossRef CAS; Z. Wu, B. Fang, A. Bonakdarpour, A. Sun, D. Wilkinson and D. Wang, Appl. Catal., B, 2012, 125, 59 CrossRef.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y.-L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457 CrossRef CAS PubMed; P. Gono, J. Wiktor, F. Ambrosio and A. Pasquarello, ACS Catal., 2018, 8, 5847 CrossRef.
- Z. Lu, H. Wang, D. Kong, K. Yan, P.-C. Hsu, G. Zheng, H. Yao, Z. Liang, X. Sun and Y. Cui, Nat. Commun., 2014, 5, 4345 CrossRef CAS PubMed; L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, J. Am. Chem. Soc., 2012, 134, 17253 CrossRef PubMed; W. T. Hong, M. Risch, K. A. Stoerzinger, A. Grimaud, J. Suntivich and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 1404 RSC; L. Han, S. Dong and E. Wang, Adv. Mater., 2016, 28, 9266 CrossRef PubMed; R. D. L. Smith, M. S. Prévot, R. D. Fagan, S. Trudel and C. P. Berlinguette, J. Am. Chem. Soc., 2013, 135, 11580 CrossRef PubMed; M. Fekete, R. Hocking, S. Chang, C. Italiano, A. Patti, F. Arena and L. Spiccia, Energy Environ. Sci., 2013, 6, 2222 RSC; A. Bergmann, I. Zaharieva, H. Dau and P. Strasser, Energy Environ. Sci., 2013, 6, 2745 RSC; M. Risch, F. Ringleb, M. Kohlhoff, P. Bogdanoff, P. Chernev, I. Zaharieva and H. Dau, Energy Environ. Sci., 2015, 8, 661 RSC; F. Yan, C. Zhu, S. Wang, Y. Zhao, X. Zhang, C. Li and Y. Chen, J. Mater. Chem. A, 2016, 4, 6048 RSC.
- B. You, N. Jiang, M. Sheng, M. W. Bhushan and Y. Sun, ACS Catal., 2015, 6, 714 CrossRef CAS; M. Ledendecker, S. Krick Calderón, C. Papp, H. P. Steinrück, M. Antonietti and M. Shalom, Angew. Chem., Int. Ed., 2015, 54, 12361 CrossRef PubMed; X. Fang, L. Jiao, R. Zhang and H.-L. Jiang, ACS Appl. Mater. Interfaces, 2017, 9, 23852 CrossRef PubMed; A. Dutta and N. Pradhan, J. Phys. Chem. Lett., 2016, 8, 144 CrossRef PubMed; X. Yu, Y. Feng, B. Guan, X. W. D. Lou and U. Paik, Energy Environ. Sci., 2016, 9, 1246 RSC; X. Yu, Y. Feng, B. Guan, X. Lou and U. Paik, Energy Environ. Sci., 2016, 9, 1246 RSC; Z. Yin, C. Zhu, C. Li, S. Zhang, X. Zhang and Y. Chen, Nanoscale, 2016, 8, 19129 RSC; X. Yu, S. Zhang, C. Li, C. Zhu, Y. Chen, P. Gao, L. Qi and X. Zhang, Nanoscale, 2016, 8, 10902 RSC.
- L. Liang, H. Cheng, F. Lei, J. Han, S. Gao, C. Wang, Y. Sun, S. Qamar, S. Wei and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 12004 CrossRef CAS PubMed; J. Zhang, Y. Hu, D. Liu, Y. Yu and B. Zhang, Adv. Sci., 2017, 4, 1600343 CrossRef PubMed; L.-L. Feng, G. Yu, Y. Wu, G.-D. Li, H. Li, Y. Sun, T. Asefa, W. Chen and X. Zou, J. Am. Chem. Soc., 2015, 137, 14023 CrossRef PubMed; M.-R. Gao, Y.-F. Xu, J. Jiang, Y.-R. Zheng and S.-H. Yu, J. Am. Chem. Soc., 2012, 134, 2930 CrossRef PubMed; R. Xu, R. Wu, Y. Shi, J. Zhang and B. Zhang, Nano Energy, 2016, 24, 103 CrossRef; C. Tang, N. Cheng, Z. Pu, W. Xing and X. Sun, Angew. Chem., Int. Ed., 2015, 127, 9483 CrossRef; Y. Wang, Y. Sun, F. Yan, C. Zhu, P. Gao, X. Zhang and Y. Chen, J. Mater. Chem. A, 2018, 6, 8479 RSC.
- D. McAteer, I. J. Godwin, Z. Ling, A. Harvey, L. He, C. S. Boland, V. Vega-Mayoral, B. Szydłowska, A. A. Rovetta and C. Backes, Adv. Energy Mater., 2018, 8, 1702965 CrossRef CAS; Y. Rao, Y. Wang, H. Ning, P. Li and M. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 33601 CrossRef PubMed; J. Chi, H. Yu, B. Qin, L. Fu, J. Jia, B. Yi and Z. Shao, ACS Appl. Mater. Interfaces, 2017, 9, 464 CrossRef PubMed; F. Song, K. Schenk and X. Hu, Energy Environ. Sci., 2016, 9, 473 RSC.
- S. Dresp, F. Luo, R. Schmack, S. Kühl, M. Gliech and P. Strasser, Energy Environ. Sci., 2016, 9, 2020 RSC; M. Wang, J. Jiang and L. Ai, ACS Sustainable Chem. Eng., 2018, 6, 6117 CrossRef CAS; S. Fu, J. Song, C. Zhu, G.-L. Xu, K. Amine, C. Sun, X. Li, M. H. Engelhard, D. Du and Y. Lin, Nano Energy, 2018, 44, 319 CrossRef.
- M. Qian, S. Cui, D. Jiang, L. Zhang and P. Du, Adv. Mater., 2017, 29, 1704075 CrossRef CAS PubMed; J. Song, C. Zhu, B. Z. Xu, S. Fu, M. H. Engelhard, R. Ye, D. Du, S. P. Beckman and Y. Lin, Adv. Energy Mater., 2017, 7, 1601555 CrossRef; W. Li, X. Gao, X. Wang, D. Xiong, P.-P. Huang, W.-G. Song, X. Bao and L. Liu, J. Power Sources, 2016, 330, 156 CrossRef.
- B. Dong, X. Zhao, G.-Q. Han, X. Li, X. Shang, Y.-R. Liu, W.-H. Hu, Y.-M. Chai, H. Zhao and C.-G. Liu, J. Mater. Chem. A, 2016, 4, 13499 RSC; J. Yin, Y. Li, F. Lv, M. Lu, K. Sun, W. Wang, L. Wang, F. Cheng, Y. Li and P. Xi, Adv. Mater., 2017, 29, 1704681 CrossRef CAS PubMed.
- A. T. Marshall and R. G. Haverkamp, Electrochim. Acta, 2010, 55, 1978 CrossRef CAS.
- H. N. Nong, H. S. Oh, T. Reier, E. Willinger, M. G. Willinger, V. Petkov, D. Teschner and P. Strasser, Angew. Chem., Int. Ed., 2015, 54, 2975 CrossRef CAS PubMed; H.-S. Oh, H. N. Nong, T. Reier, A. Bergmann, M. Gliech, J. Ferreira de Araújo, E. Willinger, R. Schlögl, D. Teschner and P. Strasser, J. Am. Chem. Soc., 2016, 138, 12552 CrossRef PubMed; K. A. Stoerzinger, L. Qiao, M. D. Biegalski and Y. Shao-Horn, J. Phys. Chem. Lett., 2014, 5, 1636 CrossRef PubMed; G. Gardner, J. Al-Sharab, N. Danilovic, Y. B. Go, K. Ayers, M. Greenblatt and G. Charles Dismukes, Energy Environ. Sci., 2016, 9, 184 RSC.
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang and J. K. Norskov, Science, 2016, 353, 1011 CrossRef CAS PubMed.
- C. Zhao, Y. E and L. Fan, Microchim. Acta, 2012, 178, 107 CrossRef CAS; L.-Y. Jiang, X.-X. Lin, A.-J. Wang, J. Yuan, J.-J. Feng and X.-S. Li, Electrochim. Acta, 2017, 225, 525 CrossRef; E. Antolini, ACS Catal., 2014, 4, 1426 CrossRef; Y. Li, P. Hasin and Y. Wu, Adv. Mater., 2010, 22, 1926 CrossRef PubMed.
- J. Du, Z. Zou, C. Liu and C. Xu, Nanoscale, 2018, 10, 5163 RSC; Y. Li, L. Hu, W. Zheng, X. Peng, M. Liu, P. K. Chu and L. Y. S. Lee, Nano Energy, 2018, 52, 360 CrossRef CAS.
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765 CrossRef CAS; C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977 CrossRef PubMed.
- N. Danilovic, R. Subbaraman, K. C. Chang, S. H. Chang, Y. Kang, J. Snyder, A. P. Paulikas, D. Strmcnik, Y. T. Kim and D. Myers, Angew. Chem., Int. Ed., 2014, 53, 14016 CrossRef CAS PubMed; T. D. Nguyen, G. G. Scherer and Z. J. Xu, Electrocatalysis, 2016, 7, 420 CrossRef; Z. Jian, P. Liu, F. Li, P. He, X. Guo, M. Chen and H. Zhou, Angew. Chem., Int. Ed., 2014, 53, 442 CrossRef PubMed; P. Lettenmeier, L. Wang, U. Golla-Schindler, P. Gazdzicki, N. A. Canas, M. Handl, R. Hiesgen, S. S. Hosseiny, A. S. Gago and K. A. Friedrich, Angew. Chem., Int. Ed., 2016, 55, 742 CrossRef PubMed; D. Y. Kuo, J. K. Kawasaki, J. N. Nelson, J. Kloppenburg, G. Hautier, K. M. Shen, D. G. Schlom and J. Suntivich, J. Am. Chem. Soc., 2017, 139, 3473 CrossRef PubMed; Z. Ma, Y. Zhang, S. Liu, W. Xu, L. Wu, Y.-C. Hsieh, P. Liu, Y. Zhu, K. Sasaki, J. N. Renner, K. E. Ayers, R. R. Adzic and J. X. Wang, J. Electroanal. Chem., 2018, 819, 296 CrossRef.
- J. Xu, T. Jarlborg and A. Freeman, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 7939 CrossRef CAS.
- T. Q. Qiu and C. L. Tien, J. Heat Transfer, 1993, 115, 835 CrossRef CAS.
- J. M. Kahk, C. G. Poll, F. E. Oropeza, J. M. Ablett, D. Céolin, J. P. Rueff, S. Agrestini, Y. Utsumi, K. D. Tsuei, Y. F. Liao, F. Borgatti, G. Panaccione, A. Regoutz, R. G. Egdell, B. J. Morgan, D. O. Scanlon and D. J. Payne, Phys. Rev. Lett., 2014, 112, 117601 CrossRef CAS PubMed; V. Pfeifer, T. E. Jones, J. J. V. Velez, R. Arrigo, S. Piccinin, M. Haevecker, A. Knop-Gericke and R. Schloegl, Chem. Sci., 2017, 8, 2143 RSC.
- A. K. Shukla, A. M. Kannan, M. S. Hegde and J. Gopalakrishnan, J. Power Sources, 1991, 35, 163 CrossRef CAS.
- M. Chauhan, K. P. Reddy, C. S. Gopinath and S. Deka, ACS Catal., 2017, 7, 5871 CrossRef CAS.
- J. S. Kim, B. Kim, H. Kim and K. Kang, Adv. Energy Mater., 2018, 8, 1702774 CrossRef CAS; Y. Cheng and S. P. Jiang, Prog. Nat. Sci.: Mater. Int., 2015, 25, 545 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nr10163a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |