
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Annealing multicomponent supramolecular gels†
Ana M.
Fuentes-Caparrós
 a,
Francisco
de Paula Gómez-Franco
ab,
Bart
Dietrich
a,
Claire
Wilson
a,
Christopher
Brasnett
c,
Annela
Seddon
cd and
Dave J.
Adams
*a
a,
Francisco
de Paula Gómez-Franco
ab,
Bart
Dietrich
a,
Claire
Wilson
a,
Christopher
Brasnett
c,
Annela
Seddon
cd and
Dave J.
Adams
*a
aSchool of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: dave.adams@glasgow.ac.uk
bDepartment of Chemical Engineering, Faculty of Sciences, University of Granada, 18071 Granada, Spain
cSchool of Physics, HH Wills Physics Laboratory, Tyndall Avenue, University of Bristol, Bristol, BS8 1TL, UK
dBristol Centre for Functional Nanomaterials, HH Wills Physics Laboratory, Tyndall Avenue, University of Bristol, Bristol, BS8 1TL, UK
First published on 30th January 2019
Abstract
Annealing is widely used as a means of changing the physical properties of a material. The rate of heating and cooling used in the annealing process controls the final properties. Annealing can be used as a means of driving towards the, or at least a, thermodynamic minimum. There is surprisingly little information on annealing kinetically-trapped supramolecular gels. Here, we show that annealing multicomponent gels can be used to prepare materials with tunable mechanical properties. We show that annealing in a two-component gel leads to a self-sorted network, which has significantly different mechanical properties to the as-prepared gels. Whilst the fibres are self-sorted, we show that the annealing of this system leads to significant change in the network level of assembly, and it is this that leads to the increase in storage modulus. We also show that it is possible to selectively anneal only a single component in the mixture.
Introduction
Gels are widely used and extremely useful materials. One interesting class, low molecular weight gels (LMWG) has applications in areas as diverse as biomaterials and optoelectronics.1–5 These gels arise from the self-assembly of small molecules into fibre networks. These LMWG are often not at equilibrium, but rather the gels represent kinetically-trapped states. The process by which the gels are prepared often controls the final properties and hence it is possible to make different gels under the same final conditions by varying how the gel is formed.6–8 This means that it can be difficult to prepare reproducible gels if the conditions used are not sufficiently well controlled, but this also opens up opportunities. For example, instead of preparing many molecules to find one that gives gels with the required properties, one can more easily vary the process of gelation instead.8,9One of the key issues with using LMWG is that it is very difficult to predict which molecules will form gels and which will not.4 On top of this, predicting what the properties will be of a gel formed from a particular LMWG is essentially impossible. Hence, a more pragmatic approach is to find robust LMWG and develop methods for controlling, modulating and modifying the properties of the gels that can be formed from robust LMWG.
Many materials are prepared such that a kinetically-trapped state is formed.6,7,10 Kinetically-trapped states provide opportunities to prepare a range of interesting and useful materials from a single set of components, as long as the energy well is not too deep.10,11 From the same starting material, different properties can be accessed by varying how the different states are accessed. It can be possible to move between states, hence allowing tunable and triggerable materials to be accessed.
Considering that LMWG are often kinetically-trapped, and many gels are formed using a heat-cool approach, there has been surprisingly little work carried out on annealing them. Where it has been reported, it is for single component systems.7,12–15 LMWG are a result of non-covalent interactions and so there is the potential to change between states if the energy barriers are suitably low.
Using multicomponent systems where both components can independently form gels can be an effective strategy for preparing gels with properties that cannot be achieved with either individual component.16–21 Multicomponent systems offer even higher possibilities for tuning than single component systems, but the complexity is significantly greater. Whilst annealing has been examined for a small number of single component systems, to the best of our knowledge there are no examples for multicomponent systems, which are significantly more complicated, but also provide added opportunities.
Results and discussion
In this paper, we discuss a multicomponent system of two gelators,22–25 based around a dipeptide or an amino acid functionalized at the N-terminus (1 and 2, Fig. 1a).26,27 We use a solvent-triggered approach where the gelator is initially dissolved in a water-miscible solvent such as DMSO, followed by the addition of water.9,28–32 This drives a phase separation that results in spherulitic domains of fibers that entangle to form a self-supporting gel.30,33,34 The gels formed by this method are kinetically trapped. Applying a multicomponent gelator approach here, we used gelation conditions which were optimized for mixing experiments; the concentrations of the gelators were chosen such that both 1 and 2 formed gels at the same final ratio of DMSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water (a ratio of 3:7 was used throughout) and that the gel melting temperatures were in a suitable range for analysis (see below). We stress that we have optimized the system here to demonstrate the principle; the concentrations and ratios of gelators and the solvent composition have been chosen to allow distinct melting points to be compared. We have done this as we aim to show clearly the effects of annealing; it is extremely difficult to deconvolute this information when there are simultaneous processes occurring.
:water (a ratio of 3:7 was used throughout) and that the gel melting temperatures were in a suitable range for analysis (see below). We stress that we have optimized the system here to demonstrate the principle; the concentrations and ratios of gelators and the solvent composition have been chosen to allow distinct melting points to be compared. We have done this as we aim to show clearly the effects of annealing; it is extremely difficult to deconvolute this information when there are simultaneous processes occurring.
 | ||
| Fig. 1 (a) Chemical structure of 1 and 2. (b) Photograph of (left to right) a gel of 1 before annealing, a gel of 1 after annealing, a gel of 2 before annealing, and a gel of 2 after annealing. The scale bar represents 1 cm. (c) Rheological data for a gel of 1 on heating and cooling at a rate of 1 °C min−1. (d) Rheological data for a gel of 2 on heating and cooling at a rate of 1 °C min−1. For (c), and (d), closed symbols represent G′ and open symbols represent G′′. The black data are for the heating cycle, and the red data for the cooling cycle. | ||
1 forms transparent gels at this ratio of DMSO:water and a concentration of 4 mg mL−1 (Fig. 1b). The gels are reproducible (Fig. S10†), and typical of a LMWG. The gel is stable until a strain of ∼4% before G′ starts to deviate from linearity (Fig. S10a†). The storage modulus (G′) and loss modulus (G′′) are relatively independent of frequency (Fig. S10b†). We have previously shown that heating and cooling gels formed by 1 results in the gel melting, and then re-forming.30 On heating, G′ and G′′ slowly decrease until a plateau at around 65 °C (Fig. 1c). On re-cooling, G′ and G′′ only start to increase again at around 36 °C, with a gel of similar transparency formed to the original. At 15 °C, the values of both moduli are slightly higher than for the original gel.
The increase in moduli by annealing is explained by a change in the gel microstructure (Fig. 2a), from spherulitic domains to a more uniform distribution of fibers.
 | ||
| Fig. 2 (a) Confocal images of gels of 1 before (left) and after (right) annealing. (b) Confocal images of gels of 2 before (left) and after (right) annealing (scale bars represent 20 μm). The contrast has been enhanced to better show the structures; the originals are in Fig. S9.† The gels were stained with Nile Blue. | ||
The thermal behaviour was backed up by the dropping ball method, which showed similar melting and re-gelation temperatures (Fig. S11†). The hysteresis between the melting and re-gelling temperature can be explained by the need to form a network. On melting, the gel gets weaker as 1 dissolves. On cooling, 1 starts to re-assemble, but a gel is only formed when the network spans the entire sample. NMR experiments show a symmetrical solubility on heating and cooling of 1 (Fig. S12†), showing that this hysteresis is not due to asymmetric solubility. Such hysteresis has been observed elsewhere,35,36 although we note that not all LMWG show this effect.12,13 After annealing, the gels are again reproducible, and are essentially frequency-independent (Fig. S13†). The gels break at a very similar strain as before annealing, although the absolute values of the moduli are higher.
Gels can also be formed at this ratio of DMSO:water using 2 (Fig. 1a). To re-iterate, we optimized the conditions such that the gels melted at a different temperature to those of 1. Hence, gels were formed from 2 at 8 mg mL−1. The single component gel of 2 was turbid (Fig. 1b). The gels were again reproducible (Fig. S14†). G′ and G′′ were essentially frequency independent, with the gels having a similar breakdown to 1 under increasing strain. On heating, G′ and G′′ slowly decrease up to 80 °C (Fig. 1d). On cooling, G′ and G′′ increase once again at around 62 °C, with higher moduli than for the original gel (Fig. 1d). After annealing, the gels of 2 are reproducible (Fig. S15†). The annealed gel is a little more transparent than the original (Fig. 1b) with small crystals in the gel (Fig. S16†). A significant change in microstructure also occurs on annealing (Fig. 2b). Using a very slow cooling rate, it is possible to grow crystals from the gel which are suitable for crystal X-ray diffraction study (Fig. S16 and S17†). A hydrogen bond links the carboxylate OH (O2) and amide oxygen (O11′), forming a chain which propagates parallel to the b-axis as shown in Fig. S17.† However, we have shown a number of times that the crystal structure of crystals obtained even from a gel phase and the diffraction pattern obtained from the gel phase show little correlation,37,38 leading us to suggest that it is likely that there is little link here between the packing in the gel and the crystal phase.
We then examined the multicomponent system, denoted (1 + 2). The amount of each gelator used was the same as in the single component (4 mg mL−1 of 1 and 8 mg mL−1 of 2). Compared to the individual components, the rate of assembly is different as shown by turbidity measurements (Fig. S18†). 1 alone forms a gel via a process where highly scattering structures are initially formed, before a transparent gel phase is formed.30,392 alone forms a highly scattering gel almost immediately. In the mixture (1 + 2), there is an initial process which is reminiscent of the assembly of 1, followed by an increase in turbidity associated with the gelation of 2. Such changes in the rate of assembly have been shown in multicomponent systems previously.29
A gel formed from (1 + 2) is shown in Fig. 3a. The properties of the mixed gels are again typical of a LMWG. The gels are frequency independent, with the value of G′ being higher than that of either 1 or 2 alone, as expected on the basis of the higher overall gelator concentration (Fig. 3b). However, the strain sweep shows a profile which is much more similar to that of the gels of 2 alone as opposed to that of 1 (Fig. 3c).
 | ||
| Fig. 3 (a) Photograph of multicomponent gel as formed (left) and after annealing (right). The scale bar representing 1 cm. (b) Frequency sweeps for gels of (1 + 2); black data are before annealing and red data are after annealing. (c) Strain sweeps for gels of (1 + 2); black data are before annealing and red data are after annealing. (d) Rheological data comparison of gels 1, 2 and (1 + 2) on heating and cooling at a rate of 1 °C min−1. For (b)–(d), the closed symbols represent G′ and open symbols represent G′′. The black data are for the heating cycle, and the red data for the cooling cycle. For (d), purple and orange lines refer to 1 and 2 respectively. The dashed lines refer to the melting temperature on heating, and the dotted line to the temperature at which the gels start reforming on cooling. | ||
When annealing the gel from (1 + 2) (Fig. 3d), G′ and G′′ slowly decrease before reaching a plateau at around 65 °C, following the profile of 1 alone, before decreasing again as for 2. Sequential melting points in a self-sorted system have been previously described,35 but not linked to the rheological data. On cooling, there is a short plateau before G′ and G′′ increase, stabilizing around 50 °C, before a second increase in G′ and G′′. The temperatures at which both increases in G′ and G′′ occur closely match those for the single component gels. This strongly implies self-sorting is occurring.25 After annealing, the gels are two orders of magnitude stiffer than originally and are frequency-independent (Fig. 3b). There is a significantly different breakdown under strain as compared to before heating, with two points in the strain sweep where the moduli decrease indicating two types of network (Fig. 3c). Hence, annealing affects both the absolute stiffness and also the breakage strain. We stress here that the effects observed are due to the annealing and not due to time (Fig. S23†).
To explain these differences on annealing, we used small angle X-ray scattering (SAXS) and confocal microscopy. SAXS probes the primary structures forming the gels, whilst confocal microscopy probes the microstructure. As such, we cover the important length scales with these techniques. Critically, both techniques allow us to probe the gel without the need for drying. Drying can lead to artefacts,40 especially in gels from binary solvent mixtures. The scattering data for the gels formed from 1 alone are very similar before and after annealing, showing that the primary structures leading to gelation are essentially the same (Fig. 4a; fits, table of fitting parameters, and further data are shown in Fig. S19†). The data are typical of this class of LMWG30 and can be fitted to a flexible cylinder model combined with a power law to take into account the scattering at low Q. The cylinders have a radius of 3.7 ± 0.04 nm before annealing, and 3.9 ± 0.06 nm afterwards, with similarly small changes in the Kuhn length. Confocal microscopy shows a significant difference in the microstructure (Fig. 5a). Hence, for 1, we can interpret that the primary structures leading to gelation are essentially the same, but their distribution in space is different, leading to the different gel properties.
 | ||
| Fig. 4 SAXS data before (black) and after (red) annealing for (a) 1, (b) 2 and (c) 1 + 2. The baseline for 2 drops after annealing as there is crystallisation and a small amount of precipitation, meaning that there is less sample in the beam after annealing. | ||
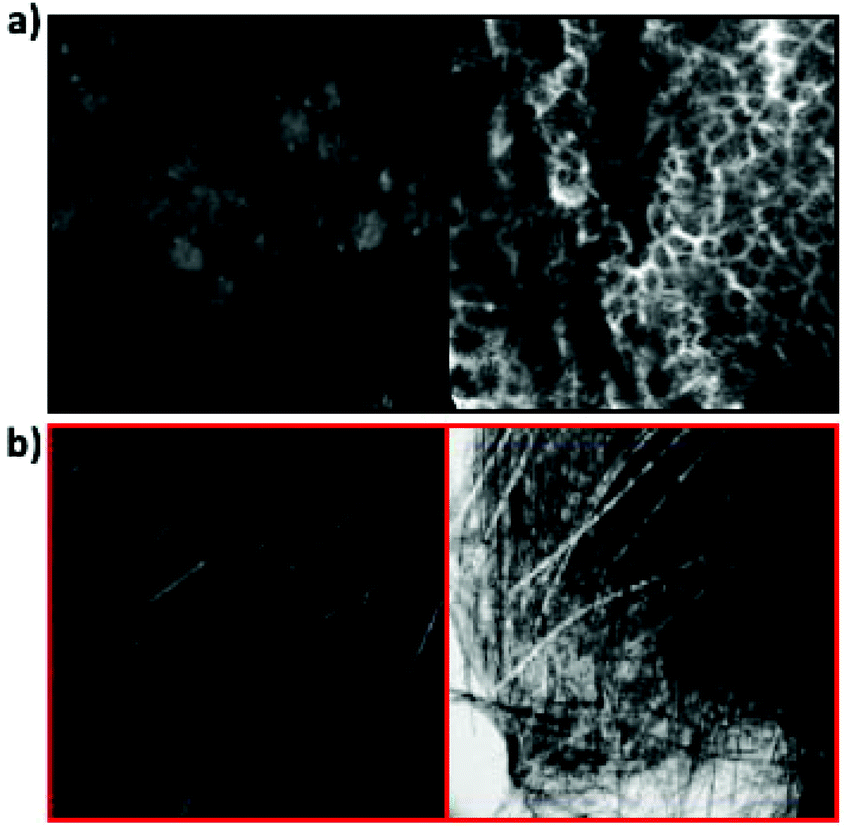 | ||
| Fig. 5 (a) Image of multicomponent gel as formed in confocal (left) and under polarised light (right). (b) Image of multicomponent gel after annealing in confocal (left) and under polarised light (right). The contrast has been enhanced in the confocal images to better show the structures; the originals are shown in Fig. S9.† For the confocal data, the gels were stained with Nile Blue. | ||
For gels formed from 2 alone, the SAXS data are different before and after heating, agreeing with the observations that the annealing leads to some crystallization. The scattering is dominated by a power law (Fig. 4b), which is consistent with the structures that are scattering being outside the accessible Q-range over which the data were collected in agreement with the confocal microscopy (Fig. 2b). The annealing has led to the structures becoming significantly larger, with a propensity to crystallize.
For the multicomponent system, the SAXS data are significantly different before and after annealing. Before heating, the data are reminiscent of that of 2 alone. After annealing, the data are very similar to that of 1 alone, and the data fits well to a flexible cylinder model with a radius of 4.3 ± 0.07 nm. The confocal microscopy however shows structures after annealing that are most similar to those of 2 (Fig. 3). This apparent contradiction can be explained by the fact that, on cooling, 2 forms structures first so entraps the dye added as stain (Nile Blue). When 1 assembles, there is little dye still available and hence the structures formed by 1 are not detected by confocal microscopy. However, under polarized light, there are limited indications of crystallinity (Fig. 3b and S20†), implying that the structures formed by 2 have been modified by being formed in the presence of 1. This is backed up by NMR data; whilst the signal intensity for 1 in the mixture is essentially the same as for the single component, the solubility of 2 is affected by the presence of 1 (Fig. S21†).
The changes in the gel structure in the gels on annealing can also be rationalized from the power law gradient of the SAXS data.41,421 has a gradient of −2, which would be expected from a mass fractal network such as the kind of gel described here. 2 has a gradient of −4, which, as with the fit of the data to a power law suggests objects too large to be resolved by the SAXS. Prior to annealing, (1 + 2) has a gradient of −3, which suggests a mass fractal with a tight, interconnected structure. However, after annealing, the gradient changes to −2, showing that the structure has returned to a network resembling gel 1. The process for the single components and the mixture is shown schematically in Fig. 6.
 | ||
| Fig. 6 Cartoon of the assembly networks before and after annealing. For (a) and (b) scale bars in confocal and under polarized light images represent 20 μm and 200 μm respectively. | ||
Finally, the distinct melting temperatures show that the networks are independent on initial formation and we have a self-sorted network after annealing. If this is the case, it should be possible to selectively melt and anneal only one of the networks. This is indeed possible. Heating the (1 + 2) gel to 65 °C leads to a decrease in the moduli in line with the melting of the gel of 1 only. Subsequent cooling leads to the expected increase in moduli consistent with our data (Fig. S22†).
Conclusions
Most reports on gels describe static systems where a gel is formed and used as is. In terms of tunable materials, there are many examples of simple gel-to-sol transitions in single component systems, for example triggered by light or by heating. There has also been significant recent work on the formation of transient assemblies,43,44 but their use is limited to the few applications require the networks to only exist for short times. Instead, we believe that there is more utility in being able to prepare tunable and responsive networks, where specific input from the user results in a significant and useful change in properties.Annealing can be applied to many different materials as a means of driving from kinetically trapped structures to those at the (or at least a local) thermodynamic minimum. Whilst routinely prepared using heat-cool cycles, annealing is not common in supramolecular gels. We have shown how annealing can be used to change the underlying microstructure in both single and multicomponent gels. Annealing in a two-component gel leads to a self-sorted network, which has significantly different mechanical properties to the as-prepared gels. A key question is why there is an increase in mechanical properties. Showing and proving self-sorting at the fibre level does not necessarily provide information on the network level. We suggest that annealing of this system leads to significant change in the network level of assembly, resulting in the increase in storage modulus.
Such changes in mechanical properties are likely to not only affect the stiffness, but also diffusion and transport in the gel. This could be of use in controlled release systems for example. It is also possible to selectively anneal only a single component in the mixture. Whilst we show here a specific example, we believe that this approach will be widely applicable and so opens up new opportunities to control gel networks on demand to provide tunable, triggerable materials. We have shown that annealing a multicomponent system results in gels with properties that are not simply an addition of the properties of what might be expected from the single components.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
AMFC thanks the University of Glasgow for funding. FPGF acknowledges an Erasmus traineeship. DJA thanks the EPSRC for a Fellowship (EP/L021978/1), which funded BD. MarvinSketch 16.11.28.0 was used for naming chemical structures. This work benefitted from SasView software, originally developed by the DANSE project under NSF award DMR-0520547. SasView also contains code developed with funding from the EU Horizon 2020 programme under the SINE2020 project Grant No. 654000. The X-ray scattering apparatus was purchased under (EP/K035746/1). We thank Finlay Walton for help with the confocal microscope. We thank Emily R. Draper for many helpful discussions and Emily R. Cross and Michael C. Nolan for their time and help throughout the project.Notes and references
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3160 CrossRef CAS PubMed.
- A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS PubMed.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1218 CrossRef CAS PubMed.
- R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef CAS PubMed.
- M. Zhou, A. M. Smith, A. K. Das, N. W. Hodson, R. F. Collins, R. V. Ulijn and J. E. Gough, Biomaterials, 2009, 30, 2523–2530 CrossRef CAS PubMed.
- J. Raeburn, A. Zamith Cardoso and D. J. Adams, Chem. Soc. Rev., 2013, 42, 5143–5156 RSC.
- F. Tantakitti, J. Boekhoven, X. Wang, R. V. Kazantsev, T. Yu, J. Li, E. Zhuang, R. Zandi, J. H. Ortony, C. J. Newcomb, L. C. Palmer, G. S. Shekhawat, M. O. de la Cruz, G. C. Schatz and S. I. Stupp, Nat. Mater., 2016, 15, 469 CrossRef CAS PubMed.
- J. Boekhoven, J. M. Poolman, C. Maity, F. Li, L. van der Mee, C. B. Minkenberg, E. Mendes, J. H. van Esch and R. Eelkema, Nat. Chem., 2013, 5, 433 CrossRef CAS PubMed.
- C. Colquhoun, E. R. Draper, R. Schweins, M. Marcello, D. Vadukul, L. C. Serpell and D. J. Adams, Soft Matter, 2017, 13, 1914–1919 RSC.
- Y. Yan, J. Huang and B. Z. Tang, Chem. Commun., 2016, 52, 11870–11884 RSC.
- R. Alberstein, Y. Suzuki, F. Paesani and F. A. Tezcan, Nat. Chem., 2018, 10, 732–739 CrossRef CAS PubMed.
- L. Wang, X. Shi, Y. Wu, J. Zhang, Y. Zhu and J. Wang, Soft Matter, 2018, 14, 566–573 RSC.
- C. M. Rubert Perez, L. A. Rank and J. Chmielewski, Chem. Commun., 2014, 50, 8174–8176 RSC.
- E. C. Barker, A. D. Martin, C. J. Garvey, C. Y. Goh, F. Jones, M. Mocerino, B. W. Skelton, M. I. Ogden and T. Becker, Soft Matter, 2017, 13, 1006–1011 RSC.
- H. Kumari, S. E. Armitage, S. R. Kline, K. K. Damodaran, S. R. Kennedy, J. L. Atwood and J. W. Steed, Soft Matter, 2015, 11, 8471–8478 RSC.
- E. R. Draper, E. G. B. Eden, T. O. McDonald and D. J. Adams, Nat. Chem., 2015, 7, 848 CrossRef CAS PubMed.
- J. Buendia, E. Matesanz, D. K. Smith and L. Sanchez, CrystEngComm, 2015, 17, 8146–8152 RSC.
- H. Shigemitsu, T. Fujisaku, W. Tanaka, R. Kubota, S. Minami, K. Urayama and I. Hamachi, Nat. Nanotechnol., 2018, 13, 165–172 CrossRef CAS PubMed.
- D. J. Cornwell, O. J. Daubney and D. K. Smith, J. Am. Chem. Soc., 2015, 137, 15486–15492 CrossRef CAS PubMed.
- D. A. Tómasson, D. Ghosh, Z. Kržišnik, L. H. Fasolin, A. A. Vicente, A. D. Martin, P. Thordarson and K. K. Damodaran, Langmuir, 2018, 34, 12957–12967 CrossRef PubMed.
- A. Sandeep, V. K. Praveen, K. K. Kartha, V. Karunakaran and A. Ajayaghosh, Chem. Sci., 2016, 7, 4460–4467 RSC.
- L. E. Buerkle and S. J. Rowan, Chem. Soc. Rev., 2012, 41, 6089–6102 RSC.
- J. Raeburn and D. J. Adams, Chem. Commun., 2015, 51, 5170–5180 RSC.
- B. O. Okesola and A. Mata, Chem. Soc. Rev., 2018, 47, 3721–3736 RSC.
- E. R. Draper and D. J. Adams, Chem. Soc. Rev., 2018, 47, 3395–3405 RSC.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150–8177 RSC.
- A. Mahler, M. Reches, M. Rechter, S. Cohen and E. Gazit, Adv. Mater., 2006, 18, 1365–1370 CrossRef CAS.
- R. Orbach, L. Adler-Abramovich, S. Zigerson, I. Mironi-Harpaz, D. Seliktar and E. Gazit, Biomacromolecules, 2009, 10, 2646–2651 CrossRef CAS PubMed.
- L. Chen, J. Raeburn, S. Sutton, D. G. Spiller, J. Williams, J. S. Sharp, P. C. Griffiths, R. K. Heenan, S. M. King, A. Paul, S. Furzeland, D. Atkins and D. J. Adams, Soft Matter, 2011, 7, 9721–9727 RSC.
- N. A. Dudukovic, B. C. Hudson, A. K. Paravastu and C. F. Zukoski, Nanoscale, 2018, 10, 1508–1516 RSC.
- W. Liyanage and B. L. Nilsson, Langmuir, 2016, 32, 787–799 CrossRef CAS PubMed.
- N. A. Dudukovic and C. F. Zukoski, Soft Matter, 2014, 10, 7849–7856 RSC.
- J. Raeburn, G. Pont, L. Chen, Y. Cesbron, R. Levy and D. J. Adams, Soft Matter, 2012, 8, 1168–1174 RSC.
- M. M. Smith and D. K. Smith, Soft Matter, 2011, 7, 4856–4860 RSC.
- E. Ressouche, S. Pensec, B. Isare, G. Ducouret and L. Bouteiller, ACS Macro Lett., 2016, 5, 244–247 CrossRef CAS.
- E. R. Draper, K. L. Morris, M. A. Little, J. Raeburn, C. Colquhoun, E. R. Cross, T. O. McDonald, L. C. Serpell and D. J. Adams, CrystEngComm, 2015, 17, 8047–8057 RSC.
- D. J. Adams, K. Morris, L. Chen, L. C. Serpell, J. Bacsa and G. M. Day, Soft Matter, 2010, 6, 4144–4156 RSC.
- R. Orbach, I. Mironi-Harpaz, L. Adler-Abramovich, E. Mossou, E. P. Mitchell, V. T. Forsyth, E. Gazit and D. Seliktar, Langmuir, 2012, 28, 2015–2022 CrossRef CAS PubMed.
- L. L. E. Mears, E. R. Draper, A. M. Castilla, H. Su, Zhuola, B. Dietrich, M. C. Nolan, G. N. Smith, J. Doutch, S. Rogers, R. Akhtar, H. Cui and D. J. Adams, Biomacromolecules, 2017, 18, 3531–3540 CrossRef CAS PubMed.
- H. D. Bale and P. W. Schmidt, Phys. Rev. Lett., 1984, 53, 596–599 CrossRef CAS.
- J. Teixeira, J. Appl. Crystallogr., 1988, 21, 781–785 CrossRef.
- J. H. van Esch, R. Klajn and S. Otto, Chem. Soc. Rev., 2017, 46, 5474–5475 RSC.
- S. A. P. van Rossum, M. Tena-Solsona, J. H. van Esch, R. Eelkema and J. Boekhoven, Chem. Soc. Rev., 2017, 46, 5519–5535 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nr09423c |
| This journal is © The Royal Society of Chemistry 2019 |