
Shape-control of one-dimensional PtNi nanostructures as efficient electrocatalysts for alcohol electrooxidation†

Abstract
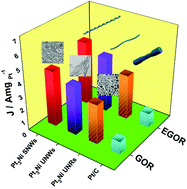
Bimetallic one-dimensional (1D) nanostructures such as nanowires (NWs) and nanorods (NRs), serving as high-efficiency anode electrocatalysts, have attracted extensive attention in the past decade. However, the precise design and synthesis of 1D Pt-based nanocrystals with tunable morphology and size still remain an arduous challenge. Driven by this, we report a facile yet efficient strategy for the first time to prepare PtNi ultrafine NWs (UNWs), sinuous NWs (SNWs) and ultrashort NRs (UNRs) by adjusting the amount of citric acid, ascorbic acid and glucose. Detailed analysis of their electrocatalytic properties has indicated that the as-obtained PtNi SNWs exhibit the most outstanding electrocatalytic activity toward ethylene glycol oxidation reaction (EGOR) and glycerol oxidation (GOR), 4.5 and 4.3 times higher in mass activity as well as 4.3 and 3.9 times higher in specific activity compared with the commercial Pt/C catalyst. The as-prepared PtNi SNWs are also more stable than the commercial Pt/C catalyst after successive durability tests. The proposed method provides insight into more rational designs of bimetallic nanocatalysts with 1D architectures and the as-synthesized PtNi catalysts with improved electrocatalytic performance assist in promoting the further development of direct alcohol fuel cells (DAFCs).
- This article is part of the themed collection: Editor’s Choice: Controlling anisotropy in nanomaterials
 Please wait while we load your content...
Please wait while we load your content...

