
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCreating a synthetic platform for the encapsulation of nanocrystals with covalently bound polymer shells†
Rieke
Koll‡
 a,
Lisa Sarah
Fruhner‡
bc,
Hauke
Heller
a,
Jürgen
Allgaier
b,
Wim
Pyckhout-Hintzen
b,
Margarita
Kruteva
*b,
Artem
Feoktystov
d,
Ralf
Biehl
b,
Stephan
Förster
bc and
Horst
Weller
*a
a,
Lisa Sarah
Fruhner‡
bc,
Hauke
Heller
a,
Jürgen
Allgaier
b,
Wim
Pyckhout-Hintzen
b,
Margarita
Kruteva
*b,
Artem
Feoktystov
d,
Ralf
Biehl
b,
Stephan
Förster
bc and
Horst
Weller
*a
aInstitute of Physical Chemistry, Fraunhofer CAN, Grindelallee 117, 20146 Hamburg, Germany. E-mail: weller@chemie.uni-hamburg.de
bJülich Centre for Neutron Science (JCNS-1) and Institute for Complex Systems (ICS-1), Forschungszentrum Jülich GmbH, Leo-Brandt-Straße, 52425 Jülich, Germany. E-mail: m.kruteva@fz-juelich.de
cInstitute of Physical Chemistry, RWTH Aachen University, Landoltweg 2, 52056 Aachen, Germany
dForschungszentrum Jülich GmbH, Jülich Centre for Neutron Science (JCNS) at MLZ, Lichtenbergstrasse 1, 85748 Garching, Germany
First published on 4th February 2019
Abstract
We present a platform for the encapsulation of superparamagnetic iron oxide nanocrystals (SPIONs) with a highly stable diblock copolymer shell allowing a homogeneous dispersion of the nanocrystals into a polymer matrix in the resulting nanocomposites. High polymer shell stability was achieved by crosslinking the inner polydiene shell for example in a persulfate based redox process. The advantage of this crosslinking reaction is the avoidance of heat and UV light for the initiation, making it suitable for heat or UV sensitive systems. In addition, we were able to minimize the ligand excess needed for the encapsulation and showcased a variation of molecular weight and composition as well as different ligands which lead to stable micelles. The encapsulated nanocrystals as well as the nanocomposite materials were characterized by transmission electron microscopy (TEM) and small angle scattering (SAXS and SANS).
Introduction
Polymer encapsulation is a key technological step for nanocrystal (NC) applications ranging from high performance nanocomposites1–4 over quantum converter films in display and lighting5,6 to contrast agents and drug delivery systems7–13 in nanomedicine. The role of the encapsulation is twofold: it allows solubility adjustment and ensures stability and compatibility in the respective environment. A very powerful method is based on amphiphilic diblock copolymers. Here, hydrophobic NCs act as nucleation centres for micelle formation of the copolymers around the NCs. For the system polyisoprene-block-poly(ethylene oxide) (PI-b-PEO) it was shown that radical initiated crosslinking of the polyisoprene core significantly enhances the biocompatibility due to shielding effects of the NCs against the surrounding medium.14 An important parameter is the control of the number of NCs within one micelle, in which in many applications just one NC per micelle is the target structure. In nanocomposites, moreover, it is mostly desirable to achieve a homogeneous dispersion of individually encapsulated NCs within the surrounding matrix material as the nanocomposite properties strongly depend on the local organization of the NCs.15 Here we report a combined transmission electron microscopy (TEM) and small angle X-ray and neutron scattering (SAXS and SANS) study on structure formation during the encapsulation process of superparamagnetic iron oxide nanocrystals (SPIONs) in a poly(ethylene oxide) matrix. We have chosen 1,2-polybutadiene-block-poly(ethylene oxide) (PB-b-PEO) of various molar masses for the encapsulation and compare different crosslinking techniques of the hydrophobic micellar core. Furthermore, we present how a homogeneous distribution of the SPIONs can be achieved by adjustment of PEO block lengths of the diblock copolymers and the PEO matrix material, respectively. The excess of polymer ligand needed for encapsulation was minimized by choosing a NC specific anchoring group, facilitating a future upscale of the process. In addition, a SANS contrast variation study was employed to examine the details of the polymer shell.Results and discussion
Encapsulation of SPIONs
In order to avoid agglomeration and to get a homogeneous distribution of the nanocrystals inside the PEO matrix, the SPIONs have to be stabilized. We used SPIONs with a TEM determined diameter of 15.4 nm ± 5%. The stabilization of the SPIONs was achieved by encapsulation with PB-b-PEO and crosslinking of the inner PB shell. The encapsulation of SPIONs serves as a model system due to their well-known synthesis of high quality nanocrystals. Nevertheless, the presented procedure is adaptable to various types of nanocrystals.14,16–20 The encapsulation was achieved in three steps; a schematic illustration is presented in Scheme 1. The first step is the exchange of the native oleic acid ligands by a polymeric ligand in an equilibrium process. As ligands the 2,2′-diamino-diethylamine functionalized polybutadiene (PB-DETA) and phosphonic acid functionalized polybutadiene (PB-PA) were used. The polymer ligand serves as seed for the following micelle formation. Afterwards, the polymer ligand stabilized SPIONs were mixed with PB-b-PEO and were transferred into water. The diblock copolymer acts in this process as emulsifying agent. In a final step, the double bonds of the inner PB shell were crosslinked. In order to remove empty micelles the encapsulated SPIONs were purified using a magnetic column. The purified encapsulated SPIONs, dispersed in water, were then mixed with an aqueous PEO solution, lyophilized and melted at 65 °C to yield the nanocomposite. | ||
| Scheme 1 Schematic illustration of the encapsulation process. First, the native oleic acid ligands are exchanged against a polymer ligand. Then, the SPIONs are mixed with a diblock copolymer and transferred into water, where the micelle formation takes place. Finally, the double bonds of the inner polymer shell are crosslinked. | ||
Examination of different crosslinking pathways
For the crosslinking of the inner polymer shell, several different pathways are possible. We already introduced the thiol–ene click reaction with 1,6-hexanedithiol (HDT) to be a suitable method to crosslink and therefore stabilize the polymer shell in order to obtain homogeneously dispersed nanocrystals in a PEO matrix.21 The thiol–ene clickreaction can be initiated with either a thermal radical initiator like 2,2′-azobis(2-methylpropionitrile) (AIBN) or with a photoinitiator like 2,2-dimethoxy-2-phenyl-acetophenone (DMPA). Both initiation techniques are unsuitable for heat or UV sensitive materials. An alternative crosslinking method is provided by a common redox reaction using potassium persulfate as a water soluble radical initiator. The persulfate decomposition at room temperature is catalyzed by a redox system comprising sodium metabisulfite and iron(II) sulfate heptahydrate.22,23 Information about the crosslinking reactions are found in Scheme 1S–3S.† An additional advantage of the persulfate based redox process is the direct crosslinking of the polymer without integration of crosslinker into the shell. This is the case for example for the thiol–ene click reactions, where large disulfide moieties are incorporated between the polymer chains. The fixation of micellar structures formed by amphiphilic diblock copolymers via the persulfate based redox reaction has already been published,24 but in combination with nanocrystals it is still unknown.The completeness of the encapsulation process was examined by TEM. In these experiments the PEO shell was selectively stained with phosphotungstic acid, leading to a dark corona around the SPIONs. Combining TEM imaging with this staining agent is a suitable method to study the completeness of the encapsulation process due to possible etching of the SPIONs by the staining agent. Etching is only prevented in case of a homogeneous and stable polymer shell.21Fig. 1 shows representative TEM images of encapsulated SPIONs, crosslinked with persulfate. Further TEM images of differently crosslinked samples are given in the ESI (Fig. 1S†). As can be seen from the Fig. 1 and 1S,† all crosslinking procedures lead to closed polymer shells of a high quality.
 | ||
| Fig. 1 Exemplarily TEM images of dried samples with two different magnifications of encapsulated SPIONs, where the PEO shell was stained with phosphotungstic acid. The polymer shell consists of PB3k-PEO4k, crosslinked in a persulfate based redox process. | ||
The encapsulated SPIONs, crosslinked with the persulfate based redox reaction or the thiol–ene clickreaction, were dispersed in a polymer matrix and the resulting nanocomposites were analyzed with SAXS. Fig. 2a shows the scattering curves for the different crosslinked samples. All scattering curves exhibit a form factor of spherical SPIONs with well-defined minima indicating the narrow size distribution of the used SPIONs. The form factor oscillations can be described by a SPIONs radius of 8.2 ± 0.4 nm, which is in a good agreement to the TEM determined radius of 7.7 nm, as the SAXS radius is weight-averaged and the TEM radius is number-averaged. To get information about the number of individually encapsulated SPIONs and therefore the dispersion state, the scattering curves were fitted with the pearl-necklace model.21,25 This gives the number of encapsulated SPIONs N forming a “chain” (agglomeration number). The distribution of N is presented in Fig. 2b. Both the persulfate based redox reaction and the thermally and photochemically initiated thiol–ene clickreaction lead to a stable polymer shell, which enables a homogeneous distribution of the SPIONs inside the PEO matrix with a ratio of single SPIONs of around 90%. The remaining SPIONs form mainly dyads and to a marginal amount triads. We already clarified the origin of dyads and triads coming from a multiple encapsulation during the phase transfer step from organic solvent into water.21 Slight differences in the relative amount of single SPIONs arise from different encapsulation processes during the phase transfer step. In contrast, the sample crosslinked photochemically without the photoinitiator DMPA shows only 75% of single SPIONs, which indicates insufficient polymer shell stability. This can be explained by a lack of radical polymerization of the PB double bonds when DMPA is missing. The same effect has been shown when using the radical initiator AIBN with and without HDT.21
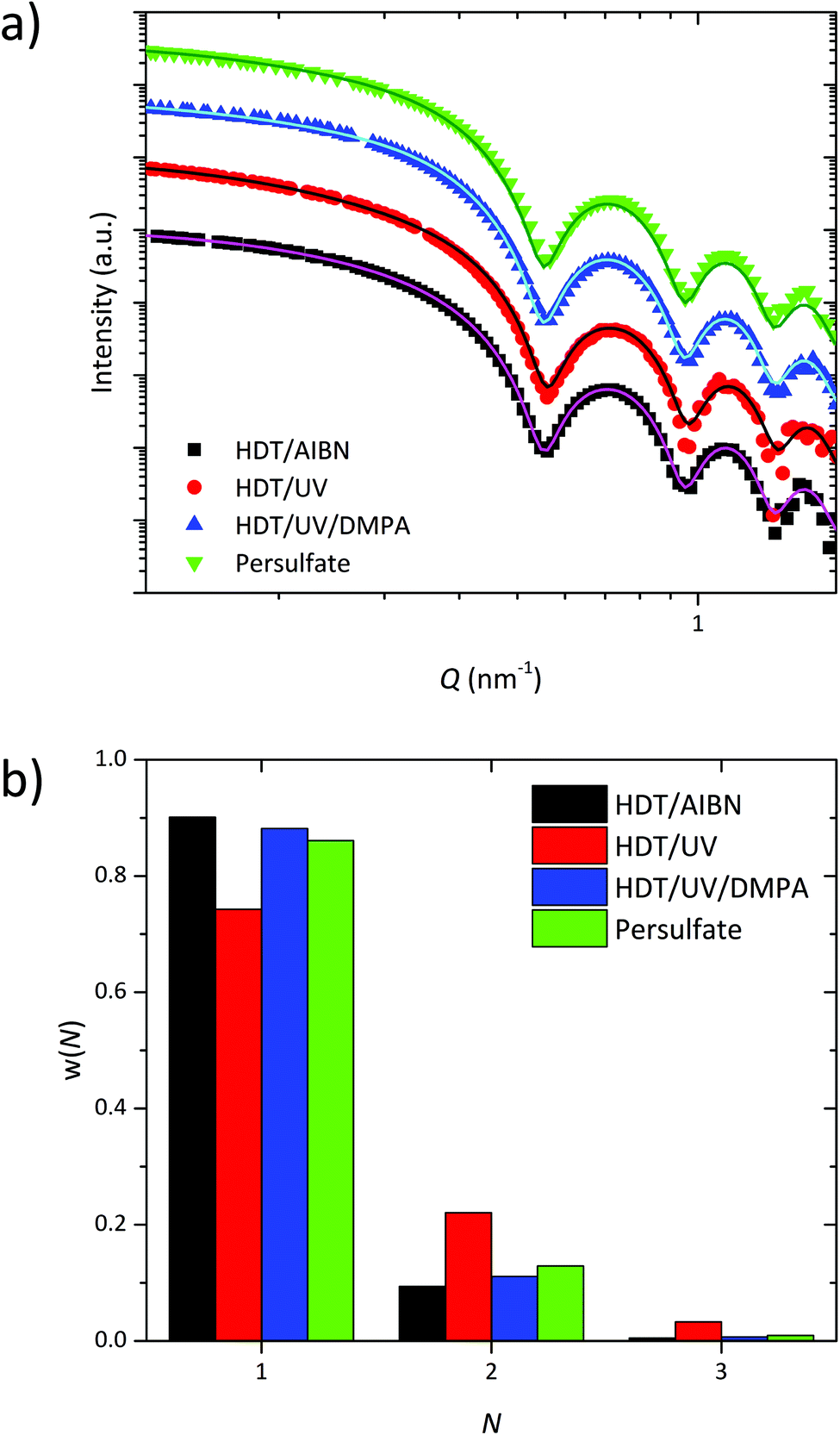 | ||
| Fig. 2 (a) SAXS curves (arbitrarily shifted) of differently crosslinked samples. (b) Distribution of the agglomeration number N for each sample. | ||
Variation of the diblock copolymer chain length
For the SPION encapsulation we varied the chain length and the composition of the diblock copolymers. PB5k-PEO10k, PB3k-PEO4k and PB2k-PEO5k were used, while the chain length of the polymer ligand PB-DETA was kept constant at 2k. The polymer shells were crosslinked with HDT/AIBN and analyzed with TEM. Furthermore, we dispersed the encapsulated SPIONs in PEO and investigated the dispersion state with SAXS measurements. To obtain further knowledge about the polymer shells, SANS experiments of SPIONs encapsulated with two different diblock copolymers were performed.Typical TEM images of samples with a PB5k-PEO10k and a PB2k-PEO5k polymer shell are presented in Fig. 3. The PEO shell was stained with phosphotungstic acid. Independent from the diblock copolymer chain length, all samples indicate a PB shell thickness of about 3 nm, which is represented by the unstained yet slightly darkened area around the SPIONs. This area darkens in additional TEM images, where the PB was selectively stained with osmium tetroxide (see Fig. 2S†). This shell primarily consists of diblock PB and to a smaller extent of PB-DETA. Nevertheless, its thickness is independent of the PB length in the diblock. Hence, the grafting density of the PEO corona decreases with increasing PB block length. This was confirmed by thermogravimetric analysis (TGA) measurements, where the mass loss due to the polymer depended only slightly on its chain length (see Fig. 3S†). In further experiments, where the PB-DETA molecular weight was increased from 2k to 5k, the PB shell thickness stayed constant at 3 nm (see Fig. 7S†). However, the total size of the micelles increases with increasing diblock copolymer size which was ascertained with dynamic light scattering (DLS) measurements (see Fig. 4S†).
 | ||
| Fig. 3 TEM images after drying of PB-b-PEO encapsulated SPIONs with a (a) PB5k-PEO10k and a (b) PB2k-PEO5k polymer shell. The scale bars in the inserts correspond to 25 nm. | ||
The polymer shell thickness was exemplarily analyzed with SANS for the PB2k-PEO5k and PB5k-PEO10k. In the case of PB2k-PEO5k a contrast variation study was conducted using four different contrast scenarios which are illustrated in Fig. 4: (1) hPB-dPEO in dPEO3k; (2) hPB-hPEO in dPEO3k; (3) hPB-dPEO in D2O; (4) hPB-hPEO in D2O. For PB5k-PEO10k, only the second contrast (in dPEO10k) was measured to compare the polymer shell dimensions for different block copolymer lengths. As the scattering length density (SLD) of iron oxide is very close to the values for the deuterated matrices, the highest contribution to the scattering intensity arises from the hydrogenous polymers. Therefore, the pearl-necklace model was modified by using a spherical core–shell–shell form factor implying a step profile for the SLDs (see SI). The value for the core radius of 8.2 nm was taken from the SAXS data evaluation. Due to the low contrast between SPION core and matrix, the influence of the SPION polydispersity is small and can be neglected. The SLD of the matrix ρm and the inner PB shell ρPB were fixed to calculated values of ρPB = 0.4 × 1010 cm−2 and ρm = 6.0 × 1010 cm−2 for deuterated PEO and ρm = 6.3 × 1010 cm−2 for deuterated water. Experimental SANS curves with corresponding fits are displayed in Fig. 5.
 | ||
| Fig. 4 SANS contrast scenarios for SPIONs encapsulated with hPB-dPEO in dPEO (1), hPB-hPEO in dPEO (2), hPB-dPEO in D2O (3) and hPB-hPEO in D2O (4) and calculated values of scattering length densities. | ||
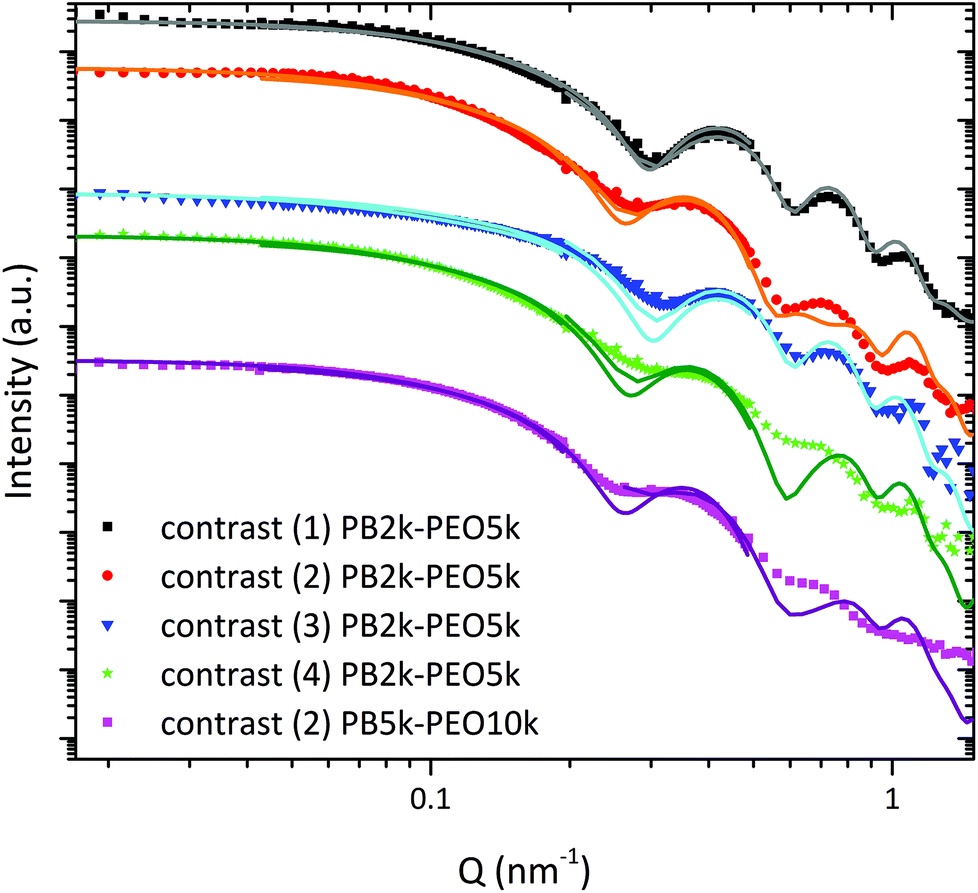 | ||
| Fig. 5 SANS curves with corresponding fits for hPB-dPEO in dPEO (1), hPB-hPEO in dPEO (2), hPB-dPEO in D2O(3) and hPB-hPEO in D2O (4), obtained from three different detector distances with corresponding resolution functions. | ||
We used contrast (1) to determine the SLD of the SPION core ρc and the thickness tPB of the PB shell, resulting in values of ρc = 6.7 ± 0.1 × 1010 cm−2 which conforms to the theoretical value for maghemite and tPB = 4.3 ± 0.1 nm. This corresponds well to the values for the PB shell thickness found via TEM and complementary methods.21 The well pronounced minima indicate a narrow distribution of the PB shell thickness. The PEO shell is invisible under these contrast conditions. The fraction of singly encapsulated SPIONs was determined to be 76%.
For the contrast (3) the data could be described by the model and the predetermined values quite well. The PEO shell contributes to the scattering curve due to a small difference between SLD of D2O and PEO. As a consequence, the minima are less pronounced.
Fixing tPB, ρc and N from contrast (1) for the examination of contrast (2) and (4) with h-PEO shells, we were able to obtain reliable values for the thickness tPEO of the outer PEO shell. For contrast (2) tPEO was equal to 4.5 ± 0.1 nm and for contrast (4) in D2O the value was 5.3 ± 0.3 nm. The PEO shell thicknesses can be compared with the sizes of the free PEO chains by taking their end-to-end distances Ree as a measure. For PEO5k in a theta solvent Ree is about 5 nm using the equation 〈Ree2〉/M = 0.8.26 For small molecular weights, it is known that chain dimensions are similar in good and theta solvents. Therefore, the PEO5k shell results of tPEO, 4.5 nm in the nanocomposite and 5.3 nm in water indicate rather dense PEO shells whereby the PEO chains are only marginally stretched. Although the molecular weight of the PEO is doubled when using PB5k-PEO10k, the dimension of the PEO shell in the melt increases only slightly to 4.9 ± 0.1 nm compared to the calculated value of Ree = 7 nm.26 The fraction of singly encapsulated SPIONs was determined to be 83%. From prior investigations it is known that for SPIONs encapsulated with the procedure presented here, a grafting density of PEO chains on the PB surface is approximately one chain per 4.2 nm2.21 One notes, that the quality of the fits becomes worse with increasing scattering contribution of the PEO shell due to the used simplified step profile. For this reason, the thickness of the PEO shell may be underestimated in our study. Nevertheless, we have to assume a rather dense PEO shell. This is confirmed by the absence of individual chain scattering (PEO form factor) characterized by the power law I(Q) ∼ Q−1.6 in the high Q-range, which is normally seen in micellar systems.27
Additional parameters of the fits are the SLDs of the PEO shells ρPEO. The values for PB2k-PEO5k and contrast (2) is 3.4 ± 0.2 × 1010 cm−2 and contrast (4) 3.5 ± 0.5 × 1010 cm−2. Together with the SLD of pure protonated PEO of 0.5 × 1010 cm−2 this translates into a volume fraction of PEO (φPEO) of 49% for contrast (2) and 33% for contrast (4). These are high values if compared for example with the swelling degree of free PEO5k at the overlap concentration c* which is 21%.28 For PB5k-PEO10k the situation is similar with a volume fraction of PEO in the shell of 44%.
Combining the PEO content of the outer shell with the PB and PEO shell thicknesses, the composition of the polymer shell can be calculated from the SANS data (Table 3S†). The values are almost identical with the ones obtained from our previous elemental analysis investigation.21
All the parameters are listed in Table 2S.†
We further varied the molecular weight ratio of the grafted PEO chains to the free matrix PEO chains. The encapsulated SPIONs with the different diblock copolymer shells were dispersed in a PEO10k or a PEO3k matrix. The molecular weight ratio R of grafted to free chains ranged from 0.5 to 3.33. The nanocomposites were analyzed by SAXS. Several groups15,29–32 identified R to be an important parameter for the dispersion state of the NCs in a polymer matrix. For R smaller than 1 the interpenetration of free chains into the PEO corona is conformationally unfavored, leading to an expulsion from the grafted chains and therefore to attractive interactions between the NCs. In the case of R of 1 or larger than 1 the smaller free chains act as a solvent for the grafted chains, which causes repulsive interactions between the NCs and induces an individual dispersion. We also observed this parameter to be a suitable way of adjusting the solubilization state of the SPIONs inside the nanocomposite (Fig. 6). The original SPION samples encapsulated with PB2k-PEO5k and PB5k-PEO10k contained mainly singly encapsulated SPIONs (see Fig. 5S). The dispersion in PEO caused a visible agglomeration if SPIONs encapsulated with PB2k-PEO5k were mixed into a PEO10k matrix (R = 0.5). In contrast, no additional agglomeration appeared if PB2k-PEO5k encapsulated SPIONs were mixed with PEO3k or PB5k-PEO10k SPIONs were mixed with both PEO3k and PEO10k (R ≥ 1). The samples with R = 0.5 and R = 1.7, that were phase transferred and crosslinked with the same PB2k-PEO5k diblock copolymer, necessarily had the same amount of singly encapsulated SPIONs and the same polymer shell stability. Therefore, multiple encapsulation and shell stability can be excluded as causes for the increase in agglomeration in the case of R = 0.5. Insofar, our findings are in good agreement with prior results.
 | ||
| Fig. 6 (a) SAXS curves (arbitrarily shifted) and (b) distribution for N for nanocomposites with different grafted to free chain length ratios. | ||
Investigation of the ligand exchange step
The ligand exchange step was examined by varying the amount of polymeric ligand and using two different head groups. PB-DETA contains a 2,2′-diaminodiethylamine head group and PB-PA is equipped with a phosphonic acid head group, both polymers having a molecular weight of 2k. In the past experiments a high ligand amount of about 3000 PB-DETA chains per SPION was used. This value translates into 4.0 PB-DETA chains per nm2 of iron oxide surface considering the diameter to be 15.4 nm. Starting without polymer ligand we increased the amount of PB-DETA to determine the minimum quantity for full encapsulation. The same procedure was applied for PB-PA. The SPIONs encapsulated with PB3k-PEO4k were analyzed with TEM using phosphotungstic acid as staining reagent. TEM images showed that the encapsulation without polymer ligand lead to an irregular polymer shell with SPIONs partially etched by the staining agent, phosphotungstic acid (Fig. 7a). The same was observed if only 0.1 PB-DETA chains per nm2 of iron oxide surface were added (Fig. 7b). Increasing the amount of PB-DETA to 0.3 chains per nm2 iron oxide surface resulted in completely closed polymer shells where the SPIONs are protected against the etching by phosphotungstic acid (Fig. 7c). For PB-PA already the addition of 0.1 chains per nm2 iron oxide surface was sufficient to gain closed polymer shells and to protect the encapsulated SPIONs against etching. This is understandable as phosphonic acid has a higher affinity to the NC surface than DETA.33 In addition, PB-DETA may form an ion pair with the released oleic acid leading to a decreased amount of free PB-DETA, which increases the required ligand excess.34 As the maximum grafting density of polymeric ligands is about 1 chain per nm2.35,36 A complete exchange of the native oleic acid ligands is not required in order to obtain closed polymer shells. Additional TEM images of PB2k-PEO5k and PB5k-PEO10k encapsulated SPIONs are presented in Fig. 6S† where the results are similar to these of PB3k-PEO4k. | ||
| Fig. 7 TEM images after drying of SPIONs encapsulated with PB3k-PEO4k, stabilized with (a) oleic acid, (b) a 100-fold PB-DETA excess (0.1 chains per nm2), (c) a 250-fold PB-DETA excess (0.3 chains per nm2) and (d) a 100-fold PB-PA excess (0.1 chains per nm2). | ||
The SPIONs encapsulated with different ligand quantities were dispersed in PEO and the distribution was analyzed by SAXS. The scattering curves were fitted with the pearl necklace model to calculate the fraction of single SPIONs inside the nanocomposite. The scattering curves and the distribution of N for PB3k-PEO4k encapsulated SPIONs with different ligands and ligand excess are shown in Fig. 8. The scattering curves and the ratio of single SPIONs inside the nanocomposite for PB2k-PEO5k and PB5k-PEO10k encapsulated SPIONs are presented in Fig. 8S and 9S.† Regardless of the ligand and the ligand amount, all samples exhibit a fraction of single SPIONs of 80% to nearly 100%.
 | ||
| Fig. 8 (a) SAXS curves (arbitrarily shifted) and (b) distribution for N for nanocomposites with PB3k-PEO4k encapsulated SPIONs, stabilized with different ligands and excess. | ||
Experimental
Materials
The following chemicals were used as received. Tetrahydrofuran (THF, 99.7%), n-hexane (96%) and ethanol (99.98%) were purchased from VWR. 1,6-Hexanedithiol (HDT, 97%) was obtained from Alfa Aesar. 2,2′-Azobis(2-methyl-propionitrile) (AIBN, 98%), 2,2-dimethoxy-2-phenylaceto-phenone (DMPA, 99%), potassium persulfate (K2S2O8, 99.0%), iron(II) sulfate heptahydrate (FeSO4·7H2O, 99%), osmium tetroxide solution (OsO4, 4% in H2O) and poly(ethylene oxide) (Mn ∼ 2.05 kDa, 3.1 kDa and 10.4 kDa) were purchased from Sigma-Aldrich. Sodium metabisulfite (Na2S2O5, 98.0%) was obtained from Merck. Water was purified using an ELGA PURELAB® flex 2 system (18.2 MΩ).SPIONs were synthesized according to Yu et al.37 and were initially stabilized with oleic acid. The syntheses and characterizations of the polymers are presented in the ESI.†
DLS data were collected with a Zetasizer Nano ZS system (Malvern).
Encapsulation of SPIONs and nanocomposite synthesis
To exchange the oleic acid ligands, SPIONs were incubated with different quantities of PB-DETA or PB-PA in n-hexane for more than 20 h. Before encapsulation, the SPIONs were precipitated with ethanol and centrifuged. The polymer-coated SPIONs were dispersed in THF and mixed with a 3000-fold excess of PB-b-PEO in THF to yield a 0.8 μM solution.For HDT/AIBN crosslinking HDT (ratio HDT to butadiene units present in the diblock copolymer of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12) and AIBN (ratio AIBN to butadiene units present in the diblock copolymer of 1:4) was added to the SPION-polymer mixture. The mixture was transferred into the 10-fold volume of water with a programmable flow system20 and heated up to 80 °C for 4 h. Finally, the SPIONs were purified using a magnetic column.
:12) and AIBN (ratio AIBN to butadiene units present in the diblock copolymer of 1:4) was added to the SPION-polymer mixture. The mixture was transferred into the 10-fold volume of water with a programmable flow system20 and heated up to 80 °C for 4 h. Finally, the SPIONs were purified using a magnetic column.
For HDT/UV and HDT/UV/DMPA crosslinking the SPION-polymer solution was mixed with HDT (ratio HDT to butadiene units present in the diblock copolymer of 1:4) and DMPA (ratio DMPA to HDT of 1:7), transferred into the 10-fold volume of water and irradiated with UV light (four PL-S 9W/2P BLB UV lamps from Philips with 9 W power each and 366 nm wavelength) for 4 h. Then, the encapsulated SPIONs were purified using a magnetic column.
For crosslinking with the redox reaction, the SPION-polymer mixture was transferred into the 10-fold volume of water. K2S2O8 (ratio K2S2O8 to butadiene units present in the diblock copolymer of 1:2.5) was added and the mixture was stirred at room temperature. After 3 h Na2S2O5 (ratio Na2S2O5 to K2S2O8 of 1:1.4) and FeSO4·7H2O (ratio FeSO4·7H2O to K2S2O8 of 1:49) were added successively. After 45 min the encapsulated SPIONs were purified using a magnetic column.
To get the nanocomposites, the encapsulated SPIONs, dispersed in water, were mixed with an aqueous PEO solution, lyophilized and melted at 65 °C.
Transmission electron microscopy (TEM)
TEM experiments were performed using a Jeol JEM-1011 microscope (100 keV) at the University of Hamburg. For TEM images, the aqueous SPIONs solution was partially dried on a carbon-coated copper grid and excess solution was removed. An aqueous phosphotungstic acid solution (10%) was added and removed after 30 s. Afterwards, the sample on the grid was washed with water two times. For osmium tetroxide staining, encapsulated SPIONs deposited on a carbon-coated copper grid were stained with osmium tetroxide vapor for 1 h using an osmium tetroxide solution (4% in H2O).Small angle X-ray scattering (SAXS)
SAXS experiments were performed in the Q-range from 0.12 to 1.7 nm−1 at the Gallium Anode Low-Angle X-ray Instrument (GALAXI)38 at Forschungszentrum Jülich. The incident wavelength is 1.34 Å and the detector distance was set to 3.5 m. All samples were sealed in glass capillaries of 2 mm inner diameter. Nanocomposite samples were measured above the PEO glass transition temperature. The concentrations of the SPIONs in each sample were 0.1 vol% to avoid interparticle interaction and to get a sufficient scattering signal. The data were background corrected and calibrated to absolute intensities. Data analysis has been done using the Python based project jscatter.39Small angle neutron scattering (SANS)
SANS experiments were performed at the instrument KWS-140,41 at the MLZ in Garching, Germany. The incident neutron wavelength λ was 7 Å (Δλ/λ = 10%). The data were obtained from three different detector and collimation distances: the detector distances of 1.5 m and 8 m with a collimation distance of 8 m and the detector distance of 20 m with 20 m collimation distance leading to a total Q range of 0.017 to 1.5 nm−1. The samples were measured in quartz cells with beam path of 2 mm. For the samples in deuterated water, the measurements were done at room temperature, for the nanocomposite samples, the measurements were done at 70 °C, e.g. above the melting temperature of PEO. The size of the sample aperture was set as 6 × 6 mm. The data presented here were converted to an absolute intensity unit of cm−1 taking into account the sample thickness, transmission, the scattering from a standard sample and the background from electronic noise, the solvent and the quartz cell. Data reduction has been done using the QtiKWS software,42 data analysis has been done using the Python based project jscatter39 including resolution smearing dependent on the measurement geometry.43Conclusions
Superparamagnetic iron oxide nanocrystals (SPIONs) were successfully encapsulated with a highly stable diblock copolymer shell. The inner part of the shell was crosslinked using a thermally or photochemically initiated thiol–ene click reaction or a persulfate based redox process at room temperature. The major advantage of the latter crosslinking reaction is the avoidance of heat or UV light for the initiation, making it suitable for heat or UV sensitive systems and facilitating a future upscaling of the process. The presented procedure is adaptable to various types of nanocrystals, whereas the encapsulation of SPIONs serves as a model system due to their well-known synthesis of high quality nanocrystals.We could show by TEM, SAXS and SANS that the size of the polymer shell is not considerably influenced by the polymer molecular weight and composition. By minimizing the ligand excess during the encapsulation, the process was further improved with regard to a future upscaling whereby PB-PA allows an even smaller ligand excess than PB-DETA.
The exchange of oleic acid by the polymeric ligand is necessary to obtain a closed polymer shell around the SPIONs. This is of great importance for example in the case of biomedical applications. However, a high fraction of single SPIONs in the PEO matrix can also be achieved if the ligand exchange is omitted.
The SANS examination performed with different contrasts indicates that (i) the inner shell contains only PB and has a size comparable to prior results and (ii) the PEO shell is rather dense.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by SPP 1681 (projects KR 3929/2-1 and KR 3929/2-2) of the German Research Foundation (DFG). The authors acknowledge Artur Feld for support with iron oxide nanocrystals.References
- A. S. Robbes, F. Cousin, F. Meneau, F. Dalmas, F. Boué and J. Jestin, Macromolecules, 2011, 44, 8858–8865 CrossRef CAS
.
- S. K. Kumar, N. Jouault, B. Benicewicz and T. Neely, Macromolecules, 2013, 46, 3199–3214 CrossRef CAS
- D. Maillard, S. K. Kumar, B. Fragneaud, J. W. Kysar, A. Rungta, B. C. Benicewicz, H. Deng, L. C. Brinson and J. F. Douglas, Nano Lett., 2012, 12, 3909–3914 CrossRef CAS PubMed
- C. Yang, H. Wei, L. Guan, J. Guo, Y. Wang, X. Yan, X. Zhang, S. Wei and Z. Guo, J. Mater. Chem. A, 2015, 3, 14929–14941 RSC
- F. Jin, M.-L. Zheng, M.-L. Zhang, Z.-S. Zhao and X.-M. Duan, RSC Adv., 2014, 4, 33206–33214 RSC
- C. Woelfle and R. O. Claus, Nanotechnology, 2007, 18, 1–9 CrossRef
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS PubMed
- S. Kalia, S. Kango, A. Kumar, Y. Haldorai, B. Kumari and R. Kumar, Colloid Polym. Sci., 2014, 292, 2025–2052 CrossRef CAS
- L. Dykman and N. Khlebtsov, Chem. Soc. Rev., 2012, 41, 2256–2282 RSC
- K. Ferji, I. Hamouda, C. Chassenieux, B. Nadal, B. Dubertret, C. Gaillard and E. Nicol, J. Colloid Interface Sci., 2016, 476, 222–229 CrossRef CAS PubMed
- M. S. Nikolic, M. Krack, V. Aleksandrovic, A. Kornowski, S. Förster and H. Weller, Angew. Chem., 2006, 118, 6727–6731 CrossRef
- Z. Chen, Z. Liu, Z. Li, E. Ju, N. Gao, L. Zhou, J. Ren and X. Qu, Biomaterials, 2015, 39, 15–22 CrossRef CAS PubMed
- Z. Chen, Z. Li, Y. Lin, M. Yin, J. Ren and X. Qu, Chem. – Eur. J., 2013, 19, 1778–1783 CrossRef CAS PubMed
- E. Pöselt, C. Schmidtke, S. Fischer, K. Peldschus, J. Salamon, H. Kloust, H. Tran, A. Pietsch, M. Heine, G. Adam, U. Schumacher, C. Wagener, S. Förster and H. Weller, ACS Nano, 2012, 6, 3346–3355 CrossRef PubMed
- C. Chevigny, F. Dalmas, E. Di Cola, D. Gigmes, D. Bertin, F. Boué and J. Jestin, Macromolecules, 2011, 44, 122–133 CrossRef CAS
- M. S. Nikolic, M. Krack, V. Aleksandrovic, A. Kornowski, S. Förster and H. Weller, Angew. Chem., Int. Ed., 2006, 6577–6580 CrossRef CAS PubMed
- C. Schmidtke, E. Pöselt, J. Ostermann, A. Pietsch, H. Kloust, H. Tran, T. Schotten, N. G. Bastus, R. Eggers and H. Weller, Nanoscale, 2013, 7433–7444 RSC
- J. Dimitrijevic, L. Krapf, C. Wolter, C. Schmidtke, J.-P. Merkl, T. Jochum, A. Kornowski, A. Schüth, A. Gebert, H. Gereon, T. Vossmeyer and H. Weller, Nanoscale, 2014, 6, 10413–10422 RSC
- J. Merkl, C. Wolter, S. Flessau, C. Schmidtke, J. Ostermann, A. Feld, A. Mews and H. Weller, Nanoscale, 2016, 8, 7402–7407 RSC
- C. Schmidtke, R. Eggers, R. Zierold, A. Feld, H. Kloust, C. Wolter, J. Ostermann, J. P. Merkl, T. Schotten, K. Nielsch and H. Weller, Langmuir, 2014, 30, 11190–11196 CrossRef CAS PubMed
- A. Feld, R. Koll, L. S. Fruhner, M. Krutyeva, W. Pyckhout-Hintzen, C. Weiß, H. Heller, A. Weimer, C. Schmidtke, M. S. Appavou, E. Kentzinger, J. Allgaier and H. Weller, ACS Nano, 2017, 11, 3767–3775 CrossRef CAS PubMed
- L. H. Peebles Jr., J. Appl. Polym. Sci., 1973, 17, 113–128 CrossRef
- A. S. Sarac, Prog. Polym. Sci., 1999, 24, 1149–1204 CrossRef CAS
- Y. Y. Won, K. Paso, H. Ted Davis and F. S. Bates, J. Phys. Chem. B, 2001, 105, 8302–8311 CrossRef CAS
- R. Schweins and K. Huber, Macromol. Symp., 2004, 211, 25–42 CrossRef CAS
- L. J. Fetters, D. J. Lohse, S. T. Milner and W. W. Graessley, Macromolecules, 1999, 32, 6847–6851 CrossRef CAS
-
B. Smarsly, M. Groenewolt and M. Antonietti, Scattering Methods and the Properties of Polymer Materials, Springer, Berlin, Heidelberg, New York, 2005 Search PubMed
-
M. Rubinstein and R. H. Colby, Polymer Physics, Oxford University Press, 2003 Search PubMed
- A. S. Robbes, F. Cousin, F. Meneau, F. Dalmas, R. Schweins, D. Gigmes and J. Jestina, Macromolecules, 2012, 45, 9220–9231 CrossRef CAS
- D. Sunday, J. Ilavsky and D. L. Green, Macromolecules, 2012, 45, 4007–4011 CrossRef CAS
- D. F. Sunday and D. L. Green, Macromolecules, 2015, 48, 8651–8659 CrossRef CAS
- S. Srivastava, P. Agarwal and L. a. Archer, Langmuir, 2012, 28, 6276–6281 CrossRef CAS PubMed
- K. Davis, B. Qi, M. Witmer, C. L. Kitchens, B. A. Powell and O. T. Mefford, Langmuir, 2014, 30, 10918–10925 CrossRef CAS PubMed
- J. De Roo, Y. Justo, K. De Keukeleere, F. Van den Broeck, J. C. Martins, I. Van Driessche and Z. Hens, Angew. Chem., Int. Ed., 2015, 54, 1–5 CrossRef PubMed
- S. Ehlert, S. M. Taheri, D. Pirner, M. Drechsler, H.-W. Schmidt and S. Förster, ACS Nano, 2014, 8, 6114–6122 CrossRef CAS PubMed
- K. Gharbi, F. Salles, P. Mathieu, C. Amiens, V. Collière, Y. Coppel, K. Philippot, L. Fontaine, V. Montembault, L. S. Smiri and D. Ciuculescu-Pradines, New J. Chem., 2017, 41, 11898–11905 RSC
- W. W. Yu, J. C. Falkner, C. T. Yavuz and V. L. Colvin, Chem. Commun., 2004, 2306–2307 RSC
- E. Kentzinger, M. Krutyeva and U. Rücker, J. Large-Scale Res. Facilities, 2016, 2, 1–5 Search PubMed
- R. Biehl, Jscatter, a Program for Evaluation and Analysis of Experimental Data, https://zenodo.org/record/1470307.
- Heinz Maier-Leibnitz Zentrum, J. Large-Scale Res. Facilities, 2015, 1, 1–4 CrossRef
- A. V. Feoktystov, H. Frielinghaus, Z. Di, S. Jaksch, V. Pipich, M. S. Appavou, E. Babcock, R. Hanslik, R. Engels, G. Kemmerling, H. Kleines, A. Ioffe, D. Richter and T. Brückel, J. Appl. Crystallogr., 2015, 48, 61–70 CrossRef CAS
- V. Pipich, QtiKWS, http://www.qtikws.de.
- J. S. Pedersen, D. Posselt and K. Mortensen, J. Appl. Crystallogr., 1990, 23, 321–333 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available: Syntheses and characterization of polymers, crosslinking reactions, additional TEM and SAXS analysis, SANS fit model and fit results, TGA and DLS measurements. See DOI: 10.1039/c8nr10018g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |