
Recent development of carbon quantum dots regarding their optical properties, photoluminescence mechanism, and core structure†
Keenan J.
Mintz
 ,
Yiqun
Zhou
and
Roger M.
Leblanc
*
,
Yiqun
Zhou
and
Roger M.
Leblanc
*
Department of Chemistry, University of Miami, Coral Gables, Florida 33146, USA. E-mail: rml@miami.edu
First published on 28th February 2019
Abstract
Carbon quantum dots (CDs) are a relatively new class of carbon nanomaterials which have been studied very much in the last fifteen years to improve their already favorable properties. The optical properties of CDs have drawn particular interest as they display the unusual trait of excitation-dependent emission, as well as high fluorescence quantum yields (QY), long photoluminescence (PL) decay lifetimes, and photostability. These qualities naturally lead researchers to apply CDs in the field of imaging (particularly bio-imaging) and sensing. Since the amount of publications regarding CDs has been growing nearly exponentially in the last ten years, many improvements have been made in the optical properties of CDs such as QY and PL lifetime. However, a great deal of confusion remains regarding the PL mechanism of CDs as well as their structural properties. Therefore, presented in this review is a summary and discussion of the QYs and PL lifetimes reported in recent years. The effect of method as well as precursor has been evaluated and discussed appropriately. The current theories regarding the PL mechanism of CDs are discussed, with special attention to the concept of surface state-controlled PL. With this knowledge, the improvement of preparation and applications of CDs related to their optical properties will be easily accomplished. Further improvements can be made to CDs through the understanding of their structural and optical properties.
1. Introduction
Carbon quantum dots (CDs) were discovered in 2004 when Xu et al. were attempting to prepare single walled carbon nanotubes. However, during characterization of carbon nanotubes they found a group of “fluorescent Nanoparticles”.1 Since then, great strides have been made in the preparation of different types of CDs with much improved photoluminescence (PL) properties compared to when CDs were first studied, though the mechanism of PL is still not well understood.2 However, the favorable optical properties of CDs, such as excitation-dependent PL, high fluorescence quantum yield (QY), and long PL lifetime, have prompted much attention toward the development of CDs for bio-imaging and sensing.3–7 As seen in Scheme 1, the number of citations regarding QY and PL lifetime has grown exponentially over the last ten to fifteen years. This has led to a vast amount of literature for CDs, with many papers using different methods and precursors, which has added to the confusion that surrounds CDs PL mechanism, as many different explanations regarding the phenomena surrounding CDs are different. Comparison of these methods is needed to show the greatest improvements that have been made in the literature in recent years.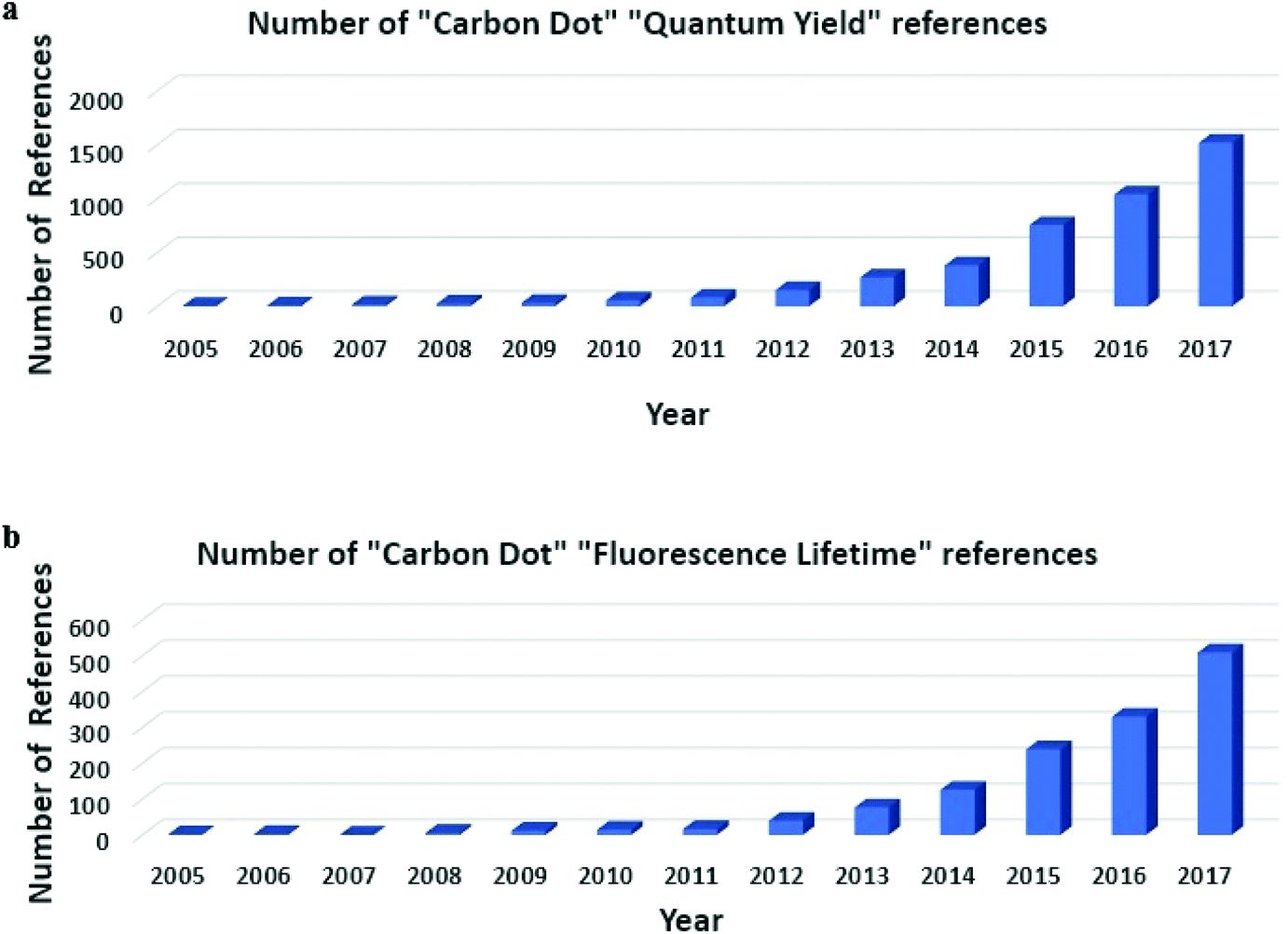 | ||
| Scheme 1 (a) Histogram showing number of references related to CDs and QY per year. (b) Histogram showing number of references related to CDs and fluorescence lifetime per year (Note: while not the correct technical term, fluorescence lifetime is the term used most often in the literature to describe the PL decay lifetime of CDs). | ||
Since they were discovered, CDs have been viewed as a less toxic alternative to traditional quantum dots and very promising for the use in bio-imaging and sensing.8,9 CDs preparation is classified in two ways: top-down and bottom-up approaches. Top-down approaches utilize large carbon structures such as charcoal or carbon powder and cut them down to nanometer sized particles through methods such as laser ablation and chemical oxidation.10,11 Bottom-up approaches use small organic molecules or salts as precursors and under harsh conditions (e.g. high temperatures or microwave) the precursors will be carbonized to form CDs.5,12 When prepared, as previously mentioned, CDs are often characterized by their optical characteristics. A great deal of research has been made to improve the PL of CDs, particularly their QY. Several different factors can affect the optical properties of CDs upon production such as: method, precursor, passivation, and heteroatom doping. Common methods include laser ablation, microwave, and hydrothermal.13 These methods generally provide different results, although no study has been done to compare them under similar conditions. The precursors presented in CDs literature are extensive, as virtually any carbon source could be used as a CDs precursor. Passivation of the as-prepared CDs is often achieved with polymers and/or small organic molecules containing heteroatoms (N, S, P, etc.).14,15 It has been shown that different types of precursors, surface passivation, or doping with elements such as nitrogen, sulfur, or boron can all result in higher QY and PL decay lifetime for CDs.14,16–19
QY study is an important characterization for CDs because of their potential to be used as imaging agents in biological samples. In view of the great effort devoted on increasing the QY of CDs to improve their performance in this area,20,21 a systematic review of the progress made is needed. Different precursors, synthetic methods, and doping methods have been designed for this goal of increasing QY. In addition, the PL decay lifetime is an intrinsic property of CDs, which is usually on the low nanosecond scale. However, it can be tuned in different pH or temperature conditions.22,23 Also, the length of lifetime can be used to analyze the PL mechanism of CDs.24 In addition, the increase of PL decay lifetime of CDs is beneficial for the in vivo bioimaging as well as the expansion of a broad application of CDs. Therefore, it is of great significance of the theory study of the PL and structure of CDs.
There are two main points related to CDs which are not well understood. These are the mechanism by which they emit light and their core structure.2,25,26 Many articles have been published endeavoring to elucidate these two points. Regarding the PL mechanism of CDs different theories have been published including: surface state,27,28 quantum confinement,29,30 and molecular state.31,32 Distinguishing between these phenomena is important to further the understanding of CDs as well as to aid in the rational design of CDs for specific applications.
Understanding the core structure is vital to understand the structural dynamics in CDs. Many applications rely on the surface of CDs (e.g. drug delivery) which is reasonably well-defined. However, to fully understand the interactions in CDs as well as the potential interactions of CDs in their application, the entire structure of CDs must be defined. Some have suggested graphitic cores,33 carbon nitride,34 or polymeric structures.35 Determining the structural features of CDs is once again necessary for the rational design of CDs and for them to realize their full potential in a broad spectrum of applications.
There are currently several reviews for CDs in the literature. Zuo et al.13 reviewed CDs bioanalytical applications. Wang et al.36 presented CDs biological applications. Qu and coworkers37 summarized heteroatom doping and bioimaging in CDs. Namdari et al.38 focused on the biomedical applications of CDs. Zhou and coworkers39 highlighted the quenching of CDs PL for sensing applications. Yan et al.40 provided an overview of CDs’ surface modification and functionalization. Huang and coworkers41 discussed the sensing application of CDs. This review will present an overview of the current CDs preparation methods and precursors, and specifically how these two factors influence the QY and PL lifetime, and their applications in bio-imaging and sensing. For quick reference, a table is presented in ESI (Table S1†) which summarizes the properties of select CDs preparations. Then the current theories regarding the PL mechanism and core structure will be presented. The current literature expresses the unique ability of CDs due to their high QYs, long PL lifetimes and photostability, and compels further research to refine and better define this fast-growing field.
2. Developments in carbon dots’ quantum yield based on precursor and preparation method
Quantum yield of PL is most commonly calculated through the use of a reference standard, although there are means to determine the absolute QY values. These require specialized and expensive instrumentation, while the use of a reference requires only traditional UV/vis and fluorescence spectrophotometers.42,43 QY (Φ) is defined according to eqn (1), which states that it is the ratio of emitted photons to absorbed photons.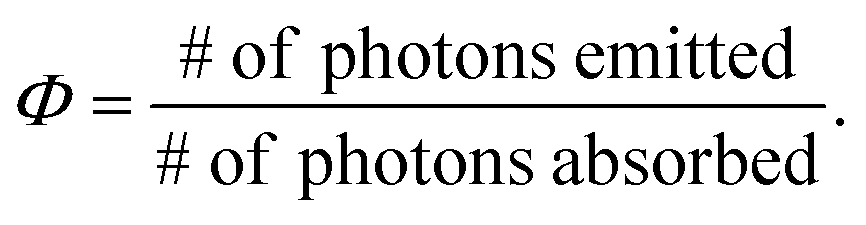 | (1) |
The most common way in which the value of QY for CDs is obtained, is according to eqn (2):
 | (2) |
2.1 Laser ablation preparations
In 2008, Sun. et al.44 applied laser ablation to carbon Nanoarticles to obtain CDs. Then they treated the as-prepared CDs with nitric acid and doped them through the addition of Zn(CH3COO)2 with NaOH or Na2S to obtain ZnO doped CDs and ZnS doped CDs, respectively. Both products showed similar absorption spectra, and the QY was calculated upon excitation at 440 nm for both, using quinine sulfate as a reference. The ZnS-CDs showed QY result at 50%, whereas the ZnO-CDs showed a slightly lower QY at 45%. The difference between the QY of the two CDs is negligible based on these results. Although laser ablation has been shown to generate photoluminescent CDs, there has not been many CDs obtained from laser ablation that have been applied in the field of bio-imaging. CDs obtained with this method are generally used in sensing applications.24,452.2 Microwave-mediated preparations
Another common method of synthesizing CDs is using microwave to pyrolyze the precursors. In 2014, Qu et al.46 prepared CDs by mixing urea and citric acid in two ratios, 0.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (CNP1-carbon Nanoarticle) and 2:1 (CNP2), in water, and microwaving the mixture at 650 W for 5 min. The obtained solid was dissolved in water and ethanol and centrifuged to remove larger particles. CNP1 aqueous solution showed a QY of 15% with maximum emission at 440 nm emission (360 nm excitation), and CNP2 aqueous solution showed a QY of 18% (maximum emission at 540 nm with 420 nm excitation). When the QY was measured in an ethanol solution, the value for CNP1 was unchanged, but CNP2 showed a slightly blue-shifted emission at 526 nm (420 nm excitation) with a QY of 36%. The increased QY of CNP2 as well as the green emission show the possible effects dopants (in this case urea as nitrogen dopant) can have on CDs. Another microwave-assisted synthetic method that has been used was reported by Choi et al.17 in 2016, and can give more information on nitrogen doping as well as doping with heteroatoms (boron and nitrogen). They synthesized 4 types of CDs. The first (BN-CDs) was made from mixing boric acid, citric acid, and 1,2-ethylenediamine (EDA) in water and microwaving at 700 W for 2 min. The second (N-CDs) was obtained with the same synthetic method without the presence of boric acid. The third (B-CDs) and fourth (CDs) were prepared using citric acid with and without, respectively, boric acid by a hydrothermal approach at 180 °C for 6 h because they were not able to obtain the desired CDs from the microwave-mediated synthesis of just citric acid. The highest QY was shown by BN-CDs with 80.8 ± 5.1% and N-CDs showed 40.2 ± 1.8% with emission at 455 nm (excitation 350 nm). B-CDs and CDs showed a QY of 1.2 and 2.1% respectively.17 Nitrogen doping is well known to increase the QY of CDs as shown here,47,48 but the positive effect of adding an additional dopant of boron is shown as well as the QY jumps to over 80%. The comparison between CDs and B-CDs may not be reliable as QY for both are not apparently different (1–2%), however the increase in QY due to heteroatom doping can be still clearly seen in the microwave-mediated preparations.
:1 (CNP1-carbon Nanoarticle) and 2:1 (CNP2), in water, and microwaving the mixture at 650 W for 5 min. The obtained solid was dissolved in water and ethanol and centrifuged to remove larger particles. CNP1 aqueous solution showed a QY of 15% with maximum emission at 440 nm emission (360 nm excitation), and CNP2 aqueous solution showed a QY of 18% (maximum emission at 540 nm with 420 nm excitation). When the QY was measured in an ethanol solution, the value for CNP1 was unchanged, but CNP2 showed a slightly blue-shifted emission at 526 nm (420 nm excitation) with a QY of 36%. The increased QY of CNP2 as well as the green emission show the possible effects dopants (in this case urea as nitrogen dopant) can have on CDs. Another microwave-assisted synthetic method that has been used was reported by Choi et al.17 in 2016, and can give more information on nitrogen doping as well as doping with heteroatoms (boron and nitrogen). They synthesized 4 types of CDs. The first (BN-CDs) was made from mixing boric acid, citric acid, and 1,2-ethylenediamine (EDA) in water and microwaving at 700 W for 2 min. The second (N-CDs) was obtained with the same synthetic method without the presence of boric acid. The third (B-CDs) and fourth (CDs) were prepared using citric acid with and without, respectively, boric acid by a hydrothermal approach at 180 °C for 6 h because they were not able to obtain the desired CDs from the microwave-mediated synthesis of just citric acid. The highest QY was shown by BN-CDs with 80.8 ± 5.1% and N-CDs showed 40.2 ± 1.8% with emission at 455 nm (excitation 350 nm). B-CDs and CDs showed a QY of 1.2 and 2.1% respectively.17 Nitrogen doping is well known to increase the QY of CDs as shown here,47,48 but the positive effect of adding an additional dopant of boron is shown as well as the QY jumps to over 80%. The comparison between CDs and B-CDs may not be reliable as QY for both are not apparently different (1–2%), however the increase in QY due to heteroatom doping can be still clearly seen in the microwave-mediated preparations.
In the same year, Yu et al.49 were also able to synthesize highly photoluminescent CDs using a microwave-assisted synthetic approach. They mixed citric acid, PEG 400, and N-(2-hydroxyethyl) ethylenediamine in water, sonicated for 2 min, and heated in a microwave oven for 20 min at 800 W. The obtained solid was purified by dialysis and underwent optical characterization. Their obtained product showed an excitation wavelength-dependent emission with a QY of 79.63% at 447 nm (351 nm excitation). The ability of nitrogen to enhance the QY of CDs is again clearly shown here. The addition of the polymer, PEG 400, facilitated the formation of a hydrogel film of CDs, which showed an ability to detect Hg2+ ions based on the quenching effect of PL of CDs. Also, they were able to show a low detection limit of 0.089 μmol L−1.
In 2017, Kudr et al.50 synthesized CDs by using a microwave oven and employed them to detect DNA damage in PC-3 cells. They dissolved citric acid and diethylenetriamine (DETA) in water and placed in the microwave oven for 3 min at 800 W. They measured the QY to be 4% with maximum emission at 480 nm (360 nm excitation). They attributed this low QY to the extended dialysis time they used to purify the CDs. They also reported the fluorescence decay lifetime to be 12.8 ns as it was significantly long for DNA damage detection. When the CDs were mixed with ethidium bromide and DNA, CDs transferred energy to ethidium bromide through a FRET process and the fluorescence observed was dependent on the DNA damage.
Another method assisted by microwave that will be discussed is from Roshni and Divya51 They used sesame seeds as precursors of CDs and used a microwave oven at 800 W for 15 min. The QY reported was 8.02% with 440 nm maximum emission (365 nm excitation). They also conducted photostability tests by constantly irradiating the sample with UV light for 6 h. After 6 h, their CDs sample showed only a 5% decrease in fluorescence intensity.
2.3 Hydrothermal or solvothermal preparations
Another common synthetic method, of which some examples have already been discussed, is the hydrothermal or solvothermal approach. This approach will be discussed to see the effect of precursors on the QY of CDs. With this method, the most common precursor used as a carbon source is citric acid.In 2017, Li et al.52 used citric acid in conjunction with poly(ethylenimine) (PEI) to synthesize CDs. They heated the precursors up to 110 °C for 2 h in a tightly-sealed autoclave. This yielded a product with a pH dependent (optimum pH = 4) QY of 48.3 ± 5.3% with a maximum emission of 459 nm (excitation of 360 nm). In 2017, Zhang et al.47 and Lin et al.48 published two articles in which citric acid and N-(b-aminoethyl)-g-aminopropyl methyldimethoxy silane (AEAPMS) were utilized as precursors for the preparation of CDs with a thermal method. The precursors were heated up to 220 °C in the presence of ethanol for 3 and 5 min, respectively. These products showed red-shifted emissions around 600 nm (580 and 610 nm, respectively) with a QY of 37% for 3 min and 19% for 5 min. Both products show an unusually long wavelength of PL for CDs formed from citric acid.53,54 The PL wavelength is red-shifted for the longer reaction time (5 min), but the QY is decreased by almost half. Both also show relatively long PL lifetimes of 12.55 ns (3 min) and 12.29 ns (5 min). Citric acid has been used with L-cysteine in a hydrothermal method to prepare CDs. This yielded an unusually short emission wavelength (418 nm), but with a high QY of 64%.55 Citric acid has also been used as a CD precursor with a eutectic mixture of salts (NaNO3/KNO3/NaNO2).56 This method yielded a QY of 20.8% which is a lower value than those which have been reported for many other citric acid-based CDs. This could indicate the benefit of organic, nitrogen-containing compounds as heteroatom dopants and surface passivating agents. Chandra et al.57 performed a study which follows the same trend. They used diammonium citrate as a CDs precursor and obtained CDs with a QY of 11.21%.57 This is further emphasized in a study performed by Yang et al. in which aqueous ammonium citrate was used as the sole CD precursor.58 This yielded a QY of 13.5% which is again lower than other citric acid/citrate based CDs which use organic nitrogen dopants.
In 2015, Wang et al. prepared three CDs from the reaction of urea with glycolic acid, malic acid, and citric acid (one, two, and three carboxylic groups, respectively).59 They saw good correlation between the number of carboxylic groups with optical properties. The emission wavelength, PL lifetime, and QY (12.9, 32.4, and 54.9%, respectively) all increased with the number of carboxylic groups. This is attributed to the strengthened ability of citric acid to conjugate with the amine groups on urea, which decreases with malic acid and then with glycolic acid. These CDs were also shown to be good bio-imaging agents in cancer cell line MG-63.
The highest QY reported for CDs to date was in 2015 from Zheng et al.22 They prepared CDs using citric acid and tris-(hydroxymethyl)aminomethane (tris) with a thermal method. This yielded CDs with emission at a short wavelength (417 nm) and an ultra-high QY of 93.3%. They attributed this high value to the nitrogen doping with tris which promotes radiative recombination for relaxation.
A common precursor that is often paired with citric acid to synthesize CDs is EDA. In 2017, Zhou et al.60,61 prepared CDs using citric acid and EDA in a 1:14 molar ratio with a solvothermal method of varying temperatures. The optimum conditions reported were heating the precursors to 160 °C for 1 h. The obtained product was a very viscous gel-like CDs which provided a QY of 33.7% with a maximum emission of 450 nm (excitation of 350 nm). After a series purification by acetone wash and separation by thin layer chromatography, four CDs fractions were obtained. Among the four fractions, there was one fraction exhibited excitation-independent PL with a QY of 55%, which was almost double the QY of the initial gel-like CDs.62 Wen et al.63 also used citric acid and EDA as CDs precursors. Using a furnace, they heated the mixture to 200 °C for 4 h and purified the raw product with size exclusion chromatography (SEC). Their two obtained fractions displayed wavelengths of emission of 460 and 530 nm, with QYs of 40.69% and 69.30%, respectively. In 2017, Parvin and Mandal64 used citric acid and EDA as precursors, but also added H3PO4 to study the effect of adding another heteroatom, namely phosphorous. They heated their mixture to 250 °C for 2 h and filtered the product to remove the precipitate from the product. Their product showed typical excitation wavelength-dependent PL and they recorded the QY for two different excitation wavelengths, 360 and 440 nm. The QY was 30 and 78%, respectively. They demonstrated the utility of the obtained CDs by testing their imaging capabilities in vitro (in cell line RAW 264.7) and in vivo (in nude mice). There have been several studies which attribute the high QY from citric acid-based CDs to organic fluorophores which are generated during CDs preparation and incorporated into the CDs.54,55,65 However, the vast majority of reports of this phenomenon are only concerning citric acid derived-CDs, and this effect cannot be extrapolated to discuss all CDs.
EDA has also been used as a precursor in preparations that do not involve citric acid. In 2015, CDs were prepared from alanine and EDA through a hydrothermal method. The resulting CDs possessed a QY of 46.2% with maximum emission of 390 nm.66 These showed potential application in cellular imaging and in sensing of NADH. In 2017, Tao et al.67 used polyacrylic acid and EDA to prepare CDs. These CDs displayed emission at 450 nm and a QY of 44.2%. Yang et al.68 used ethanediamine, an isomer of EDA, with C3N4 (carbon nitride) to prepare CDs, which showed a QY of 21% and maximum emission wavelength of 470 nm. There PL intensity also exhibited temperature dependent-behavior. This property enabled the CDs to be used as both in vitro and in vivo temperature probes in HeLa cells and mice, respectively. Tryptophan has been used with EDA to prepare CDs with QY of 48.4%.69 This value was significantly higher than the QY for the same reaction performed with urea replacing EDA (21.5%). This displays the high value of EDA as nitrogen dopant.
The importance of heteroatom doping to the QY of CDs cannot be understated and has been demonstrated by Xu et al.70,71 In 2017, they prepared nitrogen and phosphorous co-doped CDs through the use of sodium citrate and diammonium phosphate as precursors. These CDs displayed a QY value of 53.8% with excitation-dependent emission and good pH stability. Interestingly, when the ratio of sodium citrate to diammonium phosphate was decreased below 19:1, the level of heteroatoms decreased as well as the QY. The proposed explanation for this suggests that an excess of heteroatoms could block the surface defects reducing the efficiency of PL.
Metal atom doping is a strategy which is not commonly used but has been shown to increase CDs’ QY substantially. Xu et al.72 prepared CDs from sodium citrate, citric acid, and manganese carbonate which exhibited a QY of 54%. In this case citric acid was used in addition to sodium citrate to enhance the solubility of manganese carbonate. They also showed pH played an important role in the reaction, by showing the decrease of QY when larger amounts of citric acid were present in the reaction mixture. The metal carbonate bond in the CDs was believed to be the important factor for enhancing the QY of the CDs. This group was shown to be more effective for this cause than the metal–carbon or metal–oxide bond. This has been the highest QY to date for metal-doped CDs.
As was previously discussed, nitrogen doping has often been used to improve CDs’ QY. Unsurprisingly, nitrogen containing polymers have often been studied as CDs precursors. In 2013, branched polyethyleneimine (bPEI) and ammonium persulfate were used with a hydrothermal method to prepare CDs.73 This yielded CDs with the maximum emission at 460 nm and a QY of 54.3% (350 nm excitation). These CDs were promising in cell imaging and gene delivery. In 2015, Yang et al.74 prepared CDs using β-cyclodextrin, PEI, and phosphoric acid. This yielded a type of CDs with a QY of 30% with the maximum emission at 510 nm. These CDs demonstrated excellent capabilities as imaging agents and theragnostic carriers in association with hyaluronic acid, which is supported by the fluorescence bioimaging in Fig. 1. Folic acid and PEI were applied with a 180 °C hydrothermal method to prepare CDs in 2017. It yielded CDs with 450 nm emission and a QY of 42%.75 These also showed excellent capability in cellular imaging as well as the unusual CDs’ trait of being positively charged, which provided applications for the CDs in delivery through the electrostatic association with DNA. These few examples above are representatives of CDs formed from PEI, which typically have blue emission and high QY.
 | ||
| Fig. 1 Bright field and fluorescence images of: (A) H1299 cells. (B) H1299 cells incubated with CDs. (C) H1299 cells incubated with nanocomplexes of CDs and hyaluronic acid. Scale bar is 20 μm. Reproduced from ref. 74 with permissions from The Royal Society of Chemistry. | ||
Biological molecules and natural products have been used to prepare CDs as well. Yang et al. prepared CDs from glucose and monopotassium phosphate. The reaction yielded a low QY of 2.4% with 435 nm emission.76 In 2014, L-tartaric acid, lauryl chloride, and D-glucose were used to prepare CDs which emitted in the highest intensity at 460 nm with a QY of 16.5%.77 Because these CDs possessed an amphiphilic nature, they were able to be used to image cell membrane as seen in Fig. 2. In 2017, aminosalicylic acid was used with ethanol as a precursor for CDs. The resulting CDs showed a maximum emission wavelength of 516 nm with a QYof 16.4%. These CDs were used as an in vitro sensor for Fe3+ in H1299 cells.78 Xu et al.79 produced CDs from casein, a common protein found in milk. These CDs possessed a QY of 31.8% at 446 nm emission, and their capabilities were shown in cellular imaging and also for detection of Hg2+ in HeLa cells. Aspartic acid was also used by Yang et al. to prepare CDs in 2017. They showed the maximum emission wavelength of 402 nm with a QY of 41.3%.80 Ghosh et al.81 prepared CDs from bovine serum albumin (BSA). These CDs yielded the maximum emission at 460 nm with a QY of 44%. The authors demonstrated the CDs utility in promoting algal growth through converting UV light from the solar spectrum to visible blue light.
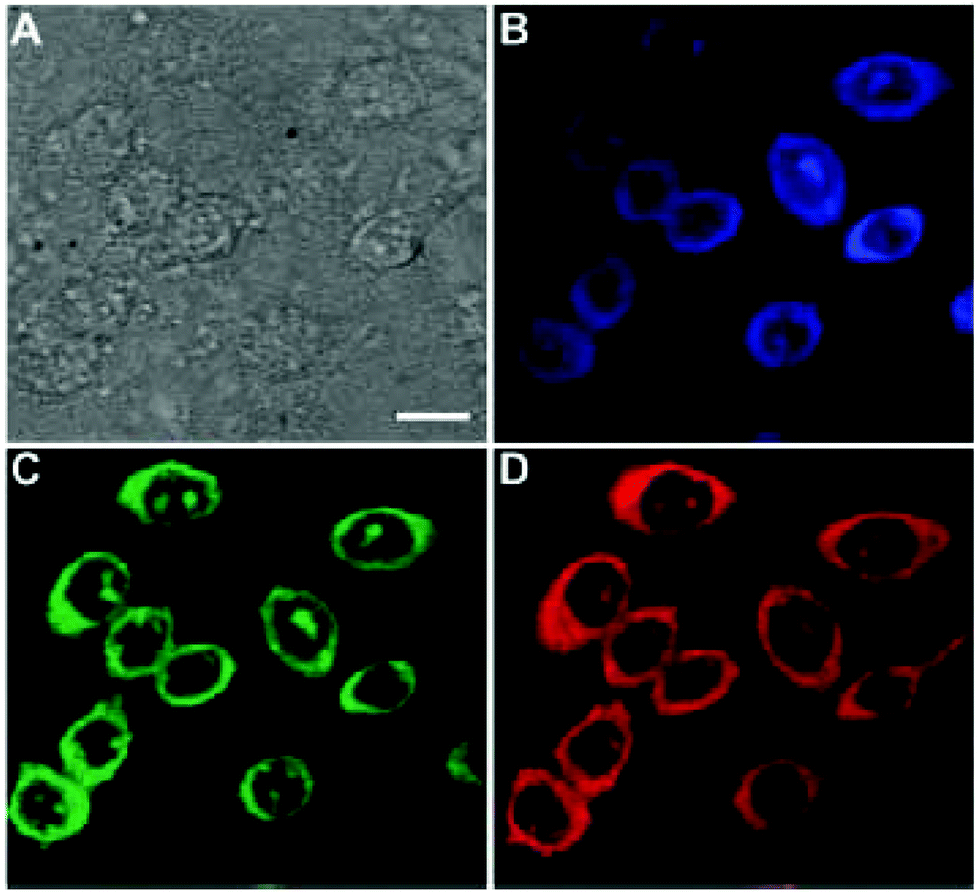 | ||
| Fig. 2 CHO cells imaged by CDs. (A) Bright field image. (B) Confocal image excited at 405 nm. (C) Confocal image excited at 488 nm. (D) Confocal image recorded at 561 nm. Scale bar is 10 μm. Ref. 77 – published by The Royal Society of Chemistry. | ||
2.4 Foodstuff precursors in preparations
As was previously discussed, sesame seeds have been used to prepare CDs. Many other foodstuffs have also been used as CDs precursors. The QY of around 8% in Roshni et al.'s study51 is similar to other QY reported for processes involving foodstuffs, such as the study reported by Zhou et al. where watermelon peel was used as a precursor. The peel was carbonized at 220 °C for 2 h, purified by dialysis to acquire CDs. They obtained a QY of 7.1% with maximum emission at 490 nm (excitation 340 nm).82 In 2013, De and Karak83 used banana juice as a CDs precursor, mixing it with ethanol and heating to 150 °C in an oven for 4 h. Their obtained QY was 8.95% with maximum emission at 460 nm (excitation 360 nm). In 2014, Wang and Zhou84 synthesized CDs from milk with a hydrothermal method at 180 °C for 2 h. Their obtained QY was 12% with maximum emission of 454 nm (excitation 360 nm). To examine the potential of their CDs in the cellular imaging they also did photostability experiments and found that after continuous excitation at 365 nm for 3 h the fluorescence intensity did not decrease. In addition, they reported that the CDs retained 90% fluorescence intensity after storage for 6 months at 4 °C. Since the CDs were stable, they labeled U87 cells and imaged the cells with confocal microscopy, which is shown in Fig. 3. | ||
| Fig. 3 Confocal image of CDs labeling the U87 cells. Adapted with permission from ref. 84. Copyright 2014 American Chemical Society. | ||
Interestingly, Mandani et al.85 discovered that CDs are naturally present in honey. These CDs possessed blue emission (456 nm) with a QY of 1.6%. Due to the low QY, these CDs are limited in application. However, this represented the first time CDs have been shown to naturally occur.
One more foodstuff precursor example was discussed in 2016 when Liu et al.86 synthesized CDs from carrot juice using a hydrothermal method at 160 °C for 6 h. Their CDs showed a QY of 5.16% with emission at 442 nm (excitation 360 nm). They then did cell viability and imaging tests with a HaCAT cell line. The viability test showed that over 85% of the cells remained viable with a high concentration of 700 μg mL−1 CDs. The imaging results that they obtained using confocal microscope are shown in Fig. 4.
 | ||
| Fig. 4 HaCaT cell confocal images without (a–c) and with (d–f) 500 μg mL−1 CDs. Images a and d are excited at 405 nm, b and e are excited at 488 nm, c and f are excited at 543 nm. Figure adapted from ref. 86 with permission Korean Carbon Society. | ||
As shown in the examples mentioned above, the “top-down” synthetic approach of CDs using foodstuff precursors did not result in a high QY. However, the CDs obtained through these methods have been widely applied in sensing and imaging.
As has been discussed, there is a vast amount of preparation methods for CDs in the literature today. Hydrothermal and microwave methods often produce higher QY values than other procedures. Concerning precursors, heteroatom doping with elements such as boron, nitrogen, and phosphorous is an important strategy to increase the QY of CDs. The amount of methods and QY values can lead to some discrepancies, but the potential of CDs as optical probes through high QYs has been established repeatedly. Now the focus will turn to another important optical property.87
3. Developments in carbon dots’ photoluminescence lifetime
PL decay lifetime of CDs indicates the time CDs spend in the excited state before returning to the ground state with the emission of photons.88 The PL lifetime can only be measured using time resolved spectrophotometers, and is commonly measured using femtosecond laser pulses.89 Considering the stable and constant PL required, the length of PL decay lifetime is also an important indicator to measure if the obtained CDs have great potential regarding application in the solar cells,90 bioimaging and sensing fields especially as intracellular probes for time-gated cellular detection.91 Sensing is an especially important application related to PL lifetime as the analyte usually shortens the lifetime for CDs.92 Besides, as was measured and reported by previous literatures, CDs possess a longer PL decay lifetime than most common fluorophores such as fluorescein, Rhodamine B, and Alexa Fluor 488 and 647,93 which confirms CDs can serve as a biocompatible alternative to the common toxic fluorophores in in vivo tests. However, as mentioned before, the QY of CDs varies depending on different synthetic approaches and post-treatment,48,94,95 the PL decay lifetime is also different based on different fabrication methods.68,96 Therefore, in order to widely apply CDs into bioimaging and sensing experiments, besides the QY, the PL decay lifetime also needs to be prolonged. Here, we summarized the PL decay lifetime of CDs in recent years with the aim of comparing different syntheses and finding the best reaction conditions.3.1 Top-down approaches for carbon dots’ preparations
In 2011, Zhou et al.82 obtained photoluminescent CDs by low-temperature carbonization (200 °C) of watermelon peel. As a typical representative of a “top-down” method, this preparation process is more facile and environmentally friendly than the other “top-down” methods considering the ease of the reaction conditions, including the low temperature and short production time as well as the availability of the precursor.10,97Fig. 5a showed the UV/vis absorption and PL emission spectra, which revealed the CDs, similar to most CDs, were excitation wavelength dependent98–100 and the PL decay lifetime was measured with an acceptable value of 5.72 ± 0.05 ns. In terms of application, the strong blue PL and sufficient PL decay lifetime ensured the CDs could work as a good imaging probe when they were incubated with HeLa cells (Fig. 5b).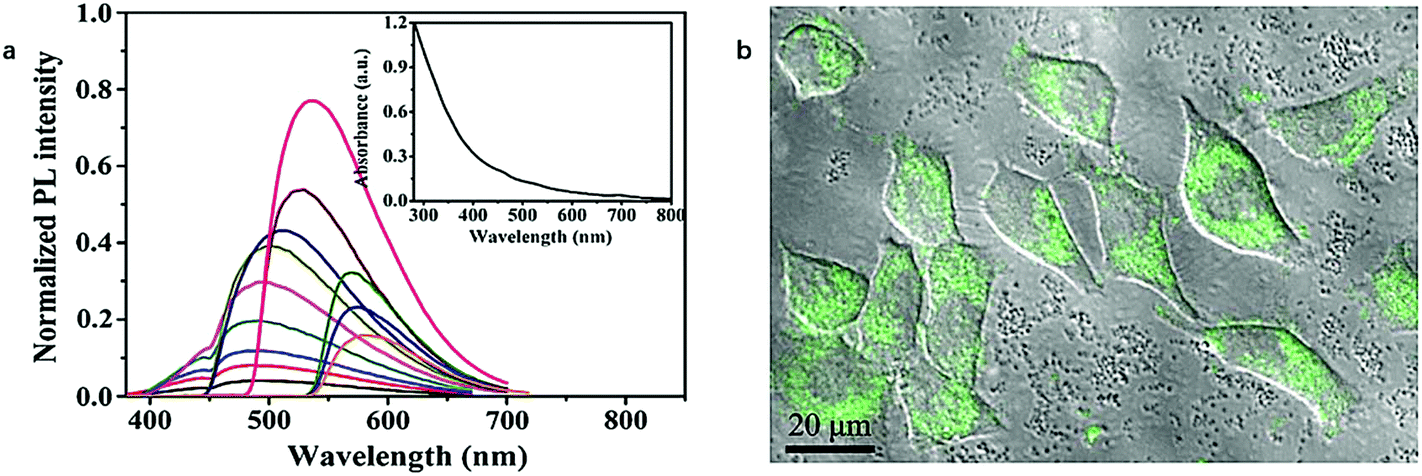 | ||
| Fig. 5 (a) Normalized PL emission spectrum with excitation wavelength increasing from 310 nm in 20 nm increment (inset: UV/vis absorption spectrum of CDs obtained); (b) confocal microscopy image of HeLa cell incubated with CDs (λex = 488 nm). Figure adapted from ref. 82 with permissions from Elsevier. | ||
Lamp or candle soot101,102 after burning is always an optimal candidate as the precursor of CDs. Liu101 and Kumar103 have successively prepared CDs with lamp or candle soot as the initial carbon source. And in order to improve the PL emission ability, Liu et al.20 dispersed the candle soot into 25 M NaOH aqueous solution to fabricate fluorescent hydroxyls-coated CDs with a PL decay lifetime of 9.5 ns, which indicates the radiative recombination nature of excitons. The advantage of hydroxyl-coated CDs over the regular carboxyl-coated CDs was the surface hydroxyls had strong electron donation ability which would be beneficial for the PL emission while the surface carboxyl has a strong electron withdrawing ability that would reduce the PL intensity and the different surface states of CDs were illustrated in Fig. 6a. In addition, due to the enriched hydroxyl surface, the obtained CDs was studied in the detection of metals and metal ions such as Hg2+, Cr3+, Al3+, and Fe3+ would easily quench the PL of CDs which provided great opportunity for measuring Cr3+, Al3+, and Fe3+ in human fluids (Fig. 6b). In Kumar and Bohidar's research paper,103 they studied the solvent dependent spectroscopy of a non-functionalized CDs made of lamp soot. The PL decay lifetime tests revealed the average lifetime didn't change in the aromatic solvents which was 4–5 ns, decreased with the polarity of hydrogen bonded solvents such as methanol and ethanol, and increase with the polarity of aprotic solvents such as acetonitrile.
 | ||
| Fig. 6 (a) The schematic illustration of PL emission of hydroxyls-coated CDs better than the regular carboxyls-coated CDs (e−: electrons, h+: holes); (b) The PL quenching effect of CDs by different metal ions (λex = 310 nm). Figure adapted from ref. 20 with permissions from Springer. | ||
In 2015, Nguyen et al.24 performed femtosecond laser ablation of graphite powder within a polyethylene glycol (PEG200 N) solution to prepare CDs. As the PL mechanism of typical CDs is always under debate, researchers proposed two different pathways of electron–hole (exciton) radiative recombination to study the carrier (electron or hole) dynamics in CDs and each pathway had a relaxation time scale. For example, the relaxation of carriers from the carbogenic core to the surface of CDs Nanoarticles owned a relatively longer PL decay lifetime (>14 ns) while the direct radiative recombination of the surface of CDs was much faster with a short decay lifetime of 1.3 ns. Also, they found when the excitation wavelength was short, the short decay lifetime was dominant which was explained by the relaxation of carriers from carbogenic core to the surface of CDs. Meanwhile, the long PL decay lifetime was measured when the excitation wavelength used was longer, which could be mainly due to the direct relaxation of carriers on the surface of CDs. The mechanism also worked to account for why some CDs are excitation wavelength dependent at longer wavelengths while independent at shorter wavelengths.
3.2 Bottom-up approach for carbon dots’ preparations
In addition to the “top-down” approaches to synthesize CDs mentioned above, the “bottom-up” approaches are the other and more prevalent branch of the preparation strategy to create CDs. Regarding the initial materials, “bottom-up” approaches usually take use of small organic molecules such as citric acid,104,105 as previously discussed, as a monomer-like unit modified by amine compounds to fabricate polymeric CDs. Even though there seem more options, compared in terms of PL decay lifetime of CDs obtained from “top-down” approach, “bottom-up” approach doesn't show any apparent improvement.With citric acid as the initial material, Yang et al.58 in 2013 reported a nitrogen-doped carbon-rich CDs synthesized from ammonium citrate via a hydrothermal treatment. Interestingly, the CDs were excitation wavelength independent when they were excited from 245 to 395 nm and the emission wavelength was 437 nm measured from the PL spectrum (Fig. 7a). The PL decay profile of the CDs shown in Fig. 7b exhibited a single exponential decay kinetic of the CDs excited at 337 nm with a PL decay lifetime of 10.6 ns, which showed a relatively long radiative recombination of the excitons with PL emission.
 | ||
| Fig. 7 PL emission spectrum of CDs excited from 245 to 395 nm (a); PL decay lifetime profile of the CDs (b). Figure adapted from ref. 58 with permission The Royal Society of Chemistry. | ||
As an excellent carbon source to prepare CDs, citric acid was again employed by Li et al.106 in 2014 to synthesize highly photoluminescent CDs. The same preparation was performed at four different temperatures (130, 160, 200 and 240 °C) and the PL properties such as QY and PL decay lifetime varied depending on the amino-passivation degree of the surface of CDs which was determined by the reaction temperature (Fig. 8a). In comparison, the CDs obtained at 160 °C owned the highest QY of 44.7% and the longest PL decay lifetime of 7.13 ns which was due to the highest amino-passivation on the surface. Another interesting difference between the CDs made at different temperatures was observed in the PL emission spectra. In the spectra, we can observe the CDs made at 160 °C are excitation wavelength independent while the CDs synthesized at 240 °C are excitation wavelength dependent (Fig. 8b). The excitation wavelength independent behavior of CDs made at 160 °C relied on the single surface state, while as for CDs made at 240 °C, when excitation energy was higher than the energy gap, emission was no longer mainly related to the energy gap transition and it was controlled by the surface state transition. Also, the metal ion test showed the best quenching effect of Fe3+ and Be2+ on the CDs, which showed the selectivity and sensitivity of the CDs to Fe3+ and Be2+, and the detection limit of toxic Be2+ was as low as 23.3 μM (Fig. 8c).
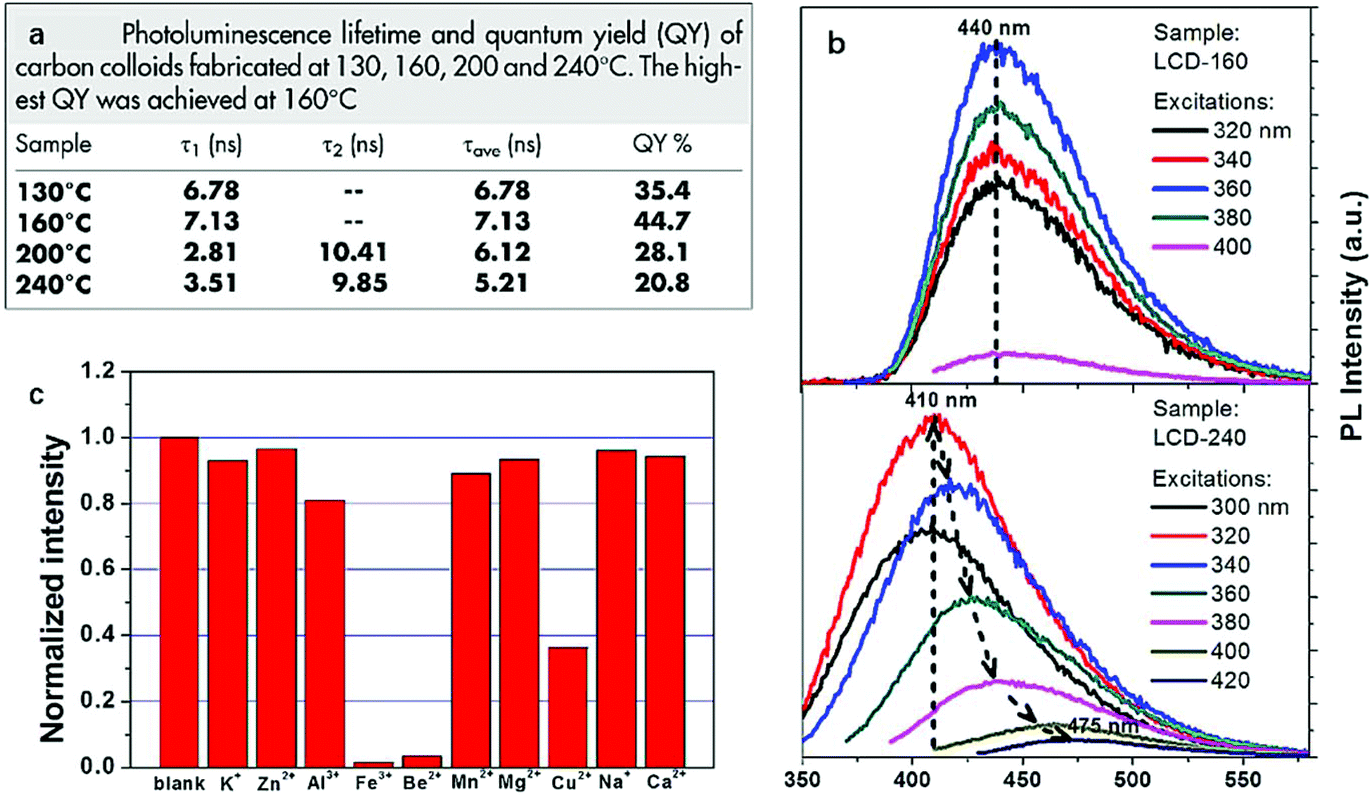 | ||
| Fig. 8 PL decay lifetime and quantum yield of CDs made at 130, 160, 200 and 240 °C (a); the PL emission spectra of CDs made at 160 and 240 °C (b); the quenching effect of different ions on the CDs. Figure adapted from ref. 106 with permissions from Nature Research. | ||
In 2015, Liu et al.105 prepared nitrogen doped CDs (N-CDs) by using citric acid and ammonium hydroxide through a hydrothermal route. Even though the PL decay lifetime and QY have not been remarkably improved which were 40.5% and 9.03 ns, respectively, the selective detection of Hg2+ in real water with a detection limit of 0.087 μM was still of great significance due to the severe environmental and health problems caused by Hg2+.107
In order to improve the PL behaviors of CDs, a great deal of research involving enormous efforts including the selection of the reaction precursors and optimization of the synthetic approach have been continually sought.108 Zheng et al.22 reported a novel pH-sensitive N-CDs with the highest QY (93.3%, as previously discussed) and longest PL decay lifetime (19.5 ns) among all the CDs reported so far. The whole synthesis took citric acid and tris-(hydroxymethyl)aminomethane (Tris) as precursors under 160 °C with the assistance of microwave digestion treatment for 10 min. The CDs in different pH environment exhibited different PL behaviors and based on different pH environments, the surface of the CDs was rich in H or OH, namely CDs-H and CDs-OH. The emission of CDs-H was found to be excitation wavelength dependent while that of CDs-OH was free of excitation wavelength dependence. Also, the PL decay lifetime was dependent on the emission wavelength for CDs-H while insensitive to emission wavelength for CDs-OH. The difference resulted from the uniform surface state of CDs-OH due to the deprotonation of carboxylic groups on the surface.
Compared between in vivo and in vitro experiment, CDs are more commonly tested as a potential probe for in vitro imaging and sensing. However, in 2016, Kalytchuk et al.23 prepared CDs with an average size of 4.5 nm in diameter with citric acid as a carbon source and L-cysteine as the nitrogen and sulfur dopant of the CDs via a hydrothermal route. The CDs exhibited temperature-dependent PL lifetimes decay (Fig. 9a) and their PL decay lifetime monotonically decreased from 11.0 to 5.3 ns as temperature increased from 2 to 80 °C. In addition, their PL lifetime was free of the wide variation of pH, concentration of CDs, and solution ionic strengths. Also, as the cell uptake and cytotoxicity experiment of CDs showed, the CDs have low toxicity and excellent biocompatibility, which laid the foundation of the CDs as an intracellular thermal sensor. As a result, the temperature, determined using the calibration curve based on the PL decay lifetime, extracted from PL transients recorded every 15 min for 24 h of the HeLa cells cultured in CDs aqueous solution agreed with that measured with a reference thermometer, and the absolute average accuracy of temperature detection was 0.27 °C (Fig. 9b–e). Therefore, due to the low toxicity and sole temperature-dependent PL lifetime behavior, the CDs are a great Nanorobe of the intracellular temperature.
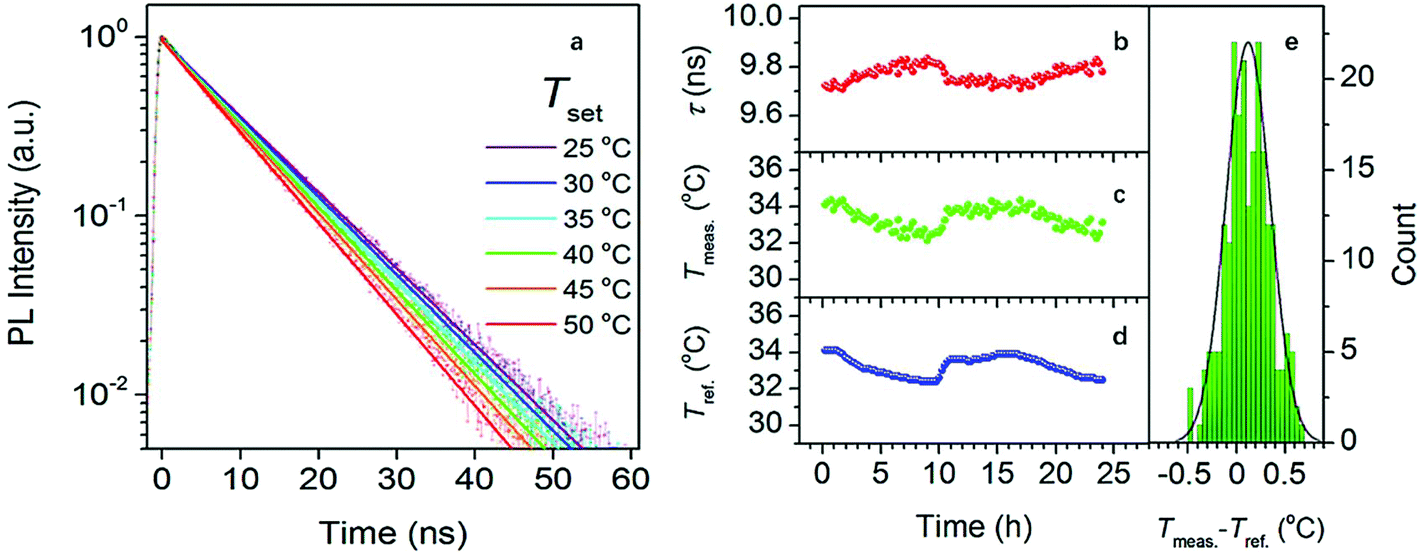 | ||
| Fig. 9 (a) PL emission decays of HeLa cells cultured in CDs aqueous solution with a concentration of 500 μg mL−1 at different temperatures; (b) PL decay lifetimes extracted from the PL transients recorded every 15 min for 24 h of HeLa cells cultured in CDs aqueous solution (500 μg mL−1); (c) temperatures obtained using the calibration curve based on a calibration curve described by an equation: T = 330.59 − 94.99τ + 11.87τ2 − 0.54τ3, Radj2 = 0.998, T, temperature; τ, lifetime; (d) temperatures measured by a reference thermometer; (e) histogram revealing the deviation between (c) and (d), the solid line is the distribution curve. Figure adapted from ref. 23 with permissions from the American Chemical Society. | ||
3.3 Development of phosphorescence in carbon dots
Recently the exciting development of ultralong-lifetime, room-temperature phosphorescence has been observed in CDs. Lin and coworkers109,110 used phosphoric acid with either ethanolamine or EDA to generate CDs which displayed phosphorescence to the naked eye for up to 10 s. They showed the lifetime to be 1.46 and 1.39 s, respectively for the CDs made with ethanolamine and EDA. These CDs were used for security feature printing. As would be expected, the phosphorescence was quenched in solution and was only observed in the solid state. This phosphorescence was attributed to the doping of phosphorous into the CDs structure which produced the long lifetime emission.Phosphorescence in CDs has also been achieved without doping phosphorous. Long et al.111 used a solvothermal method with glucose and triethylamine trihydrofluoride to prepare CDs which exhibited phosphorescence with a lifetime of 1045 ms XPS data showed 14% nitrogen and 7% fluorine. They believed the phosphorescence was due to the low energy difference between the singlet and triplet states for the C–N/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds. Additionally, the semi-ionic nature of the C–F bond stabilized the triplet state and reduced quenching by oxygen. Similarly, Gao et al.112 used glucose and aspartic acid to generate CDs with a phosphorous lifetime of 747 ms. Here they claim the poly-aspartic acid chains on the surface of the CDs creates a barrier to air and moisture under ambient conditions which disallows the quenching of emission in the solid state. Both previous examples allow phosphorescence through doping with heteroatoms which are not phosphorous. Other studies have shown that hydrogen bonding is an important factor in the phosphorescence of CDs.
N bonds. Additionally, the semi-ionic nature of the C–F bond stabilized the triplet state and reduced quenching by oxygen. Similarly, Gao et al.112 used glucose and aspartic acid to generate CDs with a phosphorous lifetime of 747 ms. Here they claim the poly-aspartic acid chains on the surface of the CDs creates a barrier to air and moisture under ambient conditions which disallows the quenching of emission in the solid state. Both previous examples allow phosphorescence through doping with heteroatoms which are not phosphorous. Other studies have shown that hydrogen bonding is an important factor in the phosphorescence of CDs.
In 2018, Li et al.113 used citric acid and urea to generate CDs which were then passivated with cyanuric acid. These CDs surprisingly exhibited phosphorescence in an aqueous suspension with a lifetime of 687 ms. This lifetime was much higher than the lifetime for the dry powder which was 253 ms. This was attributed to the increased rigidity of the system introduced by the hydrogen bonding of water and cyanuric acid with the CO bond of CDs. Hydrogen bonding also played an important role in a study by Yue and coworkers.114 CDs prepared from citric acid and folic acid displayed a phosphorescence lifetime of 705 ms in pH 11.5. This basic pH displayed a longer lifetime than did neutral or acidic aqueous solutions. The basic pH enhanced the phosphorescence through deprotonating the carboxylic groups and increasing the conjugation of the electronic system. This in addition to the intraparticle hydrogen bonding greatly increased the lifetime and QY of phosphorescence.
4. Developments in understanding of carbon dots’ photoluminescence mechanism
4.1 Current theories for PL mechanism of carbon dots
Soon after the discovery of CDs in 2004, researchers began to speculate regarding the nature of CDs’ PL and why they possessed excitation-dependent emission.87 Three main theories emerged, namely: quantum confinement, molecular state, and surface state.30 The theory of quantum confinement in CDs was a natural inquiry to pursue, as metal-based quantum dots are well known to possess emission based on this phenomenon.115 While there are several papers published using this explanation to explain the PL mechanism of CDs, it is not the most common as many CDs systems simply do not have the data to support this theory. Another common theory suggests the synthesis of different molecular fragments which are attached to the surface of CDs in the preparation process. This explanation is referred to as a “molecular state”. There are many papers with strong evidence to support this theory, but the scope of this explanation is limited to certain CDs preparations. Several CDs preparations using citric acid have used this explanation for the PL mechanism.116,117 Additionally, there has been some studies, which used a fluorescent precursor in the preparation method, to explain their CDs PL using molecular state.31 These claimed the precursor/precursor fragment is on the surface of CDs. While there is strong evidence for this theory in some CDs systems, it is certainly limited based on the precursor and cannot completely explain the excitation-dependent emission CDs usually possess. The third and most common theory that is used is the surface state-controlled PL. The frequent use of this explanation has caused the general acceptance of this theory for the PL mechanism of CDs.30 However, very infrequently is the origin of this theory traced back to solid-state physics and semiconductors. Additionally, when surface-state controlled PL is offered as an explanation for CDs PL, it is not stated if the concept can be directly applied to CDs or if modifications are necessary. In view of this, a greater understanding of surface states is needed to understand CDs’ PL.4.2 Surface states in semiconductors
The concept of surface state controlled electronic properties/structure has been developed by solid-state physicists to describe the electronic properties of semiconductors.118,119 This concept was first applied to CDs in 2006.8 Surface states in semiconductors are often classified into intrinsic and extrinsic surface states. Intrinsic surface states are electronic states which result in the termination of the elemental lattice at the semiconductor/vacuum interface. Intrinsic surface states are modeled computationally as Shockley or Tamm states. Shockley surface states are modeled using a “nearly-free electron” model and can be used accurately for narrow gap semiconductors (bandgaps approximately 1–2 eV). Tamm states are modeled using the “tight-bound” approach and are expressed as a linear combination of atomic orbitals (LCAO). These states are valuable for broad gap semiconductors (2–4 eV bandgaps).120Extrinsic surface states in semiconductors can arise from defects in the crystal lattice at the surface, adsorbates to the semiconductor, and interfaces between two materials. Extrinsic surface states are much more difficult to characterize and model than intrinsic surface states. Additionally, these surface states are often unique for a particular system, depending on the atoms and structure present, and the defects in that structure.121 While it is not always explicitly stated, extrinsic surface states are claimed to control the PL of CDs in many papers. To understand what has been reported for CDs’ PL mechanism, attention will now be turned to the application of this concept to CDs.
4.3 Recent developments in understanding carbon dots photoluminescence
Surface state-controlled PL in CDs was first suggested by Sun et al.8 in 2006 for CDs prepared by laser ablation and passivated by PEG1500 N. They proposed surface energy traps which are stabilized by passivation with PEG1500 N. Since then there has been many developments and variations of this idea. In 2015, Ding et al.122 produced CDs from p-phenylenediamine and urea and separated different fractions through column chromatography. They found the wavelength of PL increased with the increasing degree of oxidation (as seen by FTIR and XPS). They attribute the PL of CDs to conjugated sp2 carbons on the surface of CDs whose bandgap can reduced by the degree of oxidative surface defects as shown in Fig. 10. Modeling of graphene oxide through density functional theory supports the distortion of the electronic environment by oxidation on sp2 carbons.123 | ||
| Fig. 10 Image of different CDs fractions under UV light and the modeling of their bandgap based on surface oxidation. Figure adapted from ref. 122 with permissions from the American Chemical Society. | ||
Zhang et al.124 also attributed excitation-dependent emission to surface oxidation. They prepared CDs (CD1) using an electrochemical preparation from urea, graphite, and NaOH. They also prepared CDs (CD2) from citric acid and trisaminomethane using a microwave preparation. Both CD1 and CD2 displayed blue PL, but CD1 possessed excitation wavelength-independent emission, whereas CD2 was dependent on excitation wavelength. XPS atomic ratio showed that CD2 possessed much higher surface oxygen levels. The authors conclude CD2 has many different oxygen functionalities in the n–π* region of CDs absorption (ca. 350 nm) which creates different surface states resulting in the shifting emission of CD2. Both preceding studies use surface oxidation to explain excitation-dependent emission as well as the PL color of CDs. However, the shifting which results from Ding et al.122 work spans a much larger region (blue to red emission) than does the work by Zhang et al.124 (just blue emission). This could indicate both the complexity of the PL process in CDs as well as the heavy dependence of CDs’ optical properties on precursor and preparation method.
Other atoms have been hypothesized to influence CDs PL color. Han et al.125 used a hydrothermal method with hydroquinone and EDA to generate CDs which could be separated via silica-gel chromatography to give blue, green, and yellow emissive CDs fractions. They determined the PL color to be related to the surface state due to the influences of pH and solvent on emission of the three fractions. Through XPS analysis of the N 1s binding energies, they were able to show the CN percentage increased with the increasing wavelength of emission. Other nitrogen groups (–NH2 and C–N–C) displayed no correlation with PL color of CDs. As further evidence for imine-controlled emission, the excitation spectrum of CDs overlapped with n–π* transition for CN in the absorbance spectrum. Their concluding hypothesis states that imine groups on the surface of CDs creates surface defects which introduces energy levels into the bandgap of CDs, thus reducing the energy of the emitted PL.126
Recently, Yuan et al.127 compared the optical and surface properties of 4 different CDs (Fig. 11a). The color of each CDs image in Fig. 11a represents the PL spectra of the produced CDs. When they compared the nitrogen content of the four CDs (Fig. 11d), they could see clear differences for type and amount of nitrogen functionality in each sample. They attribute the red emission to the distortion of p-phenylenediamine. When ethylenediamine (EDA) is introduced into the preparation the amount of pyrrolic and amino nitrogen decreases and the amount of pyridinic nitrogen increases. This leads to the conclusion that pyridinic nitrogen is responsible for the green emission. When PEI is used as a precursor, blue emission is again seen and the pyrrolic nitrogen increases, which suggests the blue emission and pyrrolic nitrogen are related. These relationships are further confirmed by the presence of red and blue emission in CDs prepared from proline. Based on these observations, the authors suggest a representative structure for each CDs and create an energy level diagram to illustrate the effect of surface nitrogen functionalization on the bandgap of CDs (Fig. 11B and C).
 | ||
| Fig. 11 (A) Summary of preparation of CDs. (B) Proposed representative structure for each CDs. (C) Proposed energy level diagram for surface states of CDs. (D) Nitrogen percentages in CDs samples. Figures adapted from ref. 127 with permissions from The Royal Society of Chemistry. | ||
Liu et al.128 used a microwave reaction with formamide and ortho, meta, and para-phenylenediamine separately to form o-CDs, m-CDs, and p-CDs. Surprisingly, the PL color did not follow the isomeric trend as the CDs possessed yellow, blue, and orange PL emission, respectively. All CDs also displayed excitation-independent emission, which they attribute to uniformity of structure. From surface analysis of the CDs through XPS they see varying degrees of heteroatoms present. It was found that as CO/CONH content increased so did the wavelength of emission. This was once again attributed to oxidative surface defects reducing the bandgap in the CDs. Another trend was also observed between non-amino nitrogen content (pyridinic and pyrrolic nitrogen) and QY. The determined QY for p-CDs, m-CDs, and o-CDs was 7.5, 14.3, and 45.0%, respectively. Coinciding with this trend they saw an increase in the non-amino nitrogen content, which they attribute to the increase in the conjugated structure at the CDs surface which promoted radiative recombination. As previously discussed, nitrogen doping is well known to increase the QY for CDs, but this study provides more information on the specific groups which increase QY.
Due to the increasing success of surface state theory to explain CDs PL properties it has become much more common than quantum confinement and molecular state. Quantum confinement is occasionally used to explain the PL of CDs, but it is often used in conjunction with another concept such as surface state.2,29,129,130 In 2018, Liu et al.130 prepared CDs from a perylene derivative and triethylamine and separated the resulting mixture using column chromatography. This yielded 25 fractions with emission ranging from blue to the near infrared. Their characterization showed a rough correlation between size, polarity, and emission wavelength. This led the authors to conclude that the emission of the CDs is roughly adjusted by quantum confinement effects and more subtly adjusted by surface states. In recent years, most researchers have not used quantum confinement as the sole factor for CDs PL mechanism and many studies’ data do not support a size-dependent emission for CDs.
Molecular state is also used occasionally, but it is clear that the concept cannot be applied to all CDs systems. CDs prepared from citric acid and a nitrogen dopant and also precursors which already possess fluorescence are the limit of molecular states’ ability to explain CDs PL.31,131,132 Based on the trends in recent CDs literature, it appears that a surface state controlled PL will be investigated more deeply in the future.
5. Developments in elucidation of carbon dots’ core structure
Similar to graphene quantum dots (GQDs) and polymer dots (PDs), CDs are known for core–shell structure. However, in term of chemical structure, they are distinct. To be specific, GQDs’ core consists of simply single or few layers of graphene, which are connected to chemical groups on the edge to form the shell of GQDs.133 PDs that result from the assembly/aggregation or cross-link of non-conjugated polymers are composed of carbon core and polymer chains as the shell.2 In comparison, CDs’ structure is the combination of a sp3-hybridized matrix of oxygen/nitrogen-containing surface functional groups (defect states) and core (intrinsic states).134 The formation of CDs is a nucleation process with a gradual growth of core and a “self-passivated” shell comprising functional groups.135 In 2018, Ren et al. reported that both core and surface electronic states of CDs contributed to the optical properties and electronic acceptor levels while the chemical nature of the surface groups determined the hydrogen bonding behavior of the CDs.136 It arouses much attention to the functions of the core and the surface chemistry of the shell of CDs.The surface chemistry of CDs shell consists of the chemical structure and functions of the shell. As to the chemical structure, in the shell of CDs, there are various connected or modified functional groups such as oxygen-, amino-based groups or polymer chains, etc., which depends on the starting materials or dopant species and were usually characterized by FTIR and XPS.2 In addition, there ought to be abundant CC and CO conjugate structures that were often observed from UV/vis spectra of CDs.94 Regarding the function, the surface chemistry of shell controls the stability of CDs aqueous solution. The surface zeta potential can reveal the strength of electrostatic repulsion amongst CDs. Specially, the electrostatic repulsion gets weaker when the zeta potential value is smaller, which suggests the less stability of the CDs aqueous solution and ease to aggregate. In addition, the composition (functional groups) of the shell is vital to subsequent functionalization required for many applications.137–139 Furthermore, the shell involves in the unique optical properties and electron donor/acceptor of CDs. For example, CDs have pH-dependent PL, which has been widely reported, and the PL can be monitored by different pH to study the effect of the surface groups on optical properties.140,141 Also, depending on the surface moieties, the surface-related electronic acceptor levels can be modulated, which may affect PL properties of CDs.136
However, even though the shell is widely investigated since the discovery of CDs due to the excellent PL, the core study was not begun until 2008.142 In 2008, Bourlinos et al. put forward that single CD should have a carbogenic core, which is composed of carbonized intermediates with a highly defected structure of co-existing aromatic and aliphatic regions.142 However, due to the highly photoluminescent nature, further characterization regarding the structure was limited then and is still a problem presently. The core is often considered carbogenic without deeper understanding even in many recent research articles.143 Therefore, it is necessary to summarize the development in the understanding of the core to date as well as the role it plays in the PL of CDs.
Owing to the recent wide investigation, current theories points out that there are two types of cores of CDs, graphitic crystalline and amorphous cores, based on the degree of the presence of sp2 carbon in the core.94 Among them, the graphitic crystalline core has been more reported,144,145 and the cores are small in size (2–3 nm) with a lattice spacing of ∼0.2 nm.146 However, sometimes the core exists in the form of two types of cores. For example, Galan's group reported a type of CDs through a 3 min synthesis contained an sp3-enriched crystalline core and the abundant sp3 carbon induced the amorphousness.147 Moreover, the type of cores depends on the synthetic approaches and can be converted between each other.148 In general, reaction temperatures above 300 °C lead to significant graphitization while those below lead to amorphous core,149 unless sp2/sp hybridized carbon was present in the precursor.150 For example, many early synthetic approaches of CDs contained the fragmentation of the macroscopic carbon source, which caused or maintained the crystalline organization in the core of synthesized CDs.151
However, until now, there is still no direct observable result of the core in CDs and the present theory merely results from the evidence according to various characterization techniques such as NMR, XPS, TEM, XRD, and Raman spectroscopy.
TEM, XRD and Raman spectroscopy are direct characterization methods. High-resolution TEM provides a straightforward route to confirm the presence of CDs. Some researchers observed graphitic crystalline structure in Nanoarticles by high-resolution TEM and it was believed to be the core of CDs.152 XRD is also an important technique to help detect the morphology of the CDs core. For example, Martindale et al.148 mentioned in their work the powder XRD pattern of CDs core shows a broad peak centered at 27.0° 2θ consistent with a nanocrystalline graphitic structure. As to Raman spectroscopy, a G band indicates the first-order Raman band of all sp2 hybridized carbon materials while a D band is a defect activated band in sp2 hybridized carbon materials. Herein, in the core study, the ratio of spectral intensities of D and G bands (ID/IG) reflects the defect density of the CDs core. Hola et al.144 synthesized CDs from lauryl (dodecyl) gallate (LG27), propyl gallate (PG27), and methyl gallate (MG27) with a decreasing ID/IG, which was explained by the effect of decreasing alkyl-chain length on the carbogenic core. In addition, upon observing the identical emission spectra of CDs from LG27 and LG 270, they hypothesized a direct excitonic recombination from the core rather than the surface is the major PL emission source. However, the PL mechanism is always under debate and the conclusion above probably only works for their own CDs since it contradicts many other research results as previously discussed. For example, Nguyen et al.153 supported that the PL of CDs resulted from the abundant surface functional groups rather than the core.
In addition, NMR, XPS and TGA are used as the supplementary characterization methods to determine the general structure and properties of the core of CDs.154 In 2013, Giannelis and co-workers155 performed the structural analysis of the surface modifier on three types of CDs by using 1D or 2D high resolution NMR spectroscopy in solution. However, they didn't observe any 1H NMR signal that could be assigned to the CDs core, which was consistent with the core carbonization hypothesis from Bourlinos et al.142,156 So, they proposed a solid-state NMR is needed in the future to fully confirm the core carbonization hypothesis and study the chemical structure of the core. Based on high-resolution XPS measurement, for N-doped CDs the core is not purely composed of carbon.157 Doped-N was also a contributor to the formation of core in cyclic form including pyrrolic, graphitic and pyridinic nitrogen resulting in a strong electron-withdrawing ability within the conjugated C plane. This significance of nitrogen in the construction of CDs was supported by Hill et al. In Hill et al.'s work,147 the morphology of the core of CDs can be tuned by surface passivating agent. The longer linker could help increase the size of core but also might induce the disorder and lower graphitization degree. And the incorporation of nitrogen could also improve the PL by enhancing the core emission, which was later reported by Gregorkiewicz and co-workers.143 Also, in Weijian et al.'s work,157 they performed TGA on both CDs and N-doped CDs. Meanwhile, the TGA measurements exhibited distinct stages for the surface and core of CDs.
The core is closely correlated to the optical properties of CDs. As is known the UV/vis spectra of typical CDs have two typical absorption peaks at around 250 and 350 nm, which could be assigned to π–π* and n–π* transitions of CC and CO, respectively, in the core of CDs.158,159 As discussed previously, the PL of CDs is usually believed to originate from the radiative recombination of excitons in core (carbon-core states) and surface electronic states, molecular states, surface functional groups and surface energy traps. Among them molecular states and carbon-core states were demonstrated for the first time by Giannelis and co-workers160 in CDs prepared from citric acid and ethanolamine, and the radiative recombination of excitons in the surface states are more tunable.161 Later, the presence of three different emission centers including molecular states, aromatic domain states, and carbon-core states was shown by Shamsipur et al.134 in CDs synthesized through pyrolysis of citric acid and ethylenediamine for the first time. And they discussed that the main PL of citric acid-based CDs derives from the molecular states rather than the carbon-core states, which results in lower photostability and higher PL reduction. Meanwhile, compared to the molecular states, the carbon-core states usually emit at shorter wavelengths and exhibit much lower PL QY.160 Also, more uniform functional groups on a highly crystalline core result in higher PL QY. On the contrary, lower QY can be observed for CDs with more traps and fewer surface functional groups.153 Therefore, even though the surface state theory is a dominant theory for the PL mechanism, we can't ignore the contribution of the core in the PL emission especially in the short-wavelength region. According to Gregorkiewicz and co-workers,122 the core related PL emission results from the π–π* transition of sp2 clusters assisted by the quantum confinement effect and the emission at longer wavelengths is known to be related to the hybridized oxygen functional groups with the core.162
The PL mechanism can also be interpreted by the electronic transition involving the coupling of the core and surface states.145 As is concluded from Yu et al.'s work, both surface states and carbon core are critical for the regulation of PL emission since the band gap of surface states (4.5 eV) is narrower than that of the core (5.0 eV).163 In addition, the oxygen and nitrogen elements and related chemical bonds will produce impurity levels in the band gap, which leads to the change of excitation and emission spectra of CDs. As shown in Fig. 12, there may be an energy transfer process between the carbon core and the surface state. The CDs display five emission bands centered at 305, 355, 410, 445, and 500 nm, which are correlated with the electron transition at intrinsic C (4.1 eV), graphitic N (3.5 eV), pyridine N (3.0 eV), amino N (2.8 eV), and CO (2.5 eV) related levels, respectively. With the development of characterization methods, probably one day we will be able to clearly observe and accurately separate the core from the shell. Then many ambiguous questions can get a solid answer, such as the PL mechanism and the structure of the core.
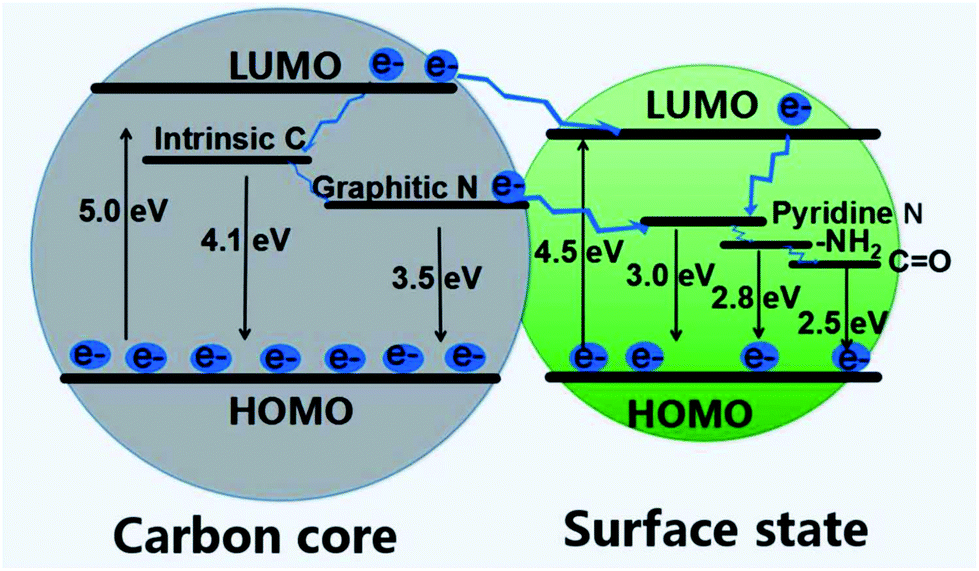 | ||
| Fig. 12 A scheme of energy band structure and possible PL process for CDs. Figure adapted from ref. 163 with permission from MDPI. | ||
6. Conclusion and future perspectives
Some important optical properties have been discussed regarding CDs, namely QY and PL decay lifetime. These are important parameters in assessing the utility of CDs in application such as imaging and sensing. Carbon sources used for preparing CDs have ranged from graphite, proteins, small molecules, and food products. Citric acid is the most commonly used precursor at the moment and has produced CDs with high QYs and long PL decay lifetime, but many different carbon sources have shown promising properties. A more significant factor appears to be heteroatom doping with nitrogen, sulfur, boron, or phosphorous. The effect of heteroatom doping has been shown many times to have a positive effect on both QY and PL lifetime and appears to be a significant factor in determining the optical properties. Additionally, the method used to prepare CDs influences the optical properties of CDs. Thermal methods are most often used and often give favorable properties, but microwave methods have also given rise to CDs with high QYs and long PL lifetimes as well. Top-down methods such as laser ablation or electrochemical methods do not often produce CDs which are used in applications such as sensing and imaging, as the QY from these CDs tends to be lower. In contrast, the obtained PL decay lifetime doesn't have an obvious enhancement regarding either “top-down” or “bottom-up” approach.Additionally, the PL mechanism of CDs has been discussed, mainly for the surface state-controlled PL. These have been shown to involve oxygen or nitrogen functionalities which create surface defects on the surface of CDs. These defects can modulate the color of PL as well as create an heterogenous electronic environment resulting in excitation wavelength-dependent emission.
The core of CDs remains unclear, but many works have been presented to support graphitic, crystalline, and amorphous cores. The discord in the literature reflects the variation in CDs properties based on preparation methods. New characterization methods are needed to provide clarity in this area.
Moving forward it is important to ensure that all literature values for QY of CDs are reliable. As previously stated, CDs possess favorable optical properties and much work is currently being done to realize their full potential. Proper selection of a QY standard and correct experimental techniques are essential to correctly reporting QYs. When this is not done, and exaggerated values are reported, reproducibility issues often occur which casts doubt on any high QYs reported in literature for CDs. Also, PL decay life is an important property that should not be missing for the analysis to understand the PL mechanism of CDs and broadening their application.
Further improvement is needed in the QYs of CDs with long wavelength emission. High values, above 90%, have been reported for emission in the blue region of light, but QY values measured at longer wavelengths are often low or are compromised due to dubious QY standard choice.122,164 There is great potential for applications in bio-imaging to avoid the autofluorescence of tissues and other areas for CDs which possess emission above 650 nm, in the red or infrared region. Compared to the QY measurement, PL decay lifetime hasn't been outlined and it is always under 20 ns, which remains to be enhanced.
As mentioned for the optical parameters above, the wide diversity of preparation methods and precursors creates difficulty in illuminating the PL mechanism and core structure of CDs. In the future it will be important to develop new characterization in these areas, as well as to rigorously compare new data with what has been previously reported. At the moment it is unclear if there are different processes occurring for different preparation methods (top-down versus bottom-up) or different precursors. New results on these topics should be compared to previous results for similar systems.
The great improvements made in the optical properties of CDs in recent years have been extensively discussed. In the areas of PL decay lifetime, QY reproducibility, long wavelength emission of CDs, and the understanding of PL mechanism and core structure, there is still more room for improvement. However, CDs have shown great potential in the areas of imaging and sensing, and their applications will continue to expand in the coming years.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding sources (R. M. L.) include the National Science Foundation through grant 1809060 and the National Institute of Health through grant R21AR072226.References
- X. Xu, R. Ray, Y. Gu, H. J. Ploehn, L. Gearheart, K. Raker and W. A. Scrivens, J. Am. Chem. Soc., 2004, 126, 12736–12737 CrossRef CAS PubMed.
- S. Zhu, Y. Song, X. Zhao, J. Shao, J. Zhang and B. Yang, Nano Res., 2015, 8, 355–381 CrossRef CAS.
- S.-T. Yang, L. Cao, P. G. Luo, F. Lu, X. Wang, H. Wang, M. J. Meziani, Y. Liu, G. Qi and Y.-P. Sun, J. Am. Chem. Soc., 2009, 131, 11308–11309 CrossRef CAS PubMed.
- S.-T. Yang, X. Wang, H. Wang, F. Lu, P. G. Luo, L. Cao, M. J. Meziani, J.-H. Liu, Y. Liu and M. Chen, J. Phys. Chem. C, 2009, 113, 18110–18114 CrossRef CAS PubMed.
- K. Qu, J. Wang, J. Ren and X. Qu, Chem. Eur. J, 2013, 19, 7243–7249 CrossRef CAS PubMed.
- Z. Lin, W. Xue, H. Chen and J.-M. Lin, Anal. Chem., 2011, 83, 8245–8251 CrossRef CAS PubMed.
- C. Ding, A. Zhu and Y. Tian, Acc. Chem. Res., 2013, 47, 20–30 CrossRef PubMed.
- Y.-P. Sun, B. Zhou, Y. Lin, W. Wang, K. S. Fernando, P. Pathak, M. J. Meziani, B. A. Harruff, X. Wang and H. Wang, J. Am. Chem. Soc., 2006, 128, 7756–7757 CrossRef CAS PubMed.
- P. G. Luo, S. Sahu, S.-T. Yang, S. K. Sonkar, J. Wang, H. Wang, G. E. LeCroy, L. Cao and Y.-P. Sun, J. Mater. Chem. B, 2013, 1, 2116–2127 RSC.
- S. Li, L. Wang, C. C. Chusuei, V. M. Suarez, P. L. Blackwelder, M. Micic, J. Orbulescu and R. M. Leblanc, Chem. Mater., 2015, 27, 1764–1771 CrossRef CAS.
- A. Małolepszy, S. Błonski, J. Chrzanowska-Giżyńska, M. Wojasiński, T. Płocinski, L. Stobinski and Z. Szymanski, Appl. Phys. A: Mater. Sci. Process., 2018, 124, 282–288 CrossRef.
- Y. Wang, K. Wang, Z. Han, Z. Yin, C. Zhou, F. Du, S. Zhou, P. Chen and Z. Xie, J. Mater. Chem. C, 2017, 5, 9629–9637 RSC.
- P. Zuo, X. Lu, Z. Sun, Y. Guo and H. He, Microchim. Acta, 2016, 183, 519–542 CrossRef CAS.
- Q. Xu, P. Pu, J. Zhao, C. Dong, C. Gao, Y. Chen, J. Chen, Y. Liu and H. Zhou, J. Mater. Chem. A, 2015, 3, 542–546 RSC.
- S. Yang, J. Sun, X. Li, W. Zhou, Z. Wang, P. He, G. Ding, X. Xie, Z. Kang and M. Jiang, J. Mater. Chem. A, 2014, 2, 8660–8667 RSC.
- Y. Dong, H. Pang, H. B. Yang, C. Guo, J. Shao, Y. Chi, C. M. Li and T. Yu, Angew. Chem., Int. Ed., 2013, 52, 7800–7804 CrossRef CAS PubMed.
- Y. Choi, B. Kang, J. Lee, S. Kim, G. T. Kim, H. Kang, B. R. Lee, H. Kim, S.-H. Shim and G. Lee, Chem. Mater., 2016, 28, 6840–6847 CrossRef CAS.
- D. Qu, M. Zheng, L. Zhang, H. Zhao, Z. Xie, X. Jing, R. E. Haddad, H. Fan and Z. Sun, Sci. Rep., 2014, 4, 5294–5304 CrossRef CAS PubMed.
- X. Li, S. Zhang, S. A. Kulinich, Y. Liu and H. Zeng, Sci. Rep., 2014, 4, 4976–4983 CrossRef CAS.
- L. Liu, Y. Li, L. Zhan, Y. Liu and C. Huang, Sci. China: Chem., 2011, 54, 1342–1347 CrossRef CAS.
- Y. Dong, H. Pang, H. B. Yang, C. Guo, J. Shao, Y. Chi, C. M. Li and T. Yu, Angew. Chem., Int. Ed., 2013, 52, 7800–7804 CrossRef CAS PubMed.
- C. Zheng, X. An and J. Gong, RSC Adv., 2015, 5, 32319–32322 RSC.
- S. Kalytchuk, K. Poláková, Y. Wang, J. P. Froning, K. Cepe, A. L. Rogach and R. Zbořil, ACS Nano, 2017, 11, 1432–1442 CrossRef CAS PubMed.
- V. Nguyen, J. Si, L. Yan and X. Hou, Carbon, 2015, 95, 659–663 CrossRef CAS.
- Y. Song, S. Zhu, J. Shao and B. Yang, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 610–615 CrossRef CAS.
- S. A. Hill, D. Benito-Alifonso, S. A. Davis, D. J. Morgan, M. Berry and M. C. Galan, Sci. Rep., 2018, 8, 12234–12243 CrossRef PubMed.
- D. Chen, H. Gao, X. Chen, G. Fang, S. Yuan and Y. Yuan, ACS Photonics, 2017, 4, 2352–2358 CrossRef CAS.
- Y. Shen, Y. Liang, Y. Wang, C. Liu and X. Ren, J. Nanopart. Res., 2018, 20, 229–238 CrossRef.
- W. Liu, C. Li, X. Sun, W. Pan, G. Yu and J. Wang, Nanotechnology, 2017, 28, 485705–485717 CrossRef PubMed.
- S. Zhu, Y. Song, J. Wang, H. Wan, Y. Zhang, Y. Ning and B. Yang, Nano Today, 2017, 13, 10–14 CrossRef CAS.
- X. Pan, Y. Zhang, X. Sun, W. Pan, G. Yu, Q. Zhao and J. Wang, J. Lumin., 2018, 204, 303–311 CrossRef CAS.
- P. M. Gharat, J. Chetodhil, A. P. Srivastava, P. Praseetha, H. Pal and S. D. Choudhury, Photochem. Photobiol. Sci., 2019, 18, 110–119 RSC.
- B. C. Martindale, G. A. Hutton, C. A. Caputo, S. Prantl, R. Godin, J. R. Durrant and E. Reisner, Angew. Chem., Int. Ed., 2017, 56, 6459–6463 CrossRef CAS PubMed.
- L. Sciortino, A. Sciortino, R. Popescu, R. Schneider, D. Gerthsen, S. Agnello, M. Cannas and F. Messina, J. Phys. Chem. C, 2018, 122, 19897–19903 CrossRef CAS.
- S. Lu, L. Sui, M. Wu, S. Zhu, X. Yong and B. Yang, Adv. Sci., 2019, 6, 1801192–1801199 CrossRef PubMed.
- J. Wang and J. Qiu, J. Mater. Sci., 2016, 51, 4728–4738 CrossRef CAS.
- J. Zhou, H. Zhou, J. Tang, S. Deng, F. Yan, W. Li and M. Qu, Microchim. Acta, 2017, 184, 343–368 CrossRef CAS.
- P. Namdari, B. Negahdari and A. Eatemadi, Biomed. Pharmacother., 2017, 87, 209–222 CrossRef CAS PubMed.
- F. Zu, F. Yan, Z. Bai, J. Xu, Y. Wang, Y. Huang and X. Zhou, Microchim. Acta, 2017, 184, 1899–1914 CrossRef CAS.
- F. Yan, Y. Jiang, X. Sun, Z. Bai, Y. Zhang and X. Zhou, Microchim. Acta, 2018, 185, 424 CrossRef PubMed.
- M. L. Liu, B. B. Chen, C. M. Li and C. Z. Huang, Green Chem., 2019, 21, 449–471 RSC.
- S. Fery-Forgues and D. Lavabre, J. Chem. Educ., 1999, 76, 1260 CrossRef CAS.
- J. Olmsted, J. Phys. Chem., 1979, 83, 2581–2584 CrossRef CAS.
- Y.-P. Sun, X. Wang, F. Lu, L. Cao, M. J. Meziani, P. G. Luo, L. Gu and L. M. Veca, J. Phys. Chem. C, 2008, 112, 18295 CrossRef CAS PubMed.
- H. Gonçalves, P. A. Jorge, J. Fernandes and J. C. E. da Silva, Sens. Actuators, B, 2010, 145, 702–707 CrossRef.
- S. Qu, X. Liu, X. Guo, M. Chu, L. Zhang and D. Shen, Adv. Funct. Mater., 2014, 24, 2689–2695 CrossRef CAS.
- Y. H. Yuan, Z. X. Liu, R. S. Li, H. Y. Zou, M. Lin, H. Liu and C. Z. Huang, Nanoscale, 2016, 8, 6770–6776 RSC.
- J. Wang, P. Zhang, C. Huang, G. Liu, K. C.-F. Leung and Y. X. J. Wáng, Langmuir, 2015, 31, 8063–8073 CrossRef CAS PubMed.
- S. Yu, K. Chen, F. Wang, Y. Zhu and X. Zhang, Luminescence, 2017, 32, 970–977 CrossRef CAS PubMed.
- J. Kudr, L. Richtera, K. Xhaxhiu, D. Hynek, Z. Heger, O. Zitka and V. Adam, Biosens. Bioelectron., 2017, 92, 133–139 CrossRef CAS PubMed.
- V. Roshni and O. Divya, Curr. Sci., 2017, 112, 385–390 CrossRef CAS.
- J.-Y. Li, Y. Liu, Q. W. Shu, J.-M. Liang, F. Zhang, X.-P. Chen, X.-Y. Deng, M. T. Swihart and K. J. Tan, Langmuir, 2017, 33, 1043–1050 CrossRef CAS PubMed.
- Y. Dong, J. Shao, C. Chen, H. Li, R. Wang, Y. Chi, X. Lin and G. Chen, Carbon, 2012, 50, 4738–4743 CrossRef CAS.
- Y. Song, S. Zhu, S. Zhang, Y. Fu, L. Wang, X. Zhao and B. Yang, J. Mater. Chem. C, 2015, 3, 5976–5984 RSC.
- L. Shi, J. H. Yang, H. B. Zeng, Y. M. Chen, S. C. Yang, C. Wu, H. Zeng, O. Yoshihito and Q. Zhang, Nanoscale, 2016, 8, 14374–14378 RSC.
- L. Li, C. Lu, S. Li, S. Liu, L. Wang, W. Cai, W. Xu, X. Yang, Y. Liu and R. Zhang, J. Mater. Chem. B, 2017, 5, 1935–1942 RSC.
- S. Chandra, T. K. Mahto, A. R. Chowdhuri, B. Das and S. K. Sahu, Sens. Actuators, B, 2017, 245, 835–844 CrossRef CAS.
- Z. Yang, M. Xu, Y. Liu, F. He, F. Gao, Y. Su, H. Wei and Y. Zhang, Nanoscale, 2014, 6, 1890–1895 RSC.
- H. Wang, P. Gao, Y. Wang, J. Guo, K.-Q. Zhang, D. Du, X. Dai and G. Zou, APL Mater., 2015, 3, 086102–086108 CrossRef.
- Y. Zhou, A. Desserre, S. K. Sharma, S. Li, M. Marksberry, C. Chusuei, P. Blackwelder and R. M. Leblanc, ChemPhysChem, 2017, 18, 890–897 CrossRef CAS PubMed.
- Y. Ji, Y. Zhou, E. Waidely, A. Desserre, M. H. Marksberry, C. C. Chusuei, A. A. Dar, O. A. Chat, S. Li and R. M. Leblanc, Inorg. Chim. Acta, 2017, 468, 119–124 CrossRef CAS.
- Y. Zhou, P. Liyanage, D. Geleroff, Z. Peng, K. Mintz, S. Hettiarachchi, R. Pandey, C. Chusuei, P. Blackwelder and R. Leblanc, ChemPhysChem, 2018, 19, 2589–2597 CrossRef CAS PubMed.
- Z.-H. Wen and X.-B. Yin, RSC Adv., 2016, 6, 27829–27835 RSC.
- N. Parvin and T. K. Mandal, Microchim. Acta, 2017, 184, 1117–1125 CrossRef CAS.
- M. J. Krysmann, A. Kelarakis, P. Dallas and E. P. Giannelis, J. Am. Chem. Soc., 2011, 134, 747–750 CrossRef PubMed.
- W.-J. Niu, Y. Li, R.-H. Zhu, D. Shan, Y.-R. Fan and X.-J. Zhang, Sens. Actuators, B, 2015, 218, 229–236 CrossRef CAS.
- S. Tao, Y. Song, S. Zhu, J. Shao and B. Yang, Polymer, 2017, 116, 472–478 CrossRef CAS.
- Y. Yang, W. Kong, H. Li, J. Liu, M. Yang, H. Huang, Y. Liu, Z. Wang, Z. Wang, T.-K. Sham, J. Zhong, C. Wang, Z. Liu, S.-T. Lee and Z. Kang, ACS Appl. Mater. Interfaces, 2015, 7, 27324–27330 CrossRef CAS PubMed.
- K. J. Mintz, G. Mercado, Y. Zhou, Y. Ji, S. D. Hettiarachchi, P. Y. Liyanage, R. R. Pandey, C. C. Chusuei, J. Dallman and R. M. Leblanc, Colloids Surf., B, 2019, 176, 488–493 CrossRef CAS PubMed.
- Q. Xu, T. Kuang, Y. Liu, L. Cai, X. Peng, T. S. Sreeprasad, P. Zhao, Z. Yu and N. Li, J. Mater. Chem. B, 2016, 4, 7204–7219 RSC.
- Q. Xu, B. Li, Y. Ye, W. Cai, W. Li, C. Yang, Y. Chen, M. Xu, N. Li and X. Zheng, Nano Res., 2017, 11, 3691–3701 CrossRef.
- Q. Xu, R. Su, Y. Chen, S. Theruvakkattil Sreenivasan, N. Li, X. Zheng, J. Zhu, H. Pan, W. Li and C. Xu, ACS Appl. Nano Mater., 2018, 1, 1886–1893 CAS.
- L. Hu, Y. Sun, S. Li, X. Wang, K. Hu, L. Wang, X.-J. Liang and Y. Wu, Carbon, 2014, 67, 508–513 CrossRef CAS.
- C. Yang, R. P. Thomsen, R. Ogaki, J. Kjems and B. M. Teo, J. Mater. Chem. B, 2015, 3, 4577–4584 RSC.
- X. Yang, Y. Wang, X. Shen, C. Su, J. Yang, M. Piao, F. Jia, G. Gao, L. Zhang and Q. Lin, J. Colloid Interface Sci., 2017, 492, 1–7 CAS.
- Z.-C. Yang, M. Wang, A. M. Yong, S. Y. Wong, X.-H. Zhang, H. Tan, A. Y. Chang, X. Li and J. Wang, Chem. Commun., 2011, 47, 11615–11617 CAS.
- S. Nandi, R. Malishev, K. P. Kootery, Y. Mirsky, S. Kolusheva and R. Jelinek, Chem. Commun., 2014, 50, 10299–10302 RSC.
- Y. Song, C. Zhu, J. Song, H. Li, D. Du and Y. Lin, ACS Appl. Mater. Interfaces, 2017, 9, 7399–7405 CrossRef CAS PubMed.
- S. Xu, Y. Liu, H. Yang, K. Zhao, J. Li and A. Deng, Anal. Chim. Acta, 2017, 964, 150–160 CrossRef CAS PubMed.
- J. Yang, W. Chen, X. Liu, Y. Zhang and Y. Bai, Mater. Res. Bull., 2017, 89, 26–32 CAS.
- S. A. Ghosh, V. B. Kumar, D. Fixler, Z. Dubinsky, A. Gedanken and D. Iluz, Algal Res., 2017, 23, 161–165 CrossRef.
- J. Zhou, Z. Sheng, H. Han, M. Zou and C. Li, Mater. Lett., 2012, 66, 222–224 CrossRef CAS.
- B. De and N. Karak, RSC Adv., 2013, 3, 8286–8290 RSC.
- L. Wang and H. S. Zhou, Anal. Chem., 2014, 86, 8902–8905 CrossRef CAS PubMed.
- S. Mandani, D. Dey, B. Sharma and T. K. Sarma, Carbon, 2017, 119, 569–572 CrossRef CAS.
- Y. Liu, Y. Liu, M. Park, S.-J. Park, Y. Zhang, M. R. Akanda, B.-Y. Park and H. Y. Kim, Carbon Lett., 2017, 21, 61–67 CrossRef.
- S. N. Baker and G. A. Baker, Angew. Chem., Int. Ed., 2010, 49, 6726–6744 CrossRef CAS PubMed.
- M. Elangovan, R. Day and A. Periasamy, J. Microsc., 2002, 205, 3–14 CAS.
- J. Ryu, U. Kang, J. Kim, H. Kim, J. H. Kang, H. Kim, D. K. Sohn, J.-H. Jeong, H. Yoo and B. Gweon, Biomed. Opt. Express, 2018, 9, 3449–3463 CrossRef PubMed.
- R. Narayanan, M. Deepa and A. K. Srivastava, J. Mater. Chem. A, 2013, 1, 3907–3918 CAS.
- C. Rahul, R. Sebastian, M. Mark, M. S. Yuri, G. Ignacy, B. Julian, G. Zygmunt and F. Rafal, Methods Appl. Fluoresc., 2016, 4, 047001–047008 CrossRef PubMed.
- H. Li, C. Sun, R. Vijayaraghavan, F. Zhou, X. Zhang and D. R. MacFarlane, Carbon, 2016, 104, 33–39 CAS.
- M. Y. Berezin and S. Achilefu, Chem. Rev., 2010, 110, 2641–2684 CAS.
- Y. Zhou, S. Sharma, Z. Peng and R. Leblanc, Polymers, 2017, 9, 67–86 CrossRef.
- Z. Yang, Z. Li, M. Xu, Y. Ma, J. Zhang, Y. Su, F. Gao, H. Wei and L. Zhang, Nano-Micro Lett., 2013, 5, 247–259 CrossRef.
- M. S. Ghamsari, A. M. Bidzard, W. Han and H.-H. Park, Nano, 2016, 10, 026028–026028 Search PubMed.
- Y. Zhou, A. Desserre, S. K. Sharma, S. Li, M. H. Marksberry, C. Chusuei, P. L. Blackwelder and R. M. Leblanc, ChemPhysChem, 2017, 18, 890–897 CrossRef CAS PubMed.
- Q. Wang, C. Zhang, G. Shen, H. Liu, H. Fu and D. Cui, J. Nanobiotechnol., 2014, 12, 58–69 CrossRef PubMed.
- D. Chowdhury, N. Gogoi and G. Majumdar, RSC Adv., 2012, 2, 12156–12159 RSC.
- A. Jaiswal, S. S. Ghosh and A. Chattopadhyay, Chem. Commun., 2012, 48, 407–409 RSC.
- H. Liu, T. Ye and C. Mao, Angew. Chem., Int. Ed., 2007, 46, 6473–6475 CrossRef CAS PubMed.
- B. Mohanty, K. V. Anita, P. Claesson and H. B. Bohidar, Nanotechnology, 2007, 18, 445102–445110 CrossRef.
- P. Kumar and H. B. Bohidar, J. Lumin., 2013, 141, 155–161 CrossRef CAS.
- H. Wang, P. Sun, S. Cong, J. Wu, L. Gao, Y. Wang, X. Dai, Q. Yi and G. Zou, Nanoscale Res. Lett., 2016, 11, 27–32 CrossRef PubMed.
- J. Liu, X. Liu, H. Luo and Y. Gao, RSC Adv., 2014, 4, 7648–7654 RSC.
- X. Li, S. Zhang, S. A. Kulinich, Y. Liu and H. Zeng, Sci. Rep., 2014, 4, 4976–4983 CrossRef CAS.
- K. M. Rice, E. M. Walker, M. Wu, C. Gillette and E. R. Blough, J. Prev. Med. Public Health, 2014, 47, 74–83 CrossRef PubMed.
- A. Sachdev, I. Matai and P. Gopinath, RSC Adv., 2014, 4, 20915–20921 RSC.
- K. Jiang, Y. Wang, X. Gao, C. Cai and H. Lin, Angew. Chem., Int. Ed., 2018, 57, 6216–6220 CrossRef CAS PubMed.
- K. Jiang, Y. Wang, C. Cai and H. Lin, Adv. Mater., 2018, 30, 1800783–1800790 CrossRef PubMed.
- P. Long, Y. Feng, C. Cao, Y. Li, J. Han, S. Li, C. Peng, Z. Li and W. Feng, Adv. Funct. Mater., 2018, 28, 1800791–1800800 CrossRef.
- Y. Gao, H. Han, W. Lu, Y. Jiao, Y. Liu, X. Gong, M. Xian, S. Shuang and C. Dong, Langmuir, 2018, 34, 12845–12852 CrossRef CAS PubMed.
- Q. Li, M. Zhou, M. Yang, Q. Yang, Z. Zhang and J. Shi, Nat. Commun., 2018, 9, 734–738 CrossRef PubMed.
- J. Tan, Y. Ye, X. Ren, W. Zhao and D. Yue, J. Mater. Chem. C, 2018, 6, 7890–7895 RSC.
- A. P. Alivisatos, Science, 1996, 271, 933–937 CrossRef CAS.
- S. Zhu, X. Zhao, Y. Song, S. Lu and B. Yang, Nano Today, 2016, 11, 128–132 CrossRef CAS.
- N. Dhenadhayalan, K.-C. Lin, R. Suresh and P. Ramamurthy, J. Phys. Chem. C, 2016, 120, 1252–1261 CrossRef CAS.
- J. L. Moll, Physics of Semiconductors, McGraw Hill, 1964 Search PubMed.
- J. P. McKelvey, Solid state and semiconductor physics, Harper and Row, 1966 Search PubMed.
- J.-P. Colinge and C. A. Colinge, Physics of semiconductor devices, Springer Science & Business Media, 2005 Search PubMed.
- S. Dimitrijev, Principles of semiconductor devices, Oxford University Press, USA, 2006 Search PubMed.
- H. Ding, S.-B. Yu, J.-S. Wei and H.-M. Xiong, ACS Nano, 2015, 10, 484–491 CrossRef PubMed.
- C. T. Chien, S. S. Li, W. J. Lai, Y. C. Yeh, H. A. Chen, I. S. Chen, L. C. Chen, K. H. Chen, T. Nemoto and S. Isoda, Angew. Chem., Int. Ed., 2012, 51, 6662–6666 CrossRef CAS PubMed.
- Y. Zhang, Y. Hu, J. Lin, Y. Fan, Y. Li, Y. Lv and X. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 25454–25460 CrossRef CAS PubMed.
- L. Han, S. G. Liu, J. X. Dong, J. Y. Liang, L. J. Li, N. B. Li and H. Q. Luo, J. Mater. Chem. C, 2017, 5, 10785–10793 RSC.
- H. Nie, M. Li, Q. Li, S. Liang, Y. Tan, L. Sheng, W. Shi and S. X.-A. Zhang, Chem. Mater., 2014, 26, 3104–3112 CrossRef CAS.
- K. Yuan, X. Zhang, R. Qin, X. Ji, Y. Cheng, L. Li, X. Yang, Z. Lu and H. Liu, J. Mater. Chem. C, 2018, 6, 12631–12637 RSC.
- C. Liu, R. Wang, B. Wang, Z. Deng, Y. Jin, Y. Kang and J. Chen, Microchim. Acta, 2018, 185, 539–546 CrossRef PubMed.
- F. Yuan, Z. Wang, X. Li, Y. Li, Z. A. Tan, L. Fan and S. Yang, Adv. Mater., 2017, 29, 1604436–1604441 CrossRef PubMed.
- Z. Liu, H. Zou, N. Wang, T. Yang, Z. Peng, J. Wang, N. Li and C. Huang, Sci. China: Chem., 2018, 61, 490–496 CrossRef CAS.
- P. M. Gharat, J. M. Chethodil, A. P. Srivastava, P. Praseetha, H. Pal and S. D. Choudhury, Photochem. Photobiol. Sci., 2019, 18, 110–119 RSC.
- Z. Shen, C. Zhang, X. Yu, J. Li, Z. Wang, Z. Zhang and B. Liu, J. Mater. Chem. C, 2018, 6, 9636–9641 RSC.
- Q. Xu, Q. Zhou, Z. Hua, Q. Xue, C. Zhang, X. Wang, D. Pan and M. Xiao, ACS Nano, 2013, 7, 10654–10661 CrossRef CAS PubMed.
- M. Shamsipur, A. Barati, A. A. Taherpour and M. Jamshidi, J. Phys. Chem. Lett., 2018, 9, 4189–4198 CrossRef CAS PubMed.
- L. Tang, R. Ji, X. Cao, J. Lin, H. Jiang, X. Li, K. S. Teng, C. M. Luk, S. Zeng, J. Hao and S. P. Lau, ACS Nano, 2012, 6, 5102–5110 CrossRef CAS PubMed.
- J. Ren, F. Weber, F. Weigert, Y. Wang, S. Choudhury, J. Xiao, I. Lauermann, U. Resch-Genger, A. Bande and T. Petit, Nanoscale, 2019, 11, 2056–2064 RSC.
- L. Xiao and H. Sun, Nanoscale Horiz., 2018, 3, 565–597 RSC.
- P. Y. Liyanage, R. M. Graham, R. R. Pandey, C. C. Chusuei, K. J. Mintz, Y. Zhou, J. K. Harper, W. Wu, A. H. Wikramanayake, S. Vanni and R. M. Leblanc, Bioconjugate Chem., 2019, 30, 111–123 CrossRef CAS PubMed.
- Y. Zhou, Z. Peng, E. S. Seven and R. M. Leblanc, J. Controlled Release, 2018, 270, 290–303 CrossRef CAS PubMed.
- V. Strauss, J. T. Margraf, C. Dolle, B. Butz, T. J. Nacken, J. Walter, W. Bauer, W. Peukert, E. Spiecker, T. Clark and D. M. Guldi, J. Am. Chem. Soc., 2014, 136, 17308–17316 CrossRef CAS PubMed.
- S. Wang, I. S. Cole, D. Zhao and Q. Li, Nanoscale, 2016, 8, 7449–7458 RSC.
- A. B. Bourlinos, A. Stassinopoulos, D. Anglos, R. Zboril, M. Karakassides and E. P. Giannelis, Small, 2008, 4, 455–458 CrossRef CAS PubMed.
- M. C. Ortega-Liebana, N. X. Chung, R. Limpens, L. Gomez, J. L. Hueso, J. Santamaria and T. Gregorkiewicz, Carbon, 2017, 117, 437–446 CrossRef CAS.
- K. Hola, A. B. Bourlinos, O. Kozak, K. Berka, K. M. Siskova, M. Havrdova, J. Tucek, K. Safarova, M. Otyepka, E. P. Giannelis and R. Zboril, Carbon, 2014, 70, 279–286 CrossRef CAS.
- A. Sciortino, E. Marino, B. V. Dam, P. Schall, M. Cannas and F. Messina, J. Phys. Chem. Lett., 2016, 7, 3419–3423 CrossRef CAS PubMed.
- W. Zhang, S. F. Yu, L. Fei, L. Jin, S. Pan and P. Lin, Carbon, 2015, 85, 344–350 CrossRef CAS.
- S. A. Hill, D. Benito-Alifonso, D. J. Morgan, S. A. Davis, M. Berry and M. C. Galan, Nanoscale, 2016, 8, 18630–18634 RSC.
- B. C. M. Martindale, G. A. M. Hutton, C. A. Caputo, S. Prantl, R. Godin, J. R. Durrant and E. Reisner, Angew. Chem., Int. Ed., 2017, 56, 6459–6463 CrossRef CAS PubMed.
- A. Cayuela, M. L. Soriano, C. Carrillo-Carrión and M. Valcárcel, Chem. Commun., 2016, 52, 1311–1326 RSC.
- B. J. Moon, Y. Oh, D. H. Shin, S. J. Kim, S. H. Lee, T.-W. Kim, M. Park and S. Bae, Chem. Mater., 2016, 28, 1481–1488 CrossRef CAS.
- Y.-P. Sun, B. Zhou, Y. Lin, W. Wang, K. A. S. Fernando, P. Pathak, M. J. Meziani, B. A. Harruff, X. Wang, H. Wang, P. G. Luo, H. Yang, M. E. Kose, B. Chen, L. M. Veca and S.-Y. Xie, J. Am. Chem. Soc., 2006, 128, 7756–7757 CrossRef CAS PubMed.
- H. Li, X. He, Z. Kang, H. Huang, Y. Liu, J. Liu, S. Lian, C. H. A. Tsang, X. Yang and S.-T. Lee, Angew. Chem., Int. Ed., 2010, 49, 4430–4434 CrossRef CAS PubMed.
- V. Nguyen, L. Yan, J. Si and X. Hou, Opt. Mater. Express, 2016, 6, 312–320 CrossRef CAS.
- H. Tetsuka, R. Asahi, A. Nagoya, K. Okamoto, I. Tajima, R. Ohta and A. Okamoto, Adv. Mater., 2012, 24, 5333–5338 CrossRef CAS PubMed.
- A. Philippidis, A. Spyros, D. Anglos, A. B. Bourlinos, R. Zbořil and E. P. Giannelis, J. Nanopart. Res., 2013, 15, 1777–1785 CrossRef.
- A. B. Bourlinos, A. Stassinopoulos, D. Anglos, R. Zboril, V. Georgakilas and E. P. Giannelis, Chem. Mater., 2008, 20, 4539–4541 CrossRef CAS.
- L. Weijian, L. Chun, S. Xiaobo, P. Wei, Y. Guifeng and W. Jinping, Nanotechnology, 2017, 28, 485705 CrossRef PubMed.
- K. J. Mintz, B. Guerrero and R. M. Leblanc, J. Phys. Chem. C, 2018, 122, 29507–29515 CrossRef CAS.
- Z. Peng, E. H. Miyanji, Y. Zhou, J. Pardo, S. D. Hettiarachchi, S. Li, P. L. Blackwelder, I. Skromne and R. M. Leblanc, Nanoscale, 2017, 9, 17533–17543 RSC.
- M. J. Krysmann, A. Kelarakis, P. Dallas and E. P. Giannelis, J. Am. Chem. Soc., 2012, 134, 747–750 CrossRef CAS PubMed.
- P. Roy, P.-C. Chen, A. P. Periasamy, Y.-N. Chen and H.-T. Chang, Mater. Today, 2015, 18, 447–458 CrossRef CAS.
- S. H. Jin, D. H. Kim, G. H. Jun, S. H. Hong and S. Jeon, ACS Nano, 2013, 7, 1239–1245 CrossRef CAS PubMed.
- J. Yu, C. Liu, K. Yuan, Z. Lu, Y. Cheng, L. Li, X. Zhang, P. Jin, F. Meng and H. Liu, Nanomaterials, 2018, 8, 233–244 CrossRef PubMed.
- Y. Zhou, K. Mintz, C. Oztan, S. Hettiarachchi, Z. Peng, E. Seven, P. Liyanage, S. De La Torre, E. Celik and R. Leblanc, Polymers, 2018, 10, 921–932 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nr10059d |
| This journal is © The Royal Society of Chemistry 2019 |