
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural nanotechnology: three-dimensional cryo-EM and its use in the development of nanoplatforms for in vitro catalysis
Mark V.
de Ruiter
a,
Robin
Klem
a,
Daniel
Luque
b,
Jeroen J. L. M.
Cornelissen
 *a and
José R.
Castón
*c
*a and
José R.
Castón
*c
aDepartment of Biomolecular Nanotechnology, MESA+ Institute for Nanotechnology, University of Twente, 7500 AE Enschede, The Netherlands. E-mail: j.j.l.m.cornelissen@utwente.nl
bCentro Nacional de Microbiología/ISCIII, Majadahonda, Madrid, Spain
cDepartment of Structure of Macromolecules, Centro Nacional de Biotecnología (CNB-CSIC), Campus Cantoblanco, Madrid, Spain. E-mail: jrcaston@cnb.csic.es
First published on 18th February 2019
Abstract
The organization of enzymes into different subcellular compartments is essential for correct cell function. Protein-based cages are a relatively recently discovered subclass of structurally dynamic cellular compartments that can be mimicked in the laboratory to encapsulate enzymes. These synthetic structures can then be used to improve our understanding of natural protein-based cages, or as nanoreactors in industrial catalysis, metabolic engineering, and medicine. Since the function of natural protein-based cages is related to their three-dimensional structure, it is important to determine this at the highest possible resolution if viable nanoreactors are to be engineered. Cryo-electron microscopy (cryo-EM) is ideal for undertaking such analyses within a feasible time frame and at near-native conditions. This review describes how three-dimensional cryo-EM is used in this field and discusses its advantages. An overview is also given of the nanoreactors produced so far, their structure, function, and applications.
 Mark V. de Ruiter (middle), Robin Klem (right) and Jeroen J. L. M. Cornelissen (left) | Mark V. de Ruiter was born in Rhenen, the Netherlands in 1990. He obtained his BSc in Applied physics (2012) and MSc in chemistry (2014 – cum laude) from the University of Twente. His research involves viral protein cages as artificial organelles. Robin Klem was born in Wageningen, the Netherlands in 1991. He obtained his BSc and MSc (cum laude) in Biopharmaceutical Sciences in 2012 and 2014, respectively. He engineers encapsulins as catalytic protein nanocages. They both work as PhD candidates at the MESA+ institute under the supervision of Prof. Dr Jeroen J. L. M. Cornelissen. He is Professor in Biomolecular Nanotechnology at the University of Twente, Enschede, the Netherlands. He studied chemistry at the Eindhoven University of Technology and Supramolecular Chemistry and Catalysis at the University of Nijmegen. He received his PhD (cum laude) from the latter university in 2001 for research carried out under the supervision of Prof. R. J. M. Nolte. After post-doctoral work at the IBM Almaden Research Center in San Jose, USA he returned to Nijmegen, where he was appointed as an Assistant Professor until 2009. His current research interests are in well-defined polymer architectures, hybrid systems of synthetic macromolecules and biopolymers and the use of viruses as building blocks in functional materials. |
 Daniel Luque (left) and José R. Castón (right) | Daniel Luque is a biochemist and group leader since 2009 of the Electron and Confocal Microscopy Unit of the Instituto de Salud Carlos III in Madrid. His BSc in Biochemistry was followed by a PhD in Molecular Biology at the UAM with Dr Castón. As a postdoctoral researcher at the CNB, he characterized various viral systems by cryo-EM and image processing. His current research focuses mainly on viral pathogens in human health using a multidisciplinary approach, which includes state-of-the-art cryo-EM approaches to obtain near-atomic resolution structures. |
José R Castón is a biologist and group leader since 2006 of the Viral Molecular Machines Laboratory at the National Center for Biotechnology (CNB) of the Spanish National Research Council (CSIC) in Madrid (Spain). He graduated in Biology from the Universidad Autónoma de Madrid (UAM), followed by a master's degree and a PhD in Biochemistry from the UAM. His postdoctoral research on cryo-EM of viruses was carried out at the National Institutes of Health (Bethesda, MD, USA). In his research, he studies structure–function–evolutionary relationships of viral macromolecular complexes or nanomachines using three-dimensional cryo-EM. Structural characterization of these viral assemblies offers new alternatives for altering their function and could allow their development as nanoreactors to be used in nanotechnology. |
1. Introduction
Enzymes, and the metabolic processes in which they participate, are often compartmentalized in different organelles. Eukaryotic cells predominantly organize their metabolic pathways within organelles with lipid membranes,1e.g., endosomes, the Golgi apparatus, or the cell nucleus. Like prokaryotes, they also contain protein-based cages to confine their enzymes.2,3 Both cell types use these latter structures to control substrate and product flux, to improve enzyme stability, and to create defined microenvironments that presumably afford some biological advantage for the biochemistry that occurs within them. Viral capsids can also be considered protein-based cages.4Man-made nanoreactors based on these cages might have biomedical applications, be useful in the search for new materials, and provide new vehicles for catalysis.5 The properties of protein-based cages correlate strongly with their structure, and developing nanoreactors from natural protein-based cages therefore requires detailed knowledge of their three-dimensional (3D) structure. X-ray crystallography, and to a lesser extent nuclear magnetic resonance (NMR) spectroscopy, have long been used examine the structure of proteins and nucleic acids at near-atomic resolution. NMR is limited by the size of the macromolecules under study, however, and X-ray crystallography by the difficulty in crystallizing large, conformationally flexible complexes and membrane proteins. Large quantities of protein-based cages are also needed for crystallization trials, and these are not easy to obtain. In the past few years, 3D cryo-electron microscopy (cryo-EM) has revolutionized the field of structural biology.6,7 The 2017 Nobel Prize for chemistry, awarded to Jacques Dubochet (University of Lausanne), Joachim Frank (Columbia University), and Richard Henderson (MRC Laboratory of Molecular Biology), recognized the impact of cryo-EM in disciplines across biology and chemistry.8 Perhaps not surprisingly, this technique has been used to examine the structure of protein-based cages and will likely be central to the development of man-made nanoreactors.
Various strategies can be used to direct enzymes into the interior of protein-based cages to manufacture nanoreactors. These methods can be summarized in six loading categories: covalent, charge-based, affinity tag, statistical, supramolecular, and scaffold-assisted loading (Fig. 1). In covalent loading, the cargo is linked by a covalent bond to the interior-facing residues of the protein monomers that form the cage; they can be chemically or enzymatically fused, or produced in fused form via genetic manipulation.9–11 In charge-based loading, enzymes are attracted into a protein cage when its interior is charged. This requires that the cargo enzyme charge be opposite to that of the protein cage.12 Affinity tag loading involves a tag fused to the enzyme that binds at high affinity to the cage interior, e.g., a peptide tag or a specific hairpin nucleotide sequence.13 In statistical loading, enzymes are added in high concentration to the cages during their assembly. A proportion of those formed will contain the desired cargo. Supramolecular loading takes advantage of non-covalent interactions for cargo-directed assembly e.g., using coil–coil interactions or other supramolecular affinity tags; commonly, one of the binding partners is fused to the enzyme and the other to the cage protein, usually followed by an incubation/cage assembly step.10 In scaffold loading, the enzyme is fused, often by genetic means, to a scaffolding protein used in the assembly of the protein cage.
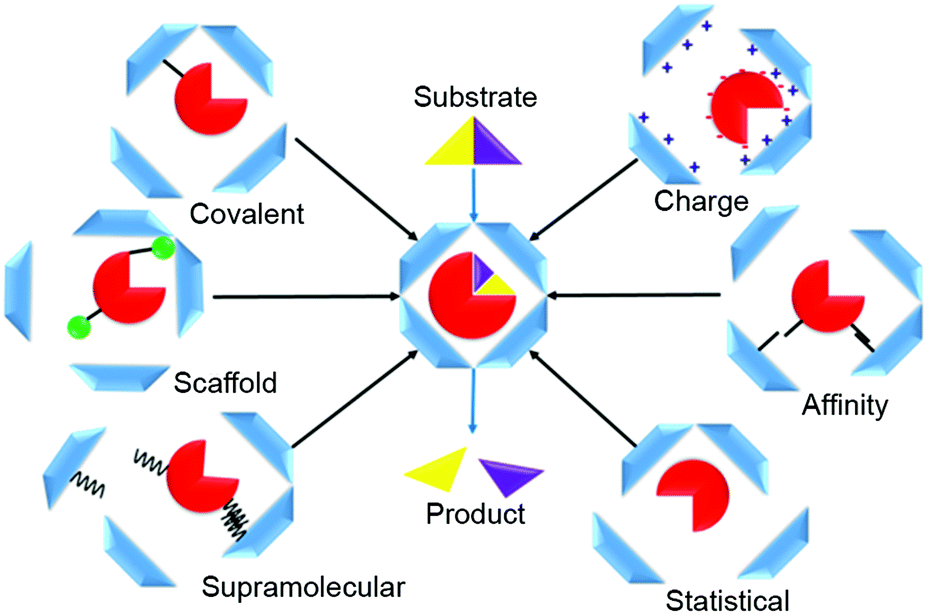 | ||
| Fig. 1 Overview of various strategies used to encapsulate enzymes in protein nanocages. These include covalent-, charge-, affinity tag-, statistical-, supramolecular-, and scaffold-assisted assembly of enzyme-based protein nanoreactors. Container proteins are shown in blue, the enzyme in red, scaffold proteins in green, and the different tags in black. | ||
This review focuses on the 3D cryo-EM examination of protein-based cages and nanoreactors designed to promote in vitro catalysis. The nanoreactors reported in the literature, and the structural and chemical bases for developing such catalytic nanoplatforms, are discussed.
2. Three-dimensional cryo-EM analysis of protein cages
3D cryo-EM provides an effective means to determine the structure of many macromolecular assemblies at near-atomic resolution. The development of direct electron detectors (DED) for recording images was a key factor driving the “resolution revolution”,6,14 as were improvements in sample preparation methods, the development of field emission gun (FEG)-equipped, high-resolution automated electron microscopes with a 300 kV acceleration voltage, and computational methods to deal with structural/compositional heterogeneity.153D cryo-EM requires only a small amount of sample; this is particularly advantageous when sample material is in short supply or precious. Unlike other techniques, cryo-EM can be used to study a broad range of assemblages, from single proteins16,17 to large macromolecular complexes such as virus particles.18,19 Cell organelles, entire eukaryotic cells, bacteria and tissue sections can be analyzed by an analogous method known as cryo-electron tomography.20,21
2.1. Specimen preparation and imaging
Specimens are commonly prepared using the negative staining technique; they are dehydrated and embedded in a heavy metal salt cast, which replicates the specimen's shape.22 This quick, easy method gives high contrast images, and is used to assess sample homogeneity and quality. The resolution is limited by the stain granularity to ∼20 Å.23For cryo-EM, rapid freezing of the sample in a buffer is required (to −180 °C) so that the water molecules are immobilized in an amorphous state. The so-called vitreous ice formed avoids specimen disruption by ice crystal formation.24 To prepare these frozen samples, an aliquot of the specimen in its buffer is applied to an electron microscopy grid coated with a hole-containing carbon support film. It is then blotted with filter paper to leave a very thin film of particle suspension in the holes. The grid is plunged into liquid ethane cooled by liquid nitrogen.25,26 To improve reproducibility, this is done using semi-automated plunge-freezer aids such as a Vitrobot (FEI), Cryo-plunge (Gatan), or EM GP (Leica) device.
The concentration of the buffered sample is critical for obtaining a thin film with even specimen distribution after blotting (usually confirmed by negative staining analysis). The presence of glycerol, sucrose, or cesium chloride, used to purify samples by density gradient ultracentrifugation, can reduce vitrification efficiency. These compounds bubble when exposed to the electron beam and are thus incompatible with high-resolution imaging.15
Sample stability and homogeneity must be checked prior to cryo-EM analysis.27 Size-exclusion chromatography separates specimen subpopulations based on the hydrodynamic radius of their particles, and is an appropriate method for preparing homogeneous samples.28,29 In addition to the presence of contaminant complexes, specimen heterogeneity can result from a sample found in distinct compositional or conformational states. The GraFix technique can also be used to help ‘purify’ the sample.30 In this method, a chemical crosslinker such as glutaraldehyde is added to a glycerol density gradient before centrifugation, to stabilize and separate complexes from dissociated elements and aggregates. The high-throughput Proteoplex method can be used to screen the stability of multisubunit complexes in different buffers in the presence of small fluorescent molecules, via analysis of thermal unfolding transitions.31
2.2. Image acquisition
Specimen sensitivity to radiation can be a major problem in electron microscopy. Use of low electron doses to limit exposure nonetheless leads to images with a very poor signal-to-noise ratio (SNR) for subsequent image processing. Hence, many different views of the same macromolecular complex are averaged to enhance the SNR and calculate a 3D reconstruction.Direct electron detectors (DED), such as the Falcon camera (from FEI ThermoFisher), the K camera (from Gatan), and the DE camera (from Direct Electron Inc.), were key in improving resolution. DED have a high detective quantum efficiency (DQE, a measure of detection efficiency as a function of spatial frequency),32,33 which increases the SNR. DED can operate in integration or counting mode. In integration mode, the signal is integrated over the entire exposure time or is dose-fractionated into multiple movie frames; in counting mode, single electron scattering events are detected.
DED have a rapid readout rate and images are collected as movies, which allows dose fractionation into multiple frames per second34,35 (each frame has an extremely low electron dose, ∼1 e− per pixel). More importantly, the beam-induced specimen drift that results in motion-induced image blurring can be tracked and corrected by computational alignment of a series of low-dose frames taken in one area before averaging them.36 In addition, subframes with optimized doses can be selected for subsequent image processing37 (later frames would be of lower quality due to radiation damage). The combination of dose fractionation and motion correction greatly improves data acquisition efficiency; nearly all images are of a quality suitable for recovering high resolution information.38
2D projection images are modulated by the contrast transfer function (CTF) of the microscope, and image defocus correction is necessary for accurate interpretation of specimen structure. During data acquisition, images are recorded at a range of defocus settings to enhance the specimen's different features and to fill in missing information when they are combined into a 3D reconstruction. Averaged movie quality can be analyzed by inspection of the CTF zeros or Thon rings in the image computed diffraction patterns. Large symmetrical assemblies can be imaged at small defocus levels with good image contrast and preserved high-resolution signals. For small (100–300 kDa) and asymmetric assemblies, high defocus settings are needed, which limit the resolution that can be achieved. Cryo-EM using a Volta phase plate greatly improves phase contrast and enables in-focus phase-contrast imaging.39,40
A number of automated data collection systems, such as SerialEM41 and Leginon42 (both free), as well as the commercial EPU (FEI), JADAS (JEOL) and Latitude (Gatan) systems are available for collecting large data sets for reliable statistical analysis.
2.3. Structure determination by single particle cryo-EM
The general workflow of single particle analysis includes quality assessment of motion-corrected average images, particle picking, image CTF correction, particle classification, estimation of particle orientation and refinement, image reconstruction and refinement, resolution assessment, and map validation43 (Fig. 2). A number of classical image processing packages perform these tasks, provide classification of multiple conformational or compositional states, and sort images into different 2D and 3D image classes. These software packages include RELION,44 EMAN2,45 Xmipp46 and FREALIGN.47 SCIPION48 offers a framework for integrating software packages via a workflow-based procedure designed for less experienced users as well as user-friendly graphic interfaces. Additional software packages like CryoSPARC,49 cisTEM50 and SPHIRE51 also have workflows useful for processing cryo-EM images and obtaining high-resolution 3D reconstructions from them.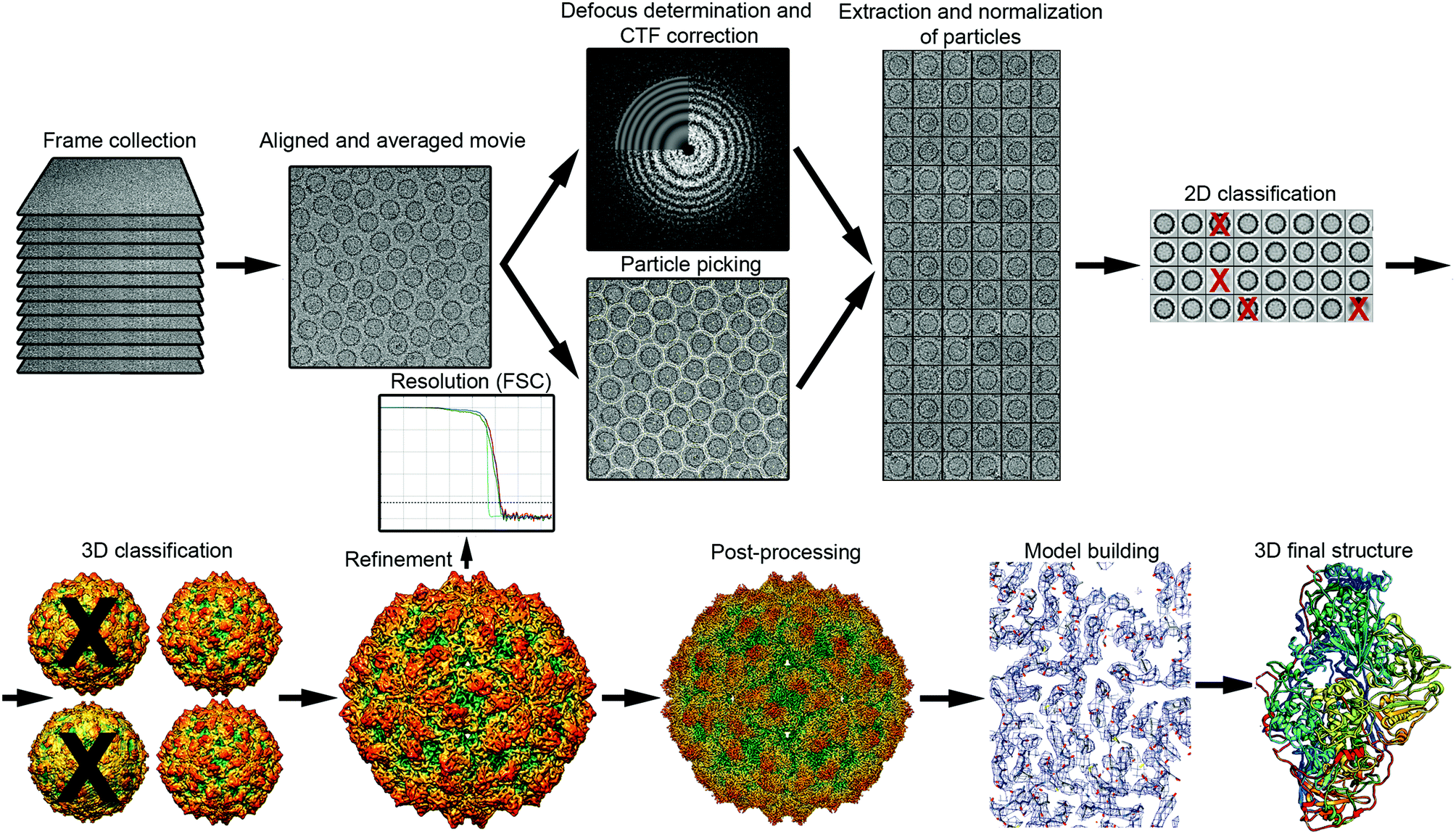 | ||
| Fig. 2 Overview of single-particle cryo-EM workflow, from data collection to 3D model. The vitrified sample is imaged by collecting movie frames that are aligned and averaged. Using motion-corrected images, image defocus is calculated for CTF correction, and individual particles (such as protein cages) are picked. Particles are extracted, cleaned (removal of bad projections) and normalized, and then subjected to 2D classification and averaging, to obtain class averages according to similarity. Class averages with improved SNR can be used to obtain an ab initio 3D model. This low-resolution model is used as a reference for 3D classification, a necessary step for identifying distinct conformations, macromolecular heterogeneities, or particle subsets with different structural integrity. Orientation refinement is performed iteratively until the structure converges, as indicated in the resolution analysis by the FSC method. The final map is sharpened by applying an estimated B-factor that reveals high resolution details. Finally, the protein sequence is fitted into the 3D near-atomic map to build a de novo 3D model of the protein. 2D images and 3D maps correspond to the cryo-EM structure determined at 3.7 Å resolution of the fungal virus Rosellinia necatrix quadrivirus 1.192 | ||
Most of these packages continuously implement new approaches with better automatic particle-picking algorithms, more precise and faster CTF determinations, and improvements in beam-induced motions correction, local-resolution estimation, 2D and 3D classification methods, as well as alignment and reconstruction methods. The implementation in RELION of Ewald sphere curvature correction, together with Bayesian-based beam-induced motion correction, allows resolution improvements of 0.2–0.7 Å.52 This continuous software development implies a myriad of possibilities for combining distinct programs and their different parameters.
Single-particle cryo-EM provides the 3D structure of the specimens examined by computationally merging images of many (easily exceeding tens or hundreds of thousands) individual macromolecules in identical (or similar) conformations. Each particle image is a randomly orientated 2D projection image that contains all the structural detail of the 3D specimen. Angular orientation parameters are determined by comparing the 2D projections with spatially defined reprojections of an initial 3D model filtered at low resolution. A new 3D map of the macromolecular complex is then calculated from these 2D projections by “back-projection”, i.e., combining all views into a single 3D map.53 This projection-matching process is performed iteratively to obtain new 3D reconstructions with improved resolution until no further improvement is possible. Finally, the known protein sequence is fitted into the 3D map, positioning the bulky amino acid side chains first. The 3D atomic model built is then further refined.
Owing to the very low electron doses used to avoid radiation damage, biological specimens yield low contrast, extremely noisy images. Resolution can also be affected by the conformational and/or compositional heterogeneity of the sample, in which multiple states of a macromolecular assembly coexist in the same sample. Sophisticated 3D classification methods are therefore becoming essential for high-resolution analysis of single particle cryo-EM data.54–56 These methods reconstruct 3D structures by combining many images of a homogeneous subset of particles that have been aligned with each other at high precision. One of the promising developments is the imaging of short-lived states (10–1000 ms) by time-resolved cryo-EM.57 This development would be especially relevant for 3D analysis of nanoplatforms for in vitro catalysis.
2.4. Resolution estimation, model building and validation
Defining the resolution of a cryo-EM map remains problematic. When generating such maps, it is now standard to randomly split the dataset into two independent (even–odd) halves and, by iterative refinement and reconstruction, to compute two independent maps from each half-set. These two reconstructions are then correlated as a function of the spatial frequency to determine the extent to which structural features have been reliably established. This is termed the “gold standard” Fourier shell correlation (FSC) method.58 As resolution is often non-isotropic in different regions of the map, visual inspection of map quality in key regions is needed to establish local conformations and their functional implications.59 Local variations in the resolution can be assessed using Resmap60 or MonoRes61 programs.Although cryo-EM can potentially determine ab initio protein structures in the 2–4 Å range, the true nature of many specimens can remain elusive. Many large protein-based cages modified with specific cargos must be classified into different 3D map classes because of their heterogeneity (they may be intact, incomplete, truncated or damaged), or because they have different conformations or functional states. The structures of protein-based cages are typically solved at subnanometer or even modest resolutions. Atomic structures obtained by X-ray crystallography and NMR are then fitted into the cryo-EM map.
3. Protein-based nanoreactors
Nanoreactors made from bacterial amyloids,62 nano-sized crosslinked enzymes, or encapsulated enzymes in other protein matrices63 are not discussed here. We focus on well-defined natural protein nanocages and how they can be modified to include non-natural enzymes. The natural protein-based nanocompartments based on viruses are first described, followed by protein-based nanocages of prokaryotic origin, and finally, nanoreactors found in eukaryotes. The structures of the native and synthetic protein-based nanoreactor particles are provided, most determined by 3D cryo-EM.3.1. Virus-like particles
Viruses consist of a few building blocks that self-assemble into precisely defined nanosized structures; these can be icosahedral, rod-like, or some other shape, and fall into the nano to micron size range.64 These properties have led to exploration of possible virus uses in nanotechnological applications.65 Their capsids show remarkable parallels with the micro-nanosized66 protein-based cages of bacteria, in some cases including the icosahedral symmetry of the latter. This has resulted in virus use as nanoreactors that mimic these bacterial cages, to help understand the effect of confinement by the protein shell on cargo enzyme reactions.A variety of viral capsids are reported to be capable of encapsulating a protein cargo. These include those of cowpea chlorotic mottle virus (CCMV), bacteriophages P22, Qβ, and MS2, murine polyomavirus, bluetongue virus, lentivirus, and simian virus 40.67–70 Not all viral capsids are confirmed to have a porous shell, however, or can be used to pack enzymes. The following lines thus focus on the origin, structure, and use of five viruses that have been used successfully to generate nanoreactors.
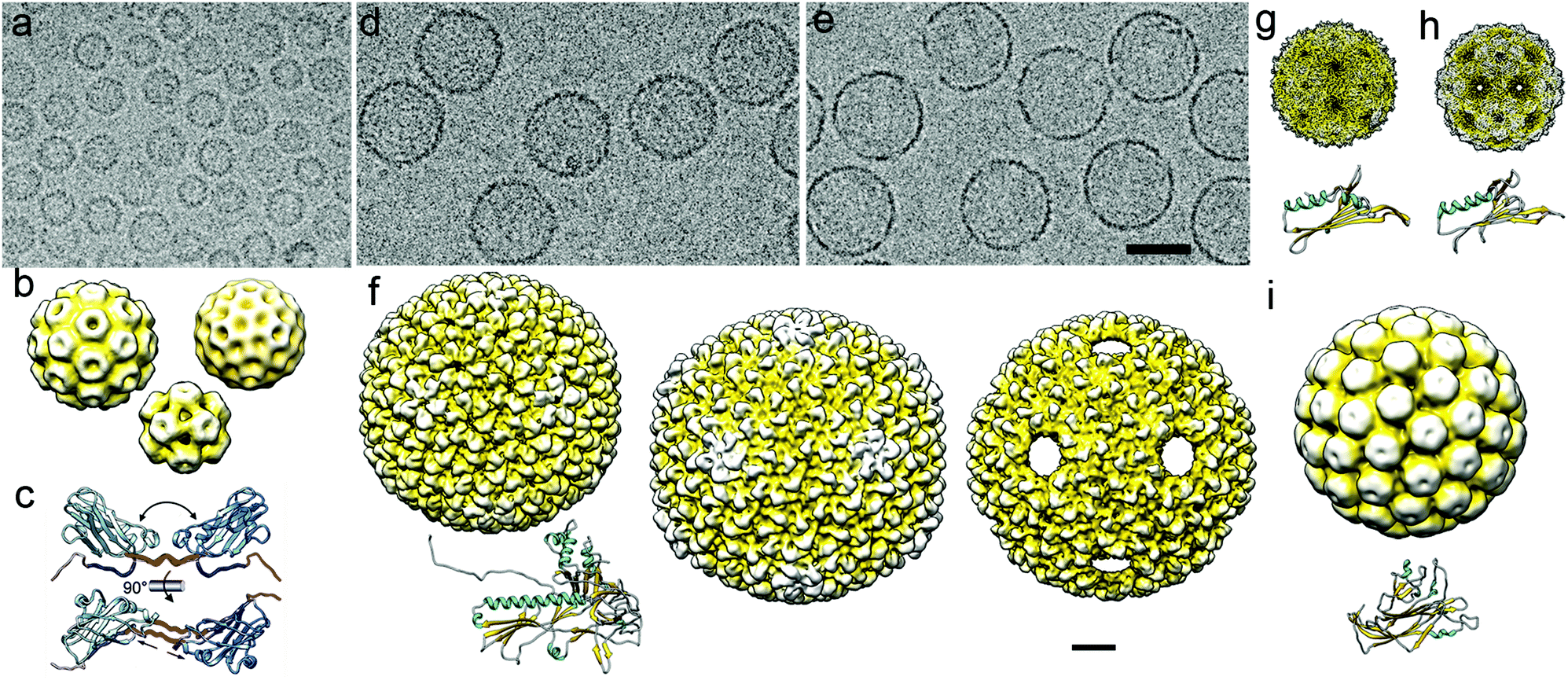 | ||
| Fig. 3 Cryo-EM of viral capsids used as nanoreactors and atomic models of their major capsid proteins. (a) Cryo-EM image of a CCMV sample formed by three classes of assembled particles. (b) Cryo-EM 3DR of the T = 3 (diameter 280 Å; top left), T = 2 (252 Å; top right), and T = 1 (204 Å; bottom center) CCMV capsids. (c) Capsid protein (PDB 1CWP) dimers show variability in the hinge dihedral angle (from ∼40° to ∼60°, top) and in dimeric contacts (bottom). (d, e) Cryo-EM images of empty P22 procapsids (d) and P22 wiffle ball capsids (e). Bar = 500 Å (a, d, e). (f) Surface-shaded representation of the outer surface of P22 procapsid (EMD-3171, left), mature or expanded (EMD-3164, center) and wiffle ball (EMD-4388, right) capsid. Atomic model of the mature P22 capsid protein (PDB 5UU5). (g) Cryo-EM map of the T = 3 capsid of bacteriophage MS2 and its monomer atomic coordinates (EMD-8397, PDB T5C1). (h) Cryo-EM map of the T = 3 capsid of bacteriophage Qβ and its monomer atomic coordinates (EMD-8708, PDB 5VLY). (i) Cryo-EM map of the T = 7 capsid of SV40 and its monomer atomic coordinates (EMD-5187, PDB 5BWQ). Cryo-EM maps in (b)–(i) are viewed along an icosahedral twofold axis, in the same scale. Bar = 100 Å. | ||
The first artificial nanoreactor reported was, in fact, based on CCMV.73 It was made using a statistical loading method, the cargo mixed into the solution of CP that were reassembled at pH 5. Horseradish peroxidase and cytochrome p450 were successfully encapsulated and retained their activity, providing nanoreactors that can be used for pro-drug activation and assessment of drug metabolism.73,74 Atlhough encapsulation efficiency was low and the nanoreactors were unstable at neutral pH, improvements were made using supramolecular loading. Coiled-coil interactions, in which one coil is attached to the CP N terminus and the other to the enzyme, were used to controllably load the enzyme lipase B of Pseudozyma Antarctica.75 The ‘stability at neutral pH’ problem was solved through chimeric expression in E. coli with an elastin-like polypeptide attached to the CP N terminus, which stabilizes the VLP in strong salt solutions.9 In other work, T4 lysozyme was attached covalently to the CP N terminus using sortase A as a catalyst.76 CP assembly resulted in encapsulation of the enzyme, since the N terminus now pointed inwards.
In attempts to introduce a negative charge to the enzymes, appropriately charged polystyrene sulfonate or single stranded DNA polymers have been linked to the cargo enzyme. This allows carriage of native or commercial enzymes and their expression in chimeric hosts, at the cost of a slight reduction in catalytic efficiency.12,77 This method has been used to encapsulate single enzymes such as horseradish peroxidase,77 and multiple enzymes such as a DNA-heme-based DNAzyme and glucose oxidase.12 These negatively charged chains resemble the native cargo and stabilize the nanoreactor at physiological conditions, thus enabling CCMV to be used as an artificial nanocage.78 The structure of the glucose oxidase-filled CCMV, as resolved by cryo-EM, confirmed effective encapsulation of the enzyme and a two-state polymorphism for the CCMV T = 1 capsid. One of these states is the expanded form of the other, and with larger pores it can be used in catalysis. When enzyme cascades were thus encapsulated, an increase in Kcat/Km was seen, indicating enhanced substrate channeling as a result of successful sequential catalytic reactions.
CCMV-based nanoreactors, loaded with enzymes or inorganic catalysts, have also been applied in thin film assemblages to create effective catalysts for in vivo use.77
Enzymes are generally loaded into P22 capsids by their genetic fusion with the N-terminal truncated SP, and co-expressing them with the CP in E. coli. This results in the directed encapsulation of up to 300 copies of the enzyme per capsid. Alcohol dehydrogenase D (AdhD) from the hyperthermophile Pyrococcus furiosus (useful for ketone/alcohol reduction and oxidation) can be encapsulated in this way,80 as can phosphotriesterase (PTE) from Brevundimonas diminuta (useful for breakdown of organophosphates, including chemical warfare agents and commercial insecticides). The stability of these encapsulated enzymes with respect to temperature, proteases, and other factors is much greater than that of non-encapsulated enzymes, highlighting a clear advantage of using protein-based cages.
The enzymes Cas9 (CRISPR-associated protein 9 from Streptococcus pyogenes [SpCas9]) and hydrogenase 1 (EcHyd-1), a very active [NiFe]-hydrogenase, have also been encapsulated in the P22 capsid.81–83 These nanoreactors show promise in eukaryotic genome engineering (by allowing cell-specific delivery) and in the production of hydrogen, respectively. In another example, NADH oxidase from Pyrococcus furiosus was fused to the capsid protein, resulting in a nanoreactor with antimicrobial properties.84 Cytochrome P450 has also been encapsulated in P22, and might have use in pro-drug activation in eukaryotic cells.85–87
P22-based nanoreactors are not only functional as separate units, but can be turned into larger assemblages that retain their enzymatic activity; this has been done with β-glucosidase. These nanoreactor-based frameworks are built using a combination of sixth generation poly-(amidoamine) dendrimers and cysteine-modified homo-trimeric capsid decoration proteins, bound to the P22 exterior.88 Dendrimers have also been used to form ordered face-centered cubic lattice structures, using nanoreactors containing ketoisovalerate decarboxylase and alcohol dehydrogenase A.89 Both enzymes retained their activity.
P22 capsids have also been applied to co-encapsulate AdhD from P. furiosus and rhodium (a catalyst).90 Using SP fusion, enzyme cascades have also been successfully encapsulated, e.g., the sugar metabolism cascade of P. furiosus. The sequential reaction involved β-glucosidase CelB91 (which hydrolyzes a wide variety of beta-linked disaccharides), ATP-dependent galactokinase, a phosphotransferase (which phosphorylates galactose), and ADP-dependent glucokinase (which catalyzes glucose to glucose-6-phosphate).92 All four enzymes produce essential intermediates for entry into glycolysis, making them particularly interesting for biofuel production. Retention of enzyme cargos within the P22 capsid was studied using cryo-EM and multiangle light scattering and small-angle X-ray scattering. While cargo size is an important consideration for its retention inside the P22 VLP, electrostatic interactions also play an essential role in both cargo retention and its release rate.89
The MS2 capsid can be filled in vivo using a CnaB2-based SpyTag/SpyCatcher system.95 The short SpyTag peptide binds covalently to the capsid and the SpyCatcher protein, which can be used to couple various complexes to proteins. Using this system, the enzymes pyridoxal phosphate (PLP)-dependent tryptophanase TnaA, flavin mononucleotide (FMN), and nicotinamide adenine dinucleotide phosphate (NADPH)-dependent monooxygenase FMO have been encapsulated in MS2 capsids in E. coli; together these enzymes synthesize deep blue indigo dye from L-tryptophan. Yield and stability of this caged enzyme complex are increased compared to non-encapsulated forms of the enzymes. This exemplifies a truly effective artificial nanocompartment that can be used to generate new non-native substances from common metabolic components in a cell.
3.2. Protein-based cages in prokaryotes
Protein-based cages are found throughout the bacterial kingdom99,100 (Fig. 4). They have crucial functions in metabolic pathways and enable growth in specific habitats.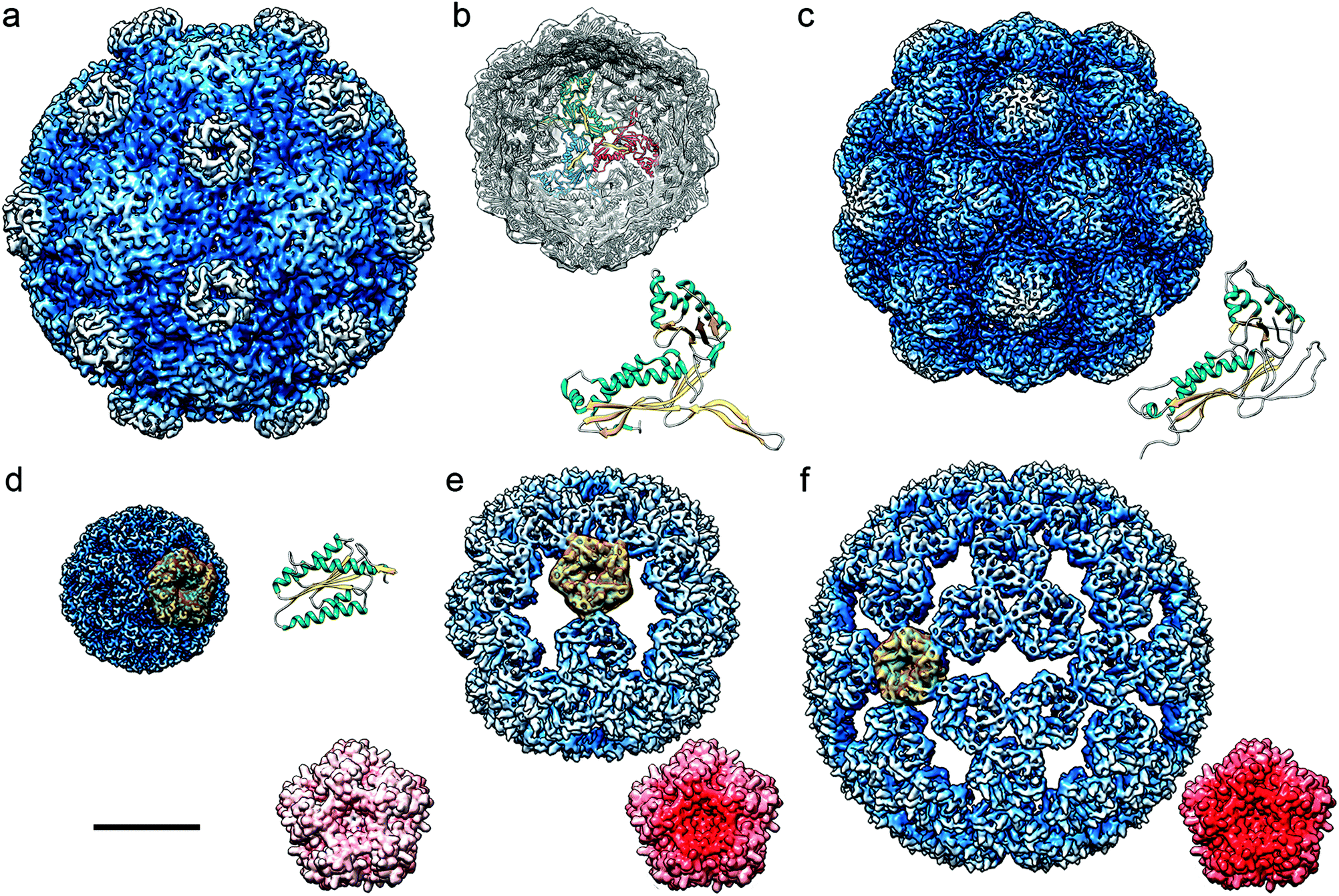 | ||
| Fig. 4 Cryo-EM based structures of bacterial nanocompartments shown as radially color-coded, surface-shaded models. Cryo-EM maps are in the same scale, bar = 100 Å. (a) BMC from H. ochraceum (EMD-8747). (b) DyP-encapsulin map from B. linens (EMD-3613) viewed along a three-fold axis, with docked T. maritima encapsulin. Encapsulin monomers at the 3-fold axis are depicted in red, green, and blue, and their FLP C-terminal ends in yellow. The T. maritima encapsulin atomic structure is shown (PDB 3DKT). (c) Encapsulin from M. xanthus (EMD-5917; PDB 4PT2). (d) Lumazine synthase from A. aeolicus (AaLS-wt, EMD-3538; PDB 5MPP). (e) AaLS-neg, an AaLS with four glutamates introduced (R83E, T86E, T120E, and Q123E) (EMD-3543, PDB-5MQ3). (f) AaLS-13, an AaLS-neg with seven additional mutations (EMD-3543, PDB 5MQ7). (d–f) A pentameric building block is highlighted in yellow for each AaLs assembly. Accessible inner surfaces of pentameric AaLS-wt (d), AaLS-neg (e) and AaLs-13 (f) represented with electrostatic potentials, showing the distribution of negative (red) and positive (blue) charges (bottom right). | ||
Similar to eukaryotic protein-based cages, those of prokaryotes reduce the crosstalk between pathway metabolites, toxic intermediates and inhibitory products. The shell is selective with respect to the molecules it allows through. For example, polar molecules are allowed to pass, while less polar and nonpolar molecules are not.100 Moreover, the pores can be modified to change their selectivity.105 This is important for substrate channeling and effective catalysis.
Prokaryotic protein-based cages can be divided into two types, the anabolic carboxysomes, involved in carbon fixation (they encapsulate carbonic anhydrase and ribulose-1,5-bisphosphate carboxylase/oxygenase [RuBisCO]),2,106 and catabolic cages, known as metabolosomes. The latter metabolize compounds depending on the enzymes encapsulated; these are commonly dehydratase, ethanol–amine–ammonialyase or glycyl–radical enzyme,103 but also aldehyde dehydrogenase, alcohol dehydrogenase, and phosphotransacylase.3
Formation of these cages is mediated by a peptide sequence that also directs cargo into the cage after assembly.107 The fact that the cargo carried can be changed renders these cages of interest for engineering nanororeactors.108,109 So far, nanoreactors have been made that are able to carry β-galactosidase,110 pyruvate decarboxylase, and alcohol dehydrogenase (which catalyzes the transformation of pyruvate into ethanol, of interest for biofuel production),111 and polyphosphate kinases112 (for polyphosphate accumulation). Large enzyme aggregates have also been encapsulated in BMC, including glycerol dehydrogenase, dihydroxyacetone kinase, methylglyoxal synthase and 1,2-propanediol oxidoreductase, for glycerol conversion to 1,2-propanediol.113
Man-made carboxysome cages have been used to improve CO2 fixation, and indeed have been expressed in chloroplasts to increase crop yields.114
Encapsulins are genetically encoded in bacteria, often in an operon also containing their cargo protein. Once expressed they self-assemble, forming monodisperse nanocages with their cargo inside. By introducing plasmids containing the operon for the cargo and encapsulin into E. coli, they can be expressed heterogeneously.13,118 Depending on the encapsulin source, they conform a T = 1 or T = 3 icosahedral structure (Fig. 4b and c), with 60 or 180 subunits that form a nanocage ranging from 20 to 40 nm in diameter.13 The structure is generally robust; indeed, encapsulins are stable at high and low pH, various salt concentrations, and at high temperatures.119
Only a few encapsulin crystal structures have been resolved to date, but most show protein shell pores approximately 5 Å across. These pores are large enough for small molecules to pass through.13 Although all encapsulins have a fold homologous to a structure first discovered in the HK97 phage,120 cages from different bacteria do differ. For example, encapsulins from Brevibacterium linens and Rhodococcus jostii naturally contain a dye-decolorizing peroxidase (DyP), while those of Thermotoga maritima and Pyrococcus furiosus contain a ferritin-like protein (FLP).13Mycobacterium tuberculosis encapsulins package three enzymes: a DyP, a bacterioferritin (BfrB or FLP) and a dihydroneopterin aldolase (FolB). These threeenzymes all have antioxidant properties, suggesting a role in oxidative stress responses.
To direct the cargo into the interior of encapsulin cages, an affinity peptide sequence (often attached to the C terminus of the cargo protein) is needed, except for P. furiosus, in which the cargo is fused to the encapsulin protein. This tag binds to the cage, but the exact binding mechanism and the encapsulin assembly mechanism are yet to be fully understood. When encapsulins are expressed in E. coli, the cargo can be altered by genetically removing the sequence for the native cargo and introducing a new cargo sequence. This has been done with B. linens encapsulin, into which fluorescent proteins were introduced.
The encapsulin provides a robust shell to the enzyme cargo, with many pores to allow solute flux. Cryo-EM analysis has indicated that the structural integrity of non-loaded encapsulins is better than that of encapsulins loaded with their natural cargo (DyP) or fluorescent proteins.119 This structural flexibility was also shown for T. maritima encapsulin.121
Encapsulin-based nanoreactors (engineered to carry non-native cargos) were first made from Rhodococcus erythropolis N771 encapsulin.122 The native DypB peroxidase was replaced with firefly luciferase fused to the DypB C-terminal affinity tag. The luciferase was still able to carry out bioluminescence reactions in the cavity.
Encapsulins have more recently been used to produce nanoreactors. The fluorescent protein cargo mNeonGreen was protected from proteasome-based degradation in the yeast Saccharomyces cerevisiae.123 In addition, encapsulin-based nanoreactors co-packaged with the split Venus components Ven-N and Ven-C became fluorescent, with high loading yields. This was further explored by encapsulating Aro10p, an enzyme involved in tyrosine catabolism that might allow pro-drugs to be activated in the body when in this form. The 5 Å pores allowed small molecule diffusion, and the enzyme showed the anticipated decarboxylation activity. The cargo was protected from protease degradation.
Myxococcus xanthus encapsulin has been used to create orthogonal nanoreactors in mammalian HEK293 T cells.124 A tag of eight amino acids was used to co-package a photo-active fluorescent protein (mEos4b) or the two halves of a split luciferase or mCherry. The strategy using the split proteins verified that they were indeed co-packaged in the same cage. The cages were then loaded with tyrosinase, which catalyzes the transformation of tyrosine into the light-absorbing polymer melanin. The results could have applications in multispectral optoacoustic tomography. The encapsulated melanin was associated with almost no toxicity. The nanoreactors thus effectively mimicked the cell's natural membrane-bound melanosomes.
An engineered peroxidase, polymerized diaminobenzidine (APEX2), was also packaged in encapsulin-based nanoreactors.124 This enzyme can be used for cellular EM imaging and proximity labeling. The system was expanded to encapsulate cystathionine γ-lyase124 which, in the presence of L-cysteine, catalyzes the conversion of cadmium acetate in aqueous solution into cadmium sulfide nanocrystals. These nanocrystals were confined in the nano-sized interior of the encapsulins, and generated a photoluminescence signal under UV.
These examples of the use of encapsulin-based nanoreactors show the wide range of applications for genetically controlled compartmentalization.
The wild-type form of the above cage (AaLS-wt) has been modified via point mutations to introduce Glu (AaLS-neg; 36 pentamers and ∼30 nm) and then further ‘directionally evolved’ to include seven additional mutations (AaLS-13; 72 pentamers and ∼40 nm). These cages have interiors of different negative charges, and cryo-EM shows they have large, open pores (Fig. 4d–f).125 The AaLS-neg cage was subjected to further engineering to increase the packaging capacity for a toxic protease derived from human immunodeficiency virus (HIV), which notably reduced the toxicity of the enzyme in E. coli.126 This was achieved by adding a positively charged peptide tag to the enzyme. In another approach, a cationic supercharged (+36) fluorescent protein was genetically linked to the enzyme cargo and used to load a variety of enzymes —kemp eliminase, β-lactamase, cyclohexylamine oxidase, catalase-peroxidase, NADH oxidase, and aldehyde dehydrogenase— into the AaLS-13 cage.127 Approximately 45 total enzyme copies could be encapsulated per cage. In general, encapsulated enzymes retained their activity inside the cage, but for most, the Kcal/Km was lower compared to the free enzyme.
The specificity of a carried enzyme for its substrates may be altered by the cage interior.128 After encapsulating a modified, sequence-specific protease from the tobacco etch virus, tests using peptides with distinct charges showed that the negatively charged cage promoted uptake and hydrolysis of those that were positively charged, but excluded negatively charged competitors. Hilvert's group made these observations by encapsulating ascorbate peroxidase for polymerizing poly 3,3-diaminobenzidine within the cage.129 They also tried to mimic natural carboxysome compartments using lumazine synthase cages co-packaged with RuBisCO and carbonic anhydrase.130 These cages did not enhance pathway efficiency, however, probably because of the much more open structure of AaLS-13 cages compared to carboxysomes, which limits their retention of intermediate substrates. Work such as this nonetheless clearly indicates the potential of lumazine synthases cages as nanoreactors in vitro and in vivo.
3.3. Natural protein-based cages in eukaryotes
Most ferritin cages, including bacterio-ferritin cages, are composed of 24 subunits with octahedral symmetry (Fig. 5a), as determined at atomic resolution.136 With an external diameter of ∼12 nm and an internal diameter of their negatively charged cavity of ∼8 nm,137 their structure includes eight hydrophilic channels involved in iron transport across the protein shell, and six hydrophobic channels that probably transport protons.138 They are therefore important for transferring substrates and products across the protein shell. Mini-ferritin cages, which have 12 subunits and tetrahedral symmetry, are likely to be too small for effective encapsulation of enzymes.139
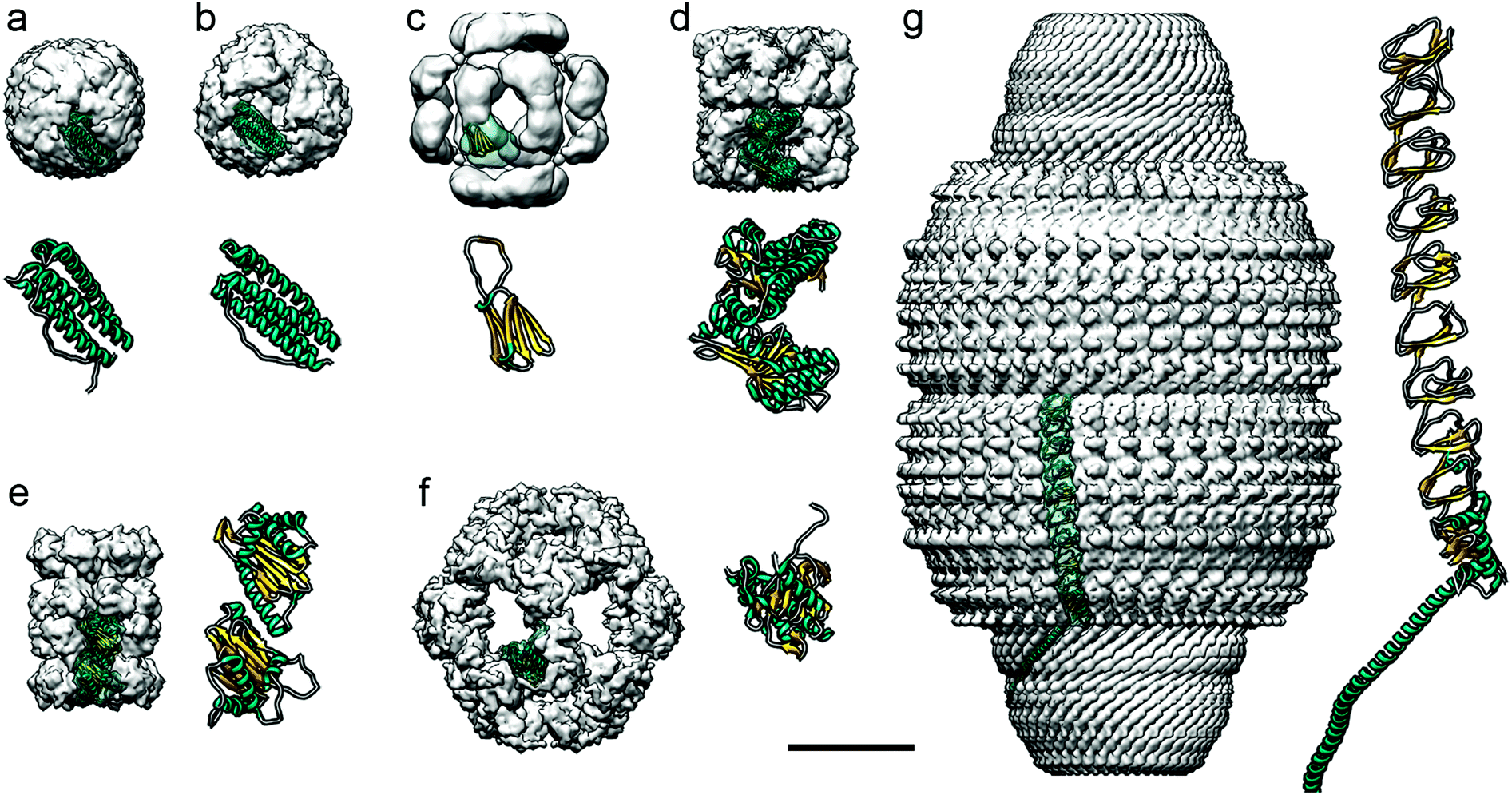 | ||
| Fig. 5 Diverse protein-based nanocompartments found in eukaryotes have been resolved at near-atomic resolution using cryo-EM. Cryo-EM maps are in the same scale (bar = 100 Å). For clarity, cryo-EM reconstructions, visualized as surface-shaded representations of the outer surface with the docked monomer atomic coordinates, are filtered out at ∼10 Å resolution. Magnified views of the monomers are shown at the same scale (the α-helical N-terminal region of the major vault protein is omitted in g). (a) Human apoferritin (Electron Microscopy Data Bank accession code EMD-0144; PDB ID code 5N27). (b) Archaeoglobus fulgidus ferritin, an X-ray crystallography-derived map (PDB 1SQ3). (c) Hsp26 from bakers’ yeast (EMD-1221; PDB 2H50). (d) Class I chaperonin GroEL from E. coli (EMD-8750; PDB 5 W0S). (e) Class II chaperonin from T. acidophilum (EMD-8741; PDB 5VY3). (f) E2 inner core of the human pyruvate dehydrogenase complex (EMD-7610; PDB 6CT0). (g) Major vault ribonucleoprotein from rat (EMD-7125; PDB 6BP7). | ||
Ferritin cages are structurally stable, which hinders their loading with large cargos such as proteins,131 and it has been difficult to turn them into enzyme-carrying nanoreactors. The archaeal ferritin cage from Archaeoglobus fulgidus is less stable, however, and can reversibly disassemble into dimers at neutral pH and in low ionic strength solutions, allowing certain enzymes to be incorporated.137 This type of cage has a tetrahedral rather than the canonical octahedral symmetry, with four large triangular openings 4.5 nm across.136 These differences in structure can be visualized using cryo-EM (Fig. 5b). Three enzymes –human carbonic anhydrase II, artificial (retro)aldolase RA95.5-8F, and Kemp eliminase HG3.17– were loaded into the cages by fusing them to green fluorescent protein with 36 positive charges (GFP + 36).137 The cages protected the protein cargo when incubated with blood plasma protease factor Xa. Archaeoglobus fulgidus ferritin cages have also been engineered to encapsulate horseradish peroxidase and Renilla luciferase,140 which increased the functional folding of the enzyme some 100-fold. These cages increased the thermal stability of the encapsulated enzymes.
A subgroup of cylindrical chaperones, known as chaperonins, encapsulate proteins to ensure their correct folding, often consuming ATP in the process. They can be divided into two classes. Class I chaperonins consist of two stacked rings, with a smaller protein on top of the rings; these include Hsp60 and GroEL (Fig. 5d), which are found in the mitochondria of eukaryotes and in prokaryotes, respectively. They have been used to encapsulate iron-based hemins to create an artificial enzyme inside the cage. Class II chaperonins, such as the thermosome Thermoplasma acidophilum found in archaea (Fig. 5e), are hetero-oligomers that usually contain built-in protrusions that act as a “lid” structure. These chaperonins have two cavities with a diameter of approximately 16 and 18 nm in the fully closed and open conformations, respectively. They are large enough to accommodate proteins of up to 50 kDa. This type of cage has 5.4 nm-diameter pores in the open conformation, useful when attempting to use these cages as polymerization nanoreactors, since the cage interior defines the degree of polymerization and polydispersity of the polymer poly(ethylene glycol) methyl ether acrylate.149 Knowing the 3D structure of the protein cage allowed introduction of a cysteine into the interior of the thermosome, followed by attachment of a multiamine ligand to immobilize Cu2+. This complex functions as a catalyst for atom transfer radical polymerization (ATRP). A nanoreactor was also made that incorporated horseradish peroxidase by covalently attaching the enzyme to the cysteine.150 Horseradish peroxidase can be used to catalyze ATRP, and to form the polymer. Like the Cu2+-immobilizing system, the resulting polymers had a small molecular weight distribution compared to the products of bulk reactions, highlighting the benefit of protein cages for catalysis.
Pyruvate dehydrogenase (PDH) complexes are generally composed of dihydrolipoyl acetyltransferase (E2), a pyruvate decarboxylase (E1), a dihydrolipoyl dehydrogenase (E3), and sometimes an E3-binding protein. In the native PDH complex of Bacillus stearothermophilus, 60 copies of E2 self-associate, forming an icosahedral assemblage. Around the exterior of this complex lie 42–48 copies of a tetrameric α2β2 E1, and 6–12 copies of tightly bound homodimeric E3, forming a second protein shell.152 Cryo-EM has been used to determine the precise conformation of these protein shells.
The arrangement of two concentric cages separated by an annular gap provides for effective active site shuttling. Although this system has not yet been modified to create a nanoreactor, it has potential application to the treatment of metabolic disorders.153
These vaults have been engineered to encapsulate non-native proteins in their interior.154,157 For this, a targeting domain of the telomerase-associated protein able to bind the major vault protein was identified. This domain was fused to luciferase and, after coexpression in insect cells, was included in the vault interior, as verified by cryo-EM.158 Charged molecules such as ATP are slowly transferred into the cavity; such selectivity can be a useful trait for a nanoreactor.
3.4. Other protein-based cages
Other protein-based cages, such as proteasomes,159 stressosomes,160 inflammasomes161 peroxiredoxins,162 clathrin cages,163 and other viral capsids of various shapes and sizes might also be used in nanoreactor production. Recently developed de novo protein cages, that is, artificially designed and constructed cages,164,165 might also have their uses. These de novo cages include tetrahedrons and other origami structures made via coiled-coil assembly,166,167 tetrahedral cages built by controlled genetic fusion of natural proteins,168 and protein cages with envelopes.169 Other protein-based cages, viruses, and nanocagess could yet be discovered,139 providing further candidates for nanoreactor creation. The field is expected to expand notably in the coming years.3.5. Advantages of protein nanoreactors over other nanoreactors
A shell around an enzyme can protect it from harsh environments and degradation.170 The increased size of the enzyme-cage complex compared with the enzyme alone can also facilitate reuse of enzymes in industry.171 Examples of non-protein-based nanosized cages are polymers,172,173 polymersomes,174,175 liposomes,176,177 giant amphiphiles,178 nanogels,179 layer-by-layer structures,180 nanodroplets,181 nanosized metal organic frameworks (MOF),182 silica nanoparticles183 and other inorganic cages.184,185 Most of these systems are especially applicable when stability is needed in extreme environments, as in the case of water-free systems, or when high (thermal) stability is required. These encapsulants nonetheless often lack control over their size and shape, suffer from biocompatibility problems, and require several steps to synthesize. These questions do no arise in protein cages given their defined structures and, often, ability to self-assemble. Other extensively studied nanoreactor systems are based on peptides or DNA (origami).186–190 DNA origamis can consist of various shapes such as tube-like honeycomb structures made from DNA that are combined into a large structure. A cavity within these structures can contain enzymes, for example, which are coupled to the DNA origami using a short complementary DNA strand. Protein cages are more economical than DNA origami, however, and are not affected by nucleases.1914. Conclusion and future perspectives
This review shows how 3D cryo-EM can be used to analyze the structure of protein-based cages, and provides an overview of the nanoreactors that have been made from them. Enzyme encapsulation in a protein cage provides clear benefits over the use of the free enzyme, including reduced toxicity, increased substrate specificity, substrate channeling, easier enzyme recovery after a catalytic reaction, and increased stability. Protein-based cages are available in various sizes and shapes and can be expressed in various heterogeneous systems to make nanoreactors. They can also be modified genetically and chemically, and engineered to include different, non-native enzymatic cargos.The study of nanoreactors using cryo-EM and other techniques has not only increased our understanding of natural protein-based cages, but has led to design of nanoreactors for industrial catalysis. The increased stability and substrate specificity allows use of cargo enzymes in more extreme reaction conditions, results in lower energy consumption, higher product yields, and greater product purity. Nanoreactors also show promise in medicine, and might be used to target enzyme delivery. This could be beneficial in treating metabolic disorders in which certain enzymes are not (or are insufficiently) produced. They might also be used to generate or degrade drugs or metabolites within cells, and for pro-drug activation. Nanoreactors might also be used in the production of functional nanomaterials, sensors, in metabolic engineering, or in enhanced bio-CO2 fixation. For all that, we have not yet reached the stage at which nanoreactors can be used routinely. Their properties, bio-uptake, biopositioning, and immunogenicity need to be better understood, applicable nanoreactors that encapsulate defined enzymes need to be built, and the associated costs must be reduced. 3D cryo-EM will undoubtedly play a role in this.
As an example, deciphering the structure of a natural nanoreactor such as DyP-encapsulin from B. linens is crucial for advances in comprehension of its function and properties (Fig. 6). DyP is assembled as a trimer of dimers (a 240 kDa hexamer), and is connected at one three-fold axis by three C-terminal extensions, which target the enzyme to specific pockets on the encapsulin interior surface.13 3D cryo-EM analysis has shown three channels around the three-fold axis that can act as direct gates for substrates and/or products to be interchanged between the cytoplasm and the confined enzyme.119 DyP-encapsulin is nevertheless a symmetry-mismatched complex.
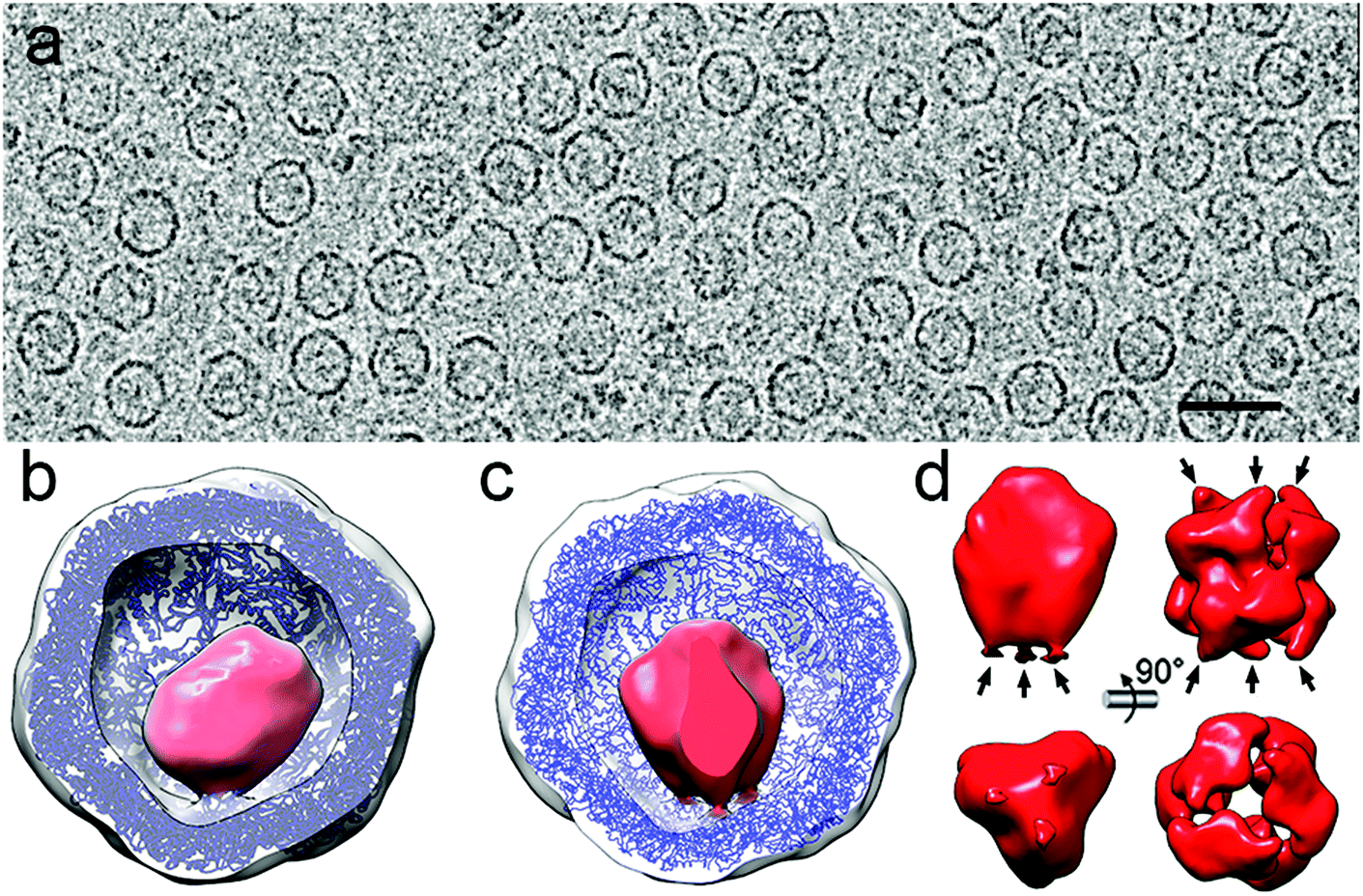 | ||
| Fig. 6 Two-dimensional movie average after translational alignment of a vitrified sample of DyP-loaded encapsulin (DyP-E). (b) 3DR with no symmetry of DyP-E with the front half of the protein shell removed. Docked T. maritima encapsulin is blue and DyP density is red. (c) DyP-E map with C3 symmetry from inside. (d) DyP cryo-EM density extracted from the map shown in c; arrows indicate connections to the inner encapsulin surface (left). DyP density calculated from negative staining electron microscopy13 (right). | ||
Whereas applying icosahedral symmetry after merging a few tens of thousands of images will eventually lead to a near-atomic structure of the icosahedral encapsulin shell, a structure solution for the cargo requires more elaborate methods. First, a larger dataset of images from high-quality purified DyP-encapsulin samples must be aligned to obtain an asymmetric map. The enzyme cargo might acquire distinct conformations inside the cages, and better classification methods will help to identify such states. Finally, time-resolved cryo-EM would allow dissection of the temporal progress of the enzyme reaction and capture different functional states of the active nanoreactor, as has been d for functioning ribosomes. The higher SNR and contrast provided by DED, together with improved image processing approaches such as projection subtraction (a difference image calculated by subtracting the original projection from the “icosahedral” projection), local 3D classification, and localized reconstruction could allow reconstruction of the mismatched cargo at high resolution.
3D cryo-EM has emerged as a powerful technique for rapid visualization of protein-based cages/nanoreactors at high resolution, which will contribute to development of improved, more efficient nanoreactors.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by grants from the Spanish Ministry of Economy and Competitivity (BFU2017-88736-R to JRC), the Comunidad Autónoma de Madrid (P2018/NMT-4389 to JRC), and the Consolidator Grant ‘Protcage’ (616907 to JLMC) from the European Research Council (ERC).Notes and references
- C. P. Satori, M. M. Henderson, E. A. Krautkramer, V. Kostal, M. M. Distefano and E. A. Arriaga, Chem. Rev., 2013, 113, 2733–2811 CrossRef CAS PubMed
.
- C. A. Kerfeld, S. Heinhorst and G. C. Cannon, Annu. Rev. Microbiol., 2010, 64, 391–408 CrossRef CAS PubMed
- C. A. Kerfeld, C. Aussignargues, J. Zarzycki, F. Cai and M. Sutter, Nat. Rev. Microbiol., 2018, 16, 277 CrossRef CAS PubMed
- L. Schoonen and J. C. van Hest, Nanoscale, 2014, 6, 7124–7141 RSC
- E. Y. Kim and D. Tullman-Ercek, Curr. Opin. Biotechnol., 2013, 24, 627–632 CrossRef CAS PubMed
- W. Kuhlbrandt, Science, 2014, 343, 1443–1444 CrossRef PubMed
- E. H. Egelman, Biophys. J., 2016, 110, 1008–1012 CrossRef CAS PubMed
- C. Bertozzi, ACS Cent. Sci., 2017, 3, 1056 CrossRef CAS PubMed
- L. Schoonen, J. Pille, A. Borrmann, R. J. M. Nolte and J. C. M. Van Hest, Bioconjugate Chem., 2015, 26, 2429–2434 CrossRef CAS PubMed
- W. F. Rurup, F. Verbij, M. S. T. Koay, C. Blum, V. Subramaniam and J. J. L. M. Cornelissen, Biomacromolecules, 2014, 15, 558–563 CrossRef CAS PubMed
- L. Schoonen, S. Eising, M. B. van Eldijk, J. Bresseleers, M. van der Pijl, R. J. M. Nolte, K. M. Bonger and J. C. M. van Hest, Bioconjugate Chem., 2018, 29, 1186–1193 CrossRef CAS PubMed
- M. Brasch, R. M. Putri, M. V. de Ruiter, D. Luque, M. S. T. Koay, J. R. Castón and J. J. L. M. Cornelissen, J. Am. Chem. Soc., 2017, 139, 1512–1519 CrossRef CAS PubMed
- M. Sutter, D. Boehringer, S. Gutmann, S. Gunther, D. Prangishvili, M. J. Loessner, K. O. Stetter, E. Weber-Ban and N. Ban, Nat. Struct. Mol. Biol., 2008, 15, 939–947 CrossRef CAS PubMed
- E. Nogales and S. H. Scheres, Mol. Cell, 2015, 58, 677–689 CrossRef CAS PubMed
- D. Elmlund, S. N. Le and H. Elmlund, Curr. Opin. Struct. Biol., 2017, 46, 1–6 CrossRef CAS PubMed
- M. Khoshouei, M. Radjainia, W. Baumeister and R. Danev, Nat. Commun., 2017, 8, 16099 CrossRef CAS PubMed
- A. Merk, A. Bartesaghi, S. Banerjee, V. Falconieri, P. Rao, M. I. Davis, R. Pragani, M. B. Boxer, L. A. Earl, J. L. S. Milne and S. Subramaniam, Cell, 2016, 165, 1698–1707 CrossRef CAS PubMed
- W. Jiang and L. Tang, Curr. Opin. Struct. Biol., 2017, 46, 122–129 CrossRef CAS PubMed
- J. T. Kaelber, C. F. Hryc and W. Chiu, Annu. Rev. Virol., 2017, 4, 287–308 CrossRef CAS PubMed
- V. Lucic, A. Rigort and W. Baumeister, J. Cell Biol., 2013, 202, 407–419 CrossRef CAS PubMed
- W. Wan and J. A. Briggs, Methods Enzymol., 2016, 579, 329–367 CAS
-
J. R. Harris, Negative staining and cryoelectron microscopy: the thin film techniques, BIOS Scientific Publishers Ltd., Oxford, 1997 Search PubMed
- Y. Cheng, E. Wolf, M. Larvie, O. Zak, P. Aisen, N. Grigorieff, S. C. Harrison and T. Walz, J. Mol. Biol., 2006, 355, 1048–1065 CrossRef CAS PubMed
- J. Dubochet, M. Adrian, J. J. Chang, J. C. Homo, J. Lepault, A. W. McDowall and P. Schultz, Q. Rev. Biophys., 1988, 21, 129–228 CrossRef CAS PubMed
- M. J. Dobro, L. A. Melanson, G. J. Jensen and A. W. McDowall, Methods Enzymol., 2010, 481, 63–82 CAS
- R. A. Grassucci, D. J. Taylor and J. Frank, Nat. Protoc., 2007, 2, 3239–3246 CrossRef CAS PubMed
- L. A. Passmore and C. J. Russo, Methods Enzymol., 2016, 579, 51–86 CAS
- K. C. Duong-Ly and S. B. Gabelli, Methods Enzymol., 2014, 541, 105–114 CAS
- G. Skiniotis and D. R. Southworth, Microscopy, 2016, 65, 9–22 CrossRef CAS PubMed
- B. Kastner, N. Fischer, M. M. Golas, B. Sander, P. Dube, D. Boehringer, K. Hartmuth, J. Deckert, F. Hauer, E. Wolf, H. Uchtenhagen, H. Urlaub, F. Herzog, J. M. Peters, D. Poerschke, R. Luhrmann and H. Stark, Nat. Methods, 2008, 5, 53–55 CrossRef CAS PubMed
- A. Chari, D. Haselbach, J. M. Kirves, J. Ohmer, E. Paknia, N. Fischer, O. Ganichkin, V. Moller, J. J. Frye, G. Petzold, M. Jarvis, M. Tietzel, C. Grimm, J. M. Peters, B. A. Schulman, K. Tittmann, J. Markl, U. Fischer and H. Stark, Nat. Methods, 2015, 12, 859–865 CrossRef CAS PubMed
- G. McMullan, A. R. Faruqi, D. Clare and R. Henderson, Ultramicroscopy, 2014, 147, 156–163 CrossRef CAS PubMed
- G. McMullan, A. R. Faruqi and R. Henderson, Methods Enzymol., 2016, 579, 1–17 CAS
- A. F. Brilot, J. Z. Chen, A. Cheng, J. Pan, S. C. Harrison, C. S. Potter, B. Carragher, R. Henderson and N. Grigorieff, J. Struct. Biol., 2012, 177, 630–637 CrossRef CAS PubMed
- S. H. Scheres, eLife, 2014, 3, e03665 CrossRef PubMed
- X. Li, P. Mooney, S. Zheng, C. R. Booth, M. B. Braunfeld, S. Gubbens, D. A. Agard and Y. Cheng, Nat. Methods, 2013, 10, 584–590 CrossRef CAS PubMed
- T. Grant and N. Grigorieff, eLife, 2015, 4, e06980 CrossRef PubMed
- Z. A. Ripstein and J. L. Rubinstein, Methods Enzymol., 2016, 579, 103–124 CAS
- R. Danev, B. Buijsse, M. Khoshouei, J. M. Plitzko and W. Baumeister, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15635–15640 CrossRef CAS PubMed
- R. Danev and W. Baumeister, eLife, 2016, 5, e13046 CrossRef PubMed
- D. N. Mastronarde, J. Struct. Biol., 2005, 152, 36–51 CrossRef PubMed
- C. Suloway, J. Shi, A. Cheng, J. Pulokas, B. Carragher, C. S. Potter, S. Q. Zheng, D. A. Agard and G. J. Jensen, J. Struct. Biol., 2009, 167, 11–18 CrossRef CAS PubMed
- Y. Cheng, N. Grigorieff, P. A. Penczek and T. Walz, Cell, 2015, 161, 438–449 CrossRef CAS PubMed
- S. H. Scheres, Methods Enzymol., 2016, 579, 125–157 CAS
- S. J. Ludtke, Methods Enzymol., 2016, 579, 159–189 CAS
- J. M. de la Rosa-Trevin, J. Oton, R. Marabini, A. Zaldivar, J. Vargas, J. M. Carazo and C. O. Sorzano, J. Struct. Biol., 2013, 184, 321–328 CrossRef CAS PubMed
- N. Grigorieff, Methods Enzymol., 2016, 579, 191–226 CAS
- J. M. de la Rosa-Trevin, A. Quintana, L. Del Cano, A. Zaldivar, I. Foche, J. Gutierrez, J. Gomez-Blanco, J. Burguet-Castell, J. Cuenca-Alba, V. Abrishami, J. Vargas, J. Oton, G. Sharov, J. L. Vilas, J. Navas, P. Conesa, M. Kazemi, R. Marabini, C. O. Sorzano and J. M. Carazo, J. Struct. Biol., 2016, 195, 93–99 CrossRef CAS PubMed
- A. Punjani, J. L. Rubinstein, D. J. Fleet and M. A. Brubaker, Nat. Methods, 2017, 14, 290–296 CrossRef CAS PubMed
- T. Grant, A. Rohou and N. Grigorieff, eLife, 2018, 7, e35383 CrossRef PubMed
- T. Moriya, M. Saur, M. Stabrin, F. Merino, H. Voicu, Z. Huang, P. A. Penczek, S. Raunser and C. Gatsogiannis, J. Visualized Exp., 2017, e55448, DOI:10.3791/55448
- J. Zivanov, T. Nakane, B. O. Forsberg, D. Kimanius, W. J. Hagen, E. Lindahl and S. H. Scheres, eLife, 2018, 7, e42166 CrossRef PubMed
- P. A. Penczek, Methods Enzymol., 2010, 482, 1–33 Search PubMed
- S. H. Scheres, J. Struct. Biol., 2012, 180, 519–530 CrossRef CAS
- S. H. Scheres, H. Gao, M. Valle, G. T. Herman, P. P. Eggermont, J. Frank and J. M. Carazo, Nat. Methods, 2007, 4, 27–29 CrossRef CAS PubMed
- D. Lyumkis, A. F. Brilot, D. L. Theobald and N. Grigorieff, J. Struct. Biol., 2013, 183, 377–388 CrossRef CAS PubMed
- B. Chen and J. Frank, Microscopy, 2016, 65, 69–79 CrossRef PubMed
- R. Henderson, A. Sali, M. L. Baker, B. Carragher, B. Devkota, K. H. Downing, E. H. Egelman, Z. Feng, J. Frank, N. Grigorieff, W. Jiang, S. J. Ludtke, O. Medalia, P. A. Penczek, P. B. Rosenthal, M. G. Rossmann, M. F. Schmid, G. F. Schroder, A. C. Steven, D. L. Stokes, J. D. Westbrook, W. Wriggers, H. Yang, J. Young, H. M. Berman, W. Chiu, G. J. Kleywegt and C. L. Lawson, Structure, 2012, 20, 205–214 CrossRef CAS PubMed
- S. Subramaniam, L. A. Earl, V. Falconieri, J. L. Milne and E. H. Egelman, Curr. Opin. Struct. Biol., 2016, 41, 194–202 CrossRef CAS PubMed
- A. Kucukelbir, F. J. Sigworth and H. D. Tagare, Nat. Methods, 2014, 11, 63–65 CrossRef CAS PubMed
- J. L. Vilas, J. Gómez-Blanco, P. Conesa, R. Melero, J. M. de la Rosa-Trevin, J. Oton, J. Cuenca, R. Marabini, J. M. Carazo, J. Vargas and C. O. S. Sorzano, Structure, 2018, 26, 337–344 CrossRef CAS PubMed
- J. Seras-Franzoso, A. Sánchez-Chardi, E. Garcia-Fruitós, E. Vázquez and A. Villaverde, Soft Matter, 2016, 12, 3451–3460 RSC
- R. A. Sheldon, Adv. Synth. Catal., 2007, 349, 1289–1307 CrossRef CAS
- K. J. Koudelka, A. S. Pitek, M. Manchester and N. F. Steinmetz, Annu. Rev. Virol., 2015, 2, 379–401 CrossRef CAS PubMed
- J. B. Bancroft, Adv. Virus Res., 1970, 16, 99–134 CrossRef CAS
- E. Cornejo, N. Abreu and A. Komeili, Curr. Opin. Cell Biol., 2014, 26, 132–138 CrossRef CAS PubMed
- N. H. Dashti, R. S. Abidin and F. Sainsbury, ACS Nano, 2018, 12, 4615–4623 CrossRef CAS PubMed
- A. K. Kar, N. Iwatani and P. Roy, J. Virol., 2005, 79, 11487–11495 CrossRef CAS PubMed
- K. W. Lee and W. S. Tan, J. Virol. Methods, 2008, 151, 172–180 CrossRef CAS PubMed
- J. W. Wilkerson, S. O. Yang, P. J. Funk, S. K. Stanley and B. C. Bundy, New Biotechnol., 2018, 44, 59–63 CrossRef CAS PubMed
- R. F. Garmann, M. Comas-Garcia, C. M. Knobler and W. M. Gelbart, Acc. Chem. Res., 2016, 49, 48–55 CrossRef CAS
- H. Liu, C. Qu, J. E. Johnson and D. A. Case, J. Struct. Biol., 2003, 142, 356–363 CrossRef
- M. Comellas-Aragonès, H. Engelkamp, V. I. Claessen, N. A. J. M. Sommerdijk, A. E. Rowan, P. C. M. Christianen, J. C. Maan, B. J. M. Verduin, J. J. L. M. Cornelissen and R. J. M. Nolte, Nat. Nanotechnol., 2007, 2, 635–639 CrossRef
- L. Sánchez-Sánchez, R. D. Cadena-Nava, L. A. Palomares, J. Ruiz-Garcia, M. S. T. Koay, J. J. L. M. Cornelissen and R. Vazquez-Duhalt, Enzyme Microb. Technol., 2014, 60, 24–31 CrossRef
- I. J. Minten, V. I. Claessen, K. Blank, A. E. Rowan, R. J. M. Nolte and J. J. L. M. Cornelissen, Chem. Sci., 2011, 2, 358–362 RSC
- L. Schoonen, S. Maassen, R. J. M. Nolte and J. C. M. van Hest, Biomacromolecules, 2017, 18, 3492–3497 CrossRef CAS PubMed
- A. Liu, M. V. de Ruiter, W. Zhu, S. J. Maassen, L. Yang and J. J. L. M. Cornelissen, Adv. Funct. Mater., 2018, 28, 1801574 CrossRef
- M. V. de Ruiter, R. M. Putri and J. J. L. M. Cornelissen, Methods Mol. Biol., 2018, 1776, 237–247 CrossRef CAS PubMed
- S. Kang, M. Uchida, A. O'Neil, R. Li, P. E. Prevelige and T. Douglas, Biomacromolecules, 2010, 11, 2804–2809 CrossRef CAS PubMed
- A. O'Neil, C. Reichhardt, B. Johnson, P. E. Prevelige and T. Douglas, Angew. Chem., Int. Ed., 2011, 50, 7425–7428 CrossRef PubMed
- D. Patterson, E. Edwards and T. Douglas, Isr. J. Chem., 2015, 55, 96–101 CrossRef CAS
- S. Qazi, H. M. Miettinen, R. A. Wilkinson, K. McCoy, T. Douglas and B. Wiedenheft, Mol. Pharm., 2016, 13, 1191–1196 CrossRef CAS PubMed
- P. C. Jordan, D. P. Patterson, K. N. Saboda, E. J. Edwards, H. M. Miettinen, G. Basu, M. C. Thielges and T. Douglas, Nat. Chem., 2016, 8, 179–185 CrossRef CAS PubMed
- D. P. Patterson, K. McCoy, C. Fijen and T. Douglas, J. Mater. Chem. B, 2014, 2, 5948–5951 RSC
- K. Chauhan, J. M. Hernandez-Meza, A. G. Rodríguez-Hernández, K. Juarez-Moreno, P. Sengar and R. Vazquez-Duhalt, J. Nanobiotechnol., 2018, 16, 17 CrossRef
- L. Sánchez-Sánchez, A. Tapia-Moreno, K. Juarez-Moreno, D. P. Patterson, R. D. Cadena-Nava, T. Douglas and R. Vazquez-Duhalt, J. Nanobiotechnol., 2015, 13, 66 CrossRef PubMed
- A. Tapia-Moreno, K. Juarez-Moreno, O. Gonzalez-Davis, R. D. Cadena-Nava and R. Vazquez-Duhalt, Biotechnol. J., 2017, 12, 1600706 CrossRef
- K. McCoy, M. Uchida, B. Lee and T. Douglas, ACS Nano, 2018, 12, 3541–3550 CrossRef CAS PubMed
- M. Uchida, K. McCoy, M. Fukuto, L. Yang, H. Yoshimura, H. M. Miettinen, B. LaFrance, D. P. Patterson, B. Schwarz, J. A. Karty, P. E. Prevelige, B. Lee and T. Douglas, ACS Nano, 2018, 12, 942–953 CrossRef CAS PubMed
- E. Edwards, R. Roychoudhury, B. Schwarz, P. Jordan, J. Lisher, M. Uchida and T. Douglas, J. Mater. Chem. B, 2016, 4, 5375–5384 RSC
- K. McCoy, E. Selivanovitch, D. Luque, B. Lee, E. Edwards, J. R. Castón and T. Douglas, Biomacromolecules, 2018, 19, 3738–3746 CrossRef CAS PubMed
- D. P. Patterson, B. Schwarz, R. S. Waters, T. Gedeon and T. Douglas, ACS Chem. Biol., 2014, 9, 359–365 CrossRef CAS PubMed
- J. E. Glasgow, M. A. Asensio, C. M. Jakobson, M. B. Francis and D. Tullman-Ercek, ACS Synth. Biol., 2015, 4, 1011–1019 CrossRef CAS PubMed
- J. E. Glasgow, S. L. Capehart, M. B. Francis and D. Tullman-Ercek, ACS Nano, 2012, 6, 8658–8664 CrossRef CAS PubMed
- T. W. Giessen and P. A. Silver, ChemBioChem, 2016, 17, 1931–1935 CrossRef CAS PubMed
- J. D. Fiedler, S. D. Brown, J. L. Lau and M. G. Finn, Angew. Chem., Int. Ed., 2010, 49, 9648–9651 CrossRef CAS PubMed
- T. Stehle, S. J. Gamblin, Y. Yan and S. C. Harrison, Structure, 1996, 4, 165–182 CrossRef CAS PubMed
- T. Inoue, M. A. Kawano, R. U. Takahashi, H. Tsukamoto, T. Enomoto, T. Imai, K. Kataoka and H. Handa, J. Biotechnol., 2008, 134, 181–192 CrossRef CAS PubMed
- G. Drews and W. Niklowitz, Arch. Mikrobiol., 1956, 24, 147–162 CAS
- C. A. Kerfeld and O. Erbilgin, Trends Microbiol., 2015, 23, 22–34 CrossRef CAS
- M. Sutter, B. Greber, C. Aussignargues and C. A. Kerfeld, Science, 2017, 356, 1293–1297 CrossRef CAS
- T. O. Yeates, M. C. Thompson and T. A. Bobik, Curr. Opin. Struct. Biol., 2011, 21, 223–231 CrossRef CAS
- J. S. Plegaria and C. A. Kerfeld, Curr. Opin. Biotechnol., 2018, 51, 1–7 CrossRef CAS
- S. Tanaka, M. R. Sawaya and T. O. Yeates, Science, 2010, 327, 81–84 CrossRef CAS PubMed
- M. F. Slininger Lee, C. M. Jakobson and D. Tullman-Ercek, ACS Synth. Biol., 2017, 6, 1880–1891 CrossRef PubMed
- B. D. Rae, B. M. Long, M. R. Badger and G. D. Price, Microbiol. Mol. Biol. Rev., 2013, 77, 357–379 CrossRef CAS PubMed
- J. N. Kinney, A. Salmeen, F. Cai and C. A. Kerfeld, J. Biol. Chem., 2012, 287, 17729–17736 CrossRef CAS PubMed
- J. B. Parsons, S. Frank, D. Bhella, M. Liang, M. B. Prentice, D. P. Mulvihill and M. J. Warren, Mol. Cell, 2010, 38, 305–315 CrossRef CAS PubMed
- B. B. Menon, Z. Dou, S. Heinhorst, J. M. Shively and G. C. Cannon, PLoS One, 2008, 3, e3570 CrossRef PubMed
- S. Choudhary, M. B. Quin, M. A. Sanders, E. T. Johnson and C. Schmidt-Dannert, PLoS One, 2012, 7, e33342 CrossRef CAS
- A. D. Lawrence, S. Frank, S. Newnham, M. J. Lee, I. R. Brown, W. F. Xue, M. L. Rowe, D. P. Mulvihill, M. B. Prentice, M. J. Howard and M. J. Warren, ACS Synth. Biol., 2014, 3, 454–465 CrossRef CAS
- M. Liang, S. Frank, H. Lünsdorf, J. Warren Martin and B. Prentice Michael, Biotechnol. J., 2017, 12, 1600415 CrossRef
- M. J. Lee, I. R. Brown, R. Juodeikis, S. Frank and M. J. Warren, Metab. Eng., 2016, 36, 48–56 CrossRef CAS PubMed
- J. Zarzycki, S. D. Axen, J. N. Kinney and C. A. Kerfeld, J. Exp. Bot., 2013, 64, 787–798 CrossRef CAS
- T. W. Giessen and P. A. Silver, Nat. Microbiol., 2017, 2, 17029 CrossRef PubMed
- C. A. McHugh, J. Fontana, D. Nemecek, N. Cheng, A. A. Aksyuk, J. B. Heymann, D. C. Winkler, A. S. Lam, J. S. Wall, A. C. Steven and E. Hoiczyk, EMBO J., 2014, 33, 1896–1911 CrossRef CAS PubMed
- D. He, S. Hughes, S. Vanden-Hehir, A. Georgiev, K. Altenbach, E. Tarrant, C. L. Mackay, K. J. Waldron, D. J. Clarke and J. Marles-Wright, eLife, 2016, 5, e18972 CrossRef PubMed
- W. F. Rurup, J. Snijder, M. S. T. Koay, A. J. R. Heck and J. J. L. M. Cornelissen, J. Am. Chem. Soc., 2014, 136, 3828–3832 CrossRef CAS PubMed
- R. M. Putri, C. Allende-Ballestero, D. Luque, R. Klem, K. A. Rousou, A. Liu, C. H. Traulsen, W. F. Rurup, M. S. T. Koay, J. R. Castón and J. Cornelissen, ACS Nano, 2017, 11, 12796–12804 CrossRef CAS PubMed
- T. W. Giessen, Curr. Opin. Chem. Biol., 2016, 34, 1–10 CrossRef CAS PubMed
- C. Cassidy-Amstutz, L. Oltrogge, C. C. Going, A. Lee, P. Teng, D. Quintanilla, A. East-Seletsky, E. R. Williams and D. F. Savage, Biochem., 2016, 55, 3461–3468 CrossRef CAS PubMed
- T. Akio, F. Yosuke, T. Taku, F. Motoko, N. Yuki, M. Yoshihiko, N. Keiichi, Y. Masafumi and O. Masafumi, Biotechnol. Bioeng., 2015, 112, 13–20 CrossRef PubMed
- Y. H. Lau, T. W. Giessen, W. J. Altenburg and P. A. Silver, Nat. Commun., 2018, 9, 1311 CrossRef PubMed
- F. Sigmund, C. Massner, P. Erdmann, A. Stelzl, H. Rolbieski, M. Desai, S. Bricault, T. P. Wörner, J. Snijder, A. Geerlof, H. Fuchs, M. Hrabě de Angelis, A. J. R. Heck, A. Jasanoff, V. Ntziachristos, J. Plitzko and G. G. Westmeyer, Nat. Commun., 2018, 9, 1990 CrossRef PubMed
- Y. Azuma, T. G. W. Edwardson and D. Hilvert, Chem. Soc. Rev., 2018, 47, 3543–3557 RSC
- B. Wörsdörfer, K. J. Woycechowsky and D. Hilvert, Science, 2011, 331, 589–592 CrossRef PubMed
- Y. Azuma, R. Zschoche, M. Tinzl and D. Hilvert, Angew. Chem., Int. Ed., 2016, 55, 1531–1534 CrossRef CAS PubMed
- Y. Azuma, D. L. V. Bader and D. Hilvert, J. Am. Chem. Soc., 2018, 140, 860–863 CrossRef CAS PubMed
- R. Frey, T. Hayashi and D. Hilvert, Chem. Commun., 2016, 52, 10423–10426 RSC
- R. Frey, S. Mantri, M. Rocca and D. Hilvert, J. Am. Chem. Soc., 2016, 138, 10072–10075 CrossRef CAS PubMed
- P. Arosio, R. Ingrassia and P. Cavadini, Biochim. Biophys. Acta, 2009, 1790, 589–599 CrossRef CAS PubMed
- P. M. Harrison, A. Treffry and T. H. Lilley, J. Inorg. Biochem., 1986, 27, 287–293 CrossRef CAS PubMed
- K. J. Thompson, M. G. Fried, Z. Ye, P. Boyer and J. R. Connor, J. Cell Sci., 2002, 115, 2165–2177 CAS
- M. Truffi, L. Fiandra, L. Sorrentino, M. Monieri, F. Corsi and S. Mazzucchelli, Pharmacol. Res., 2016, 107, 57–65 CrossRef CAS PubMed
- D. He and J. Marles-Wright, New Biotechnol., 2015, 32, 651–657 CrossRef CAS PubMed
- E. Johnson, D. Cascio, M. R. Sawaya, M. Gingery and I. Schröder, Structure, 2005, 13, 637–648 CrossRef CAS PubMed
- S. Tetter and D. Hilvert, Angew. Chem., Int. Ed., 2017, 56, 14933–14936 CrossRef CAS PubMed
- N. Surguladze, S. Patton, A. Cozzi, M. G. Fried and J. R. Connor, Biochem. J., 2005, 388, 731–740 CrossRef CAS PubMed
- W. M. Aumiller, M. Uchida and T. Douglas, Chem. Soc. Rev., 2018, 47, 3433–3469 RSC
- S. Deshpande, N. D. Masurkar, V. M. Girish, M. Desai, G. Chakraborty, J. M. Chan and C. L. Drum, Nat. Commun., 2017, 8, 1442 CrossRef PubMed
- E. A. Craig, B.
D. Gambill and R. J. Nelson, Microbiol. Rev., 1993, 57, 402–414 CAS
- M. Shi, Y. N. Wang, N. Zhu and X. X. Chen, PLoS One, 2013, 8, e59721 CrossRef CAS PubMed
- C. Jie, K. Ai, A. Jasper, A. Andrei and L. R. Benildo, Insect Sci., 2016, 23, 548–554 CrossRef PubMed
- D. A. Parsell, A. S. Kowal, M. A. Singer and S. Lindquist, Nature, 1994, 372, 475 CrossRef CAS PubMed
- X. B. Qiu, Y. M. Shao, S. Miao and L. Wang, Cell. Mol. Life Sci., 2006, 63, 2560–2570 CrossRef CAS PubMed
- H. E. White, E. V. Orlova, S. Chen, L. Wang, A. Ignatiou, B. Gowen, T. Stromer, T. M. Franzmann, M. Haslbeck, J. Buchner and H. R. Saibil, Structure, 2006, 14, 1197–1204 CrossRef CAS PubMed
- K. K. Kim, R. Kim and S. H. Kim, Nature, 1998, 394, 595–599 CrossRef CAS PubMed
- S. H. Choi, I. C. Kwon, K. Y. Hwang, I. S. Kim and H. J. Ahn, Biomacromolecules, 2011, 12, 3099–3106 CrossRef CAS PubMed
- K. Renggli, M. G. Nussbaumer, R. Urbani, T. Pfohl and N. Bruns, Angew. Chem., Int. Ed., 2014, 53, 1443–1447 CrossRef CAS PubMed
- K. Renggli, N. Sauter, M. Rother, M. G. Nussbaumer, R. Urbani, T. Pfohl and N. Bruns, Polym. Chem., 2017, 8, 2133–2136 RSC
- J. L. S. Milne, D. Shi, P. B. Rosenthal, J. S. Sunshine, G. J. Domingo, X. Wu, B. R. Brooks, R. N. Perham, R. Henderson and S. Subramaniam, EMBO J., 2002, 21, 5587–5598 CrossRef CAS PubMed
- J. L. S. Milne, X. Wu, M. J. Borgnia, J. S. Lengyel, B. R. Brooks, D. Shi, R. N. Perham and S. Subramaniam, J. Biol. Chem., 2006, 281, 4364–4370 CrossRef CAS PubMed
- S. J. Yeaman, J. A. Kirby and D. E. Jones, Immunol. Rev., 2000, 174, 238–249 CrossRef CAS PubMed
- L. H. Rome and V. A. Kickhoefer, ACS Nano, 2013, 7, 889–902 CrossRef CAS PubMed
- N. L. Kedersha, J. E. Heuser, D. C. Chugani and L. H. Rome, J. Cell Biol., 1991, 112, 225–235 CrossRef CAS PubMed
- W. Berger, E. Steiner, M. Grusch, L. Elbling and M. Micksche, Cell. Mol. Life Sci., 2008, 66, 43 CrossRef PubMed
- M. Wang, D. Abad, V. A. Kickhoefer, L. H. Rome and S. Mahendra, Methods Mol. Biol., 2018, 1798, 25–37 CrossRef CAS PubMed
- V. A. Kickhoefer, Y. Garcia, Y. Mikyas, E. Johansson, J. C. Zhou, S. Raval-Fernandes, P. Minoofar, J. I. Zink, B. Dunn, P. L. Stewart and L. H. Rome, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4348–4352 CrossRef CAS PubMed
- L. Budenholzer, C. L. Cheng, Y. Li and M. Hochstrasser, J. Mol. Biol., 2017, 429, 3500–3524 CrossRef CAS PubMed
- J. Marles-Wright, T. Grant, O. Delumeau, G. van Duinen, S. J. Firbank, P. J. Lewis, J. W. Murray, J. A. Newman, M. B. Quin, P. R. Race, A. Rohou, W. Tichelaar, M. van Heel and R. J. Lewis, Science, 2008, 322, 92–96 CrossRef CAS PubMed
- C. A. Diebolder, E. F. Halff, A. J. Koster, E. G. Huizinga and R. I. Koning, Structure, 2015, 23, 2349–2357 CrossRef CAS PubMed
- U. Meissner, E. Schröder, D. Scheffler, A. G. Martin and J. R. Harris, Micron, 2007, 38, 29–39 CrossRef CAS PubMed
- A. Fotin, T. Kirchhausen, N. Grigorieff, S. C. Harrison, T. Walz and Y. Cheng, J. Struct. Biol., 2006, 156, 453–460 CrossRef CAS PubMed
- D. P. Patterson, A. M. Desai, M. M. B. Holl and E. N. G. Marsh, RSC Adv., 2011, 1, 1004–1012 RSC
- N. P. King, J. B. Bale, W. Sheffler, D. E. McNamara, S. Gonen, T. Gonen, T. O. Yeates and D. Baker, Nature, 2014, 510, 103–108 CrossRef CAS PubMed
- H. Gradišar, S. Božič, T. Doles, D. Vengust, I. Hafner-Bratkovič, A. Mertelj, B. Webb, A. Šali, S. Klavžar and R. Jerala, Nat. Chem. Biol., 2013, 9, 362 CrossRef PubMed
- A. Ljubetič, F. Lapenta, H. Gradišar, I. Drobnak, J. Aupič, Ž. Strmšek, D. Lainšček, I. Hafner-Bratkovič, A. Majerle, N. Krivec, M. Benčina, T. Pisanski, T. Ć. Veličković, A. Round, J. M. Carazo, R. Melero and R. Jerala, Nat. Biotechnol., 2017, 35, 1094 Search PubMed
- Y.-T. Lai, K.-L. Tsai, M. R. Sawaya, F. J. Asturias and T. O. Yeates, J. Am. Chem. Soc., 2013, 135, 7738–7743 CrossRef CAS PubMed
- J. Votteler, C. Ogohara, S. Yi, Y. Hsia, U. Nattermann, D. M. Belnap, N. P. King and W. I. Sundquist, Nature, 2016, 540, 292 CrossRef CAS PubMed
- R. Koyani, J. Pérez-Robles, R. D. Cadena-Nava and R. Vazquez-Duhalt, Nanotechnol. Rev., 2017, 6, 405–419 CAS
- P. K. Robinson, Essays Biochem., 2015, 59, 1–41 CrossRef PubMed
- B. L. Montalvo-Ortiz, B. Sosa and K. Griebenow, AAPS PharmSciTech, 2012, 13, 632–636 CrossRef PubMed
- J. Gaitzsch, X. Huang and B. Voit, Chem. Rev., 2016, 116, 1053–1093 CrossRef CAS PubMed
- N. Ben-Haim, P. Broz, S. Marsch, W. Meier and P. Hunziker, Nano Lett., 2008, 8, 1368–1373 CrossRef CAS PubMed
- S. F. M. Van Dongen, W. P. R. Verdurmen, R. J. R. W. Peters, R. J. M. Nolte, R. Brock and J. C. M. Van Hest, Angew. Chem., Int. Ed., 2010, 49, 7213–7216 CrossRef PubMed
- R. Chandrawati, M. T. J. Olesen, T. C. C. Marini, G. Bisra, A. G. Guex, M. G. de Oliveira, A. N. Zelikin and M. M. Stevens, Adv. Healthcare Mater., 2017, 6, 1700385 CrossRef PubMed
- M. Jahadi and K. Khosravi-Darani, Mini-Rev. Med. Chem., 2017, 17, 366–370 CrossRef CAS PubMed
- G. Delaittre, I. C. Reynhout, J. J. L. M. Cornelissen and R. J. M. Nolte, Chem. – Eur. J., 2009, 15, 12600–12603 CrossRef CAS PubMed
- M. Yan, J. Ge, Z. Liu and P. Ouyang, J. Am. Chem. Soc., 2006, 128, 11008–11009 CrossRef CAS PubMed
- O. S. Sakr and G. Borchard, Biomacromolecules, 2013, 14, 2117–2135 CrossRef CAS PubMed
- V. Buckin and M. C. Altas, Catalysts, 2017, 7, 336 CrossRef
- X. Lian, Y. Huang, Y. Zhu, Y. Fang, R. Zhao, E. Joseph, J. Li, J. P. Pellois and H. C. Zhou, Angew. Chem., Int. Ed., 2018, 57, 5725–5730 CrossRef CAS PubMed
- F. P. Chang, Y. P. Chen and C. Y. Mou, Small, 2014, 10, 4785–4795 CrossRef CAS PubMed
- J. Lee, S. M. Kim and I. S. Lee, Nano Today, 2014, 9, 631–667 CrossRef CAS
- P. N. Catalano, A. Wolosiuk, G. J. A. A. Soler-Illia and M. G. Bellino, Bioelectrochemistry, 2015, 106, 14–21 CrossRef CAS PubMed
- S. B. P. E. Timmermans and J. C. M. van Hest, Curr. Opin. Colloid Interface Sci., 2018, 35, 26–35 CrossRef CAS
- H. Feyzizarnagh, B. W. Park, L. Sharma, M. M. Patania, D. Y. Yoon and D. S. Kim, Sen. Bio-Sens. Res., 2016, 7, 38–41 CrossRef
- V. Linko, M. Eerikäinen and M. A. Kostiainen, Chem. Commun., 2015, 51, 5351–5354 RSC
- G. Grossi, M. Dalgaard Ebbesen Jepsen, J. Kjems and E. S. Andersen, Nat. Commun., 2017, 8, 992 CrossRef PubMed
- Z. Zhao, J. Fu, S. Dhakal, A. Johnson-Buck, M. Liu, T. Zhang, N. W. Woodbury, Y. Liu, N. G. Walter and H. Yan, Nat. Commun., 2016, 7, 10619 CrossRef CAS PubMed
- J. Hahn, S. F. J. Wickham, W. M. Shih and S. D. Perrault, ACS Nano, 2014, 8, 8765–8775 CrossRef CAS PubMed
- C. P. Mata, D. Luque, J. Gómez-Blanco, J. M. Rodríguez, J. M. González, N. Suzuki, S. A. Ghabrial, J. L. Carrascosa, B. L. Trus and J. R. Castón, PLoS Pathog., 2017, 13, e1006755 CrossRef PubMed
| This journal is © The Royal Society of Chemistry 2019 |