
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA template-free and low temperature method for the synthesis of mesoporous magnesium phosphate with uniform pore structure and high surface area†
Jörn
Hövelmann
 *a,
Tomasz M.
Stawski
a,
Rogier
Besselink
ab,
Helen M.
Freeman
ac,
Karen M.
Dietmann
d,
Sathish
Mayanna
a,
Brian R.
Pauw
e and
Liane G.
Benning
afg
*a,
Tomasz M.
Stawski
a,
Rogier
Besselink
ab,
Helen M.
Freeman
ac,
Karen M.
Dietmann
d,
Sathish
Mayanna
a,
Brian R.
Pauw
e and
Liane G.
Benning
afg
aGerman Research Centre for Geosciences (GFZ), Interface Geochemistry, 14473 Potsdam, Germany. E-mail: jhoevelm@gfz-potsdam.de
bUniversité Grenoble Alpes, CNRS, ISTerre, Grenoble 38000, France
cSchool of Chemical and Process Engineering, University of Leeds, Leeds LS2 9JT, UK
dGIR-QUESCAT, Department of Inorganic Chemistry, University of Salamanca, 37008 Salamanca, Spain
eFederal Institute for Materials Research and Testing (BAM), 12205 Berlin, Germany
fDepartment of Earth Sciences, Freie Universität Berlin, 12249 Berlin, Germany
gSchool of Earth and Environment, University of Leeds, Leeds LS2 9JT, UK
First published on 27th March 2019
Abstract
Mesoporous phosphates are a group of nanostructured materials with promising applications, particularly in biomedicine and catalysis. However, their controlled synthesis via conventional template-based routes presents a number of challenges and limitations. Here, we show how to synthesize a mesoporous magnesium phosphate with a high surface area and a well-defined pore structure through thermal decomposition of a crystalline struvite (MgNH4PO4·6H2O) precursor. In a first step, struvite crystals with various morphologies and sizes, ranging from a few micrometers to several millimeters, had been synthesized from supersaturated aqueous solutions (saturation index (SI) between 0.5 and 4) at ambient pressure and temperature conditions. Afterwards, the crystals were thermally treated at 70–250 °C leading to the release of structurally bound water (H2O) and ammonia (NH3). By combining thermogravimetric analyses (TGA), scanning and transmission electron microscopy (SEM, TEM), N2 sorption analyses and small- and wide-angle X-ray scattering (SAXS/WAXS) we show that this decomposition process results in a pseudomorphic transformation of the original struvite into an amorphous Mg-phosphate. Of particular importance is the fact that the final material is characterized by a very uniform mesoporous structure with 2–5 nm wide pore channels, a large specific surface area of up to 300 m2 g−1 and a total pore volume of up to 0.28 cm3 g−1. Our struvite decomposition method is well controllable and reproducible and can be easily extended to the synthesis of other mesoporous phosphates. In addition, the so produced mesoporous material is a prime candidate for use in biomedical applications considering that magnesium phosphate is a widely used, non-toxic substance that has already shown excellent biocompatibility and biodegradability.
1. Introduction
In the past decades, the synthesis of nanostructured materials with high surface area and porosity has become a major field of research. In particular, materials with a well-defined mesoporous structure (i.e., with pores of 2–50 nm, according to IUPAC definition1) have attracted great interest because of their extremely wide-ranging applications, e.g., in catalysis,2 sorption,3 gas-sensing,4 energy storage,5 waste water treatment6 or biomedicine.7 To date, the vast majority of mesoporous materials are synthesized through a templating route.8 Such a synthesis route relies on surfactants to form micelles in the synthesis solution that self-assemble into ordered arrangements, i.e., the template, around which the solid material is deposited. In a final step, the organic template is removed by chemical etching or thermal treatment leaving behind a rigid porous structure. Since the first template-based synthesis of mesoporous silica in 1992,9 great efforts have been devoted to extending the range of chemical compositions of mesoporous materials.Among the various non-siliceous mesoporous materials, mesoporous phosphates are gaining increasing attention, especially because of their high potential for applications in biomedicine and catalysis. Mesoporous phosphates are highly bioactive and biocompatible,10–14 the two most important pre-requisites for applications in drug delivery systems or bone tissue engineering. Moreover, the facile functionalization of PO4 and/or P-OH groups through organic or inorganic molecules could offer versatile options for tailoring their surface properties.15 Porous transition metal phosphates, in particular those of Ti and Zr, have already shown great potential for catalytical applications due to the presence of abundant active metal sites on the pore surfaces.15–18 Although the chemical diversity of mesoporous phosphates has been expanding during the last years, their template-based synthesis still bears significant problems and challenges that restrict further development. A major problem relates to the high reactivity of the inorganic phosphate precursors towards hydrolysis and condensation. This makes it difficult to control suitable interaction between micelles and the inorganic precursors, often preventing the formation of well-ordered mesostructured phosphate materials with controlled dimensions and morphologies.15 Another challenge is the conservation of the material's mesoporous structure after removal of the organic template. High temperature calcination or treatment with strong acids often results in serious degradation of the phosphate mesostructure. Milder acid-ethanol extraction, on the other hand, typically leads to incomplete removal of surfactant molecules.15 Furthermore, the presence of these often toxic residues severely restricts the application of the mesoporous phosphates in certain areas, particularly in biomedicine. Thus, the development of new strategies for synthesizing mesoporous phosphates is of great interest for both scientific research and technology development. Here, we present a template-free, low temperature method for the shape- and size-controlled synthesis of a mesoporous magnesium phosphate with a very large surface area and narrow pore size distribution. Our method involves the controlled thermal decomposition of a room temperature synthesized crystalline struvite precursor (MgNH4PO4·6H2O) at 70–250 °C. The precursor material, struvite, is a well-known mineral that readily precipitates as orthorhombic crystals from supersaturated aqueous solutions at ambient temperatures and pressures. It is well known that when heated to temperatures above 55 °C struvite starts decomposing by releasing water (H2O) and ammonia (NH3) from its structure.19–22 However, by using a combination of state-of-the art characterization techniques we show for the first time that this decomposition process results in a highly porous and ordered magnesium phosphate nanostructure that retains the original 3D morphology of the primary struvite crystal.
2. Materials and methods
2.1. Precursor material synthesis and decomposition
Reagent grade MgCl2·6H2O and (NH4)2HPO4 (Sigma Aldrich) were used as starting salts for aqueous synthesis of the struvite precursor, following the reaction: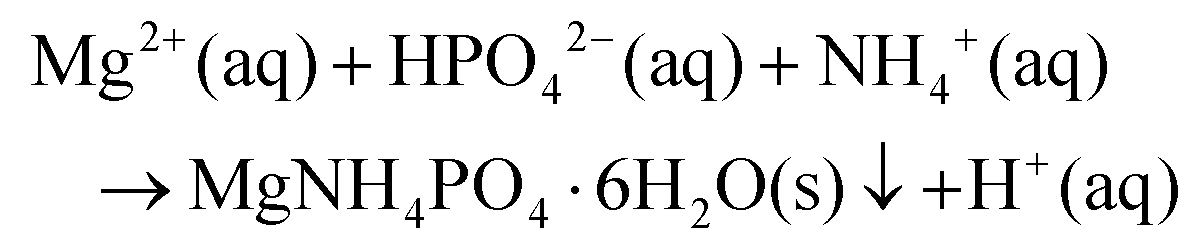 | (1) |
Solutions of each salt were prepared by dissolving proper amounts into double-deionized water (resistivity >18 MΩ cm). The typical synthesis procedure was as follows: 100 ml of a 100 mM (NH4)2HPO4 solution was poured into a 500 ml glass reactor and vigorously stirred at room temperature. Then 100 ml of a 100 mM MgCl2·6H2O solution was quickly added under continuous stirring. The mixed solution thus had and initial concentration of c(Mg, P)mix = 50 mM. Upon mixing, fine particles of a white precipitate quickly formed and the pH decreased from ca. 8 to values between 5 and 6 due to the accompanying release of protons (eqn (1)). The suspension was kept stirring for several hours to ensure that equilibrium was attained. Thereafter, the particles were collected by vacuum filtration through a 0.2 μm polycarbonate membrane and washed 3 times with 50 ml of deionized water to remove any residual salts. Finally, the powder was left to dry in air at room temperature. Syntheses were also performed at other initial concentrations and pH values (adjusted with either HCl or NaOH) to test for the effect on crystal morphology, size and phase purity (Table S1†). In addition, we also prepared larger, mm-sized struvite single crystals by using a modified counter-diffusion method. For this, 1 M solutions of (NH4)2HPO4 and MgCl2·6H2O were filled into separate 100 ml glass beakers that were placed at opposite sides of a 6 l plastic box. The plastic box was then carefully filled with deionized water until the two beakers were fully immersed. Finally, the box was closed with a lid to avoid evaporation and exchange with atmospheric CO2. Elongated, rod-like crystals started to form at the bottom of the plastic container after a few days and had been left to grow for about 2 weeks until they reached sizes of several millimetres. Thereafter, the crystals were removed from the solution, rinsed with water and dried in air at room temperature.
Following synthesis, struvite crystals were thermally treated in a drying oven at 70–250 °C for time periods ranging from 5 min to up to 180 min. Each sample aliquot was weighed before and after heat treatment to calculate the relative weight loss. Selected runs were performed in duplicate to check for reproducibility (Table 1).
| Temperature [°C] | Duration [min] | Mass loss [%] | BET surface area [m2 g−1] | BJH pore volume [cm3 g−1] | BJH avg. pore width [nm] |
|---|---|---|---|---|---|
| a Duplicate runs. | |||||
| As-prepared struvite | 1.52 ± 0.01 | — | — | ||
| 90 | 5 | 3.1 | 5.35 ± 0.24 | 0.006 | 3.97 |
| 90 | 10 | 8.9 | 31.1 ± 1.02 | 0.03 | 3.18 |
| 90 | 20 | 27.1 | 151 ± 7.89 | 0.14 | 3.16 |
| 90 | 40 | 40.2 | 274 ± 12.7 | 0.26 | 3.12 |
| 90 | 80 | 44.3 | 303 ± 12.9 | 0.28 | 3.06 |
| 90 | 120 | 44.7 | 297 ± 12.1 | 0.28 | 3.14 |
| 90 | 160 | 45 | 301 ± 13.2 | 0.28 | 3.14 |
| 90a | 160 | 44.5 | 295 ± 10.4 | 0.29 | 3.20 |
| 110 | 160 | 47.3 | 285 ± 11.4 | 0.28 | 3.34 |
| 110a | 160 | 47.5 | 288 ± 12.4 | 0.29 | 3.37 |
| 250 | 160 | 53.2 | 198 ± 2.9 | 0.23 | 3.72 |
| 250a | 160 | 53.4 | 207 ± 5.4 | 0.24 | 3.97 |
| 90/250 | 160/160 | 53.6 | 288 ± 12.7 | 0.25 | 3.29 |
| 90/250a | 160/160 | 53.7 | 293 ± 13.6 | 0.25 | 3.24 |
2.2. Material characterization
X-ray diffraction (XRD) patterns were collected on a PANanlytical Empyrean X-ray diffractometer using a Cu Kα radiation source operating at 40 kV and 40 mA. Data were collected in the 2θ range between 4.6 and 85° using a step size of 0.013° and a counting time of 60 s per step.Raman spectroscopic analyses were performed using a confocal Raman spectrometer (LabRAM HR Evolution, Horiba Jobin Yvon) operating with the 532 nm line of a frequency-doubled Nd:YAG solid state laser. Spectra in the range of 250 and 1200 cm−1 were obtained with a 100× objective lens, a 50 μm pinhole, and a grating of 1800 grooves per mm using an acquisition time 3 × 30 s. A reference spectrum of struvite was obtained from The RRUFF™ Project database.23
Scanning electron microscopy (SEM) images were recorded using an Ultra 55 Plus (Carl Zeiss SMT) field emission scanning electron microscope. A low acceleration voltage (1–3 kV) was used to limit the electron penetration depth and allow for resolving fine surface structures. All samples were coated with a 20 nm-thick layer of carbon before imaging.
Cryo-SEM investigations were performed by placing 1 ml of a freshly prepared struvite suspension into an Eppendorf tube and subsequent plunge freezing in a liquid nitrogen bath. The frozen sample was removed from the Eppendorf tube and fractured in a pre-cooled (−180 °C) polystyrene box containing a metal base. The fractured sample was placed on the cryo sample holder and then transferred to the SEM using a BALTEC cryo transfer module.
Transmission electron microscopy (TEM) images were collected on a TECNAI F20 XTWIN TEM operated at 200 kV with a field emission gun electron source, a Gatan Imaging Filter (GIF) Tridiem™, an EDAX X-ray analyzer, and a Fishione HAADF detector. Bright-field (BF) and dark-field (DF) images as well as high-resolution images were acquired as energy-filtered images. For that purpose, a 20 eV window was applied to the zero-loss peak. Samples for TEM were prepared using a focused ion beam (FIB) and supported on a holey amorphous carbon coated Cu grid.
Simultaneous measurements of weight change (TGA) and differential heat flow (DSC) were carried out on dry struvite samples using a SDT Q600 TA Instruments device. Additionally, the device was coupled to a GSD 301 T2 ThermoStar mass spectrometer (MS) in order to analyze the gases released during the thermal decomposition of the samples. All measurements were carried out under nitrogen atmosphere with a purge gas flow of 50 ml min−1. As a reference material for the heat flux DSC α-Al2O3 was used. The samples were heated from room temperature to 250 °C using a heating rate of 1 °C min−1.
Nitrogen adsorption/desorption isotherms at 77 K were measured on a Micromeritics Gemini VII 2390t surface area and porosity analyser. The specific surface areas were determined by applying the BET (Brunauer–Emmett–Teller) equation to the relative pressure range 0.05–0.3 for the adsorption branch of the isotherm. Pore volume and pore size distribution (PSD) plots were obtained by applying the BJH (Barrett–Joyner–Halenda) method. BET and BJH calculations were performed with the Gemini VII Version 3.04 software delivered with the analysis equipment.
In situ and time-resolved small- and wide-angle X-ray scattering (SAXS/WAXS) measurements were carried out at beamline I22 of the Diamond Light Source Ltd (UK). Experiments were performed using a monochromatic X-ray beam at 12.4 keV and two-dimensional scattered intensities were collected at small-angles with a Dectris Pilatus P3-2M and at wide-angles with Dectris Pilatus P3-2M-DLS-L (2D large area pixel-array detectors). Transmission was measured by means of a photodiode installed in the beam-stop of the SAXS detector. A sample-to-detector distance of 2.9 m allowed for a usable q-range of 0.005 < q < ∼0.35 Å−1 in SAXS. The scattering-range at small-angles was calibrated against silver behenate and the corresponding measured intensity was calibrated to absolute units against glassy carbon. The q-range in WAXS was calibrated with cerium(IV) oxide powder. The sample powders were loaded into borosilicate capillaries of an external diameter of 1.5 mm and a wall-thickness of 10 μm and measured in a movable capillary rack aligned with the X-ray beam. We also measured the required backgrounds, including an empty capillary, and a no-sample beamline background. The recorded 2D SAXS patterns were found to be independent of the in-plane azimuthal angle with respect to the detector (i.e., scattering patterns where circular in shape), showing that the investigated systems could be considered isotropic. In those patterns, pixels corresponding to similar q-values were averaged over all azimuthal angles, and hence the 2D patterns were reduced to 1D curves. SAXS data processing and reduction included primarily masking of undesired pixels, normalizations and correction for transmission, background subtraction and data integration to 1D for which we followed a procedure described by Pauw et al.24 The recorded 2D WAXS patterns were corrected for background in the same way as the SAXS patterns, but were not converted to 1D curves. All these steps were performed using the Data Analysis WorkbeNch (DAWN) software package (v. 2.6+) according to I22 guidelines.
Additional SAXS measurements were performed in the laboratory on an Anton Paar SAXSess slit-collimated instrument, using multilayer-mirror-monochromatized copper Kα radiation, and modified to use a Dectris Mythen photon-counting strip detector. The measurements consist of 65 repetitions of 120 10-second frames (total duration 22 hours), with the sample held at a temperature of 40 °C. The sample was mounted at the sample position inside the SAXSess vacuum chamber, uniformly distributed in a sandwich of Scotch® Magic Tape. The data have been corrected for exposure time, darkcurrent, transmission, background, absolute units scaling, and deconvoluted for slit-smearing effects. The data was scaled to absolute units using water scattering as a reference, and with the required apparent struvite sample thickness derived from the transmission factor of the struvite component, and the X-ray absorption coefficient of the sample. The transmission factor was accurately determined using the fluorescence ratio from a 10 micron iron foil in the sample-attenuated and direct beam, respectively (due to significant radiation hardening by the semi-transparent beamstop, transmission values derived from quantification of the beamstop-attenuated beam are incorrect). The q-scale was calibrated using the instrument geometry and stated detector pixel size, and has been verified using silver behenate.
3. Results and discussion
3.1. Characterization of as-synthesized struvite
The as-prepared struvite powders were highly crystalline as evidenced by the XRD patterns (Fig. S1A and B†). The obtained XRD patterns match the reference patterns for struvite (ICSD 98-001-4269) with no additional peaks demonstrating the high purity of the synthesized material. The struvite crystals synthesized from mixed solutions with high initial concentrations (c(Mg, P)mix = 50 mM) at pH 8 were 100–200 μm in size and showed mainly dendritic or X-shaped morphologies (Fig. 1A), reflecting their fast nucleation and growth.25 In contrast, the slow counter-diffusion synthesis resulted in elongated, lath-shaped crystals (up to 7 mm long and 1–2 mm wide) with well-defined faces and straight edges (Fig. 1B). These crystals were confirmed as struvite based on their Raman spectra (Fig. S1C†). Syntheses performed at lower concentrations (c(Mg, P)mix = 15–25 mM) and a pH of 6.7 also resulted in well-faceted struvite crystals showing the typical blocky or coffin-like forms (Fig. 1C–E). When observed under the SEM, the struvite crystals often showed a nanogranular morphology as well as a large number of Y- and cross-shaped gaps or cracks at the surface, ranging in width from tens to hundreds of nanometers (Fig. 1C and D). To verify whether these features were formed during crystal growth or within the SEM due to the high vacuum or electron beam damage, we performed cryo-SEM investigations of some freshly prepared struvite crystals. Under cryogenic conditions, the crystal surfaces were flat and smooth with no visible gaps, as would indeed be expected for natural growth surfaces (Fig. 1E). Hence, it is apparent that the surface features observed under conventional room temperature/high vacuum SEM imaging conditions are artefacts, most likely related to (partial) de-volatilization of the struvite. This hypothesis is supported by results from FIB-TEM investigations of one of the struvite crystals (Fig. S2†). The TEM images again revealed lots of cracks as well as a high internal porosity (Fig. S2A and B†). Interestingly, no lattice fringes were observed in high resolution TEM images (HRTEM) (Fig. S2C†) indicating that the struvite crystal imaged was likely transformed into an amorphous material in the TEM. Although, energy dispersive X-ray (EDX) analyses of the material confirmed the presence of Mg and P (Fig. S2D†), the electron energy loss spectroscopy (EELS) showed only a very weak N K-edge that disappeared completely after several minutes of beam exposure (Fig. S2E and F†). Overall, these observations indicate that struvite crystals readily decomposed even at room temperature when exposed to the high vacuum of an SEM or TEM. | ||
| Fig. 1 SEM images of struvite crystals as-synthesized at room temperature. (A) Medium sized dendritic, X-shaped struvite crystals synthesized by fast solution mixing at an initial concentration of c(Mg, P)mix = 50 mM (pH 8); inset: crystal surface imaged at higher magnification. (B) Large, millimeter-sized, lath-shaped struvite crystal synthesized by slow counter-diffusion method (see section 2.1 for details); inset: crystal surface imaged at higher magnification. (C–E) Smaller blocky, coffin-like struvite crystals synthesized at pH 6.7 by fast solution mixing using initial solution concentrations (c(Mg, P)mix) of (C) 15 mM, (D) 20 mM and (E) 25 mM. Crystals in (A–D) were imaged using conventional room temperature/high vacuum SEM conditions; they show nanogranular morphologies and abundant surface cracks. In contrast, crystals in (E) were imaged under cryogenic conditions and show flat and smooth surfaces. | ||
3.2. Thermal decomposition of struvite
Following the thermal decomposition of the precursor material with TGA/DSC (Fig. 2A) revealed that struvite started decomposing at a temperature of around 50 °C, which is in agreement with previous reports.19–22 A major mass loss step of ∼43% occurred between 50 and 90 °C. This was followed by a more gradual mass loss between 90 and 250 °C. The total (final) mass loss obtained at 250 °C was about 54%, which is close the theoretical value of 54.6% for the complete de-volatilization of struvite and the formation of Mg-pyrophosphate (Mg2P2O7) according to the reaction:| MgNH4PO4·6H2O(s) → ½ Mg2P2O7 (s) + NH3↑ + 6.5H2O↑ | (2) |
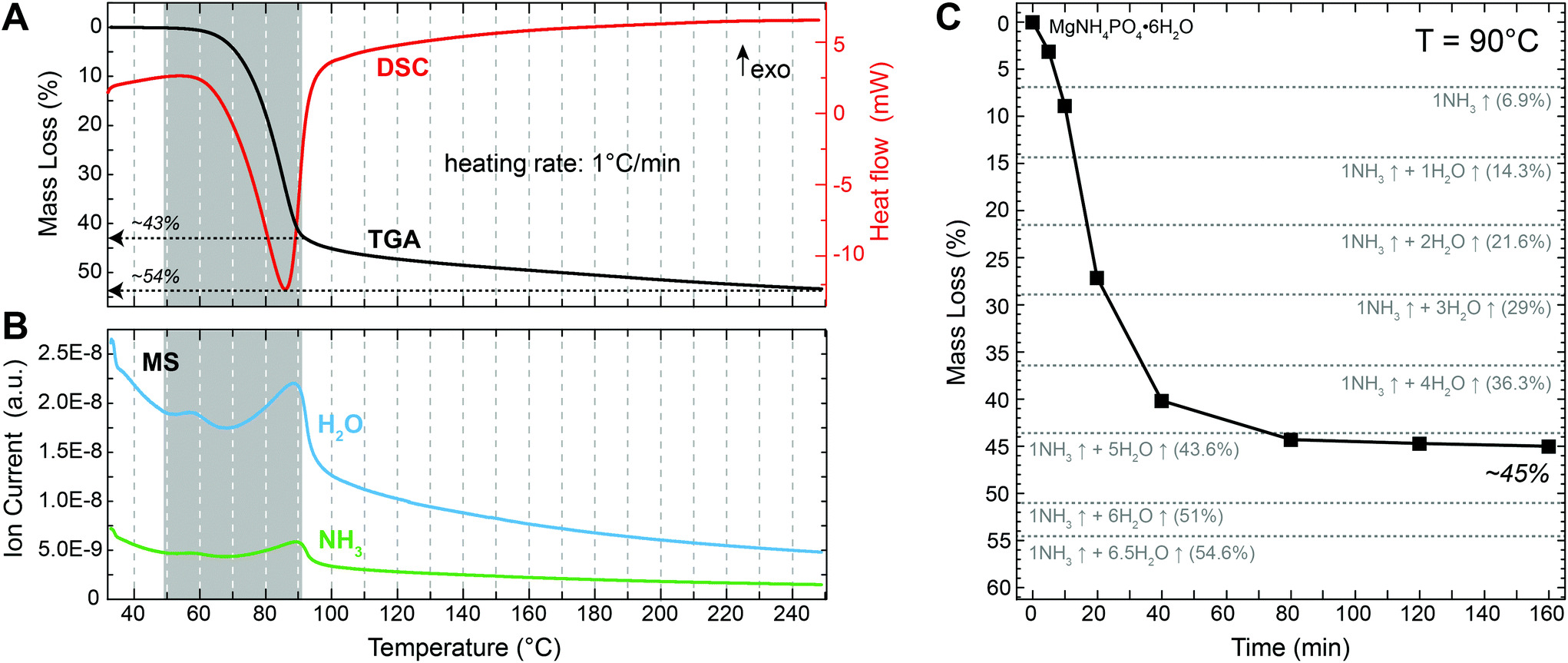 | ||
| Fig. 2 (A) TGA (black) and DSC (red) curves of struvite for a heating rate of 1 °C min−1 from 30–250 °C. (B) Corresponding MS data showing the evolution of H2O (blue) and NH3 (green) releases. Grey areas in A and B delineate the major mass loss step of 43% between 50 and 90 °C. (C) Mass evolution during isothermal struvite decomposition at 90 °C. Dotted lines represent theoretical mass losses for the release of 1 NH3 and 0–6.5 H2O molecules per structural formula. All measurements were performed on struvite crystals synthesized at room temperature by fast solution mixing at an initial concentration of c(Mg, P)mix = 50 mM and an initial pH of 8. | ||
The DSC curve shows only a single, broad endothermic peak at ca. 90 °C (Fig. 2A). This peak corresponds to the major 43% mass loss during heating. The presence of a single peak in the DSC curve indicates that water and ammonia were lost simultaneously from the struvite structure. Otherwise two distinct peaks should have been visible. This is corroborated by the corresponding MS data (Fig. 2B) where the releases of NH3 and H2O occurred simultaneously at temperatures between 50 and 90 °C. Measuring the mass evolution under isothermal conditions at 90 °C reveals that the mass loss was initially fast, reaching about 40% within the first 40 min, but then slowed down considerably and stabilized at 44–45% after about 80 min (Fig. 2C and Table 1). This mass loss is close to what would be expected for the loss of 1 NH3 and 5 H2O molecules from the initial MgNH4PO4·6H2O structure resulting in the formation of MgHPO4·H2O. However, we cannot exclude that some residual NH4 is still present in the decomposition product at this point. Nevertheless, this analysis shows that any further mass loss is temperature- rather than time-dependent.
3.3. Structural evolution during struvite decomposition
Thermal decomposition of struvite at temperatures up to 250 °C resulted in the formation of an amorphous material as shown by XRD analyses (Fig. S3†). During this transformation the initially transparent struvite crystals turned opaque white, but retained their original crystal shapes. Fig. 3A shows an SEM image of a larger, mm-sized struvite crystal that was thermally treated at 70 °C for 3 hours. Despite the heat treatment the typical faceted morphology of the original struvite crystal (cf.Fig. 1B) was preserved. However, a more detailed view revealed abundant cracks at the crystal surface, ranging between 0.1–1 μm in width and 1–10 μm in length (Fig. 3B and C). The cracks often branch out at their ends, but otherwise appear to follow a direction close to parallel to the crystal's long axis indicating that their propagation could have occurred along specific crystal planes. The surface between the cracks is characterized by a high roughness, which is in contrast to the perfectly flat growth surfaces of the original crystals (Fig. 1E). At higher magnification, it becomes evident that this roughness results from multiple overlapping circular surface depressions (Fig. 3C) suggesting that the decomposed, yet morphologically still looking like struvite crystal, experienced some shrinkage during decomposition. Hence, it seems reasonable to assume that the formation of the cracks is also related to this shrinkage process and the concomitant generation of tensile stresses within the crystal. It was however noted already above (e.g., Fig. 1D) that similar cracks were also observed on non-heat treated struvite crystals. Nevertheless, the thermally decomposed struvite crystals showed a much lower crack density than the crystals that were only decomposed under high vacuum (Fig. S4†). This possibly suggests that the decomposition in high vacuum was more abrupt leading to more vigorous gas out-bursts and giving little time for relaxation by mechanisms other than cracking. | ||
| Fig. 3 (A) SEM image of a larger, mm-sized struvite crystal after heat-treatment at 70 °C for 3 hours. (B, C) Higher magnification images showing the presence of cracks and circular depressions at the surface. (D) Higher magnification image of the area marked in (C) by a white rectangle showing a nanogranular surface texture. (E) Scanning transmission electron microscopy (STEM) image of a FIB section prepared from the same crystal. (F) Bright field TEM image of the region indicated in (E) by a white rectangle revealing the highly porous nature of the decomposed material. | ||
At a very high magnification (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×) a nanogranular surface texture that appears to be composed of small spherical particles with diameters of about 20 nm becomes evident (Fig. 3D). The interstitial space between these nanoparticles appears essentially empty implying that there should be a large amount of porosity left in the decomposed material. The highly porous nature of the decomposed crystal was confirmed by TEM investigations of a FIB section (Fig. 3E and F) showing dense arrays of near-round pores with uniform sizes of approximately 20 nm. We attempted to visualize the 3D shape and structure of these pores by acquiring multiple images of the same region in a tilt-series using tilt-steps of 1° from −70° to +70°. However, during imaging the pores widened considerably as a function of time (Fig. S5†), possibly due to an ongoing decomposition under the high vacuum or due to the sensitivity of the material to the electron beam.
000×) a nanogranular surface texture that appears to be composed of small spherical particles with diameters of about 20 nm becomes evident (Fig. 3D). The interstitial space between these nanoparticles appears essentially empty implying that there should be a large amount of porosity left in the decomposed material. The highly porous nature of the decomposed crystal was confirmed by TEM investigations of a FIB section (Fig. 3E and F) showing dense arrays of near-round pores with uniform sizes of approximately 20 nm. We attempted to visualize the 3D shape and structure of these pores by acquiring multiple images of the same region in a tilt-series using tilt-steps of 1° from −70° to +70°. However, during imaging the pores widened considerably as a function of time (Fig. S5†), possibly due to an ongoing decomposition under the high vacuum or due to the sensitivity of the material to the electron beam.
To evaluate the temporal evolution of the development of this porosity as well as their characteristics, we performed N2 gas adsorption–desorption measurements on struvite samples that were thermally treated at 90 °C for time periods between 5 and 160 min. Fig. 4A shows the N2 adsorption–desorption isotherms obtained at −196 °C. All isotherms exhibit the same general shape with four well-defined stages: (i) at low relative pressures (p/p0 ≤ 0.25) a gradual increase in N2 uptake is observed and attributed to monolayer-multilayer adsorption on the pore walls, (ii) at intermediate relative pressure (0.25 ≤ p/p0 ≤ 0.45) a prominent step occurred possibly due to capillary condensation within small pores, (iii) at high relative pressures (0.45 ≤ p/p0 ≤ 0.95) a plateau region with a slight inclination is observed indicating the absence of larger pores (i.e., the slow N2 uptake in this region is due to multilayer adsorption of N2 molecules on the external surface of the particles), (iv) at very high relative pressures (p/p0 ≥ 0.95) a further steep increase occurred that can be assigned to bulk condensation of N2. The amount of N2 adsorbed increased strongly with increasing time. For example, plateau values were below 0.2 mmol g−1 after 5 min and ∼6.5 mmol g−1 after 80 min (i.e., >30-fold increase). No significant changes in the isotherms were observed for thermal treatment times longer than ∼80 min.
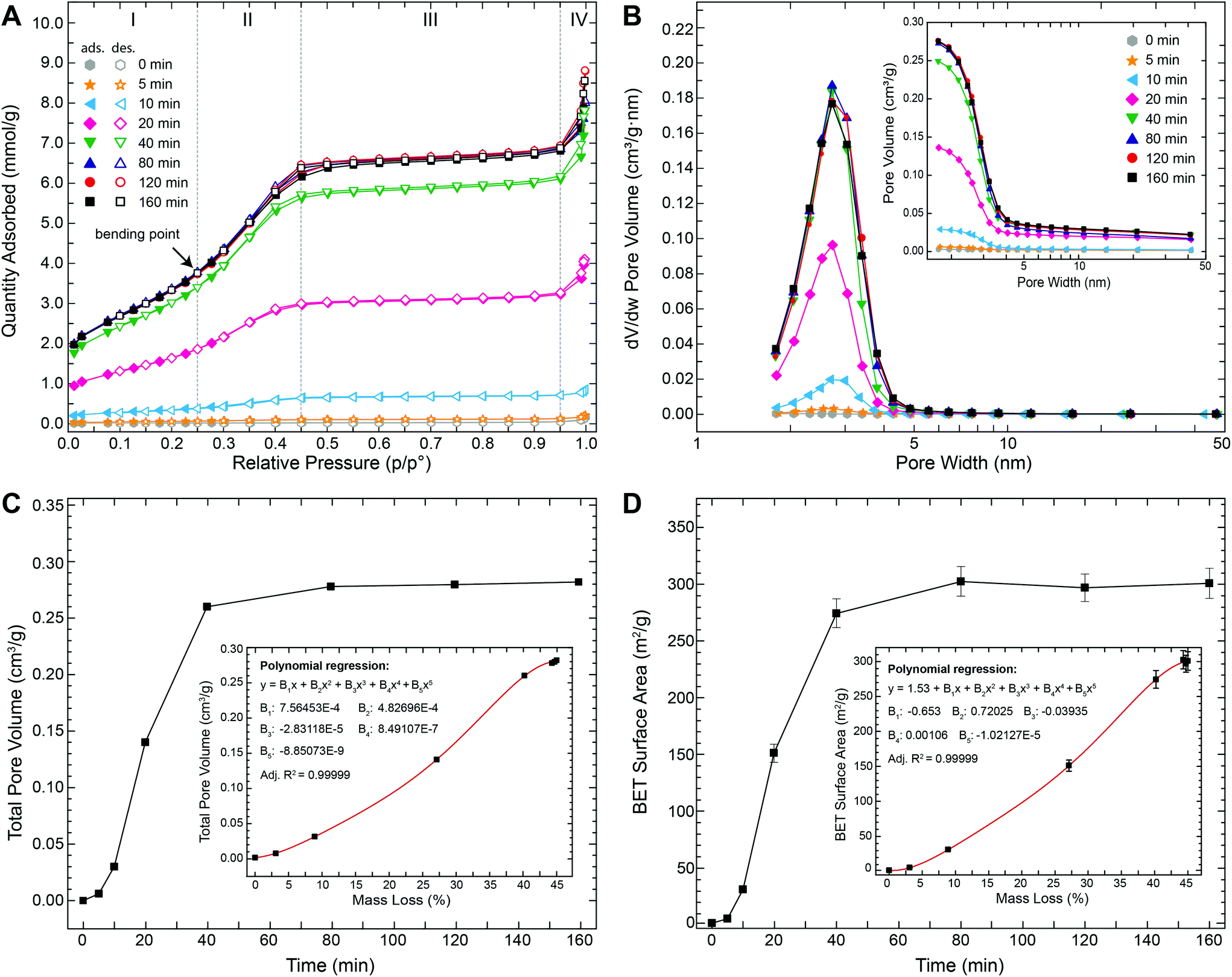 | ||
| Fig. 4 Results from N2 gas sorption analyses of struvite powders after thermal treatment at 90 °C for 5–160 min. (A) Adsorption–desorption isotherms (closed symbols: adsorption; open symbols: desorption). All isotherms have the same general shape with four well-defined stage (I–IV; delineated by vertical dotted lines) and a bending point at a relative pressure of ∼0.25 (see main text for more detailed explanations). (B) Pore size distribution plots obtained by applying the BJH method; inset: cumulative pore size distributions. (C) Total pore volume as a function of thermal treatment time; inset: correlation between total pore volume and mass loss. (D) BET surface area as a function of thermal treatment time; inset: correlation between BET surface area and mass loss. Error bars indicate measurement errors reported by the Gemini VII Version 3.04 software. | ||
The observed step on the isotherm at intermediate relative pressures (Fig. 4A) points to a type IV isotherm (in the IUPAC classification). This type of isotherm is typically observed for mesoporous materials (i.e., materials containing pores with diameters between 2 and 50 nm) with the step being indicative of capillary condensation inside the pores – a phenomenon whereby vapour condenses to a liquid-like phase at pressures below the vapour saturation pressure (p0) of the bulk liquid. In the vast majority of systems, capillary condensation is associated with a pronounced hysteresis loop starting at values of p/p0 ≈ 0.42.26,27 For example, when gas adsorption occurs in perfectly cylindrical mesopores, capillary condensation occurs via a cylindrical meniscus separating the vapour from the condensed phase. However, during desorption capillary evaporation occurs via a hemispherical meniscus. This causes pores of a specific size to be filled at higher pressures and emptied at lower pressures, hence resulting in a hysteresis. In our case, however, no or very little hysteresis was observed in the isotherms (Fig. 4A) indicating that the pores were completely filled and emptied at similar pressures. In fact, the obtained isotherms are strikingly similar to those previously reported for some commercial silica-based mesoporous materials (e.g., Mobil Composition of Matter no. 41 (MCM-41) or Santa Barbara Amorphous no. 15 (SBA-15)) with well-defined and ordered cylindrical mesopores.28–30 For these mesoporous silicas it has been shown that the ad- and desorption isotherms become reversible (i.e., no hysteresis) if the pore diameters are tuned to values between 2 and 4 nm. Nevertheless, a prominent step is still present on the isotherms of these materials demonstrating that capillary condensation is not inevitably accompanied by hysteresis. Hence, for small cylindrical mesopores (2–4 nm), there seems to exist a reversible capillary condensation regime27 that is intermediate between the regime of volume filling without condensation for very narrow pores (≲2 nm) and the irreversible capillary condensation regime for reasonably wide pores (≳4 nm). In this intermediate regime the disappearance of the hysteresis is possibly related to a tensional instability of the hemispherical meniscus caused by an increased chemical potential of the pore walls.31 Such reversible type IV isotherms are typically referred to as type IVc. Given the fact that the isotherms shown in Fig. 4A reveal the typical characteristics of type IVc isotherms, we can infer that the pores in the struvite decomposition material have a fairly uniform, channel-like shape with a narrow, nearly constant diameter between 2 and 4 nm. This conclusion was corroborated by the pore size distribution (PSD) analyses (Fig. 4B) derived by applying the Barrett–Joyner–Halenda (BJH) method. The PSDs reveal that the pore widths vary only over a relatively narrow range between 2 and 5 nm with a clear peak centred at 3.1–3.2 nm. PSDs derived from gas sorption isotherms can sometimes be affected by the so-called tensile strength effect (TSE), i.e., the spontaneous evaporation of metastable pore liquid.26,31 In such a case, isotherms typically display a clear hysteresis with a characteristic step-down26 in the desorption branch at p/p0 ≈ 0.45 and an artificial peak near 3.8 nm31 only in the desorption-derived PSD and not in the adsorption-derived PSD. In our case, we can confidently exclude a TSE contribution to the PSD, because (i) no hysteresis was observed (Fig. 4A) and (ii) both adsorption- and desorption-derived PSDs gave nearly identical results with no artificial peak (Fig. S6†). It should be noted, however, that the BJH method commonly underestimates pore sizes by up to 20–30% for pores smaller than 10 nm.26 Thus, the “real” pore size may be up to 1 nm larger than the calculated values. In any case, this pore size is more than 5 times smaller than the pores observed in TEM (Fig. 3F). The larger pore sizes in TEM images are most likely due to an artificial pore widening possibly induced by the bombardment with Ga-ions during sample preparation and high-energy electrons during TEM observation and/or a continued material decomposition in the high vacuum. Indeed, such widening of pores was also directly observed during our attempt of tilt-series imaging in TEM (Fig. S4†). In contrast, our N2 gas sorption measurements demonstrate that the average pore width remained constant at 3.1–3.2 nm for up to 160 min during the thermal decomposition at 90 °C (Fig. 4B and Table 1). The total pore volume, on the other hand, increased sharply within the first 40 min before reaching a stable maximal value of ∼0.28 cm3 g−1 after ca. 80 min (Fig. 4C and Table 1). Similarly, the BET surface area increased rapidly from 1.52 m2 g−1 (initial struvite) to 274.4 m2 g−1 with in the first 40 min and then reached a final stable value of ∼300 m2 g−1 after 80 min (Fig. 4D and Table 1). Both the total pore volume and the BET surface area show a strong positive correlation with total weight loss (insets in Fig. 4C+D).
In summary, our TGA/DSC/MS and gas sorption data demonstrate that the release of ammonia and water during the thermal decomposition of struvite led to the formation of a nanostructure featuring one of the largest surface areas (300 m2 g−1) and narrowest pore size distributions (2–5 nm) ever reported for a mesoporous phosphate.15 This nanostructure gradually formed through an initial nucleation of small pores at the crystal surfaces and their subsequent growth in depth and the formation of arrays of channel-like mesopores. The increase in total pore volume and BET surface area is solely due to a one-dimensional growth of these mesopore channels as evidenced by the development of a narrow and temporally highly constant pore size distribution. Lateral growth of pores would have caused a clear time-dependent shift in the PSDs and eventually also pore coalescence and thus a reduction in surface area (e.g., for perfectly parallel cylindrical pores the surface area would decrease by a factor of n−1/2, where n represents the number of coalescing pores). Within the experimental time frame (160 min), the nanostructural development is closely coupled to the decomposition process (i.e., the release of H2O and NH3), as evidenced by the parallel temporal evolution of pore volume, surface area and mass loss (Fig. 2B+4C and D). At 90 °C, the process is essentially completed after 80 min. The observation that the pore volume and surface area remained constant at their maximum values even after continual heating for up to 160 min, indicates no further secondary re-structuring (e.g., Ostwald ripening).
To investigate the effect of temperature on the surface area and porosity of the decomposition product we also performed thermal treatments at 110 °C and 250 °C (Table 1). As expected from our TGA data (Fig. 2A), these treatments resulted in somewhat higher mass losses (∼47.5% at 110 °C and ∼53.4% at 250 °C). N2 gas adsorption analyses also revealed a slight temperature-dependent increase in average pore widths from 3.1–3.2 nm at 90 °C to 3.3–3.4 nm at 110 °C and 3.7–4 nm at 250 °C. However, the BET surface areas and total pore volumes did not exceed the ones from the 90 °C samples. Samples treated at 110 °C had BET surface areas of 280–290 m2 g−1 and pore volumes of ∼0.28 cm3 g−1 and thus were similar to the 90 °C samples. In contrast, samples treated at 250 °C had significantly lower BET surface areas (∼200 m2 g−1) and pore volumes (∼0.24 cm3 g−1). On the other hand, when first treated at 90 °C and then at 250 °C, the BET surface areas were again ∼290 m2 g−1, while pore volumes were ∼0.25 cm3 g−1.
The nanostructural evolution during the thermal decomposition of struvite at 90 °C was also followed by small- and wide-angle X-ray scattering (SAXS/WAXS). Fig. 5A shows that the WAXS patterns collected from the 5–20 min heat treated samples contained several sharp and intense diffraction peaks in the q-range 0.8–3 Å−1 that can be assigned to crystalline struvite. After ∼20 minutes a clear decrease in intensity of the WAXS diffraction peaks with time is observed, while concomitantly two broad, gradually increasing local maxima start to develop at ∼1.6 Å−1 and 2.2 Å−1. The WAXS pattern of the (final) 160 min sample only shows a broad profile without any diffraction peaks, indicating the complete absence of long-range order (i.e., crystallinity) in the sample. From the WAXS data we can infer that the decomposition of struvite at 90 °C leads to the formation of an amorphous phase. The observed changes in the WAXS patterns (i.e., decreasing peak intensities and increasing broad local maxima heights) originate from the gradually decreasing volume fractions of struvite and the increase in the volume fraction of the amorphous phase. This struvite-to-amorphous transition does not appear to involve any intermediate crystalline phase as no diffraction peaks other than those belonging to struvite were observed at any intermediate time step.
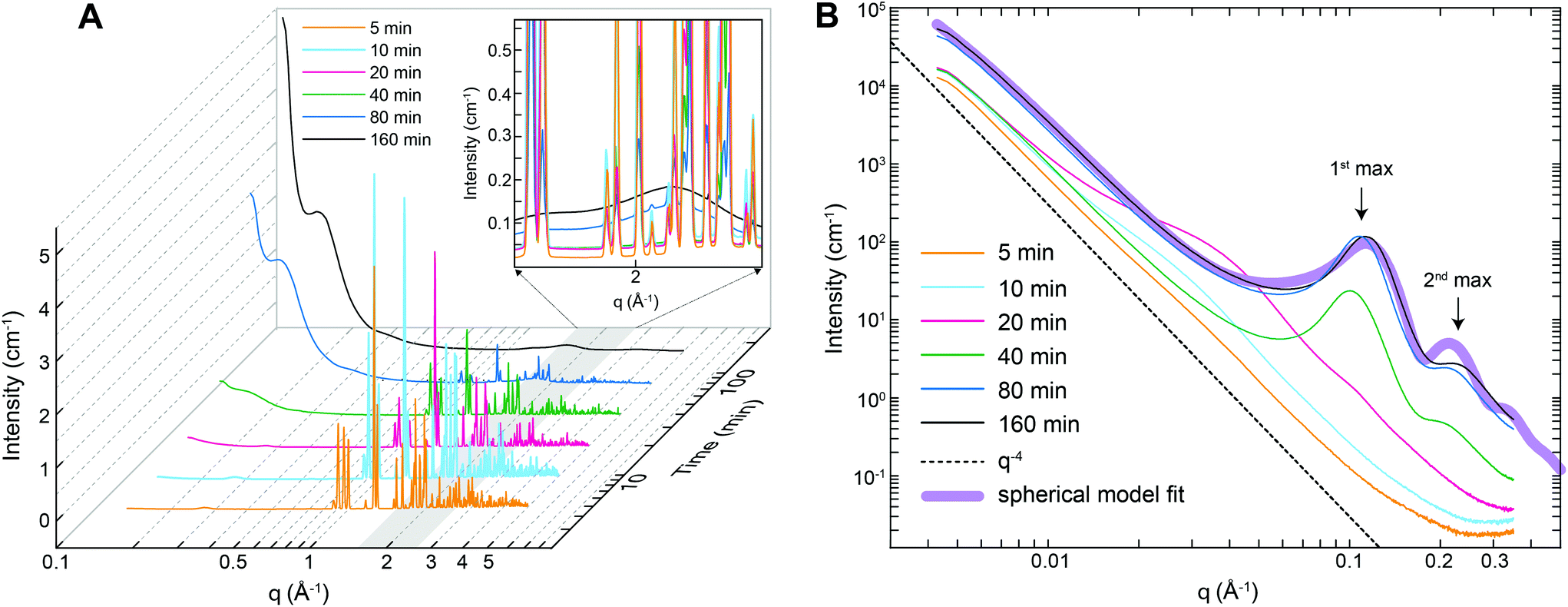 | ||
| Fig. 5 (A) WAXS and (B) SAXS patterns of struvite powders after thermal treatment at 90 °C for 5–160 min. Inset in (A) is a zoom-in into q-range 1.6–2.2 Å−1. There are some intensity fluctuations in the WAXS diffraction peaks between 5 and 20 min that are likely related to crystal orientation effects caused by an inhomogeneous packing of the capillaries. | ||
Fig. 5B shows the corresponding time-resolved SAXS patterns of the thermally treated struvite powders over the q-range where the maximum observable feature length-scale is ∼120 nm (diameter). For untreated several micron-sized struvite crystals (Fig. 1A) one would expect a scattering pattern following a I(q) ∝ Aq−4 dependence (dotted line in Fig. 5B) extending to high-q. In this relationship A is a constant proportional to the product of the surface area of the scattering features and the scattering contrast32 (i.e., the square of the electron density difference between the material and the surrounding medium/matrix). Such a scattering pattern originates from the interface between the crystal and the matrix (air). Indeed our powders that were analysed by SAXS just 5 minutes after initiation of thermal treatment were characterized by such a scattering pattern (Fig. 5B). With increasing thermal treatment time we observed that for q < 0.02 Å−1 the overall scattering intensity gradually increases by a factor of ∼5, while the scattering exponent remains constant. We attribute this overall change primarily to the progressive increase in the material's surface area. At the same time for q > 0.02 Å−1 it can be seen that the Aq−4 dependence is no longer followed and the scattering patterns contain features attributable to nano-sized morphologies. For the patterns collected after 10 and 20 min thermal treatment the observed features are difficult to interpret. In contrast, the high-q part of the curves after 20 min contains clear correlation peaks (the 1st maximum at ∼0.1 Å−1 and the 2nd at ∼0.21 Å−1, see Fig. 5B). These maxima originate from structure factor inter-feature interferences and we attribute such scattering features to nano-sized pores forming within the bulk of the material. This corroborates our observations documented above through the changes in gas adsorption characteristics (Fig. 4). The shape of the correlation peaks in the 20 min SAXS patterns point to a relatively regular pore shape, and close geometric proximity as the cause of the observed interference. Patterns collected after 40 minutes revealed a relatively constant overall shape of the scattering feature, suggesting that the regularly-shaped and relatively monodisperse pores were already formed at 40 minutes and the increase in intensity in this part of q is related to the increasing population of those pores. This matches the pore volume data from gas adsorption (Fig. 4C) where the majority of the change was also over after 40 minutes and absolutely no more change was observed after 80 minutes. From the SAXS data (Fig. 5B), the exact position of the 1st maximum, qmax, and the relationship
| d ≈ 2π/qmax | (3) |
We also carried out in situ and time-resolved SAXS measurements of a struvite sample heated to only 40 °C, but at a lower pressure of 1 mbar. Under these conditions, we only observed a single peak at q ∼ 0.2 Å−1 (Fig. S7†) suggesting a different, more disordered mesoporous structure compared to the one that formed at 90 °C and ambient pressure.
3.4 A new strategy for synthesizing mesoporous phosphates
Mesostructured phosphates have been extensively studied for their unique properties and great potential applications, particularly in biomedicine and catalysis.10–18 However, although various mesoporous phosphates have been documented in the literature, their synthesis is still fraught by several problems and limitations. Present strategies for the synthesis of mesoporous phosphates are based largely on the use of organic templates. Such templating strategies have proven to be very successful for the preparation of mesoporous silica, but present great difficulties for phosphates. Partly, these difficulties are due to the more rapid hydrolysis and condensation processes in metal–phosphate systems. This often results in fast precipitation and thus less interaction between the organic template and the inorganic metal/phosphate precursors. Another problem relates to the subsequent removal of the organic template. While calcination at high temperatures or treatment with strong acids often leads to severe degradation of the mesoporous phosphate structure, milder extraction methods usually result in incomplete removal of the often harmful organic species. For these reasons, there are only few reports on the shape and size-controlled synthesis of phosphates with well-defined mesoporous structures.15 Moreover, until now, the synthesis of porous phosphates has been confined mainly to zirconium and titanium phosphates, while other mesoporous metal phosphates have been only sporadically reported.In this study, we developed a template-free strategy for the synthesis of a mesoporous Mg-phosphate based on the pseudomorphic thermal decomposition of a crystalline struvite precursor. Our results show that this thermal decomposition strategy is well-controllable and -reproducible. Compared to current template-based synthesis routes it combines several other advantages. Firstly, the synthesis of struvite is a straight-forward process that only involves the mixing of magnesium and phosphate solutions at ambient temperature and pressure conditions. This process is also expected to have a good scale-up potential, especially considering that struvite can even be synthesized from agricultural and industrial waste waters.33 The morphology and size of the struvite precursor (and thus the final mesoporous product) can be varied by simply changing synthesis parameters such as concentration, pH or solution mixing dynamics. Here, we show that crystals with various morphologies (i.e., dendritic, X-shaped; blocky, coffin-lid shaped; elongated, lath-shaped) and sizes ranging from just a few micrometres to several millimetres can be easily prepared (Fig. 1). Thermal decomposition of this struvite precursor already starts at temperatures well below 100 °C, so that calcination at high temperatures is also not required. This also makes the process very energy efficient. Under vacuum decomposition occurs at temperatures even below 50 °C, but leads to a different, more disordered mesoporous structure (Fig. S7†). Therefore, a combined heat and low pressure treatment could thus possibly be used to tailor the pore size distributions of the final material over a broader range. Finally, this synthesis and decomposition strategy could readily be transferred to other porous phosphates since struvite can accept a wide range of substitutes within its M1M2A·6H2O structure (M1: NH4+, K+, Rb+, Cs+, Tl+; M2: Mg2+, Ni2+, Zn2+, Co2+, Cd2+, Cr3+, Mn2+, VO2+; A: PO43−, AsO43−).34–41 In this regard, the incorporation of transition metals could lead to a range of materials for applications in the field of catalysis.15–18
The material described in this work has one of the largest surface areas (300 m2 g−1) and narrowest pore size distributions (2–5 nm) ever reported for a mesoporous phosphate.15 This material could become useful for biomedical applications considering that it is composed of Mg-phosphate – a non-toxic compound that has already shown high biocompatibility and – degradability.42,43 In particular, it may be of interest for the development of new drug delivery systems where narrow pore size distributions are indispensable for homogenous drug loading and precise control of drug release rates.7
4. Summary and conclusions
Herein we show that the thermal decomposition of struvite at temperatures below 100 °C leads to a pseudomorphic transformation into a mesoporous Mg-phosphate featuring a well-defined pore structure and high surface area. Based on the presented results, the following formation process (Fig. 6) can be proposed: In the beginning, small pores start to nucleate at the external surfaces of the struvite crystal due to the removal of structurally bound water and ammonia. This is accompanied by a localized collapse of the crystalline structure (amorphization). Subsequently, the pores start to deepen without growing in width resulting in relatively uniform, channel-like pores with a very narrow size distribution between 2–5 nm. At the same time, water and ammonia are continuously released through the pore channels from a retreating crystalline–amorphous interface. This process continues until the struvite crystal is fully transformed into the amorphous Mg-phosphate phase leaving behind a porous network with a high specific surface area (∼300 m2 g−1) and pore volume (0.28 cm3 g−1). The presented synthesis procedure could provide a basis for the development of advanced functional phosphate-based materials for various applications in biomedicine and catalysis.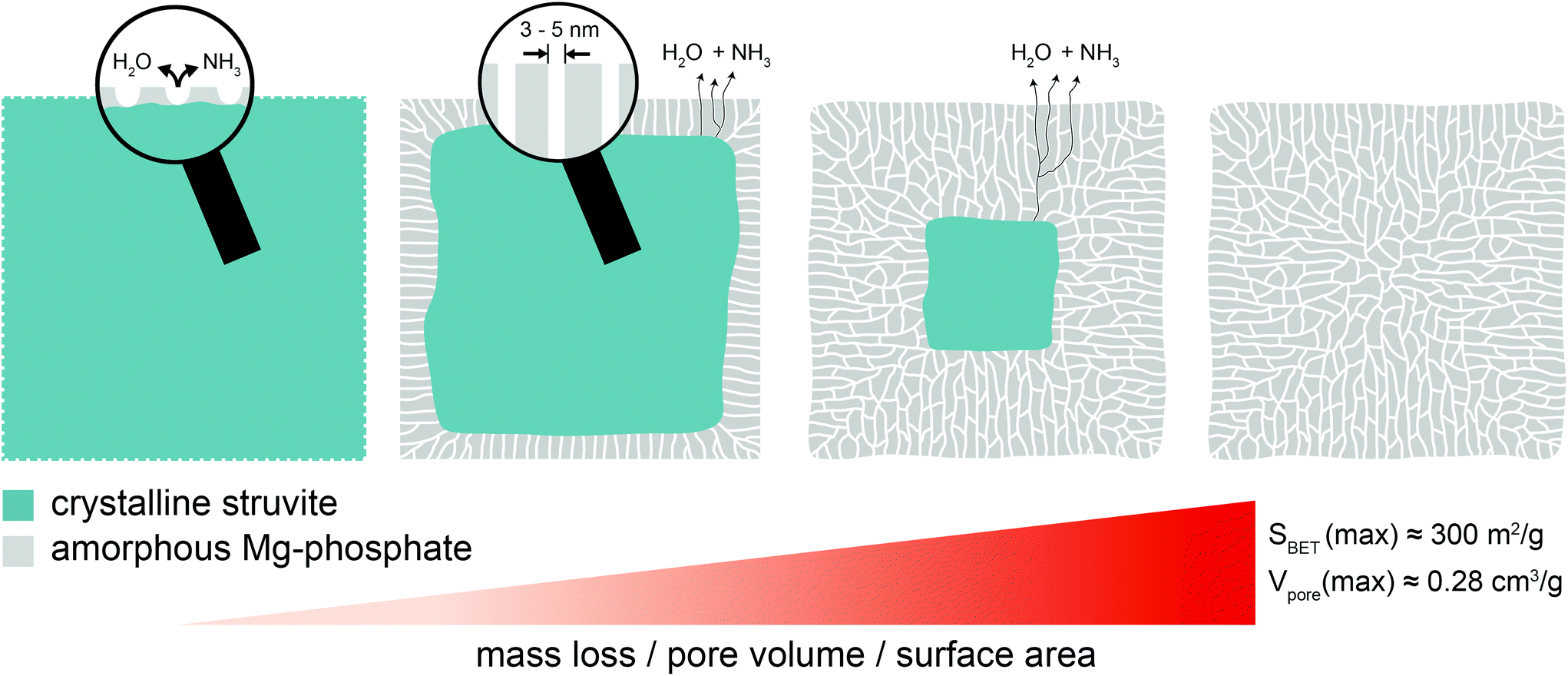 | ||
| Fig. 6 Schematic illustration of the struvite decomposition process. | ||
Note
The information contained in this paper is patent pending (European Patent Application no. EP18158825.2).Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
L. G. B., J. H., T. M. S., R. B., H. M. F. and S. M. acknowledge the financial support of the Helmholtz Recruiting Initiative (Award No. I-044-16-01). We thank Diamond Light Source for access to beamline I22 through a grant to T. M. S, L. G. B., J. H. and R. B. This research was partially supported by a Marie Curie grant from the European Commission in the framework of the NanoSiAl Individual Fellowship (Project No. 703015) to T. M. S. and a Scientific Network grant from the German Research Council (DFG) (Project No. 389508713) to H. M. F. L. G. B. and K. M. D. also acknowledge funding received through the European Union's Horizon 2020 Marie Skłodowska-Curie Innovative Training Network Metal-Aid (Project No. 675219). We thank our GFZ colleagues Judith Schicks for her help with the Raman analyses, Anja Schleicher for her help with the XRD analyses and Anja Schreiber for the preparation of the FIB sections.References
- IUPAC – International Union of Pure and Applied Chemistry, https://iupac.org/.
- A. Corma, From Microporous to Mesoporous Molecular Sieve Materials and Their Use in Catalysis, Chem. Rev., 1997, 97(6), 2373–2420 CrossRef CAS PubMed.
- Z. Wu and D. Zhao, Ordered Mesoporous Materials as Adsorbents, Chem. Commun., 2011, 47(12), 3332 RSC.
- M. Tiemann, Porous Metal Oxides as Gas Sensors, Chem. – Eur. J., 2007, 13(30), 8376–8388 CrossRef CAS PubMed.
- F. Cheng, Z. Tao, J. Liang and J. Chen, Template-Directed Materials for Rechargeable Lithium-Ion, Society, 2007, 667–681 Search PubMed.
- O. Hernandez-Ramirez and S. M. Holmes, Novel and Modified Materials for Wastewater Treatment Applications, J. Mater. Chem., 2008, 18(24), 2751–2761 RSC.
- M. Vallet-Regí, Ordered Mesoporous Materials in the Context of Drug Delivery Systems and Bone Tissue Engineering, Chem. – Eur. J., 2006, 12(23), 5934–5943 CrossRef PubMed.
- T. Asefa and V. Dubovoy, Ordered Mesoporous/Nanoporous Inorganic Materials via Self-Assembly, Elsevier, 2nd edn, 2017, vol. 9 Search PubMed.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Ordered Mesoporous Molecular Sieves Synthesized by a Liquid-Crystal Template Mechanism, Nature, 1992, 359, 710–712 CrossRef CAS.
- J. Yao, W. Tjandra, Y. Z. Chen, K. C. Tam, J. Ma and B. Soh, Hydroxyapatite Nanostructure Material Derived Using Cationic Surfactant as a Template, J. Mater. Chem., 2003, 13(12), 3053 RSC.
- T. Peltola, M. Jokinen, H. Rahiala, E. Levanen, J. B. Rosenholm, I. Kangasniemi and A. Yli-Urpo, Calcium Phosphate Formation on Porous Sol-Gel-Derived SiO2 and CaO- P2O5-SiO2 Substrates in Vitro, J. Biomed. Mater. Res., 1998, 44(1), 12–21 CrossRef.
- J. Zhang, M. Fujiwara, Q. Xu, Y. Zhu, M. Iwasa and D. Jiang, Synthesis of Mesoporous Calcium Phosphate Using Hybrid Templates, Microporous Mesoporous Mater., 2008, 111(1–3), 411–416 CrossRef CAS.
- P. J. T. Reardon, J. Huang and J. Tang, Mesoporous Calcium Phosphate Bionanomaterials with Controlled Morphology by an Energy-Efficient Microwave Method, J. Biomed. Mater. Res., Part A, 2015, 103(12), 3781–3789 CrossRef CAS PubMed.
- O. G. da Silva, M. M. Alves, I. M. G. dos Santos, M. G. Fonseca and M. Jaber, Mesoporous Calcium Phosphate Using Casein as a Template: Application to Bovine Serum Albumin Sorption, Colloids Surf., B, 2017, 158, 480–487 CrossRef PubMed.
- R. Lin and Y. Ding, A Review on the Synthesis and Applications of Mesostructured Transition Metal Phosphates, Materials, 2013, 6(1), 217–243 CrossRef CAS PubMed.
- B. Tian, X. Liu, B. Tu, C. Yu, J. Fan, L. Wang, S. Xie, G. D. Stucky and D. Zhao, Self-Adjusted Synthesis of Ordered Stable Mesoporous Minerals by Acid–base Pairs, Nat. Mater., 2003, 2(3), 159–163 CrossRef CAS PubMed.
- M. Pramanik, J. Lee, S. Tominaka, Y. Ide, J. H. Kim and Y. Yamauchi, Unique Nanocrystalline Frameworks in Mesoporous Tin Phosphate Prepared through a Hydro Fl Uoric Acid Assisted Chemical Reaction †, J. Mater. Chem. A, 2016, 1–9 Search PubMed.
- M. Pramanik, C. Li, M. Imura, V. Malgras, Y. M. Kang and Y. Yamauchi, Ordered Mesoporous Cobalt Phosphate with Crystallized Walls toward Highly Active Water Oxidation Electrocatalysts, Small, 2016, 12(13), 1709–1715 CrossRef CAS PubMed.
- M. I. H. Bhuiyan, D. S. Mavinic and F. A. Koch, Thermal Decomposition of Struvite and Its Phase Transition, Chemosphere, 2008, 70(8), 1347–1356 CrossRef CAS PubMed.
- M. V. Ramlogan and A. A. Rouff, An Investigation of the Thermal Behavior of Magnesium Ammonium Phosphate Hexahydrate, J. Therm. Anal. Calorim., 2016, 123(1), 145–152 CrossRef CAS.
- A. K. Sarkar, Hydration/Dehydration Characteristics of Struvite and Dittmarite Pertaining to Magnesium Ammonium Phosphate Cement Systems, J. Mater. Sci., 1991, 26(9), 2514–2518 CrossRef CAS.
- G. Kurtulus and A. C. Tas, Transformations of Neat and Heated Struvite (MgNH4PO 4·6H2O), Mater. Lett., 2011, 65(19–20), 2883–2886 CrossRef CAS.
- B. Lafuente, R. T. Downs, H. Yang and N. Stone, The Power of Databases: The RRUFF Project, in Highlights in Mineralogical Crystallography, Walter de Gruyter GmbH, Berlin, Germany, 2016 Search PubMed.
- B. R. Pauw, A. J. Smith, T. Snow, N. J. Terrill and A. F. Thünemann, The Modular Small-Angle X-Ray Scattering Data Correction Sequence, J. Appl. Crystallogr., 2017, 50(6), 1800–1811 CrossRef CAS PubMed.
- R. J. C. Mclean, J. Downey, L. Clapham and J. C. Nickel, A Simple Technique for Studying Struvite Crystal Growth in Vitro, Urol. Res., 1990, 18, 39–43 CrossRef CAS PubMed.
- M. Thommes, Physical Adsorption Characterization of Nanoporous Materials, Chem.-Ing.-Tech., 2010, 82(7), 1059–1073 CrossRef CAS.
- A. V. Neimark, P. I. Ravikovitch and A. Vishnyakov, Adsorption Hysteresis in Nanopores, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2000, 62(2), R1493–R1496 CrossRef CAS.
- C. G. Sonwane and S. K. Bhatia, Characterization of Pore Size Distributions of Mesoporous Materials from Adsorption Isotherms, J. Phys. Chem. B, 2000, 104(39), 9099–9110 CrossRef CAS.
- A. V. Neimark, P. I. Ravikovitch, M. Grün, F. Schüth and K. K. Unger, Pore Size Analysis of MCM-41 Type Adsorbents by Means of Nitrogen and Argon Adsorption, J. Colloid Interface Sci., 1998, 207(1), 159–169 CrossRef CAS PubMed.
- P. L. Llewellyn, Y. Grillet, F. Schüth, H. Reichert and K. K. Unger, Effect of Pore Size on Adsorbate Condensation and Hysteresis within a Potential Model Adsorbent: M41S, Microporous Mater., 1994, 3(3), 345–349 CrossRef CAS.
- J. C. Groen, L. A. A. Peffer and J. Pérez-Ramírez, Pore Size Determination in Modified Micro- and Mesoporous Materials. Pitfalls and Limitations in Gas Adsorption Data Analysis, Microporous Mesoporous Mater., 2003, 60(1–3), 1–17 CrossRef CAS.
- R. Besselink, T. M. Stawski, A. E. S. Van Driessche and L. G. Benning, Not Just Fractal Surfaces, but Surface Fractal Aggregates: Derivation of the Expression for the Structure Factor and Its Applications, J. Chem. Phys., 2016, 145(21), 211908 CrossRef CAS PubMed.
- S. Kataki, H. West, M. Clarke and D. C. Baruah, Phosphorus Recovery as Struvite from Farm, Municipal and Industrial Waste: Feedstock Suitability, Methods and Pre-Treatments, Waste Manag., 2016, 49, 437–454 CrossRef CAS PubMed.
- H. Basset and W. L. Bedwell, Studies of Phosphates. Part I. Ammonium Magnesium Phosphate and Related Compounds, J. Chem. Soc., 1933, 854–871 RSC.
- E. Banks, R. Chianelli and R. Korenstein, Crystal Chemistry of Struvite Analogs of the Type MgMPO4·6H2O (M+ = K+, Rb+, Cs+, Ti+, NH4+), Inorg. Chem., 1975, 14(7), 1634–1639 CrossRef CAS.
- V. Stefov, B. Šoptrajanov, F. Spirovski, I. Kuzmanovski, H. D. Lutz and B. Engelen, Infrared and Raman Spectra of Magnesium Ammonium Phosphate Hexahydrate (Struvite) and Its Isomorphous Analogues. I. Spectra of Protiated and Partially Deuterated Magnesium Potassium Phosphate Hexahydrate, J. Mol. Struct., 2004, 689, 1–10 CrossRef CAS.
- V. Stefov, B. Soptrajanov, M. Najdoski, B. Engelen and H. D. Lutz, Infrared and Raman Spectra of Magnesium Ammonium Phosphate Hexahydrate (Struvite) and Its Isomorphous Analogues. V. Spectra of Protiated and Partially Deuterated Magnesium Ammonium Arsenate Hexahydrate (Arsenstruvite), J. Mol. Struct., 2008, 872, 87–92 CrossRef CAS.
- R. V. S. S. N. Ravikumar, S. N. Rao, B. J. Reddy and Y. P. Reddy, Electronic Spectra of Hexa Aqua Coordinated Transition Metal Doped Zinc Struvite, Ferroelectrics, 1996, 189(1–4), 139–147 CrossRef CAS.
- R. V. S. S. N. Ravikumar, A. V. Chandrasekhar, S. N. Rao, N. Madhu, B. J. Reddy and Y. P. Reddy, Orthorhombic Site Symmetry of Cr3+ in ZnNH4 PO4 6H2 O Crystals, Cryst. Res. Technol., 1999, 34, 911–914 CrossRef CAS.
- R. V. S. S. N. Ravikumar, A. V. Chandrasekhar, B. J. Reddy, Y. P. Reddy and K. Ikeda, X-Ray Powder Diffraction, DTA and Vibrational Studies of CdNH4PO4.6H2O Crystals, Cryst. Res. Technol., 2002, 37(10), 1127–1132 CrossRef CAS.
- P. Chand and O. P. Agarwal, Electron Paramagnetic Resonance Study of Doped Synthetic Crystals of Struvite and Its Zinc Analogue, Spectrochim. Acta, Part A, 1991, 47(6), 775–783 CrossRef.
- Y. Yu, J. Wang, C. Liu, B. Zhang, H. Chen, H. Guo, G. Zhong, W. Qu, S. Jiang and H. Huang, Evaluation of Inherent Toxicology and Biocompatibility of Magnesium Phosphate Bone Cement, Colloids Surf., B, 2010, 76(2), 496–504 CrossRef CAS PubMed.
- F. Tamimi, D. L. Nihouannen, D. C. Bassett, S. Ibasco, U. Gbureck, J. Knowles, A. Wright, A. Flynn, S. V. Komarova and J. E. Barralet, Biocompatibility of Magnesium Phosphate Minerals and Their Stability under Physiological Conditions, Acta Biomater., 2011, 7(6), 2678–2685 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: XRD and Raman analyses of as-synthesized and thermally treated struvite samples; additional SEM images, TEM images, EDX and EELS spectra of the materials; comparison of BJH pore size distributions calculated from N2 adsorption vs. desorption data; additional time-resolved SAXS patterns of struvite decomposed under vacuum at 40 °C. See DOI: 10.1039/c8nr09205b |
| This journal is © The Royal Society of Chemistry 2019 |