
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceKinetics, energetics, and size dependence of the transformation from Pt to ordered PtSn intermetallic nanoparticles†
Minda
Chen
 a,
Yong
Han
b,
Tian Wei
Goh
a,
Rong
Sun
ac,
Raghu V.
Maligal-Ganesh
a,
Yuchen
Pei
a,
Chia-Kuang
Tsung
d,
James W.
Evans
*be and
Wenyu
Huang
*ae
a,
Yong
Han
b,
Tian Wei
Goh
a,
Rong
Sun
ac,
Raghu V.
Maligal-Ganesh
a,
Yuchen
Pei
a,
Chia-Kuang
Tsung
d,
James W.
Evans
*be and
Wenyu
Huang
*ae
aDepartment of Chemistry, Iowa State University, Ames, Iowa 50011, USA. E-mail: whuang@iastate.edu
bDepartment of Physics and Astronomy, Iowa State University, Ames, Iowa 50011, USA. E-mail: evans@ameslab.gov
cDepartment of Chemistry, Beijing Normal University, Beijing, China
dDepartment of Chemistry, Boston College, Chestnut Hill, MA 02467, USA
eDivision of Chemical and Biological Sciences, Ames Laboratory, U.S. Department of Energy, Ames, Iowa 50011, USA
First published on 18th February 2019
Abstract
The outstanding catalytic activity and chemical selectivity of intermetallic compounds make them excellent candidates for heterogeneous catalysis. However, the kinetics of their formation at the nanoscale is poorly understood or characterized, and precise control of their size, shape and composition during synthesis remains challenging. Here, using well-defined Pt nanoparticles (5 nm and 14 nm) encapsulated in mesoporous silica, we study the transformation kinetics from monometallic Pt to intermetallic PtSn at different temperatures by a series of time-evolution X-ray diffraction studies. Observations indicate an initial transformation stage mediated by Pt surface-controlled intermixing kinetics, followed by a second stage with distinct transformation kinetics corresponding to a Ginstling–Brounstein (G–B) type bulk diffusion mode. Moreover, the activation barrier for both surface intermixing and diffusion stages is obtained through the development of appropriate kinetic models for the analysis of experimental data. Our density-functional-theory (DFT) calculations provide further insights into the atomistic-level processes and associated energetics underlying surface-controlled intermixing.
Introduction
The importance of intermetallic compounds (IMCs) has been long recognized for their use in magnetic materials,1–3 superconducting materials,4–6 hydrogen storage,7,8 shape-memory materials,9 and heterogeneous catalysis.10–13 IMCs have been reported to show improved selectivity to hydrogenation,14,15 oxidation,12,13 steam reforming,16,17 and hydroformylation18 compared to their monometallic counterparts, providing an effective alternative to the noble metal catalysts used in the industry. In addition, the long-range-ordered nature of IMCs, as opposed to random alloys, provides more precise control of their surface structure and potentially better understanding of structure–property-relationship, and enables easier characterization with tools such as X-ray diffraction.19In heterogeneous catalysis, as well as many other fields, it is generally desirable to synthesize IMC nanoparticles (NPs) with a small size, high mono-dispersity, and a single-phase nature.20,21 However, this remains challenging despite numerous efforts made to engineer the synthesis routes of IMCs.22,23 For example, poor control over the NPs often results in either mixed phases,24,25 heterogeneous surfaces,26 or severe aggregation.12 In addition, the underlying kinetics and thermodynamics of IMC formation at the nanoscale have been rarely discussed, although an in-depth understanding could lead to profound improvements in designing the synthesis routes for these IMC NPs.27 Many methods for synthesizing IMCs involve transformation from monometallic to bimetallic material, including successive impregnation, chemical vapor deposition, metallurgical alloying, and the seeded growth method.10,28 The seeded growth method is especially useful as it often retains the structure, shape and dispersity of the parent monometallic material.14,29,30
Colloidal synthesis of PtSn IMCs has been achieved and is widely used in heterogeneous catalysis. Komatsu et al. used PtSn supported on a zeolite as an effective catalyst for the dehydroisomerization of butane into isobutene.31 Eichhorn et al. developed PtSn as an efficient CO-tolerant electrocatalyst for H2 oxidation.32 Our group previously reported the use of PtSn for the selective hydrogenation of furfural to obtain furfural alcohol.14 Recently, PtSn NPs were also reported to improve pairwise selectivity in the hydrogenation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond for parahydrogen-induced hyperpolarization NMR.33 However, there are no systematic studies yet on the temperature and size dependence of the detailed formation kinetics of PtSn at the nanoscale. Suitably tailored kinetic modeling would enable reliable extraction of key activation barriers, which could ideally be compared with ab initio density-functional-theory (DFT) analysis. We note one recent study34 considered PtSn NP formation for a single size of Pt seed and under temperature ramping, wherein a generic bulk Avrami model was used to fit kinetics. In this work, we report our study on phase transformation from Pt to PtSn IMCs, using 5 and 14 nm Pt NPs that are encapsulated in mesoporous silica (Pt@mSiO2). The mSiO2 shell not only prevents aggregation of the NPs, but also allows uninhibited access to the surrounding chemical environment. Specifically, it allows the addition of the Sn precursor to form encapsulated PtSn IMCs (PtSn@mSiO2). Previous studies extensively investigated the role of mesoporous silica during the formation of intermetallic NPs, in particular revealing that etching of its inner surface by locally concentrated inorganic ions, in this case H+ and Cl−, creates space for uninhibited NP growth.14,35 The Pt to PtSn transformation was performed under various fixed temperatures and monitored by powder X-ray diffraction (PXRD). Analysis of the deconvoluted PXRD patterns suggests that the transformation involves two distinct stages: a surface-controlled intermixing step followed by a Ginstling–Brounstein (G–B) type solid-state diffusion-controlled step. We crafted appropriate kinetic models for these two stages in order to assess the relevant activation barriers. Further DFT analysis relevant to the surface-controlled intermixing stage provides additional insight into the underlying atomistic diffusion processes and the relevant energetics and pathways.
C bond for parahydrogen-induced hyperpolarization NMR.33 However, there are no systematic studies yet on the temperature and size dependence of the detailed formation kinetics of PtSn at the nanoscale. Suitably tailored kinetic modeling would enable reliable extraction of key activation barriers, which could ideally be compared with ab initio density-functional-theory (DFT) analysis. We note one recent study34 considered PtSn NP formation for a single size of Pt seed and under temperature ramping, wherein a generic bulk Avrami model was used to fit kinetics. In this work, we report our study on phase transformation from Pt to PtSn IMCs, using 5 and 14 nm Pt NPs that are encapsulated in mesoporous silica (Pt@mSiO2). The mSiO2 shell not only prevents aggregation of the NPs, but also allows uninhibited access to the surrounding chemical environment. Specifically, it allows the addition of the Sn precursor to form encapsulated PtSn IMCs (PtSn@mSiO2). Previous studies extensively investigated the role of mesoporous silica during the formation of intermetallic NPs, in particular revealing that etching of its inner surface by locally concentrated inorganic ions, in this case H+ and Cl−, creates space for uninhibited NP growth.14,35 The Pt to PtSn transformation was performed under various fixed temperatures and monitored by powder X-ray diffraction (PXRD). Analysis of the deconvoluted PXRD patterns suggests that the transformation involves two distinct stages: a surface-controlled intermixing step followed by a Ginstling–Brounstein (G–B) type solid-state diffusion-controlled step. We crafted appropriate kinetic models for these two stages in order to assess the relevant activation barriers. Further DFT analysis relevant to the surface-controlled intermixing stage provides additional insight into the underlying atomistic diffusion processes and the relevant energetics and pathways.
Methods
Synthesis of 5 nm Pt@mSiO2
In a typical synthesis of 5 nm Pt@mSiO2, 52 mg tetradecyltrimethylammonium bromide (TTAB, ≥99%, Sigma-Aldrich) was added to an aqueous solution (20 mL, 0.750 mM) of H2PtCl6 (Acros Organics, 40% Pt) and sonicated until dissolved. The reaction mixture was heated in an oil bath at 60 °C for 10 min, and a freshly prepared aqueous solution (26 mL, 10 mM) of sodium borohydride (Alfa Aesar, 98%) was then added quickly. After stirring vigorously for 10 min, a dark brown colloidal solution of Pt NPs was observed. To the solution was added an aqueous solution (0.1 mL, 1 M) of sodium hydroxide. Next, 1 mL of 10% tetraethylorthosilicate (TEOS, Aldrich, reagent grade, 98%) solution in ethanol was added dropwise with vigorous stirring. After 6 h, the solution was allowed to cool to room temperature, and 20 mL ethanol was added. The solution was then centrifuged at 8000 rpm, and the coated NPs were collected and dispersed in methanol. The surfactant was removed via an acidic methanol refluxing session (6% hydrochloric acid solution) at 90 °C for 24 h. The platinum content in Pt@mSiO2 was measured using inductively coupled plasma mass spectrometry (ICP-MS).Synthesis of 14 nm Pt@mSiO2
14 nm Pt@mSiO2 is synthesized using a previously reported method58 with a slight modification. In a typical synthesis, an aqueous solution (5 mL, 10 mM) of K2PtCl4 (Acros Organics, 46–47% Pt) was added to an aqueous solution (12.5 mL, 400 mM) of TTAB. Additionally, 29.5 mL of distilled water was added. After stirring for 10 min at room temperature, a cloudy solution was obtained, which was then heated in an oil bath at 50 °C for another 10 min to obtain a clear solution. A freshly prepared ice-cold aqueous solution (3 mL, 500 mM) of sodium borohydride was then added. After the solution was stirred for 15 to 20 h at 50 °C, the resulting dark brown colloidal solution of Pt NPs was allowed to cool to room temperature and centrifuged at 3000 rpm four times for 30 min with the residue discarded after each session. Finally, the supernatant was centrifuged at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 15 min twice, and the solid was collected and dispersed in 35 mL of water. About 1 mL of a 0.05 M sodium hydroxide solution was added to the Pt colloidal solution to obtain a pH between 11 and 12. While stirring, 500 μL of a 10% TEOS in methanol solution was added dropwise. After 24 h, the sample was centrifuged at 12000 rpm twice, and the coated particles (Pt@mSiO2) were dispersed in methanol. The surfactant was removed via an acidic methanol refluxing session (6% hydrochloric acid solution) at 90 °C for 24 h. The platinum content in Pt@mSiO2 was identified using ICP-MS.
000 rpm for 15 min twice, and the solid was collected and dispersed in 35 mL of water. About 1 mL of a 0.05 M sodium hydroxide solution was added to the Pt colloidal solution to obtain a pH between 11 and 12. While stirring, 500 μL of a 10% TEOS in methanol solution was added dropwise. After 24 h, the sample was centrifuged at 12000 rpm twice, and the coated particles (Pt@mSiO2) were dispersed in methanol. The surfactant was removed via an acidic methanol refluxing session (6% hydrochloric acid solution) at 90 °C for 24 h. The platinum content in Pt@mSiO2 was identified using ICP-MS.
Transformation of 5 and 14 nm Pt@mSiO2 to PtSn@mSiO2
In a typical synthesis of PtSn@mSiO2, Pt@mSiO2 containing 20 mg Pt was centrifuged and re-dispersed in 39 mL of tetraethylene glycol (Alfa Aesar, 99%) in a 250 mL two-neck flask. SnCl2·2H2O (Alfa Aesar, 98%) with a molar ratio of Pt:Sn = 1:2 was dissolved in 1 mL tetraethylene glycol in a 5 mL glass vial. The vial was placed inside the 250 mL two-neck flask so that the Pt solution and the SnCl2 solution were separated but can be heated isothermally. After vacuuming the flask and refilling with argon, the flask was heated using a temperature-controlled heating mantle to the designated temperature, and the SnCl2 solution was added quickly into the Pt@mSiO2 dispersion. Samples at various reaction times were removed from the reaction mixture using a syringe, immediately quenched in a cold-water bath, and added to 30 mL acetone. Each sample was then centrifuged, washed with acetone, and dried for characterization.
Characterization
Powder X-ray diffraction (PXRD) patterns were collected at room temperature using a Bruker D8 Advance powder diffractometer with a Cu Kα1 radiation source (40 kV, 40 mA, λ = 1.5406 Å). Transmission electron microscopy (TEM) measurements were carried out using a TECNAI G2 F20 electron microscope at an acceleration voltage of 200 kV. The energy-dispersive X-ray spectroscopy (EDX) compositional profile was taken on a Titan Themis 300 probe corrected TEM with a Super-X EDX detector. ICP-MS measurements were carried out using a Thermo Fisher Scientific X Series 2 ICP-MS. Typically, powdered samples were dissolved in 5 mL of aqua regia to dissolve all metal content before the addition of around 100 to 300 μL 30% HF solution to dissolve the mesoporous silica completely. Liquid samples containing tetraethylene glycol were calcined to burn off the organic component, and the residue was dissolved in 5 mL of aqua regia. All samples were diluted with 2% nitric acid before the ICP-MS measurements.Results and discussion
Characterization
Core–shell Pt@mSiO2 NPs were synthesized with a Pt core size of 13.8 ± 1.4 or 4.8 ± 0.7 nm, as shown by TEM (Fig. 1). Mesoporous silica shells of thickness 14.1 ± 0.8 and 9.1 ± 1.0 nm, respectively, were coated on them to effectively prevent NP aggregation particularly during their subsequent conversion to intermetallic NPs. The diameter of the mesopores in the silica shells is 2.1 and 2.5 nm, respectively, which allows the solution-phase diffusion of the second metal precursor to reach and be reduced at the Pt surface.35 Such a space-confined strategy to prevent sintering is also seen in other literature studies.36–38 TEM images of PtSn NPs after 120 min transformation of 14 and 5 nm Pt NPs to incorporate Sn were taken (Fig. S1 and S2†) and mono-dispersed metal NPs without aggregation were confirmed. 14 nm NPs grew to 18.0 ± 1.5 nm and 5 nm NPs grew to 6.5 ± 0.7 nm due to the incorporation of Sn, and the growth also involved a change in the crystal structure from a face-centered-cubic (fcc) close-packed Pt to NiAs-type PtSn structure. This measured expansion is consistent with the theoretical value calculated from the corresponding change in the lattice constant.14 Sn incorporation is also confirmed by ICP-MS analysis of each powder sample and the solution after removal of NPs (Fig. S3 and S4†), and it is observed that despite the use of extra Sn during the synthesis, the Pt:Sn ratio in the NPs only reaches around 1:1.
 | ||
| Fig. 1 TEM images of (A) 14 and (B) 5 nm Pt@mSiO2. | ||
To monitor the transformation from Pt to the ordered PtSn phase, a time-evolution PXRD study was performed by quenching the reaction mixture at various times during the transformation. Fig. 2 shows the corresponding PXRD pattern of 5 and 14 nm NPs, where the various time periods of the conversion at 250 °C are indicated. PXRD patterns obtained at other temperatures are shown in Fig. S5.† Pt(111), Pt(200), PtSn(102), and PtSn(110) diffraction peaks were observed in the patterns. The Pt phase gradually diminished and PtSn phase grew, as suggested by the change in integrated peak intensity. In addition, the rate of transformation was apparently faster at the beginning of the process and gradually slowed down within the two hours. Despite the existence of other phases in the Pt–Sn phase diagram (Pt3Sn, Pt2Sn3, PtSn2, PtSn4), we did not observe their formation by PXRD. To identify the relative amount of each phase, a standard mixture of Pt and pure-phase PtSn in 1:1 mole ratio was quantified by ICP-MS and its PXRD pattern was deconvoluted into four subpeaks (Fig. 3) within the region of 34 to 52°, and the integrated intensity (A) ratio of the two most intense peaks, Pt(111) and PtSn(102), was calculated to be 1.38. It is then used as the ratio of the response factor (RF) between Pt and the PtSn phase. Standard mixtures of Pt:PtSn = 1:3 and 3:1 are also measured, as shown in Fig. S6,† and the calculated RF ratios match well with the 1:1 case. Subsequently, the amount of Pt:PtSn in each sample was calculated as:
 | (1) |
 | (2) |
 | ||
| Fig. 2 Time-evolution PXRD patterns of (A) 5 and (B) 14 nm Pt transformed to PtSn at 250 °C. | ||
 | ||
| Fig. 3 PXRD pattern of a 1:1 standard mixture of Pt and PtSn and its deconvolution, with the black circles representing PXRD raw data, the red lines representing deconvoluted peaks, the dark yellow line representing a linear baseline, and the dark blue line representing the sum of all deconvoluted peaks. | ||
Fig. 4 shows α versus reaction time t at different temperatures. As observed in Fig. 4, the transformation occurs rapidly at the beginning of the process and gradually slows down for both sizes of Pt NPs at all temperatures. Comparing 14 nm Pt@mSiO2 with 5 nm Pt@mSiO2, the smaller particle initially converts slightly slower at a fixed temperature but ultimately reaches a higher conversion after 120 min. At least the latter is expected given the smaller particle size.
 | ||
| Fig. 4 Conversion α versus time t for (A) 5 and (B) 14 nm Pt/PtSn NPs at 3 different temperatures. | ||
Since our Pt NPs are encapsulated inside mSiO2, we designed two control experiments to explore the effect, if any, of the mSiO2 on the growth of intermetallic PtSn. We first employed 5 nm Pt nanoparticles deposited onto 100 nm silica spheres (noted as Pt/SiO2) following a reported method.36 We observed similar kinetics for the conversion from Pt/SiO2 to PtSn/SiO2 as the encapsulated Pt@mSiO2 (Fig. S7†). A TEM image of the nanoparticles after conversion is shown in Fig. S8.† Next, we also tested the unsupported Pt NPs capped by myristyltrimethylammonium bromide (TTAB), but severe aggregation of the Pt NPs was observed before reaching the desired temperature (Fig. S9†). This emphasizes that the mSiO2 shell is critical to protect the NPs from aggregation. These two control experiments demonstrate that the major function of the mesoporous silica shell is to prevent the aggregation of Pt NPs, and the shell does not affect the transformation of the encapsulated Pt core to intermetallic PtSn.
Modeling of the solid-state transformation kinetics: background
Traditionally, a variety of models have been used to characterize the kinetics of solid-state transformations,37,38 specifically predicting the evolution of the conversion α versus time t. These include so-called nucleation models, fixed reaction-order models, and diffusion models.38 One might anticipate that refinement of these generic models, or potentially new tailored models need to be developed, for appropriate analysis of the formation of PtSn intermetallic NPs. For example, nucleation models based on Avrami kinetics can fit a broad range of observed kinetics by including variable nucleation and growth rates.34,39 However, in addition to assumptions regarding the time dependence of nucleation and growth rates, Avrami kinetics is based on a picture of spatially homogeneous nucleation rates in an extended medium which is not well suited to nanoscale systems.It is however perhaps instructive to perform a benchmark analysis of our data with a simple fixed reaction-order model.38 Such a model for the kinetics assumes the form
 | (3) |
Given the above assessment of at least two distinct stages for the conversion of 14 nm Pt NPs, we have refined our modeling to incorporate this feature. We describe the first stage as a “surface stage” that involves the reduction of the Sn precursor and formation of PtSn starting at the surface of the Pt NPs, anticipating that a complete intermetallic shell around the periphery of the NP has not yet formed. Furthermore, we anticipate that the kinetics will reflect the feature that a portion of the surface of the original Pt NPs remains unconverted which can facilitate reduction and intermixing of Sn. To show that the Pt surface facilitates Sn reduction, we have performed a control experiment where SnCl2 is dissolved in TEG and heated to 280 °C without the existence of Pt, and we observed no reduction of Sn2+ to form metallic Sn. On the other hand, we describe the second stage after which a complete intermetallic shell has formed as a “Ginstling–Brounshtein (G–B) diffusion stage”, and will analyze associated behavior with a suitably adapted G–B type diffusion model.40 A schematic picture of the structural evolution of NPs through these two stages is shown in Fig. 5, where further discussion is provided in the following subsections.
 | ||
| Fig. 5 Schematic structural evolution of NPs during complete conversion to IMC. Data shown in Fig. 4 does not correspond to complete conversion, but such extended data are shown in the ESI (Fig. S16B†). | ||
Kinetics in the “surface-specific intermixing stage”
For the first stage, i.e., the surface stage, it is reasonable to anticipate that the kinetics of both Sn precursor reduction and Sn–Pt intermixing will be sensitive to the fraction of the unconverted Pt NP surface. Certainly, precursor reduction will be enhanced by the unconverted Pt surface, and the intermixing kinetics will differ and plausibly also be enhanced for Sn directly intermixing with Pt rather than intermixing through an intermetallic shell. We specify that the surface stage ends when conversion α reaches a value αsurf < 1, and furthermore assume that the fraction of the unconverted Pt particle surface is roughly proportional to αsurf − α (see Fig. 5). In formulating the kinetics, one could anticipate that multiple Pt surface sites are required to facilitate Sn reduction, and possibly also Sn intermixing, a feature which would correspond to higher-order Langmuirian kinetics. Fig. S10† shows a scheme of the model. The resulting form of the kinetics will depend on whether reduction or intermixing is rate limiting. See section S14 in the ESI† for a more detailed discussion. However, in either case, the potential multiple-site requirement implies higher-order kinetics of the form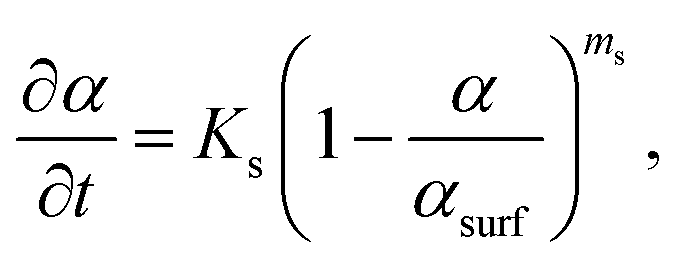 | (4) |
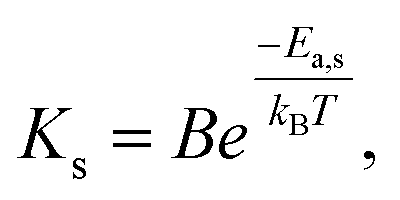 | (5) |
First, we analyze the data for the smaller 5 nm Pt NPs, where it is anticipated that the observed behavior will correspond mainly or entirely to the first surface stage. Least-squares fitting by adjusting all ms, αsurf, and Ks as free parameters produces ms = 1.78, 1.79, and 1.75 with αsurf = 0.79, 0.57, and 0.30 for T = 250, 240, and 230 °C, respectively. The fit for associated activation energy gives Ea,s ≈ 245 kJ mol−1. A key observation indicating the veracity of the model is the similar values of ms obtained for different T, noting that behavior in the surface stage is expected to be described by a single reaction order. From this perspective, it is natural to refit the data by imposing a single value of ms = 1.75, and adjusting just αsurf and Ks as free parameters. This analysis produces αsurf ≈ 0.788, 0.566, and 0.297 for T = 250, 240, and 230 °C, respectively, but preserves Ea,s = 245 kJ mol−1. These fits are shown in Fig. 4A, and the Arrhenius analysis is shown in Fig. 6A.
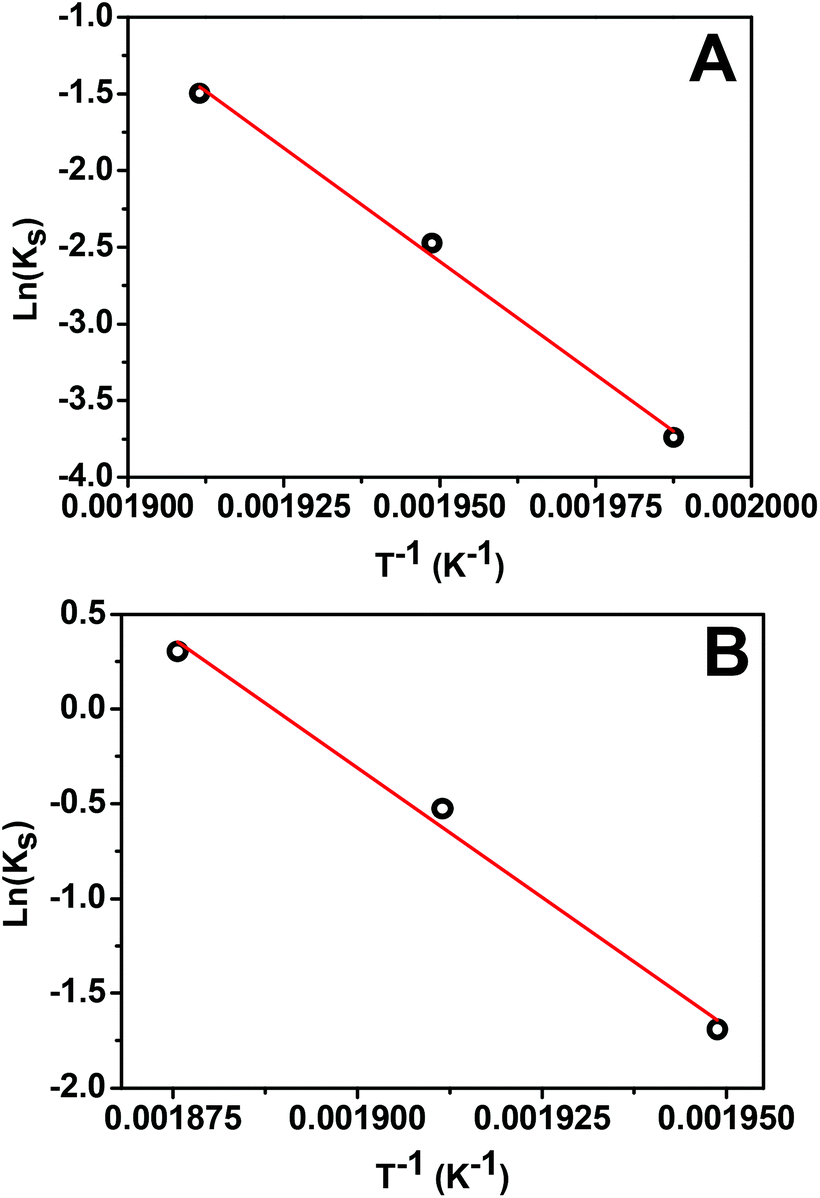 | ||
| Fig. 6 Arrhenius plot for the surface stage of: (A) 5 nm Pt NPs with ms = 1.75; (B) 14 nm Pt NPs with ms = 1.63. | ||
Second, we analyzed behavior in the first surface stage for 14 nm Pt NPs. Here, we first make a reasonable selection of the values of αsurf based on the form of the conversion curves, and then assess the associated reaction order and activation energy. Choosing αsurf = 0.81, 0.58, and 0.51, for T = 260, 250, and 240 °C, respectively, yields ms = 1.64, 1.62, and 1.64. From these choices of αsurf and ms, one obtains Ea,s ≈ 225 kJ mol−1. The similarity of the values of ms for different T, again, supports the veracity of the modeling. Furthermore, just as for the 5 nm Pt NPs, it is natural to reanalyze the data imposing a single value of ms = 1.63 which yields only a negligible modification in the values of αsurf = 0.795, 0.574, and 0.506, for T = 260, 250, and 240 °C, respectively, but preserves Ea,s ≈ 225 kJ mol−1. These fits are shown in Fig. 4B (blue curve), and the corresponding Arrhenius analysis is shown in Fig. 6B. Note that the above fitting is based on only 5, 4, and 4 data points for 260, 250, and 240 °C, respectively.
An additional expectation for a reliable model is that ms for 5 and 14 nm Pt NPs should not be greatly different. Thus, our determination of ms = 1.75 for the 5 nm Pt NPs versus ms = 1.63 for 14 nm Pt NPs seems reasonable. Similarly, one should expect somewhat similar values of Ea,s for the two different particle sizes, a feature which is produced in our modeling: Ea,s ≈ 245 kJ mol−1 for 5 nm Pt NPs versus 225 kJ mol−1 for 14 nm Pt NPs.
However, we should note that, perhaps unexpectedly, the initial rate of conversion (corresponding to Ks) is higher for larger Pt NPs than for smaller NPs (when the comparison is made at the same temperature). We interpret this trend as related to the feature that the larger NPs are much more likely to include grain boundaries. This is confirmed by additional PXRD analysis using the Scherrer equation, which consistently gives a grain size of around 11 nm for the larger Pt NPs, while TEM indicates a particle size of 14 nm. The existence of grain boundaries in the 14 nm Pt@mSiO2 is also confirmed by high resolution TEM (HR-TEM) (Fig. S11†). We anticipate that the diversity of “defect sites” along the grain boundary near the surface of the Pt NPs will facilitate the initiation of conversion to the IMC.41,42 Indeed, our DFT analysis discussed below indicates significant inhibition to the initiation of conversion on perfect Pt(100) facets.
Further characterization of the elemental distribution, especially the location of Sn during the transformation is provided by energy-dispersive X-ray spectroscopy (EDX) analysis. First, an EDX line scan analysis corresponding to the first stage (Fig. 7A) provides no clear indication of a reduced Sn shell surrounding the Pt core. It suggests that intermixing of Sn is more facile than Sn reduction, and therefore reduction would be rate limiting. In this case, Ea,s should reflect an activation barrier for reduction. However, it is possible that a thin shell of Sn has formed around the Pt NPs which is not distinguishable in the EDX analysis. In this scenario, Ea,s could be associated with intermixing. Second, a line scan corresponding to the second stage, as seen in Fig. 7B, indicates that, at 260 °C, a complete Sn shell has formed on the 14 nm NPs with an α value around 0.8. This observation implies that αsurf should not be above this value, which is consistent with the above analysis that αsurf = 0.795 at 260 °C. Additional data are available in the ESI (Fig. S12 and S13†) to show that the EDX line scan results are representative of the sample. The structure of the 14 nm NPs after a 30 min conversion at 260 °C was obtained from HR-TEM (Fig. 7C) and it shows a core of the Pt domain with (200) lattice fringes and the shell of the PtSn domain with (101) lattice fringes, which correspond to the beginning of the G–B type diffusion stage for the 14 nm NPs. This observation is consistent with our schematic picture of the NP evolution shown in Fig. 5.
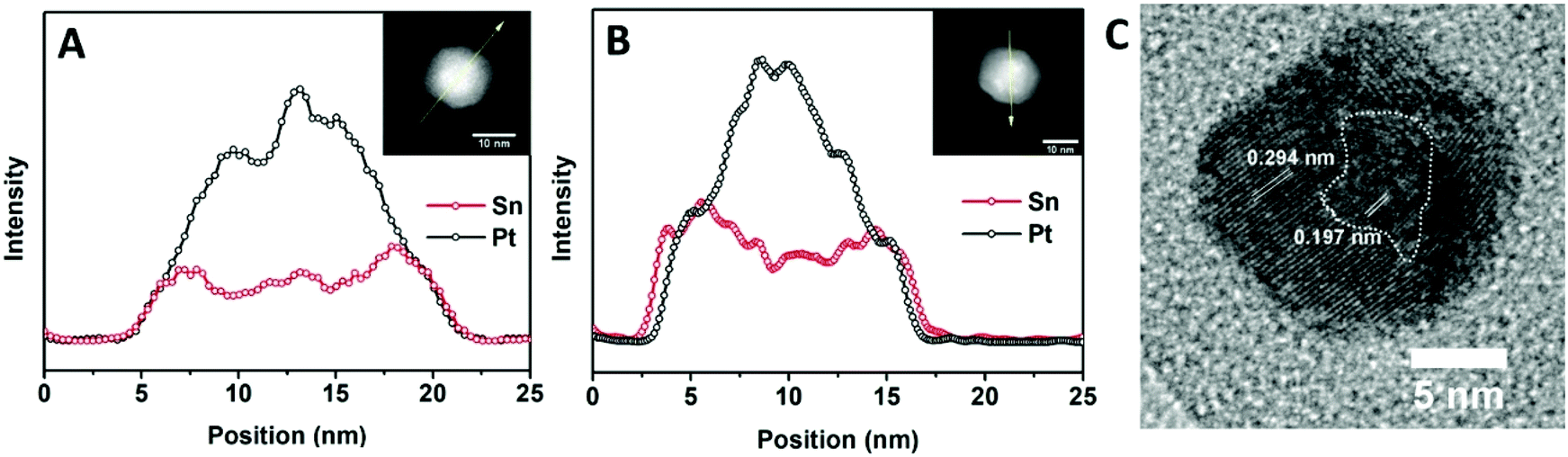 | ||
| Fig. 7 Elemental distribution by EDX line scan analysis along the direction indicated by the yellow arrow on (A) 14 nm NP after 15 min conversion at 240 °C, when α = 0.36 and (B) 14 nm NP after 30 min conversion at 260 °C, when α = 0.80. (C) High-resolution TEM image of a 14 nm NP after 30 min conversion at 260 °C, when α = 0.80. Lattice spacings for Pt(200) and PtSn(101) are indicated. | ||
Transition to and kinetics in the “G–B type diffusion stage”
For 14 nm Pt NPs, the behavior in the second stage has been analyzed within a picture where a complete intermetallic shell has formed and bulk diffusion through this shell dominates the formation of the PtSn phase. Traditional G–B diffusion modeling32 considers a uniform infinitesimally thin complete intermetallic shell being formed at the onset of the reaction, and treats this shell as retaining uniform thickness during growth. The rate of growth of the shell thickness, and thus of the conversion α, is determined by the diffusion flux through the intermetallic shell. For a spherical geometry of a NP, there is an exact analytic solution to the diffusion problem. However, this solution should also provide a reasonable assessment of behavior for other shapes.Naturally, the traditional G–B model does not apply to the surface stage where there is no complete intermetallic shell. Furthermore, just at the end of the surface stage where a complete shell is first formed, this shell will not be uniform. At this point, the Pt NP surface intermetallic shell has just formed at some locations on the NP surface, so the shell has negligible thickness. In contrast, at a different location where the intermetallic shell has formed much earlier, the shell will have a significant thickness. However, the subsequent diffusion-mediated growth of this nonuniform shell should quickly lead to a more uniform shell thickness, as indicated in Fig. 5. Such growth of the interface between the intermetallic shell and the unconverted Pt core is characterized by so-called anti-DLA shape stabilization,43–45 as thinner portions grow faster and thicker portions grow slower due to the diffusion-mediated nature of growth. Thus, after some interval time at the end of the surface stage, it is reasonable to apply a G–B model. Therefore, the standard G–B formula should be refined so that the shell thickness has an appropriate non-zero thickness (in contrast to the default assumption of zero thickness) when the model is first applied. Such refinement (see the ESI Fig. S14 and S15†) leads to the result
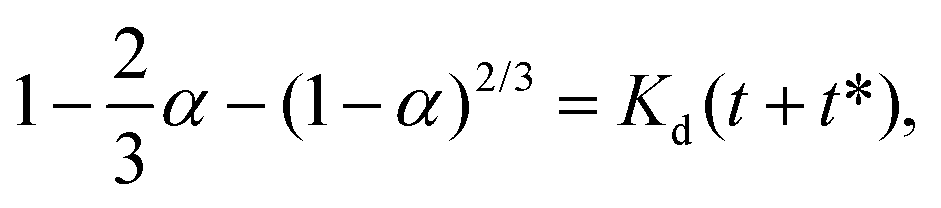 | (6) |
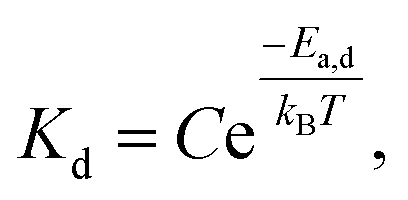 | (7) |
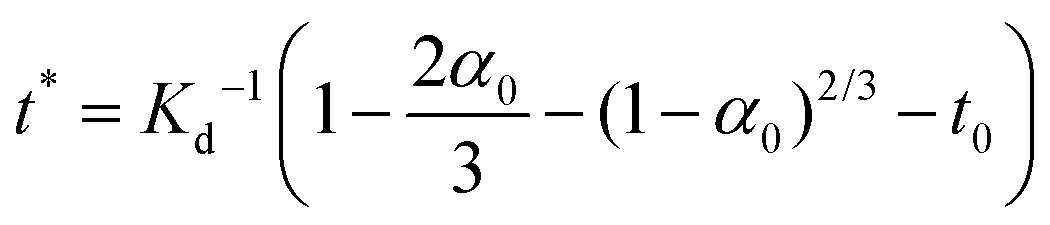 | (8) |
Finally, we believe that there are two reasons that, perhaps counterintuitively, the G–B diffusion stage is not observed during 120 min when we track the transformation kinetics for 5 nm Pt NPs, while it is observed when tracking 14 nm Pt NPs over the same time interval. First and most significantly, the initial rate of conversion is lower for the smaller 5 nm NPs, as discussed above, indicating that more time is required to reach the second stage. In addition, values of αsurf are higher for 5 than 14 nm NPs at the same temperature, further delaying the onset of the second stage for the former.
To illustrate that our model convincingly describes the transformation, we performed control experiments wherein 5 and 14 nm Pt@mSiO2 are converted to PtSn at 250 °C and 260 °C, respectively, with the reaction time extended but all other parameters following previous experiments. For the 5 nm Pt@mSiO2, the experimental conversion (Fig. S16A†) initially matches well with previous experimental results, but ultimately exceeded the surface intermixing stage limit αsurf. This confirms that for 5 nm NP, a G–B diffusion stage does occur later in the transformation. For the 14 nm Pt@mSiO2, we were able to predict before the experiment the conversion α at a given time, and the time necessary (around 930 min) for the transformation to go to completion, based on out fitting results. Experimental results (Fig. S16B†) matched well with both previous experimental result and our prediction.
DFT analysis of energetics for the surface stage of Pt–Sn intermetallic formation
While our integrated experimental and modeling analysis has determined effective barriers for intermixing, such analysis does not provide insight into the atomistic-level processes controlling these barriers. Our DFT analysis below indicates that the barrier for the first surface intermixing stage is determined by that for solid-state vacancy-mediated diffusion of Sn through crystalline fcc Pt. A second issue is whether the onset of intermixing involves the simple exchange of surface Sn with the outer layer of the Pt NP or more complex processes. This is elucidated by additional DFT analysis.Assessment of the energetics relevant for intermixing is provided by DFT analysis using the Vienna ab initio simulation package (VASP).46,47 We used the projector-augmented-wave method48 for the electron-core interactions. The analyses were performed with both the Perdew–Burke–Ernzerhof (PBE)49 and PBEsol50 generalized gradient approximation (GGA) functionals for exchange and correlation. The energy cutoff of 400 eV for the plane-wave basis was tested to be sufficient for energy convergence. For accuracy in energy minimization, the magnitude of the force acting on each atom undergoing relaxation was reduced to less than 0.1 eV nm−1. The selection of a supercell size and the corresponding Γ-centered k meshes were always tested carefully for energy convergence. We have performed benchmark calculations for bulk fcc Pt as well as both α- and β-Sn to ensure that experimentally determined lattice constants and cohesive energies are recovered. See Tables S17–S22 in the ESI† for more details. In addition, we confirm that DFT analysis for bulk Pt3Sn and PtSn alloys recovers experimental lattice constants and assures the thermodynamic stability of these alloys at T = 0 K.
Sn diffusion through fcc Pt
A key analysis particularly relevant for the surface stage of the process is the assessment of the effective barrier for the diffusion of Sn (which is formed by reduction on the surface of a Pt NP) into the interior of the Pt NP. We assumed that such diffusion is vacancy mediated, i.e., when a vacancy generated at the NP surface diffuses to a fcc site adjacent to a Sn atom embedded in the Pt NP, the Sn atom can hop into that site. Thus, the effective barrier for Sn impurity diffusion is the sum of two components:51 one is the formation energy to create a vacancy, and another one is a local hopping barrier for an atom to hop into the vacancy location (with some nuances noted below).First, we assess relevant hopping barriers using a 4 × 4 × 4 supercell with the k mesh of 5 × 5 × 5. For pure Pt containing a single vacancy, we found a barrier of Edv = 1.23 (1.41) eV from PBE (PBEsol) GGA for Pt hopping into the vacancy. These values should be compared with a previous theoretical estimate of Edv = 1.43 eV from the local-density approximation (LDA).41 For Pt containing an embedded Sn at a fcc site and a vacancy at an adjacent fcc site, we find a substantially lower barrier of Edv(Sn) = 0.68 (0.77) eV from PBE (PBEsol) GGA for Sn hopping into the vacancy site. Consequently, for hopping dynamics associated with Sn impurity diffusion, the rate-limiting process is the vacancy diffusion to a site neighboring the embedded Sn, rather than the Sn hopping to that neighboring vacancy location. For this reason, the first set of results for vacancy diffusion in pure Pt is relevant for the analysis of the effective barrier of Sn impurity diffusion.
Second, we consider relevant vacancy formation energies. To determine the vacancy formation energy Eform in a pure fcc metal, the standard procedure is as follows. One evaluates the total energy Evac for a periodic supercell of N fcc sites where N − 1 sites are populated by metal atoms, and one is a vacancy. If Ebulk denotes the total energy of the same cell populated with N metal atoms, then one has that Eform = Evac − (N − 1) Ebulk/N.41 Using this approach for fcc Pt, we obtain Eform = 0.65(0.84) eV from PBE (PBEsol) GGA. These results can be compared with previous theoretical estimates of Eform = 0.95 eV from LDA, and Eform = 0.68 eV from PBE. It is however well recognized that DFT energetics suffer from a “surface intrinsic error” for which correction procedures have been developed.52 These yields corrected estimates of Eform(corr) = 1.15 eV from LDA, and Eform(corr) = 1.18 eV for PBE.41 There are experimental estimates for Eform in Pt with a range of values from 1.25 to 1.6 eV, so the corrected estimates are on the lower end of this range.41
To supplement the above conventional analysis of the formation energy, we also determine the energy cost, Eform(Sn in Pt), for the creation of a vacancy adjacent to a single Sn impurity embedded in fcc Pt.53 To this end, we calculate the total energy Evac(Sn in Pt) of a supercell of N fcc sites with N − 2 sites occupied by Pt, one site occupied by Sn, and one vacancy adjacent to the Sn, as well as the total energy Ebulk(Sn in Pt) of a supercell of N fcc sites with N − 1 sites occupied by Pt and one site occupied by Sn. Then together with Ebulk for pure Pt mentioned above, one has that Eform(Sn in Pt) = Evac(Sn in Pt) + Ebulk/N − Ebulk(Sn in Pt). From such an analysis, we find that Eform(Sn in Pt) = 0.44 (0.62) eV for PBE (PBEsol) GGA. These formation energies are significantly below the uncorrected values for vacancy formation in pure Pt, so we reasonably assume that corrected values are also lower. This analysis indicates that the relevant formation energy controlling Sn impurity diffusion in Pt is the higher value for pure Pt of Eform(corr) ≈ 1.2 eV.
In summary, we conclude that the effective barrier, Ed(Sn in Pt), for the diffusion of Sn impurities in Pt corresponds to the effective barrier or diffusion of vacancies in pure Pt. However, this conclusion is only possible after the above comprehensive analysis. This effective barrier is given by Ed(Sn in Pt) = Eform(corr) + Edv ≈ 2.6 eV (or 250 kJ mol−1) using the higher PBEsol or LDA value for Edv, and is consistent with experimental estimates. A value of Ed(Sn in Pt) ≈ 250 kJ mol−1 is compatible with our kinetic analysis of experimental data determining the effective barrier for the surface stage of intermixing.
Intermixing of surface Sn into Pt(100)
It has been suggested that comprehensive analysis and insight into intermixing kinetics of species A into a NP of species B should assess barriers and energetics for the exchange of isolated adsorbed species A with atoms at the surface of the B NP.54 Such a process is potentially the initial step in our surface stage of intermixing after A = Sn is reduced at the surface of the B = Pt NP. Relevant energies have been tabulated for various systems based on DFT analysis.54–56 Since our Pt NP cores are predominantly cubic in shape at least for the larger NPs, we naturally focus on intermixing of Sn on Pt(100) facets. It is well recognized that exchange barriers for (100) facets of fcc metals are generally well below those for more laterally crowded hexagonal close-packed (111) facets,57 potentially making exchange competitive with surface hopping on (100) facets.From slab calculations using a 4 × 4 supercell with the k mesh of 7 × 7 × 1 (slab thickness is four single-atom Pt(100) layers plus a vacuum separation of 2.2 nm in the direction perpendicular to the slab surface), we determine that Sn prefers the four-fold-hollow (4fh) site on Pt(100), the energy at the bridge site being 0.76 (0.85) eV higher from PBE (PBEsol) GGA. Thus, the diffusion barrier for surface hopping between 4fh sites is given by Ed(Sn) ≈ 0.8 eV. In addition, upon exchange of Sn at a 4fh surface site with one of the four supporting Pt, displacing that Pt to a second nearest-neighbor 4fh site of the initial Sn 4fh site, the energy of the system actually increases by ΔE = + 0.44 (+0.59) eV from PBE (PBEsol) GGA. Thus, this process is endothermic, i.e., not thermodynamically favorable. Previous analyses indicate that it is generally not favorable for more noble (or less cohesive) metals to substitute into less noble (or more cohesive) substrates due to an energy penalty in moving the latter to the surface as adatoms.48 This picture is consistent with our analysis. We find that incorporating additional Sn adatoms which become neighbors of the Pt displaced onto the (100) surface (but are not neighbors of the Sn which substitutes into the surface layer) still does not make the exchange process exothermic.
Thus, we conclude that the onset of intermixing at a Pt(100) surface cannot be described in terms of simple single-atom processes, but must reflect more complex concerted behavior (and is presumably facilitated by the presence of defects associated with grain boundary as suggested previously).
Conclusion
In this work we have studied the formation of ordered IMCs at the nanoscale, focusing on the kinetics of transformation from Pt to PtSn NPs. With the assistance of quantitative PXRD analysis, we find that the transformation to an intermetallic NP is initiated by a surface-specific intermixing stage. The activation barrier for this stage determined from the tailored modeling of the experimental kinetics is consistent with DFT analysis of the effective barrier for vacancy-mediated diffusion of Sn through fcc Pt. Furthermore, our analysis shows that transformation kinetics for surface intermixing over the observed temperature range is effectively described by a single model or mechanism with a fixed reaction order and a single activation barrier. Also, we find that the default expectation of faster conversion of smaller NPs does not universally apply since only the larger NPs contain grain boundaries which significantly enhance their initial conversion. This initial stage is generally followed by a G–B type bulk diffusion stage where appropriate analysis of the kinetics to extract an activation barrier follows from a G–B model refined to treat the NP geometry of relevance here. In summary, our work develops a detailed picture and characterization of the kinetics of PtSn IMC formation at the nanoscale. The key aspects of this work can likely be generalized to benefit the synthesis route for the designing of other intermetallic systems, being useful in a broad spectrum of fields.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Acknowledgment is made to the Donors of the American Chemical Society Petroleum Research Fund for partial support of this research. Y. H. was supported for this work by NSF grant CHE-1507223, and his DFT calculations used resources of the National Energy Research Scientific Computing Center (NERSC), a U.S. DOE Office of Science User Facility operated under Contract No. DE-AC02-05CH11231 and used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1548562. J. W. E. was supported by the U.S. Department of Energy (USDOE), Office of Science, Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biological Sciences, and his work was performed at Ames Laboratory which is operated by Iowa State University for the USDOE under Contract No. DE-AC02-07CH11358. Partial support for this project was provided by the Presidential Interdisciplinary Research Seed (PIRS) grant program at Iowa State University.Notes and references
- A. J. Albaaji, E. G. Castle, M. J. Reece, J. P. Hall and S. L. Evans, J. Mater. Sci., 2017, 52, 13284–13295 CrossRef CAS.
- W. S. Seo, J. H. Lee, X. M. Sun, Y. Suzuki, D. Mann, Z. Liu, M. Terashima, P. C. Yang, M. V. McConnell, D. G. Nishimura and H. J. Dai, Nat. Mater., 2006, 5, 971–976 CrossRef CAS PubMed.
- Y. Shiota, T. Nozaki, F. Bonell, S. Murakami, T. Shinjo and Y. Suzuki, Nat. Mater., 2012, 11, 39–43 CrossRef CAS PubMed.
- A. Godeke, Supercond. Sci. Technol., 2006, 19, R68–R80 CrossRef CAS.
- R. J. Cava, H. Takagi, H. W. Zandbergen, J. J. Krajewski, W. F. Peck Jr., T. Siegrist, B. Batlogg, R. B. van Dover, R. J. Felder, K. Mizuhashi, J. O. Lee, H. Eisaki and S. Uchida, Nature, 1994, 367, 252–253 CrossRef CAS.
- M. Suenaga and M. Garber, Science, 1974, 183, 952–954 CrossRef CAS PubMed.
- P. Kamakoti and D. S. Sholl, J. Membr. Sci., 2003, 225, 145–154 CrossRef CAS.
- L. Z. Ouyang, J. L. Huang, H. Wang, J. W. Liu and M. Zhu, Mater. Chem. Phys., 2017, 200, 164–178 CrossRef CAS.
- M. B. Cortie, C. S. Kealley, V. Bhatia, G. J. Thorogood, M. M. Elcombe and M. Avdeev, J. Alloys Compd., 2011, 509, 3502–3508 CrossRef CAS.
- S. Furukawa and T. Komatsu, ACS Catal., 2017, 7, 735–765 CrossRef CAS.
- Q. C. Feng, S. Zhao, Y. Wang, J. C. Dong, W. X. Chen, D. S. He, D. S. Wang, J. Yang, Y. M. Zhu, H. L. Zhu, L. Gu, Z. Li, Y. X. Liu, R. Yu, J. Li and Y. D. Li, J. Am. Chem. Soc., 2017, 139, 7294–7301 CrossRef CAS PubMed.
- Z. Y. Qi, C. X. Xiao, C. Liu, T. W. Goh, L. Zhou, R. Maligal-Ganesh, Y. C. Pei, X. L. Li, L. A. Curtiss and W. Y. Huang, J. Am. Chem. Soc., 2017, 139, 4762–4768 CrossRef CAS PubMed.
- C. X. Xiao, L. L. Wang, R. V. Maligal-Ganesh, V. Smetana, H. Walen, P. A. Thiel, G. J. Miller, D. D. Johnson and W. Y. Huang, J. Am. Chem. Soc., 2013, 135, 9592–9595 CrossRef CAS PubMed.
- R. V. Maligal-Ganesh, C. X. Xiao, T. W. Goh, L. L. Wang, J. Gustafson, Y. C. Pei, Z. Y. Qi, D. D. Johnson, S. R. Zhang, F. Tao and W. Y. Huang, ACS Catal., 2016, 6, 1754–1763 CrossRef CAS.
- S. Furukawa, K. Takahashi and T. Komatsu, Chem. Sci., 2016, 7, 4476–4484 RSC.
- M. Krajci, A. P. Tsai and J. Hafner, J. Catal., 2015, 330, 6–18 CrossRef CAS.
- A. Ota, E. L. Kunkes, I. Kasatkin, E. Groppo, D. Ferri, B. Poceiro, R. M. N. Yerga and M. Behrens, J. Catal., 2012, 293, 27–38 CrossRef CAS.
- J. Arana, N. Homs, J. Sales, J. L. G. Fierro and P. R. de la Piscina, Catal. Lett., 2001, 72, 183–189 CrossRef CAS.
- R. K. Ramachandran, J. Dendooven, M. Filez, V. V. Galvita, H. Poelman, E. Solano, M. M. Minjauw, K. Devloo-Casier, E. Fonda, D. Hermida-Merino, W. Bras and G. B. Marin, ACS Nano, 2016, 10, 8770–8777 CrossRef CAS PubMed.
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron, M. Mavrikakis, M. Chi, K. L. More, Y. Li, N. M. Markovic, G. A. Somorjai, P. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed.
- K. Fominykh, M. Feckl Johann, J. Sicklinger, M. Döblinger, S. Böcklein, J. Ziegler, L. Peter, J. Rathousky, E. W. Scheidt, T. Bein and D. Fattakhova Rohlfing, Adv. Funct. Mater., 2014, 24, 3123–3129 CrossRef CAS.
- S. B. Duan and R. M. Wang, Prog. Nat. Sci.: Mater. Int., 2013, 23, 113–126 CrossRef.
- A. J. Biacchi and R. E. Schaak, ACS Nano, 2011, 5, 8089–8099 CrossRef CAS PubMed.
- M. W. Tew, H. Emerich and J. A. van Bokhoven, J. Phys. Chem. C, 2011, 115, 8457–8465 CrossRef CAS.
- N. Iwasa, N. Ogawa, S. Masuda and N. Takezawa, Bull. Chem. Soc. Jpn., 1998, 71, 1451–1455 CrossRef CAS.
- S. J. Freakley, Q. He, J. H. Harrhy, L. Lu, D. A. Crole, D. J. Morgan, E. N. Ntainjua, J. K. Edwards, A. F. Carley, A. Y. Borisevich, C. J. Kiely and G. J. Hutchings, Science, 2016, 351, 965–968 CrossRef CAS PubMed.
- C. Y. Wang, D. P. Chen, X. H. Sang, R. R. Unocic and S. E. Skrabalak, ACS Nano, 2016, 10, 6345–6353 CrossRef CAS PubMed.
- Y. Yan, S. Du Jingshan, D. Gilroy Kyle, D. Yang, Y. Xia and H. Zhang, Adv. Mater., 2017, 29, 1605997 CrossRef PubMed.
- J. C. Bauer, X. Chen, Q. S. Liu, T. H. Phan and R. E. Schaak, J. Mater. Chem., 2008, 18, 275–282 RSC.
- U. Banin, Nat. Mater., 2007, 6, 625–626 CrossRef CAS PubMed.
- T. Komatsu and H. Ikenaga, J. Catal., 2006, 241, 426–434 CrossRef CAS.
- Z. F. Liu, G. S. Jackson and B. W. Eichhorn, Angew. Chem., Int. Ed., 2010, 49, 3173–3176 CrossRef CAS PubMed.
- W. Zhao Evan, R. Maligal Ganesh, C. Xiao, T. W. Goh, Z. Qi, Y. Pei, E. Hagelin-Weaver Helena, W. Huang and R. Bowers Clifford, Angew. Chem., Int. Ed., 2017, 129, 3983–3987 CrossRef.
- L. Wu, A. P. Fournier, J. J. Willis, M. Cargnello and C. J. Tassone, Nano Lett., 2018, 18, 4053–4057 CrossRef CAS PubMed.
- Y. Pei, R. V. Maligal-Ganesh, C. Xiao, T.-W. Goh, K. Brashler, J. A. Gustafson and W. Huang, Nanoscale, 2015, 7, 16721–16728 RSC.
- B. Dong, Y. Pei, F. Zhao, T. W. Goh, Z. Qi, C. Xiao, K. Chen, W. Huang and N. Fang, Nat. Catal., 2018, 1, 135–140 CrossRef.
- M. E. Brown, D. Dollimore, A. K. Galwey, C. H. Bamford and C. F. H. Tipper, Reactions in the solid state, 1980 Search PubMed.
- A. Khawam and D. R. Flanagan, J. Phys. Chem. B, 2006, 110, 17315–17328 CrossRef CAS PubMed.
- P. Papon, J. Leblond and P. H. E. Meijer, The physics of phase transitions: concepts and applications, 2006 Search PubMed.
- A. M. Ginstling and B. I. Brounshtein, Zh. Prikl. Khim., 1950, 23, 1249–1259 Search PubMed.
- M. Schaefer, R. A. Fournelle and J. Liang, J. Electron. Mater., 1998, 27, 1167–1176 CrossRef CAS.
- H. H. Farrell, G. H. Gilmer and M. Suenaga, J. Appl. Phys., 1974, 45, 4025–4035 CrossRef CAS.
- P. Meakin and J. M. Deutch, J. Chem. Phys., 1986, 85, 2320–2325 CrossRef CAS.
- L. Paterson, Phys. Rev. Lett., 1984, 52, 1621–1624 CrossRef CAS.
- J. W. Evans, Phys. Rev. A, 1989, 40, 2868–2870 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Phys. Rev. Lett., 2008, 100, 136406 CrossRef PubMed.
- T. R. Mattsson and A. E. Mattsson, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 214110 CrossRef.
- A. E. Mattsson and W. Kohn, J. Chem. Phys., 2001, 115, 3441–3443 CrossRef CAS.
- Y. Han, D.-J. Liu and J. W. Evans, Nano Lett., 2014, 14, 4646–4652 CrossRef CAS PubMed.
- L. T. Roling and M. Mavrikakis, Nanoscale, 2017, 9, 15005–15017 RSC.
- L. Zhang, L. T. Roling, X. Wang, M. Vara, M. Chi, J. Liu, S.-I. Choi, J. Park, J. A. Herron, Z. Xie, M. Mavrikakis and Y. Xia, Science, 2015, 349, 412–416 CrossRef CAS PubMed.
- Y. Han, C. R. Stoldt, P. A. Thiel and J. W. Evans, J. Phys. Chem. C, 2016, 120, 21617–21630 CrossRef CAS.
- G. Antczak and G. Ehrlich, Surface diffusion: metals, metal atoms, and clusters, Cambridge University Press, Cambridge, 2010 Search PubMed.
- S. H. Joo, J. Y. Park, C. K. Tsung, Y. Yamada, P. D. Yang and G. A. Somorjai, Nat. Mater., 2009, 8, 126–131 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nr10067e |
| This journal is © The Royal Society of Chemistry 2019 |