
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDNA origami directed 3D nanoparticle superlattice via electrostatic assembly†
Sofia
Julin
a,
Antti
Korpi
a,
Nonappa
 b,
Boxuan
Shen
a,
Ville
Liljeström
ac,
Olli
Ikkala
b,
Adrian
Keller
d,
Veikko
Linko
*a and
Mauri A.
Kostiainen
*ab
b,
Boxuan
Shen
a,
Ville
Liljeström
ac,
Olli
Ikkala
b,
Adrian
Keller
d,
Veikko
Linko
*a and
Mauri A.
Kostiainen
*ab
aBiohybrid Materials, Department of Bioproducts and Biosystems, Aalto University, P.O. Box 16100, 00760 Aalto, Finland. E-mail: mauri.kostiainen@aalto.fi; veikko.linko@aalto.fi
bHYBER Centre of Excellence, Department of Applied Physics, Aalto University, P.O. Box 15100, 00760 Aalto, Finland
cLaboratory of Soft and Complex Matter Studies, Department of Physics, Norwegian University of Science and Technology, Høgskoleringen 5, 7034 Trondheim, Norway
dTechnical and Macromolecular Chemistry, Paderborn University, Warburger Str. 100, 33098 Paderborn, Germany
First published on 21st February 2019
Abstract
The arrangements of metal nanoparticles into spatially ordered structures is still challenging, but DNA-based nanostructures have proven to be feasible building blocks in directing the higher-ordered arrangements of nanoparticles. However, an additional DNA functionalization of the particles is often required to link them to the DNA frames. Herein, we show that ordered 3D metal nanoparticle superlattices could be formed also by plainly employing electrostatic interactions between particles and DNA nanostructures. By utilizing the negatively charged DNA origami surface, we were able to assemble 6-helix bundle DNA origami and cationic gold nanoparticles (AuNPs) into well-ordered 3D tetragonal superlattices. Further, the results reveal that shape and charge complementarity between the building blocks are crucial parameters for lattice formation. Our method is not limited to only AuNPs and the origami shapes presented here, and could therefore be used in construction of a variety of functional materials.
Introduction
Recent advances in the field of nanotechnology have given rise to a diverse toolbox of nanoobjects with arbitrary shapes, sizes and material properties. However, effective strategies for controlling and directing their arrangement at the nanoscale are still needed.1,2 Predefined, highly ordered structures are challenging and costly to construct using traditional top-down nanofabrication techniques. Nevertheless, bottom-up strategies that are inspired by nature and rely on self-assembly have emerged as attractive low-cost alternatives.3,4 Various biomolecules, such as DNA,5,6 proteins,7,8 peptides9,10 and lipids,11 have been used as self-assembling building blocks. Due to its unique chemical and biological properties, DNA is one of the most promising ones for these purposes.12,13Molecular self-assembly using DNA is a compelling approach to construct structurally versatile, well-defined and highly addressable nano- and microscale objects.13–15 In particular due to the invention of the DNA origami technique,16 custom-designed DNA-based motifs can nowadays be readily fabricated and used in various applications such as in drug delivery,17,18 plasmonics19–21 and enzymatic and chemical reaction systems.22,23 In addition, DNA nanostructures have been used to direct higher-ordered arrangements of DNA-functionalized metal nanoparticles.24,25 Recently, DNA origami frames have been used to construct also predefined three-dimensional (3D) superlattices.26–28 Spatially ordered structures of AuNPs and other metal nanoparticles have unique electronic, magnetic and optical properties, and are therefore of great interest in a variety of applications.22,29 However, the programmable arrangement of metal nanoparticles into well-ordered crystal structures is still demanding, and methods relying on nanoparticles without further modifications, such as DNA functionalization, would be highly desirable.
We have previously shown that protein cages, such as cowpea chlorotic mottle virus,30,31 tobacco mosaic virus,32 and ferritin,30 can be used to direct the formation of well-defined AuNP superstructures by employing electrostatic self-assembly. However, the structural variety of the assemblies achieved using protein cages is limited due to the predetermined shape, charge and size of the selected protein. Therefore, it is desirable to explore whether programmable and modular DNA nanostructures could be equally used to organize AuNPs into well-ordered structures. One of the most powerful techniques to assemble DNA structures is arguably DNA origami, in which a long single-stranded DNA scaffold is folded into a predefined structure through the cooperative action of dozens of complementary single-stranded oligonucleotides. The DNA origami technique can be used to construct almost any arbitrary two- or three-dimensional nanostructure.16,33,34 Further, due to the sugar-phosphate backbone of the DNA molecule, the DNA origami structures carry a high overall negative (surface) charge, which makes them suitable building blocks in electrostatic self-assembly. Recently, DNA origami structures have been used together with positively charged collagen-mimetic peptides to electrostatically assemble lamellar nanowires.35 Previous studies, in which DNA origami have been electrostatically coated with virus capsid proteins,36 cationic polymers,37–40 chitosan40 and protein–dendron conjugates,41 also demonstrate that DNA origami structures can be complexed with cationic compounds in a feasible way.
Results and discussion
In this work, we show that DNA origami nanostructures can be used to guide the higher-order arrangement of cationic AuNPs in a controlled and programmable manner. In addition, we demonstrate that the size and shape of the self-assembling building blocks are of major importance for whether an ordered crystal structure or an amorphous aggregate is formed. For this study, three different M13mp18-scaffolded DNA origami; 6-helix bundle (6HB), 24-helix bundle (24HB) and 60-helix bundle (60HB) (Fig. 1a), were folded (see ESI† for the staple strands and annealing procedures). Further, cationic AuNPs of three different sizes were synthesized, all with a narrow size distribution (see ESI† for the synthesis, characterization and data for the “extra-large” AuNP). The AuNPs were functionalized with either a covalently linked (11-mercaptoundecyl)-N,N,N-trimethylammonium bromide (MUTAB) ligand or an alkyl-oxyethylene ligand (Fig. 1b). Due to the quaternary ammonium group of the ligands, the AuNPs have a highly cationic surface over a wide range of pH values.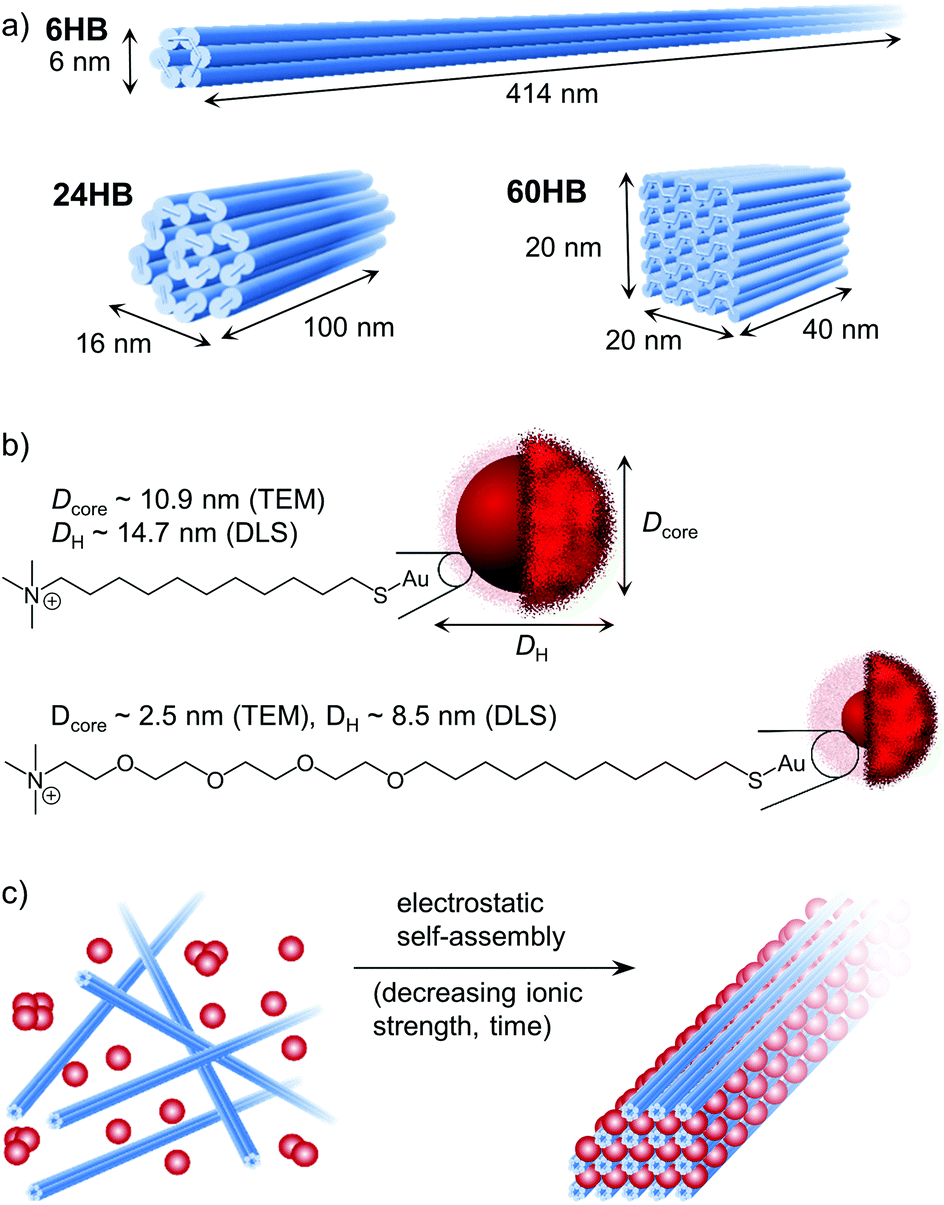 | ||
| Fig. 1 Building blocks used to study the electrostatic self-assembly of DNA origami and gold nanoparticles (AuNPs). (a) Shape and dimensions of the three DNA origami structures used in this study: 6-helix bundle (6HB), 24-helix bundle (24HB) and 60-helix bundle (60HB). (b) Presentation of the AuNPs, including the chemical structure of the cationic ligand. (c) DNA origami–AuNP crystal structures were formed upon dialysis against decreasing ionic strength. | ||
To better understand the electrostatic self-assembly of DNA origami nanostructures and AuNPs, and the formation of assemblies, different combinations of DNA origami structures and AuNPs, as well as the effect of the stoichiometric ratio between the AuNPs and the DNA origami structures (nAuNP/norigami) were studied. Assemblies could be achieved by simply mixing these two components together, but as it is known that oppositely charged objects readily form kinetically trapped structures when mixed together at low ionic strength, we chose to use dialysis to form ordered DNA origami–AuNP assemblies in a more controllable way (Fig. 1c). Initially, the components were mixed together at such a high ionic strength (cNaCl = 500–750 mM depending on the AuNP size) that the electrostatic interactions between the charged components were effectively screened. Then, by dialyzing the mixture against gradually decreasing ionic strength, we were able to tune the electrostatic interaction between the AuNPs and the DNA origami, and therefore to create micrometer-sized, ordered assemblies.
The electrostatic binding of AuNPs to the DNA origami structures was first characterized using an agarose gel electrophoretic mobility shift assay (EMSA) (Fig. 2a). A constant amount of DNA origami solution was mixed with increasing amounts of AuNP solution in order to find the amount of AuNPs needed to completely immobilize different DNA origami structures in the gel. When increasing amounts of small AuNPs (Dcore = 2.5 nm) were used (Fig. 2a, left column), a clear gradual shift in 6HB electrophoretic mobility was observed, and further, at a stoichiometric ratio of nAuNP/norigami ∼ 150, the 6HB was fully immobilized (Fig. 2a upper left panel). However, when these same AuNPs were complexed with the 24HB or the 60HB, no binding between AuNPs and DNA origami was observed within the studied concentration range (Fig. 2a, middle left and lower left panels). Larger AuNPs (Dcore = 10.9 nm), for one, were able to immobilize all of these three DNA origami at a nAuNP/norigami ∼ 30–40, indicating an efficient binding between large AuNPs and all studied DNA origami structures (Fig. 2a, right column).
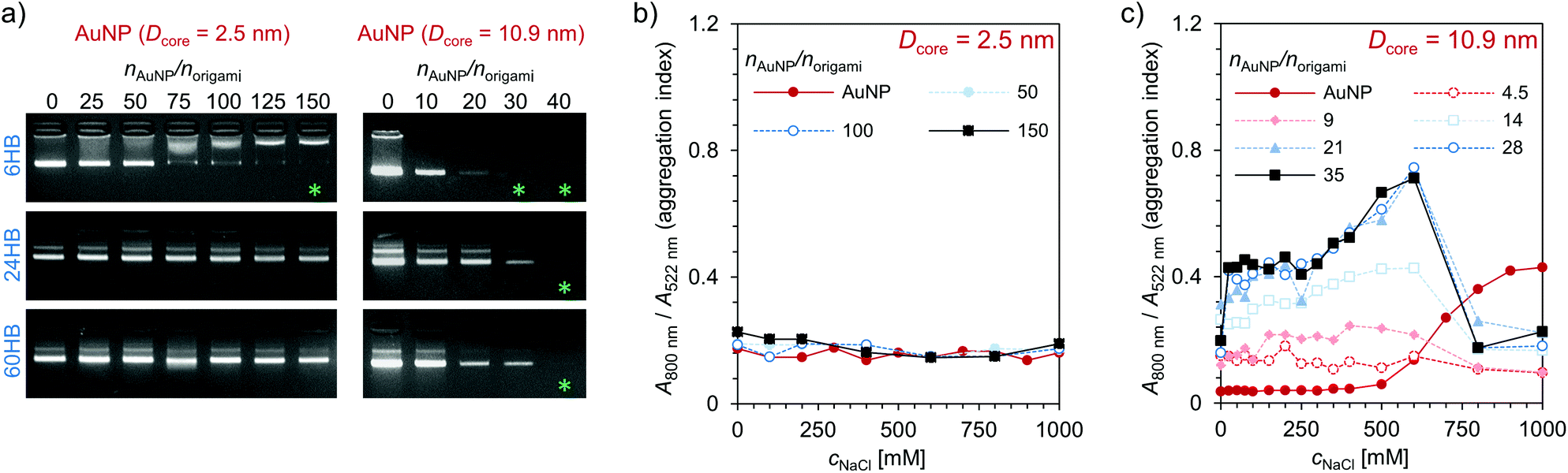 | ||
| Fig. 2 Electrostatic binding of the cationic AuNPs to the negatively charged DNA origami structures. (a) Agarose gel electrophoretic mobility shift assay (EMSA) for a constant amount of DNA origami (6HB, 24HB, or 60HB) complexed with increasing amounts of AuNPs (Dcore = 2.5 nm (left) or Dcore = 10.9 nm (right)). The samples were not dialyzed before loaded into the gel pockets. In the gel images, the green asterisk indicate successful binding of the AuNPs to the DNA origami structures. Colloidal stability of (b) small AuNPs (Dcore = 2.5 nm) and (c) large AuNPs (Dcore = 10.9 nm) when mixed with increasing amounts of DNA origami 6HB structures. | ||
In addition to the EMSA, the clustering of AuNPs was studied with UV/Vis spectroscopy. When the AuNPs bind to the DNA origami structures, the AuNPs will come into close proximity with each other, which will further cause their oscillating electric fields to interact with each other and the surface plasmons of the AuNPs to be coupled. As a result, there will be a significant shift to longer wavelengths and a broadening of the surface plasmon band.42,43 AuNPs with a diameter of ∼10 nm have an absorption peak in the region of 520 nm.44,45 Therefore, we studied the binding of the AuNPs to the DNA origami structures by following a shift in the plasmonic peak using the aggregation index A800 nm/A522 nm as a measure, where A800 nm and A522 nm are the absorbances at a wavelength of 800 nm and 522 nm, respectively. The magnitude of the plasmonic shift is highly dependent on the relative distances between the AuNPs, and it decays with large particle separation distances.46 The small AuNPs (Dcore = 2.5 nm) have a large ligand size compared to the size of their core, and therefore there will be no significant plasmonic coupling and shift in the absorption peak even though the AuNPs are closely packed (Fig. 2b). However, the larger AuNPs (Dcore = 10.9 nm) with a noticeably smaller relative ligand size show increased plasmonic coupling and aggregation with increasing DNA origami and/or NaCl concentration (see Fig. 2c and ESI†). When the DNA origami structures are present in the solution, the aggregation index is high already at low NaCl concentrations, which indicates that large AuNPs tend to bind easily to the DNA origami. For the combination of these AuNPs and 6HB, the changes in the aggregation index suggest that a nAuNP/norigami ∼ 20–30 would be optimal for formation of DNA origami 6HB–AuNP complexes. If the ionic strength is high enough, the formed assemblies will disaggregate again, which for the DNA origami–AuNP assemblies can be seen as a drop in the aggregation index at cNaCl ∼ 600 mM.
The DNA origami–AuNP assemblies were formed upon dialysis against decreasing ionic strength, and all studied combinations of DNA origami and AuNP yielded assemblies that were visible with a bare eye as a dark precipitate. To gain more insight into the structural morphology of the assemblies, small angle X-ray scattering (SAXS) measurements were carried out on aqueous samples containing different combinations of DNA origami and AuNP as well as varying stoichiometric ratios, nAuNP/norigami (Fig. 3 and ESI†). Well-ordered superlattice structures were formed in the case of 6HB and small AuNP (Dcore = 2.5 nm) (Fig. 3a), whereas all other studied combinations produced less ordered aggregates with only a short range order (see Fig. 3d and ESI†). For the assemblies of 6HB and small AuNPs, clear diffraction peaks were identified in samples having a wide range of stoichiometric ratios. Nevertheless, the best resolved diffraction peaks were obtained from the sample with nAuNP/norigami ∼ 150, while the diffraction peaks were less evident with increasing/decreasing nAuNP/norigami. The optimal nAuNP/norigami ∼ 150 is also in accordance with the obtained EMSA result.
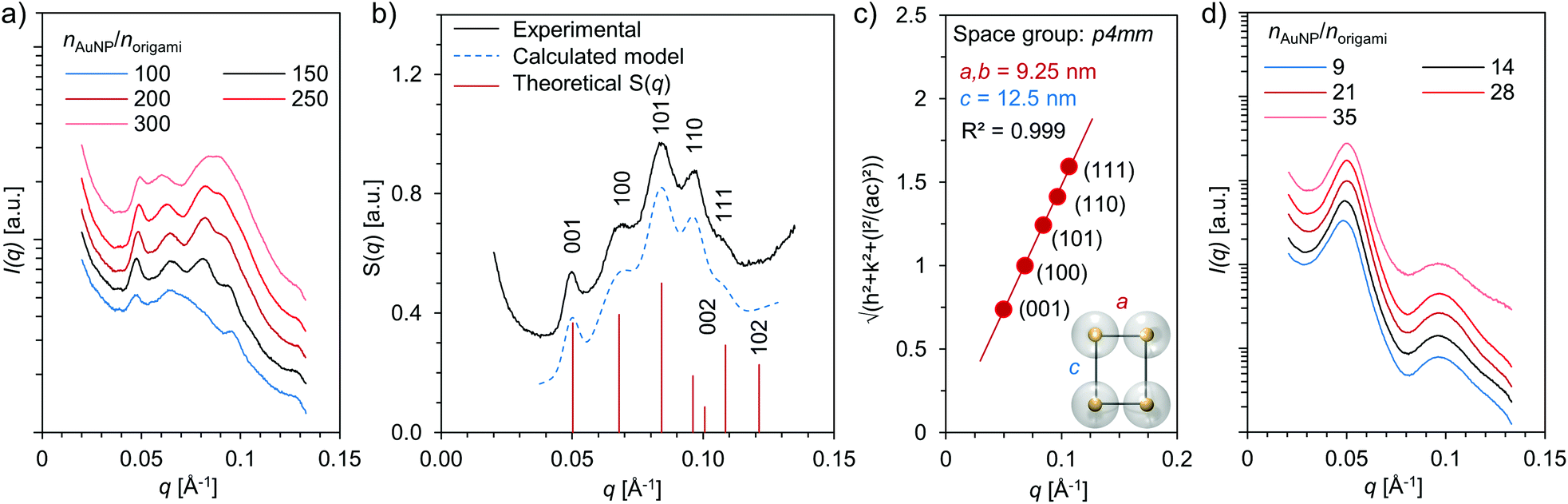 | ||
| Fig. 3 Structural characterization of DNA origami–AuNP assemblies by small-angle X-ray scattering (SAXS) (a) SAXS data measured from samples having different stoichiometric ratios between small AuNPs (Dcore = 2.5 nm) and 6HB structures (nAuNP/norigami). (b) Measured and calculated structure factor S(q) for nAuNP/norigami ∼ 150 (small AuNPs and 6HB). a = 9.25 nm and c = 12.5 nm are assumed in the calculations. (c) Quadratic Miller indices of assigned reflections plotted as a function of measured q-values (small AuNPs and 6HB, nAuNP/norigami ∼ 150) (d) SAXS data measured from samples having different stoichiometric ratios between large AuNPs (Dcore = 10.9 nm) and 6HB (nAuNP/norigami). | ||
For the combination of 6HB and small AuNPs at nAuNP/norigami ∼ 150, additional analysis of the SAXS data was conducted in order to determine the crystallographic arrangement of the AuNPs. The peak positions of the measured structure factor, S(q), were found at q = 0.050, 0.069, 0.084, 0.096 and 0.107 Å−1, which correspond to reflections from the planes with Miller indices (hkl) = (001), (100), (101), (110) and (111), respectively (Fig. 3b). These peak positions can be assigned to Bragg reflections from a 3D tetragonal lattice (space group p4mm, number 99) with lattice constants a = 9.25 nm and c = 12.5 nm (Fig. 3c). Further, the heights and peak positions of the experimental data is also in excellent agreement with the calculated theoretical SAXS model for a tetragonal system with similar dimensions.
To confirm the SAXS measurements and to visualize the formed superlattice structure, we imaged the samples with both conventional and cryogenic transmission electron microscopy (TEM and cryo-TEM) and conducted cryogenic electron tomography (cryo-ET) reconstruction of the crystalline assemblies. From the cryo-TEM images (Fig. 4a–c and e) and cryo-ET reconstruction (Fig. 4f), it is evident that 6HB and small AuNPs (Dcore = 2.5 nm) form large, micrometer-sized 3D superlattice structures. Fig. 4e shows frequency domain filtered cryo-TEM images of different projections and the expected superlattice projection viewed along the same projection axis, whereas Fig. 4g depicts a schematic view of the assembled tetragonal superlattice including the lattice parameters. The average lattice constants determined from the cryo-TEM images and cryo-ET reconstruction are a = 8.6 ± 0.9 nm (s.d.), c = 11.8 ± 1.0 nm (s.d.) (Fig. 4c and d) and a = 9.1 nm, c = 11.9 nm (Fig. 4f), respectively, which correspond well to the lattice constants obtained from the SAXS characterization. The lattice constant a is, however, slightly smaller than would be assumed from the dimensions of the building blocks, which is likely due to a compression of the rather flexible ligand of the AuNPs, observed also before in electrostatic self-assemblies formed with this AuNP.32 The lattice constant c gives the interparticle distance along the long axis of 6HB, and due to electrostatic repulsion between adjacent AuNPs (without screening effect of 6HB between the particles), c is longer than a.
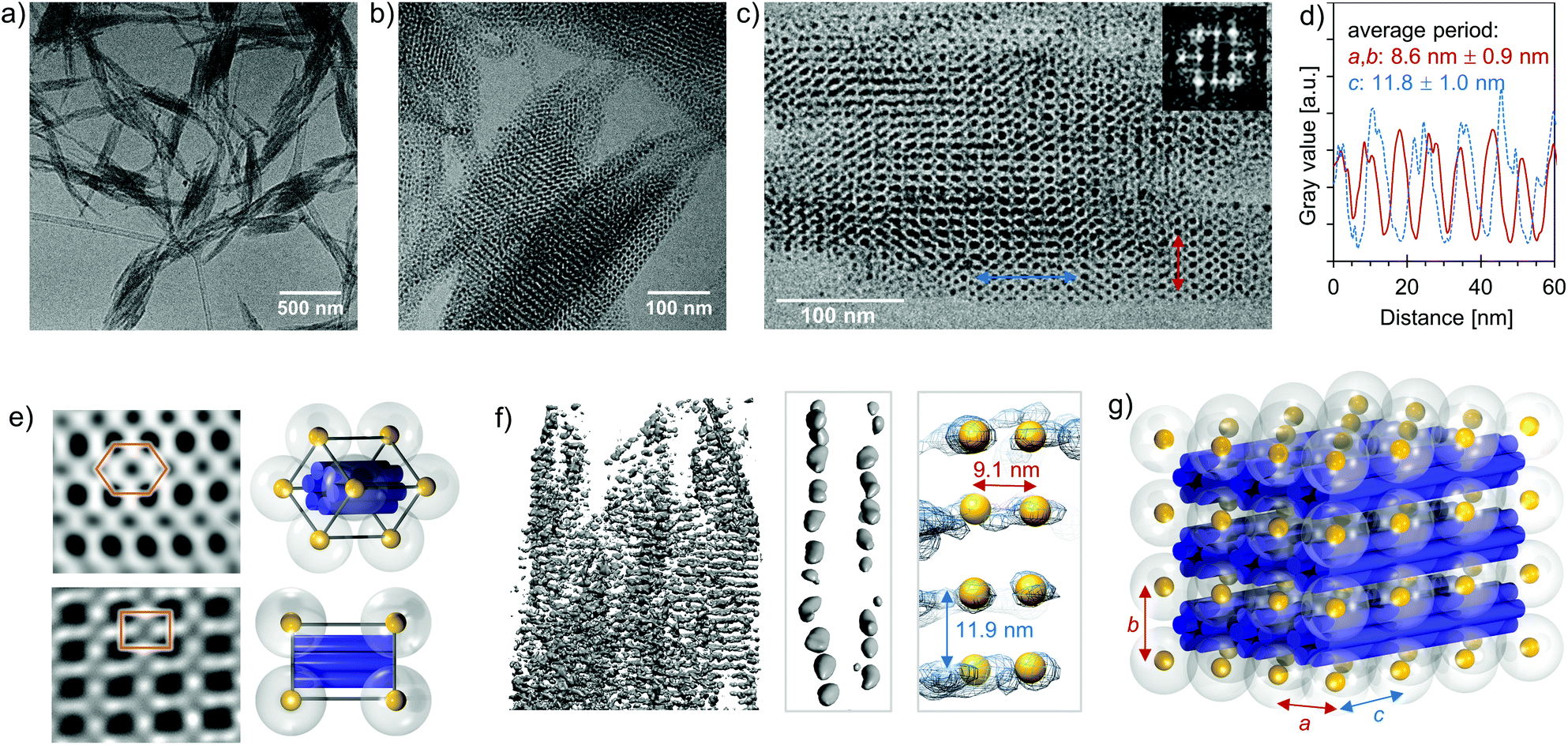 | ||
| Fig. 4 Cryo-TEM characterization of the assembled tetragonal superlattices (formed by small AuNPs and 6HB). (a)–(b) Cryo-TEM images of the superlattices at different magnification. (c) Cryo-TEM image of the superlattice viewed along the [100] projection axis. The inset shows the Fourier transform. (d) Integrated profiles along the blue and red lines indicated in image (c). (e) Inverse Fourier transform calculated with selected Fourier components from TEM images along different projection axes (top: hexagonal, bottom: [100]) and a schematic of the unit cell viewed along the same projection axis. (f) A cryo-ET reconstruction of a selected part of the assembled complexes (left) and a cryo-ET density map along a single DNA origami structure (middle and right). In the left and middle reconstruction, the grey sphere-like objects are local density maxima, and the AuNPs are denoted by yellow spheres in the right reconstruction. (g) A schematic of the 3 × 3 tetragonal unit cell assembled from small AuNPs and 6HBs, with the lattice parameters a, b and c. | ||
Electrostatic self-assembly of 6HBs and small AuNPs (Dcore = 2.5 nm) yielded highly ordered superlattices, whereas all other studied combinations resulted in aggregates with only short range order. Therefore, the results suggest that both shape and charge complementarity between the building blocks are crucial parameters for crystal formation. The 6HBs are rather flexible and when combined with highly cationic, much larger (Dorigami ∼ 6 nm vs. DAuNP ∼ 14–16 nm) AuNPs, they are prone to bend and wrap around the AuNPs instead of forming close-packed superlattices with them. The diameter of the 24HB should be optimal for formation of superlattices with all three kinds of AuNPs (see ESI† for data for the “extra-large” AuNP), but its shorter length might not provide the required cooperativity. The length of 100 nm is not necessarily long enough to guide the directional growth of the lattice, as the 24HB that binds to the formed 24HB–AuNP-pair can attach with almost any orientation. The box-like shape of the 60HB does neither exhibit a sufficient degree of anisotropy to favour lattice formation nor provide dimensional complementarity with these AuNPs. It is therefore expected that 60HB and the AuNPs assemble only into disordered structures.
Conclusions
In conclusion, we have demonstrated an assembly method based on DNA origami structures that can direct the formation of well-ordered 3D AuNP crystalline structures merely via electrostatic interactions. DNA origami design is nowadays rather user-friendly,15 and as the programmable DNA origami structures can be produced at reasonable prices,47 their use in real world applications is becoming more feasible. Our method is straightforward to employ, and in contrast to previously reported DNA origami directed AuNP superlattices,25 there is no need to functionalize the AuNPs with single-stranded DNA (ssDNA) sequences. The reported method is not limited to the AuNPs or the DNA origami shapes presented in this work. Recently, it was also observed that DNA origami structures are stable in a wide range of conditions.48 Therefore we believe our approach is suitable for many other experimental setups. By virtue of the modular DNA origami we believe that this approach in the future can open up new opportunities for the construction of a variety of functional materials with unique properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support through the Academy of Finland (267497, 286845, 308578), Jane and Aatos Erkko Foundation, Sigrid Jusélius Foundation, Walter Ahlström Foundation and Finnish Cultural Foundation is gratefully acknowledged. This work was carried out under the Academy of Finland Centers of Excellence Programme (2014–2019) and made use of the Aalto University Nanomicroscopy Center (Aalto-NMC).References
- K. J. M. Bishop, C. E. Wilmer, S. Soh and B. A. Grzybowski, Small, 2009, 5, 1600–1630 CrossRef CAS PubMed.
- Z. Nie, A. Petukhova and E. Kumacheva, Nat. Nanotechnol., 2010, 5, 15–25 CrossRef CAS PubMed.
- H. Li, J. D. Carter and T. H. LaBean, Mater. Today, 2009, 12, 24–32 CrossRef CAS.
- A. Biswas, I. S. Bayer, A. S. Biris, T. Wang, E. Dervishi and F. Faupel, Adv. Colloid Interface Sci., 2012, 170, 2–27 CrossRef CAS PubMed.
- N. C. Seeman, Nature, 2003, 421, 427–431 CrossRef PubMed.
- M. R. Jones, N. C. Seeman and C. A. Mirkin, Science, 2015, 347, 1260901 CrossRef PubMed.
- M. Uchida, M. T. Klem, M. Allen, P. Suci, M. Flenniken, E. Gillitzer, Z. Varpness, L. O. Liepold, M. Young and T. Douglas, Adv. Mater., 2007, 19, 1025–1042 CrossRef CAS.
- Q. Luo, C. Hou, Y. Bai, R. Wang and J. Liu, Chem. Rev., 2016, 116, 13571–13632 CrossRef CAS PubMed.
- J. Wang, K. Liu, R. Xing and X. Yan, Chem. Soc. Rev., 2016, 45, 5589–5604 RSC.
- D. M. Raymond and B. L. Nilsson, Chem. Soc. Rev., 2018, 47, 3659–3720 RSC.
- M. Antonietti and S. Förster, Adv. Mater., 2003, 15, 1323–1333 CrossRef CAS.
- F. A. Aldaye, A. L. Palmer and H. F. Sleiman, Science, 2008, 321, 1795–1799 CrossRef CAS PubMed.
- F. Hong, F. Zhang, Y. Liu and H. Yan, Chem. Rev., 2017, 117, 12584–12640 CrossRef CAS PubMed.
- N. C. Seeman and H. F. Sleiman, Nat. Rev. Mater., 2017, 3, 17068 CrossRef.
- S. Nummelin, J. Kommeri, M. A. Kostiainen and V. Linko, Adv. Mater., 2018, 30, 1703721 CrossRef PubMed.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- J. Li, C. Fan, H. Pei, J. Shi and Q. Huang, Adv. Mater., 2013, 25, 4386–4396 CrossRef CAS PubMed.
- V. Linko, A. Ora and M. A. Kostiainen, Trends Biotechnol., 2015, 33, 586–594 CrossRef CAS PubMed.
- A. Kuzyk, R. Schreiber, Z. Fan, G. Pardatscher, E. M. Roller, A. Högele, F. C. Simmel, A. O. Govorov and T. Liedl, Nature, 2012, 483, 311–314 CrossRef CAS PubMed.
- B. Shen, V. Linko, K. Tapio, S. Pikker, T. Lemma, A. Gopinath, K. V. Gothelf, M. A. Kostiainen and J. J. Toppari, Sci. Adv., 2018, 4, eaap8978 CrossRef PubMed.
- N. Liu and T. Liedl, Chem. Rev., 2018, 118, 3032–3053 CrossRef CAS PubMed.
- K. V. Gothelf, MRS Bull., 2017, 42, 897–903 CrossRef CAS.
- G. Grossi, A. Jaekel, E. S. Andersen and B. Saccà, MRS Bull., 2017, 42, 920–924 CrossRef CAS.
- N. C. Seeman and O. Gang, MRS Bull., 2017, 42, 904–912 CrossRef CAS.
- S. Julin, S. Nummelin, M. A. Kostiainen and V. Linko, J. Nanopart. Res., 2018, 20, 119 CrossRef PubMed.
- W. Liu, M. Tagawa, H. L. Xin, T. Wang, H. Emamy, H. Li, K. G. Yager, F. W. Starr, A. V. Tkachenko and O. Gang, Science, 2016, 351, 582–586 CrossRef CAS PubMed.
- Y. Tian, Y. Zhang, T. Wang, H. L. Xin, H. Li and O. Gang, Nat. Mater., 2016, 15, 654–661 CrossRef CAS PubMed.
- T. Zhang, C. Hartl, K. Frank, A. Heuer-Jungemann, S. Fischer, P. C. Nickels, B. Nickel and T. Liedl, Adv. Mater., 2018, 30, 1800273 CrossRef PubMed.
- Y. Ofir, B. Samanta and V. M. Rotello, Chem. Soc. Rev., 2008, 37, 1814–1825 RSC.
- M. A. Kostiainen, P. Hiekkataipale, A. Laiho, V. Lemieux, J. Seitsonen, J. Ruokolainen and P. Ceci, Nat. Nanotechnol., 2013, 8, 52–56 CrossRef CAS PubMed.
- J. Hassinen, V. Liljeström, M. A. Kostiainen and R. H. A. Ras, Angew. Chem., Int. Ed., 2015, 54, 7990–7993 CrossRef CAS PubMed.
- V. Liljeström, A. Ora, J. Hassinen, H. T. Rekola, Nonappa, M. Heilala, V. Hynninen, J. J. Joensuu, R. H. A. Ras, P. Törmä, O. Ikkala and M. A. Kostiainen, Nat. Commun., 2017, 8, 671 CrossRef PubMed.
- S. M. Douglas, H. Dietz, T. Liedl, B. Högberg, F. Graf and W. M. Shih, Nature, 2009, 459, 414–418 CrossRef CAS PubMed.
- C. E. Castro, F. Kilchherr, D. N. Kim, E. L. Shiao, T. Wauer, P. Wortmann, M. Bathe and H. Dietz, Nat. Methods, 2011, 8, 221–229 CrossRef CAS PubMed.
- T. Jiang, T. A. Meyer, C. Modlin, X. Zuo, V. P. Conticello and Y. Ke, J. Am. Chem. Soc., 2017, 139, 14025–14028 CrossRef CAS PubMed.
- J. Mikkilä, A. P. Eskelinen, E. H. Niemelä, V. Linko, M. J. Frilander, P. Törmä and M. A. Kostiainen, Nano Lett., 2014, 14, 2196–2200 CrossRef PubMed.
- J. K. Kiviaho, V. Linko, A. Ora, T. Tiainen, E. Järvihaavisto, J. Mikkilä, H. Tenhu, Nonappa and M. A. Kostiainen, Nanoscale, 2016, 8, 11674–11680 RSC.
- N. P. Agarwal, M. Matthies, F. N. Gür, K. Osada and T. L. Schmidt, Angew. Chem., Int. Ed., 2017, 56, 5460–5464 CrossRef CAS PubMed.
- N. Ponnuswamy, M. M. C. Bastings, B. Nathwani, J. H. Ryu, L. Y. T. Chou, M. Vinther, W. A. Li, F. M. Anastassacos, D. J. Mooney and W. M. Shih, Nat. Commun., 2017, 8, 15654 CrossRef CAS PubMed.
- Y. Ahmadi, E. De Llano and I. Barišić, Nanoscale, 2018, 10, 7494–7504 RSC.
- H. Auvinen, H. Zhang, Nonappa, A. Kopilow, E. H. Niemelä, S. Nummelin, A. Correia, H. A. Santos, V. Linko and M. A. Kostiainen, Adv. Healthcare Mater., 2017, 6, 1700692 CrossRef PubMed.
- J. J. Storhoff, R. Elghanian, R. C. Mucic, C. A. Mirkin and R. L. Letsinger, J. Am. Chem. Soc., 1998, 120, 1959–1964 CrossRef CAS.
- A. N. Shipway, M. Lahav, R. Gabai and I. Willner, Langmuir, 2000, 16, 8789–8795 CrossRef CAS.
- S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 8410–8426 CrossRef CAS.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- P. K. Jain, W. Huang and M. A. El-Sayed, Nano Lett., 2007, 7, 2080–2088 CrossRef CAS.
- F. Praetorius, B. Kick, K. L. Behler, M. N. Honemann, D. Weuster-Botz and H. Dietz, Nature, 2017, 552, 84–87 CrossRef CAS PubMed.
- C. Kielar, Y. Xin, B. Shen, M. A. Kostiainen, G. Grundmeier, V. Linko and A. Keller, Angew. Chem., Int. Ed., 2018, 57, 9470–9474 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nr09844a |
| This journal is © The Royal Society of Chemistry 2019 |