
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA nanofluidic device for real-time visualization of DNA–protein interactions on the single DNA molecule level
Robin
Öz
,
Sriram
KK
 and
Fredrik
Westerlund
*
and
Fredrik
Westerlund
*
Department of Biology and Biological Engineering, Chalmers University of Technology, Gothenburg, Sweden. E-mail: fredrikw@chalmers.se
First published on 4th January 2019
Abstract
Single DNA molecule techniques have revolutionized our understanding of DNA–protein interactions. Traditional techniques for such studies have the major drawback that the DNA molecule studied is attached to a bead or a surface. Stretching of DNA molecules in nanofluidic channels has enabled single-molecule studies of DNA–protein interactions without the need of tethering the molecule to a foreign entity. This in turn allows for studying reactions along the whole extension of the molecule, including the free DNA ends. However, existing studies either rely on measurements where all components are mixed before introduction into the nanochannels or where passive diffusion brings the reagents to the confined DNA molecule. We here present a new generation of nanofluidic devices, where active exchange of the local environment within the nanofluidic channel is possible, while keeping the DNA molecule stretched and in confinement. To demonstrate the functionality of this novel device we added different analytes, such as SDS, spermidine and DNase I, to YOYO-1 stained DNA and studied the response in real time. We also performed a FRET-based reaction, where two different analytes were added sequentially to the same DNA molecule. We believe that this design will enable in situ mapping of complex biochemical processes, involving multiple proteins and cofactors, on single DNA molecules as well as other biomacromolecules.
Introduction
DNA–protein interaction studies are traditionally performed in bulk experiments, where the obtained response is an average from an ensemble of molecules. By employing single DNA molecule techniques, more detailed information can be obtained on individual reaction events, thereby allowing for better understanding of molecular mechanisms behind important biological phenomena.1 Established single DNA molecule techniques include for instance atomic force microscopy (AFM) and single molecule force spectroscopy with optical and magnetic tweezers. AFM has been extensively used to visualize different DNA–protein complexes2 such as the beads-on-a-string structure of nucleosomes in chromatin3 and formation of RecA filaments on DNA.4 The possibility to obtain high-resolution images under physiological conditions makes AFM a highly potent tool for biophysical studies. Nevertheless, the fact that the DNA molecules have to be attached to a surface prior to the analysis is an unavoidable constraint of the method. For optical5 and magnetic6 tweezers, on the other hand, this constraint is surpassed as DNA molecules can be manipulated and stretched in solution. Thus, these methods allow for studies of dynamic DNA–protein interactions in real time. The most crucial requirement for such experiments is, however, to functionalize one or both DNA ends with a bead or surface, thereby obstructing possible reactions on those ends. Hence, studies of reactions that occur on DNA ends and where two DNA ends meet are inherently difficult to study on the single DNA molecule level using these methods. Furthermore, the stretching needed for reliable analysis with tweezers is generally in the pN force regime, which is significantly higher than most forces that are generated in vivo.7Nanofluidic channels were introduced ∼15 years ago as a tool to stretch long single DNA molecules8 and have during the last years also been used for studies of DNA–protein interactions on the single DNA molecule level.9 Nanofluidics enables DNA–protein interaction studies at physiological conditions without the need of attaching foreign entities to the DNA ends. The DNA molecule is stretched only due to the nanoscale confinement, with an extension that corresponds to stretching forces in the fN force regime, which is comparable to forces in living cells.7 We have previously demonstrated how nanofluidic channels can be used to study the physical properties of the stiff filaments formed by RecA10 and Rad51,11 proteins involved in DNA damage repair, upon binding to DNA. We have also shown that mutants of the bacteriophage protein Cox exhibit different modes of action when compacting DNA.12 Although our method has proven to be useful for DNA–protein interaction studies at equilibrium, there is still a need for nanofluidic techniques enabling visualization of dynamic DNA–protein interactions in real time.
Previous reports by Zhang et al. and Henkin et al. describe approaches for in situ analysis of conformational changes of DNA stretched by nanoconfinement, when exposed to various analytes such as ions, ligands or proteins.13,14 These methods are dependent on diffusion of analytes into an array of nanofluidic channels, wherein DNA is confined, and allows for direct detection of the resulting effect on the extended DNA molecule. Both are examples of real-time experiments on nanoconfined DNA. However, in order to actively add specific amounts of analyte to DNA and to study sequential reactions on single DNA molecules in situ, it is of great importance to enable repeated alteration of the local environment in the nanofluidic channels on demand. This in turn requires active exchange of buffers in a controlled manner. Capaldi et al. recently demonstrated how the local environment can be changed for DNA molecules confined in nano-slit embedded cavities.15 However, in this study the DNA was not stretched but rather entrapped by the confinement, making it difficult to visualize how different analytes are interacting with the DNA molecule. There is thus a need for a new generation of nanofluidic devices, where DNA–protein interactions can be studied in real time with controlled addition of analyte.
The main challenge for real-time experiments on DNA stretched in nanofluidic channels is to keep the DNA molecules in place when actively changing the buffer conditions. We here present a novel nanofluidic system for dynamic studies of DNA–protein interactions in situ, where individual DNA molecules can be actively exposed to analytes of interest. The device allows simultaneous experiments on several DNA molecules and it is also possible to position multiple DNA molecules in the same reaction chamber to investigate intermolecular interactions between DNA ends. As a proof of concept, we have performed a series of real-time experiments to confirm the functionality of the nanofluidic system by for instance dissociating intercalated dyes from DNA, compacting DNA by spermidine and digesting DNA with DNase I in situ.
Experimental
Fabrication of the nanofluidic
The device schematics can be seen in Fig. 1a. It consists of two parallel microchannels, connected by an array of 120 nanochannels at the centre, oriented perpendicular to the microchannels. Each nanochannel is 500 μm in length, 100 nm in depth and 100 nm in width, except for the middle region spanning 60 μm in length (that is referred to as the reaction chamber), where the width is 300 nm and the depth 130 nm. Further, the design involves two additional microchannels, one on each side of the nanochannel array. This microchannel pair connects a nano-slit (width = 30 μm, length = 500 μm, depth = 30 nm) that runs across the reaction chambers in an orthogonal fashion.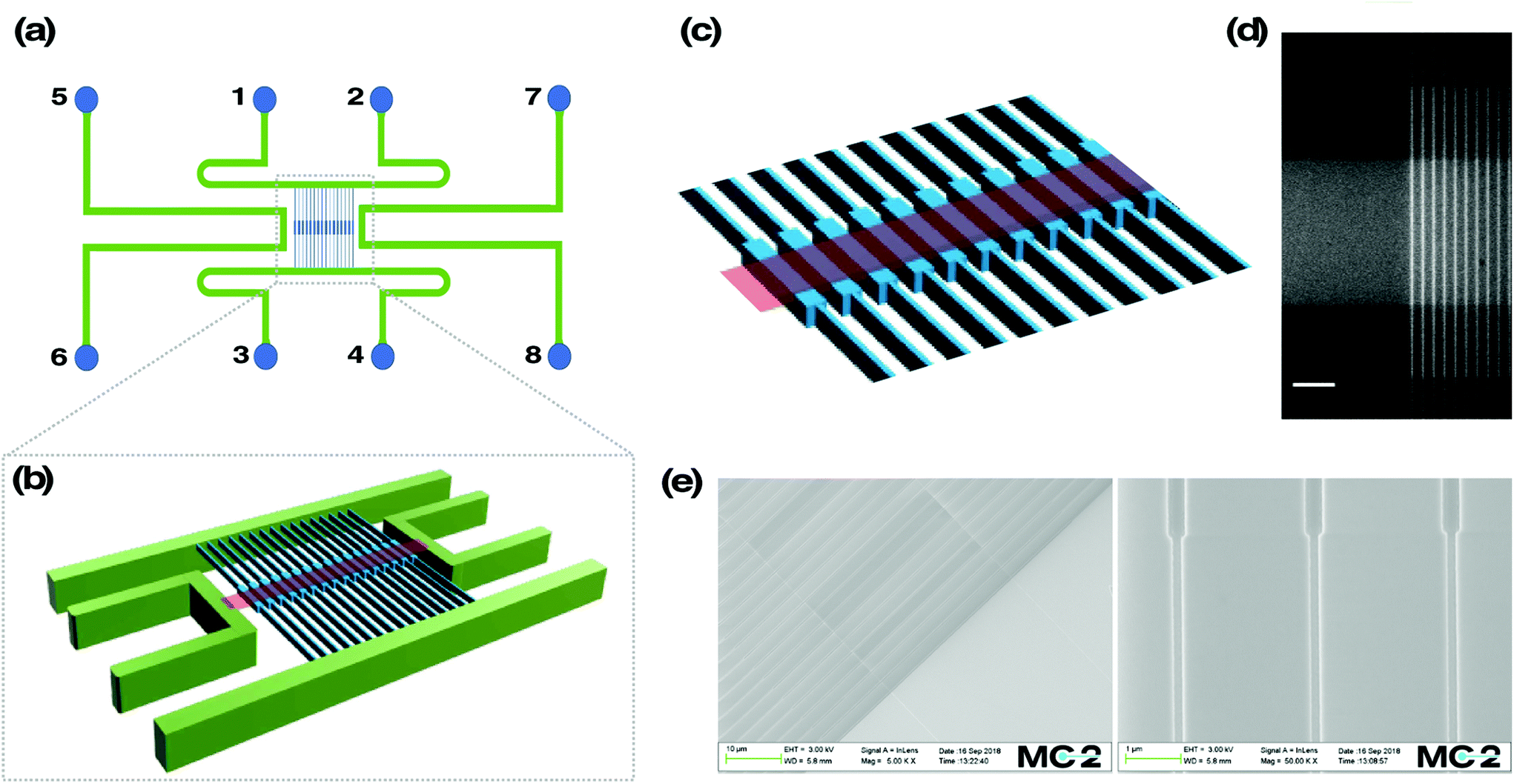 | ||
| Fig. 1 Overview of the nanofluidic chip. (a) The loading reservoirs are numbered 1–8 and there is a microchannel (green) connecting each pair of reservoirs. The vertically aligned nanofluidic channels (blue), with the reaction chambers in the center, are spanning between microchannels flanked by reservoir 1–4. The orthogonally aligned nano-slit (red) spans between microchannels flanked by reservoir 5–8. (b) Magnification of the nanochannel region. (c) A further magnification of the reaction chambers within the nanofluidic channels and (d) the corresponding fluorescence microscopy image of the same region, where fluorescein has been flushed into the system. Scale bar corresponds to 10 μm. (e) Scanning electron microscopy images showing the orthogonal nano-slit aligned on top of the reaction chambers (left), and the entropic barrier between the nanochannels and the reaction chambers (right). | ||
The fabrication scheme is as follows. First, a 2 μm thermal oxide layer was grown on a fresh 4′′ Si wafer of 500 μm thickness (N-type, Si-Mat) using an oxidation furnace (Centrotherm, 1050 °C, 780 minutes). Alignment marks to assist overlay of micro and nano structures were obtained through photolithography (PL, S1813, Shipley Inc., USA) and reactive ion etching (RIE, 50 sccm Ar, 50 sccm CHF3, 150 W RF-power, 30 mbar pressure) to obtain 900 nm deep features. This was verified using a surface profiler (Dektak D150, Veeco Instruments). Post RIE, the substrate was cleaned using piranha solution (con. H2SO4 and H2O2 at 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, 120 °C for 10 minutes), rinsed with DI-water and dried using a nitrogen gun. Subsequently, a 20 nm thick chromium layer was deposited using e-gun evaporation (AVAC HVC600), followed by sputtering of 24 nm thick SiO2 (FHR MS-150 Sputter) to form a hard mask for RIE of nanochannels. Electron beam lithography (EBL, JEOL JBX-9300FS) was carried out using AR-P 6200.13 resist (diluted in Anisole, 1:1 ratio, Allresist GmBH) and then etched using RIE (50 sccm NF3, 25 W RF-power, 8 mbar pressure) to get 100 nm deep nanochannels. Sputtered SiO2 was removed with a CF4 etch (Plasmatherm RIE, 40 sccm CF4, 100 W RF-power, 100 mT pressure) and the chromium layer was removed using a chromium etchant (SunChem AB, Sweden). The substrate was again cleaned using piranha solution, as described above. A second EBL process was carried out using the same procedure to obtain the nano-slit features (width = 30 μm, depth = 30 nm, length = 500 μm). The nano-slit depth was confirmed to be 30 nm using surface profile measurements. The substrate was again cleaned using piranha solution, rinsed with DI-water and dried using a nitrogen gun. Alternately, nanoslit fabrication can be done using standard photolithography, but we used EBL here to avoid the need for a photomask, thus allowing us to vary the design of the nanoslit to a design that works for the intended applications. Microchannels were then fabricated using PL and RIE, with the same protocol used for obtaining alignment marks. The substrate was once again cleaned using piranha solution.
:1 ratio, 120 °C for 10 minutes), rinsed with DI-water and dried using a nitrogen gun. Subsequently, a 20 nm thick chromium layer was deposited using e-gun evaporation (AVAC HVC600), followed by sputtering of 24 nm thick SiO2 (FHR MS-150 Sputter) to form a hard mask for RIE of nanochannels. Electron beam lithography (EBL, JEOL JBX-9300FS) was carried out using AR-P 6200.13 resist (diluted in Anisole, 1:1 ratio, Allresist GmBH) and then etched using RIE (50 sccm NF3, 25 W RF-power, 8 mbar pressure) to get 100 nm deep nanochannels. Sputtered SiO2 was removed with a CF4 etch (Plasmatherm RIE, 40 sccm CF4, 100 W RF-power, 100 mT pressure) and the chromium layer was removed using a chromium etchant (SunChem AB, Sweden). The substrate was again cleaned using piranha solution, as described above. A second EBL process was carried out using the same procedure to obtain the nano-slit features (width = 30 μm, depth = 30 nm, length = 500 μm). The nano-slit depth was confirmed to be 30 nm using surface profile measurements. The substrate was again cleaned using piranha solution, rinsed with DI-water and dried using a nitrogen gun. Alternately, nanoslit fabrication can be done using standard photolithography, but we used EBL here to avoid the need for a photomask, thus allowing us to vary the design of the nanoslit to a design that works for the intended applications. Microchannels were then fabricated using PL and RIE, with the same protocol used for obtaining alignment marks. The substrate was once again cleaned using piranha solution.
A 300 nm thick aluminum layer was sputtered (FHR MS-150 Sputter) to serve as a hard mask for Deep Reactive Ion Etching (DRIE) of through holes. Through hole structures (∅ = 1 mm) were fabricated using PL (AZ4533, MicroChem GmBH) and DRIE (STS-ICP, C4F8: 100 sccm, SF6: 150 sccm, O2: 10 sccm, Power: 600 Watts, platen power: 15 Watts). Aluminum was removed after DRIE using piranha solution. Fusion bonding of the substrate containing the micro- and nanofluidic features was then carried out. For this, the substrate was first subjected to oxygen plasma (40 sccm O2, 250 W RF-power, 500 mT pressure, 1 minute) and then a piranha cleaned borofloat glass (170 μm thick, double side polished, Si-Mat) was subjected to oxygen plasma (40 sccm O2, 350 W RF-power, 500 mT pressure, 2 minutes). The plasma treated surfaces were brought together and immediately formed a temporary bonding. This pair was then subjected to fusion bonding at 550 °C for 5 hours. The fusion bonding was done by gradually increasing the temperature required for bonding and slowly cooling down afterwards, to avoid any cracking due to different thermal coefficients of Si wafer and borofloat glass. Finally, individual chips of 18 × 10 mm2 were obtained using an automated dicing saw (Disco DAD3350). Each chip was glued to a custom-made acrylic frame using epoxy glue. The devices were mounted on a custom-made chuck with pressure inlets and outlets, enabling easy sample loading and application of pressure driven flow during experiments.
Scanning electron microscope (SEM) images of nanochannels and nano-slit features were obtained using Supra 60-VP EDX (Zeiss). For this, a 5 nm gold layer was sputtered (FHR MS-150 Sputter) to minimize charging effects from the SiO2 surface.
Sample preparation
DNA from phage λ (λ-DNA) was purchased from New England Biolabs and used for all experiments. The bis-intercalating DNA dye YOYO-1 was purchased from Invitrogen. 20 μM (base pairs) of λ-DNA was mixed with 4 μM YOYO-1, unless stated otherwise, in pre-mixed 5× Tris-Borate-EDTA buffer (VWR International).16 Before loading onto the nanofluidic chip, the sample was diluted 100 times with MilliQ water and 2-mercaptoethanol (Merck) was added to a final concentration of 3% (v/v) to serve as an oxygen radical scavenger. 100 μM fluorescein (Merck) solution was prepared by dissolving the powder in MilliQ water. 10% (w/w) sodium dodecyl sulphate (SDS, Merck) solution was prepared by dissolving SDS powder in MilliQ water and mixing gently. N-(3-Aminopropyl)-1,4-diaminobutane (Spermidine, Merck) was dissolved in degassed MilliQ water to a final concentration of 1 M and stored as aliquots at −20 °C. Upon use, spermidine was diluted in degassed MilliQ water to a final concentration of 500 μM before loading onto the nanofluidic chip. Ethidium bromide (EtBr) was prepared in 0.05× TBE buffer to a final concentration of 20 μM. For the Förster Resonance Energy Transfer (FRET) study, DNA was stained with a lower amount of YOYO-1 at a ratio of 1 dye molecule per 50 base pairs. Deoxyriboendonuclease I (DNase I, Merck) was diluted in 1× reaction buffer, which was provided as a 10× concentrate in the enzyme kit. 0.1 U μl−1 DNase I was loaded onto the nanofluidic chip for the digestion experiment.Nanofluidics
In all experiments, the DNA sample was loaded in one of reservoirs 1–4 (Fig. 1a), from where the DNA can be flushed in to the nanochannels. The analytes tested were loaded in one or several of reservoirs 5–8, from where they can be flushed through the orthogonal nano-slit. Pressurized nitrogen was used to control the flow of DNA and analyte within the nanofluidic system. To avoid non-specific binding of analytes to the negatively charged silicon dioxide surface, the micro- and nanochannels were passivated with a supported lipid bilayer comprising 99% 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC, Avanti) and 1% Lissamine Rhodamine B 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (DHPE-Rhodamine, Invitrogen), as described elsewhere.17Data acquisition
The real-time DNA–analyte interaction events were imaged using an epi-configured fluorescence microscope (Zeiss AxioObserver.Z1), equipped with a SPECTRA X light engine (Lumencor) and a 100× TIRF oil immersion objective (Zeiss, NA = 1.46). The excitation light for all experiments was in the 470/24 nm wavelength band. An EMCCD camera (Photometrics Evolve) was used to acquire the images. In all experiments where only YOYO-1 was used to stain DNA, a dichroic beamsplitter was used with a cut-off wavelength at 500 nm and a bandpass filter in the 530/50 wavelength region serving as an emission filter. For the FRET study, an Optosplit II (Cairn Research) was used to image the emitted light from YOYO-1 and EtBr simultaneously, using only 470/24 nm excitation light. A dichroic beamsplitter with a cut-off wavelength at 562 nm was used to separate the emission from YOYO-1 and EtBr. The emission filters used for YOYO-1 and EtBr were 530/50 nm and 617/73 nm, respectively.Results and discussion
The nanofluidic device
This study presents a nanofluidic device where single DNA molecules, stretched in nanoscale confinement, are actively exposed to analytes of interest in a controlled fashion. The single DNA molecules are stretched and confined in 130 × 300 nm2 reaction chambers, flanked by 100 × 100 nm2 nanofluidic channels (Fig. 1). A shallow slit is aligned on top of and orthogonally to the reaction chambers, enabling active buffer exchange in the local environment of the entrapped DNA molecule. The entropic barrier, created due to the width difference of the reaction chambers and the nanofluidic channels, helps keeping the DNA molecule in place when buffer is flushed through the nano-slit. The nanofluidic chip is controlled by applying variable pressures to the reservoirs.8Addition of analytes
As a first proof-of-concept of the functionality of the device, different analytes were actively flushed into reaction chambers containing single confined DNA molecules. In Fig. 2a 10% (w/w) Sodium Dodecyl Sulphate (SDS) is added to single λ-DNA molecules. SDS is a micelle forming surfactant that promotes dissociation of cationic drugs from DNA.18 As can be seen from the kymographs and the corresponding fluorescence intensity plot, YOYO-1 dissociates from the DNA molecule as the SDS reaches the reaction chamber, resulting in a rapid reduction in fluorescence intensity. Due to the higher ionic strength when SDS is added, compaction of the DNA molecule can be observed both as a decrease in extension and an increase in emission intensity, just before the YOYO-1 dissociates from the DNA.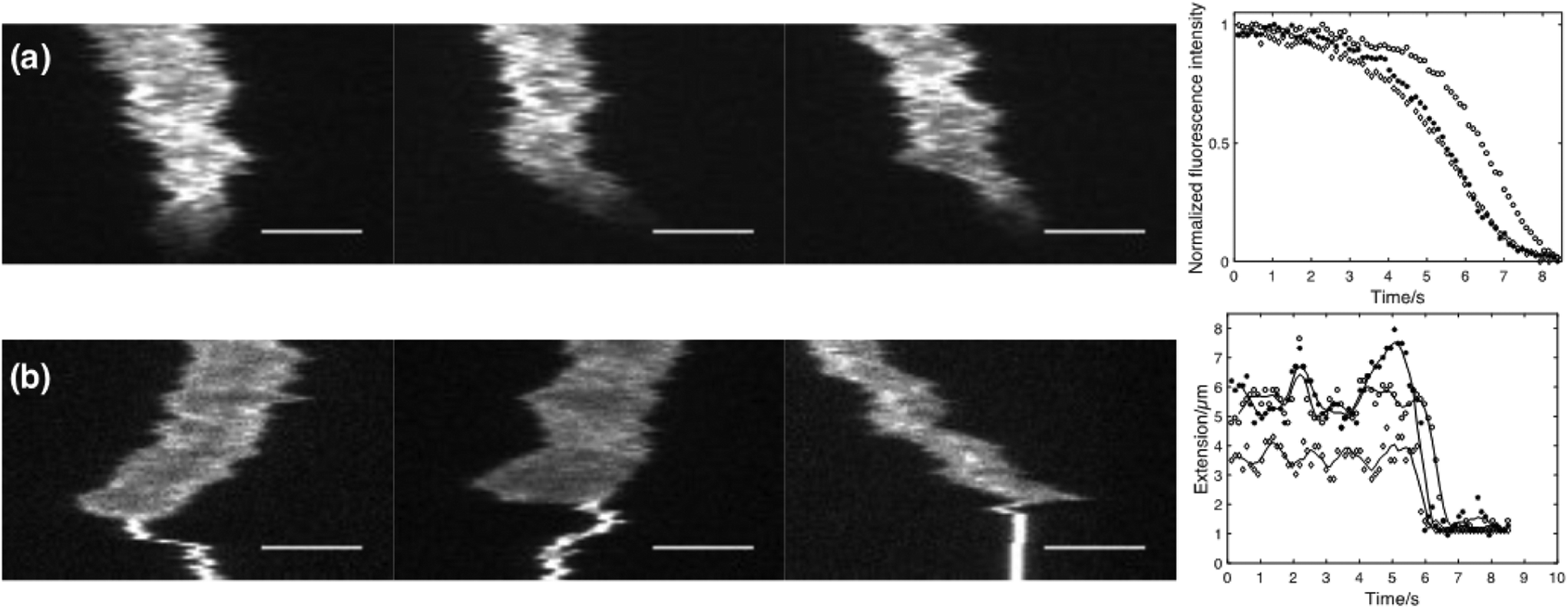 | ||
| Fig. 2 Addition of analytes to confined λ-DNA. Kymographs of YOYO-1 stained λ-DNA molecules to which (a) SDS (10% w/w) and (b) Spermidine (500 μM) is added through the slit in situ. The corresponding plots to the right show the normalized fluorescence intensity and the molecule extensions over time, respectively. Scale bars correspond to 5 μm. | ||
In Fig. 2b the polyamine spermidine (4+) is added to a confined DNA molecule. At the concentration used (500 μM), spermidine immediately compacts the DNA molecule into a dense coil. It is worth noticing that the compaction seems to start from one of the DNA ends for a vast majority of the molecules studied. This observation stresses the importance of having free and non-obstructed DNA ends as this phenomenon would otherwise have been obscured.
Repeated sequential reactions
The main feature of this novel nanofluidic device is the ability to actively change the local environment within the reaction chambers on demand. The current design allows for at least two different analytes to be added to one or several single DNA molecules at a time, meaning that reaction events can be studied in a dynamic fashion. To do so, the analytes are loaded in different reservoirs (Fig. 1a), one analyte in a reservoir on each side of the nano-slit, respectively. The analytes can then be flushed into the nanofluidic channels separately through the different feeding channels connected to the nano-slit. In order to demonstrate this functionality of the device, we performed a sequential reaction where EtBr was added to YOYO-1 stained λ-DNA followed by controlled dissociation of the dye by addition of SDS. The emission of both dyes was recorded simultaneously (see Experimental section for details). The overlapping emission spectrum of YOYO-1 and excitation spectrum of EtBr allows for energy transfer between the dyes upon excitation with blue light in the 470/24 nm wavelength band. EtBr was first added through the nano-slit to a confined λ-DNA molecule, pre-stained with YOYO-1. Simultaneous imaging of YOYO-1 and EtBr showed a decrease in YOYO-1 emission and an increase in EtBr emission when EtBr binds to DNA (Fig. 3). This suggests that EtBr has intercalated into the sparsely stained DNA molecule, giving rise to energy transfer, where the YOYO-1 emission is quenched by the presence of EtBr. The possibility that this observation is due to dissociation of YOYO-1 when EtBr binds was ruled out by subsequently adding SDS to the same DNA molecule. Upon addition of SDS, an immediate increase in YOYO-1 emission and decrease in EtBr emission was observed (Fig. 3b). This suggests that the intercalated EtBr has dissociated from the DNA molecule, leaving YOYO-1 unaffected. Given the monocationic nature of EtBr, it has a significantly lower binding affinity to DNA than YOYO-1, which has four positive charges.18 This demonstrates that by using our nanofluidic setup it is possible to perform a controlled sequential manipulation of a confined DNA molecule. The whole process was repeated several times by adding EtBr to the same DNA molecule followed by SDS, and the same response was observed. Considering the emission intensity of YOYO-1, it is clear that the maximum intensity is unaffected between each cycle, suggesting that only the EtBr is dissociated from the molecule while the number of YOYO-1 molecules is close to constant. It is possible to also dissociate YOYO-1 from DNA with SDS via longer exposure, as demonstrated above (Fig. 2a). Due to the gradual exchange of buffer within the reaction chambers the concentration of SDS will increase over time. It is apparent that in order to make EtBr dissociate from the DNA, a lower concentration of SDS is needed in comparison to the dissociation of YOYO-1.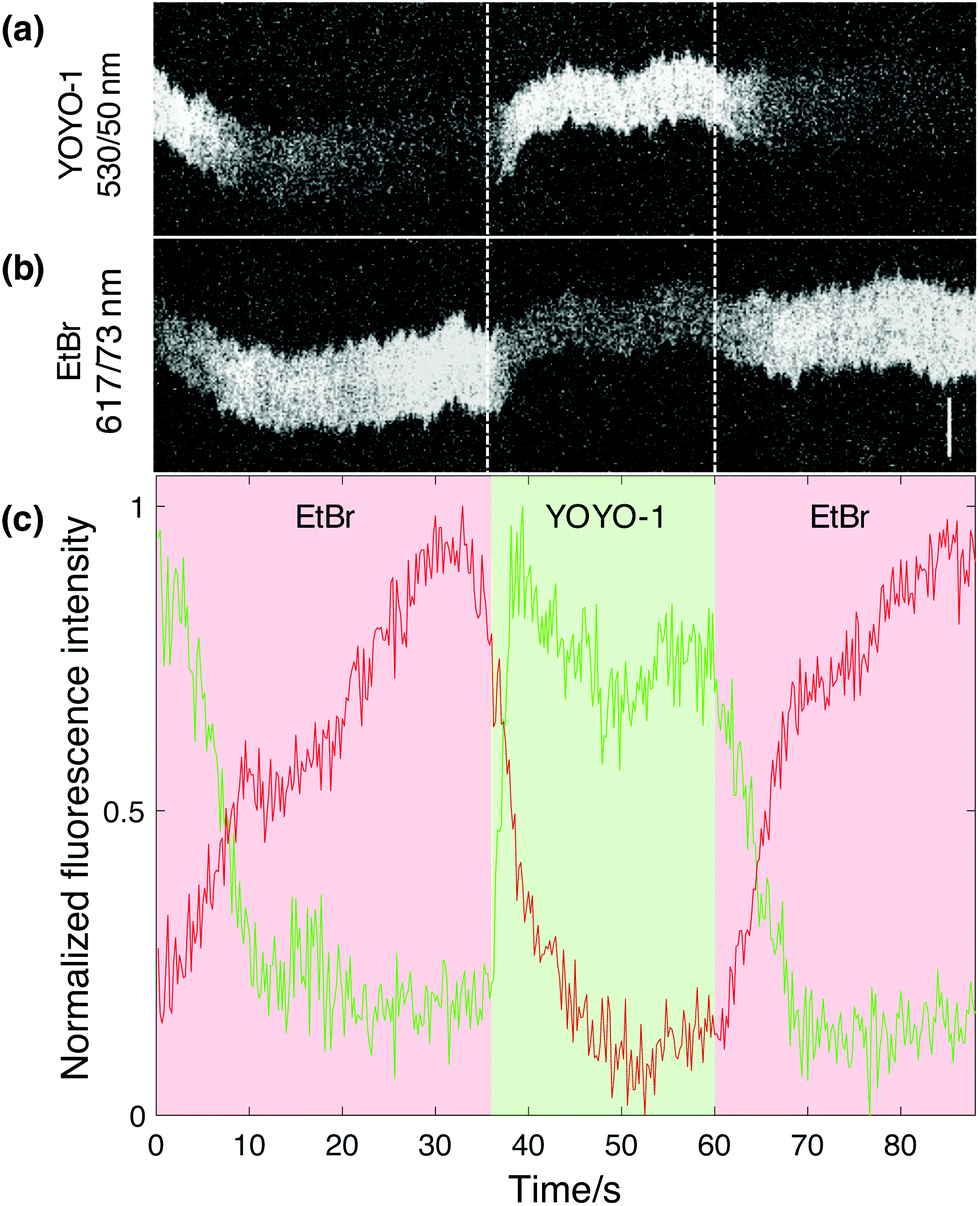 | ||
| Fig. 3 Sequential addition of EtBr and SDS to YOYO-1 stained λ-DNA. The kymographs show the emission from (a) YOYO-1 and (b) EtBr over time. (c) The plot shows the corresponding fluorescence intensity profiles for YOYO-1 (green) and EtBr (red). The color shades indicate the time points at which transitions are detected in emission patterns of the DNA molecule upon change of analyte. Scale bar corresponds to 5 μm. | ||
One interesting feature of the plot in Fig. 3c is the kinetics of the increase in EtBr and decrease in YOYO-1 emission when EtBr is added. The increase in EtBr emission seems to occur on much longer time scales than the decrease in YOYO-1 emission. This is seen in all movies collected and agrees with a model where maximum FRET occurs already at a relatively low load of EtBr. FRET is efficient on length scales of tens of basepairs, meaning that there are many empty sites for EtBr to further bind when full FRET occurs. This explains the slower increase in EtBr emission.
Endonuclease digestion of DNA
To manifest the applicability of the device in biochemical reactions, we added the nucleolytic enzyme DNase I to confined λ-DNA molecules and studied the subsequent reaction in real time. In Fig. 4 kymographs from four different λ-DNA molecules are shown, where DNase I is added in situ.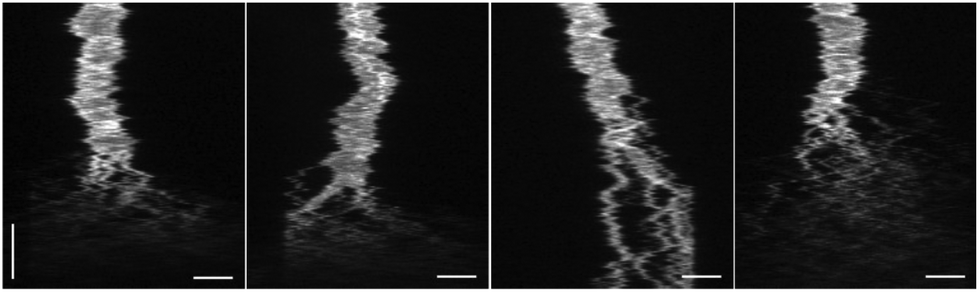 | ||
| Fig. 4 Endonuclease digestion of DNA with DNase I. Kymographs of YOYO-1 stained λ-DNA molecules to which DNase I (0.1 U μl−1) is added in situ. Vertical scale bar corresponds to 5 seconds. Horizontal scale bars correspond to 5 μm. | ||
Upon interaction with DNA, DNase I will create random nicks in the phosphate backbone of the double-stranded helix.19 Eventually, as the distance becomes shorter between two single-stranded nicks on each strand, a double-stranded break will be generated, resulting in two separate DNA fragments that diffuse away from each other. As seen in the kymographs (Fig. 4) a complete digestion of DNA occurs within seconds after the DNase I has reached the confined DNA molecule. Despite the rapid disintegration of the DNA, it is possible to distinguish individual double-stranded breaks as long as the created fragments are larger than the resolving capacity of the fluorescence microscope. This highlights an advantage of the device, where there are no external active forces acting on the DNA molecule to keep it in a stretched conformation. In contrast, methods using pulling force to stretch the DNA, such as optical and magnetic tweezers, do not allow for visualization of multiple cuts along the extension, as the DNA molecule will collapse immediately after generation of the first double-stranded break.
Additional functionalities
In addition to the functionalities mentioned above, it is worthwhile to highlight some other advantages that the design of this novel nanofluidic device is providing. One useful functionality is the inherent effect arising from the fact that loading of analyte into the nano-slit occurs from one side of the nanochannel region (Fig. 1). This means that analyte is added gradually over time, depending on how far from the entrance a specific DNA molecule is. This in turn makes it possible to study DNA–analyte interactions at different concentrations simultaneously. By flushing analyte through the nano-slit, the local environment will be changed in the reaction chambers in consecutive order, starting from the one closest to the feeding microchannel. Hence, a gradient will form, where the concentration of analyte is different in each nanochannel. In order to show the formation of a gradient with time, fluorescein was added to the nanochannels through the nano-slit (Fig. 5a). As can be seen from the snapshots at different timepoints, the concentration of fluorescein is gradually increasing due to active flushing of the dye through the nano-slit until the local environment has been fully changed in all nanochannels. By stopping the addition of analyte at a certain timepoint, it is possible to retain the concentration gradient among the nanochannels. To further demonstrate the effect of gradual addition of analyte with time, we confined YOYO-1 stained DNA molecules in several reaction chambers at different distances from the feeding microchannel, followed by addition of SDS through the nano-slit. From the dissociation curves and the corresponding kymographs (Fig. 5b) it is clear that YOYO-1 is dissociating from the DNA molecules in consecutive order, starting from the molecule closest to the feeding microchannel. This shows that there is a gradual change in the SDS concentration over time and that the micelle-sequestered dissociation of YOYO-1 is concentration dependent. This inherent functionality of the nanofluidic device can further be used to study for instance the dose dependence of different proteins or analytes when interacting with DNA.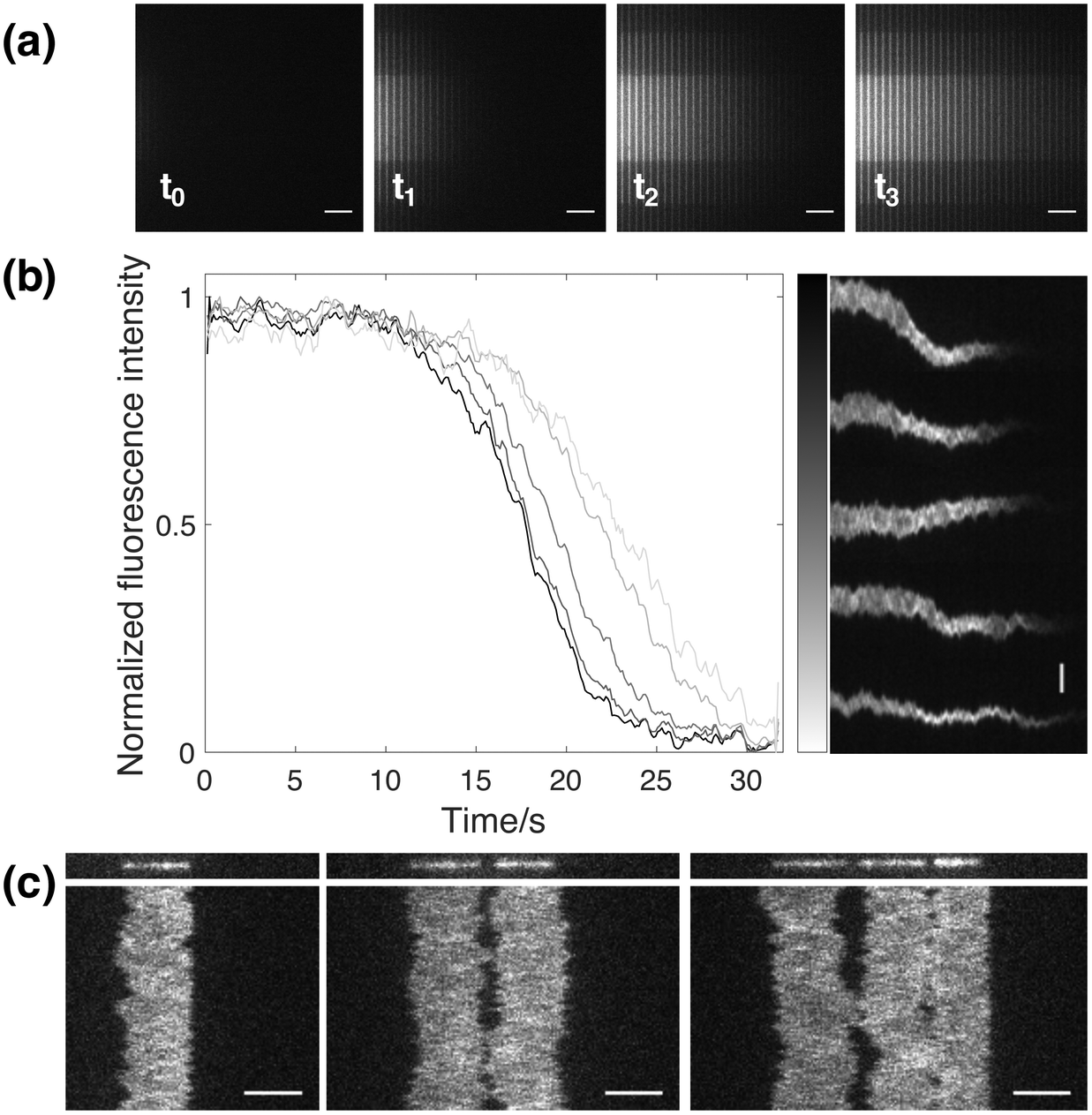 | ||
| Fig. 5 Additional features of the nanofluidic device. (a) Addition of fluorescein to the reaction chambers, showing a gradual increase in fluorescein concentration with time. t0 was set to the time at which the analyte was started to be flushed through the nano-slit. Scale bars correspond to 10 μm. (b) Micelle-sequestered dissociation of YOYO-1 from multiple DNA molecules confined in reaction chambers at different distance from the slit inlet. The normalized fluorescence intensity is plotted over time for each molecule presented in the kymographs. The grey scale bar corresponds to the distance between the molecule and the feeding microchannel, where a decreasing grey value corresponds to a longer distance. Scale bar corresponds to 5 μm. (c) One, two and three λ-DNA molecules, respectively, confined in a single reaction chamber and kept in close proximity. Scale bars correspond to 5 μm. | ||
A second useful feature of the device is the ability to confine multiple DNA molecules within the same reaction chamber (Fig. 5c). As can be seen in the kymographs, the DNA ends can be kept in close proximity over time, which in turn allows for even slow reactions to be visualized in situ. This feature enables studies of intermolecular interactions between large DNA molecules. Of particular relevance are reactions occurring on DNA ends since this device allows positioning of two DNA ends in close proximity. Reaction where two DNA ends meet are for example important in double strand break repair.20,21 Such reactions are very difficult to address with existing techniques for long DNA that require tethering of at least one end and little control of the position of the other end.
Conclusions
To conclude, we have presented a novel nanofluidic design, which enables dynamic studies of DNA–analyte interactions in situ, without the need of tethering the DNA molecule. The ability to actively change the local environment in the nanofluidic channels, while keeping the DNA molecule stretched and within the field of view, is highly important in order to be able to study complex biochemical reactions on single DNA molecules.We have demonstrated the functionality of the device by a series of experiments, where analytes, such as SDS, spermidine, EtBr and DNase I were actively added to a confined DNA molecule while studying the resulting response in situ. We have further shown that it is possible to sequentially add at least two different analytes to the same confined DNA molecule and study the resulting response in real time. By adding EtBr to a confined YOYO-1 stained DNA molecule, followed by addition of SDS to the same molecule, we also demonstrated that intercalators with different physical properties can selectively be dissociated from the DNA on demand, manifesting the controllability of the device.
We believe that the nanofluidic device presented here will enable mapping of complex biochemical processes, involving multiple proteins and co-factors, on single DNA molecules in situ. It is important to stress that the dimensions of the device can be tailored depending on the particular experiment conducted. Doing so, one needs to carefully adjust the depth of the slit, as well as the widths of the nanochannels and the reaction chambers to make sure that the entropic trapping is optimal, and to reduce the occasional escaping of DNA from the chamber upon addition of analyte. It is also important to point out that the device principle might find applications for biomacromolecules beyond DNA, where similar research questions are present and similar experiments needed.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Swedish Research Council (no. 2015–5062) and the Foundation Olle Engkvist Byggmästare. Dr Fredrik Persson and Dr Joachim Fritzsche are acknowledged for initial discussions on the design of the nanofluidic device. The MATLAB software used for some of the image analysis was developed by the group of Tobias Ambjörnsson, Lund University.References
- A. M. van Oijen, Nat. Chem. Biol., 2008, 4, 440 CrossRef CAS PubMed.
- A. Andrea and F. Paolo, Meas. Sci. Technol., 2005, 16, R65 CrossRef.
- M. J. Allen, X. F. Dong, T. E. O'Neill, P. Yau, S. C. Kowalczykowski, J. Gatewood, R. Balhorn and E. M. Bradbury, Biochemistry, 1993, 32, 8390–8396 CrossRef CAS.
- B. D. Sattin and M. C. Goh, Biophys. J., 2004, 87, 3430–3436 CrossRef CAS.
- K. R. Chaurasiya, T. Paramanathan, M. J. McCauley and M. C. Williams, Phys. Life Rev., 2010, 7, 299–341 CrossRef PubMed.
- I. D. Vlaminck and C. Dekker, Annu. Rev. Biophys., 2012, 41, 453–472 CrossRef PubMed.
- F. Persson, P. Utko, W. Reisner, N. B. Larsen and A. Kristensen, Nano Lett., 2009, 9, 1382–1385 CrossRef CAS PubMed.
- F. Persson and J. O. Tegenfeldt, Chem. Soc. Rev., 2010, 39, 985–999 RSC.
- K. Frykholm, L. K. Nyberg and F. Westerlund, Integr. Biol., 2017, 9, 650–661 CrossRef CAS PubMed.
- K. Frykholm, M. Alizadehheidari, J. Fritzsche, J. Wigenius, M. Modesti, F. Persson and F. Westerlund, Small, 2014, 10, 884–887 CrossRef CAS PubMed.
- L. H. Fornander, K. Frykholm, J. Fritzsche, J. Araya, P. Nevin, E. Werner, A. Çakır, F. Persson, E. B. Garcin, P. J. Beuning, B. Mehlig, M. Modesti and F. Westerlund, Langmuir, 2016, 32, 8403–8412 CrossRef CAS PubMed.
- K. Frykholm, R. P.-A. Berntsson, M. Claesson, L. de Battice, R. Odegrip, P. Stenmark and F. Westerlund, Nucleic Acids Res., 2016, 44, 7219–7227 CAS.
- C. Zhang, K. Jiang, F. Liu, P. S. Doyle, J. A. van Kan and J. R. C. van der Maarel, Lab Chip, 2013, 13, 2821–2826 RSC.
- G. Henkin, D. Berard, F. Stabile, M. Shayegan, J. S. Leith and S. R. Leslie, Anal. Chem., 2016, 88, 11100–11107 CrossRef CAS PubMed.
- X. Capaldi, Z. Liu, Y. Zhang, L. Zeng, R. Reyes-Lamothe and W. Reisner, Soft Matter, 2018, 14, 8455–8465 RSC.
- L. Nyberg, F. Persson, B. Åkerman and F. Westerlund, Nucleic Acids Res., 2013, 41, e184–e184 CrossRef CAS PubMed.
- F. Persson, J. Fritzsche, K. U. Mir, M. Modesti, F. Westerlund and J. O. Tegenfeldt, Nano Lett., 2012, 12, 2260–2265 CrossRef CAS.
- F. Westerlund, L. M. Wilhelmsson, B. Nordén and P. Lincoln, J. Am. Chem. Soc., 2003, 125, 3773–3779 CrossRef CAS.
- D. Suck and C. Oefner, Nature, 1986, 321, 620 CrossRef CAS PubMed.
- M. R. Lieber, Annu. Rev. Biochem., 2010, 79, 181–211 CrossRef CAS PubMed.
- S. C. Kowalczykowski, D. A. Dixon, A. K. Eggleston, S. D. Lauder and W. M. Rehrauer, Microbiol. Rev., 1994, 58, 401–465 CAS.
| This journal is © The Royal Society of Chemistry 2019 |