
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApplication of radially grown ZnO nanowires on poly-L-lactide microfibers complexed with a tumor antigen for cancer immunotherapy†
Prashant
Sharma‡
 ab,
Ji Beom
Shin‡
c,
Bum Chul
Park
c,
Jae-Won
Lee
ab,
Sang Won
Byun
c,
Na-Yoon
Jang
ab,
Yu Jin
Kim
d,
Yuri
Kim
ab,
Young Keun
Kim
*cd and
Nam-Hyuk
Cho
*abe
ab,
Ji Beom
Shin‡
c,
Bum Chul
Park
c,
Jae-Won
Lee
ab,
Sang Won
Byun
c,
Na-Yoon
Jang
ab,
Yu Jin
Kim
d,
Yuri
Kim
ab,
Young Keun
Kim
*cd and
Nam-Hyuk
Cho
*abe
aDepartment of Microbiology and Immunology, Seoul National University College of Medicine, Seoul 03080, Republic of Korea
bDepartment of Biomedical Sciences, Seoul National University College of Medicine, Seoul 03080, Republic of Korea. E-mail: chonh@snu.ac.kr
cDepartment of Materials Science and Engineering, Korea University, Seoul 02841, Republic of Korea. E-mail: ykim97@korea.ac.kr
dResearch Center for Biomedical Nanocrystals, Korea University, Seoul 02841, Republic of Korea
eInstitute of Endemic Disease, Seoul National University Medical Research Center and Bundang Hospital, Seoul 03080, Republic of Korea
First published on 27th February 2019
Abstract
Zinc oxide (ZnO)-based nanocomposites have shown promising potential for various biomedical applications, including vaccine development, owing to their multifunctionality and biocompatibility. Here, we synthesized radially grown ZnO nanowires (NWs) on poly-L-lactic acid (PLLA) microfibers with unique 3-dimensional structure and applied them as therapeutic cancer vaccines. This inorganic–organic hybrid nanocomposite has mild cellular toxicity but efficiently delivers a tumor antigen into dendritic cells, cellular bridges between innate and adaptive immunity, to stimulate them to express inflammatory cytokines and activation surface markers. We also demonstrated that the hybrid nanocomposites successfully induce tumor antigen-specific cellular immunity and significantly inhibit tumor growth in vivo. ZnO NWs on PLLA fibers systemically reduced immune suppressive TReg cells and enhanced the infiltration of T cells into tumor tissues, compared to mice immunized with PLLA fibers coated with the antigen. Our current findings open a new avenue in extending the biomedical application of inorganic metal oxide-inert organic hybrid nanocomposites as a novel vaccine platform.
Introduction
Zinc oxide (ZnO)-based nanocomposites have shown promising potential for various biomedical applications, including bioimaging, drug delivery, and vaccine development, owing to their multifunctionality and biocompatibility.1–4 In particular, it has been consistently shown that ZnO nanoparticles (NPs) possess adjuvant potential as a vaccine carrier when combined with specific antigens.1,2,5 ZnO NPs on their own can exert immunomodulatory and inflammatory effects on antigen-presenting phagocytic cells (APCs), such as macrophages and dendritic cells.2,6,7 The immune-stimulatory function of ZnO nanocomposites might be attributed to the induction of reactive oxygen species (ROSs), inflammatory cytokines, and cellular activation in APCs, which link to antigen-specific adaptive immune responses, as well as intrinsic basal toxicity in vivo.2,8,9 It is notable that ZnO NPs have the most drastic immunological effects in APCs among other inorganic NPs (TiO2, TiO2-silica, single-walled carbon nanotubes, and multi-walled carbon nanotubes).10 Dissolution of ZnO NPs in aqueous biological environment might be associated with the enhanced inflammatory effect when compared to other insoluble inorganic NPs, such as TiO2.11 Moreover, we have shown that the surface modification of ZnO nanocomposites with specific peptides that possess high affinity for metal oxide composites enables them to further enhance antigen-specific adaptive immunity, including antibody and T cell responses in vivo.1,2Recently, we also found that ZnO nanowires (NWs) can efficiently deliver associated biomacromolecules, including peptides and DNA, into eukaryotic cells when in the appropriate 3-dimensional conformation.3 Our previous findings suggest that radially grown “fan-shaped” ZnO NWs (FNWs) on a coverslip can efficiently deliver the associated peptides and DNA into eukaryotic cells via transient penetration of plasma membranes as well as cellular endocytosis, whereas densely packed vertical ZnO NWs (VNWs) might primarily deliver biomacromolecules by endocytosis and bare membrane penetration.3 Indeed, FNWs can mediate intracellular delivery of DNA into the nucleus, demonstrating intracytoplasmic penetration of the cytoplasmic membrane, whereas VNWs failed to do so. This phenomenon suggests that contact of angled NWs with plasma membrane and subsequent cellular spread can intensify membrane tension and induce transient penetration of plasma membrane, thereby enhancing biomacromolecule delivery into the cytoplasm and nucleus.3 The delivery of an antigen into host cells, especially into APCs, is a critical step of antigen processing and presentation to CD8 and CD4 T cells via the major histocompatibility complex (MHC) class I and II pathways.12 These two different antigen processing and presentation pathways are primarily dependent on antigen localization in cellular cytoplasm and intracellular vesicles, respectively.12 Intracellular delivery of a protein antigen into the cytoplasm may further enhance antigen-specific CD8 T cell responses mediated by the MHC class I antigen presentation pathway,13,14 which is linked to antigen-specific cytotoxic T lymphocyte (CTL) responses, critical for anti-tumor immunity.12,15
In this study, we investigated whether ZnO NWs, forming a unique 3-dimensional structure on poly-L-lactic acid (PLLA) microfibers, a biocompatible organic matrix, can induce tumor antigen-specific CTL response and suppress tumor growth in vivo. Radially grown ZnO NWs on PLLA fibers were complexed with a well-known tumor antigen, carcinoembryonic antigen (CEA),13 fused with ZnO-binding peptide (ZBP) to be exploited as a novel intracellular antigen delivery system. In addition, we also confirmed the immune stimulatory effect of the inorganic–organic hybrid nanocomposites in vitro using phagocytic dendritic cells, serving as a cellular bridge between innate and adaptive immunity.16 Immunization with the CEA-coated hybrid nanocomposites not only enhanced antigen-specific T cell responses, but also suppressed tumor growth significantly better than PLLA fibers mixed with CEA in an in vivo mouse model, demonstrating that incorporation of ZnO NWs into the hybrid composite has promising potential as vaccine adjuvant for antigen delivery as well as immune stimulation.
Experimental
Synthesis of PLLA fiber–ZnO nanowire composites
A solution of PLLA polymer and ZnO NPs was prepared before electrospinning. Hexadecyltrimethylammonium bromide (CTAB, CH3(CH2)15N(Br)(CH3)3, 98%, Aldrich) capped 5 nm ZnO NPs were synthesized by alkaline solution-based method according to a previous report (Fig. S1†).2 Then, PLLA polymers (M.W.: 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–160000, Aldrich) were dissolved in chloroform (15% w/v) for several hours with magnetic stirring. The ZnO NP solution at a concentration of 10 mg ml−1 in ethanol was added to the prepared PLLA solution and mixed by vortex mixer and ultrasonication. We fixed the volume ratio of PLLA and ZnO solution at 3:1. The blended solution was drawn into a 10 mL syringe mounted with a 21-gauge metal needle tip with a diameter of 0.5 mm. The distance between needle and collector was adjusted to 10 cm. PLLA fiber–ZnO NP membranes were collected by eletrospinning method at a voltage of 10 kV. Finally, PLLA fiber–ZnO NW composites were synthesized via hydrothermal method3 by which ZnO crystals are grown from ZnO NP embedded in PLLA fibers. 10 mM of zinc nitrate hexahydrate, (Zn(NO3)·6H2O, >98%, Aldrich) and 10 mM of hexamethylenetetramine (HMTA, C6H12N4, 99%, Aldrich) were dissolved in deionized water to synthesize the ZnO NWs on the PLLA fibers.
000–160000, Aldrich) were dissolved in chloroform (15% w/v) for several hours with magnetic stirring. The ZnO NP solution at a concentration of 10 mg ml−1 in ethanol was added to the prepared PLLA solution and mixed by vortex mixer and ultrasonication. We fixed the volume ratio of PLLA and ZnO solution at 3:1. The blended solution was drawn into a 10 mL syringe mounted with a 21-gauge metal needle tip with a diameter of 0.5 mm. The distance between needle and collector was adjusted to 10 cm. PLLA fiber–ZnO NP membranes were collected by eletrospinning method at a voltage of 10 kV. Finally, PLLA fiber–ZnO NW composites were synthesized via hydrothermal method3 by which ZnO crystals are grown from ZnO NP embedded in PLLA fibers. 10 mM of zinc nitrate hexahydrate, (Zn(NO3)·6H2O, >98%, Aldrich) and 10 mM of hexamethylenetetramine (HMTA, C6H12N4, 99%, Aldrich) were dissolved in deionized water to synthesize the ZnO NWs on the PLLA fibers.
Characterization of PLLA fiber–ZnO nanowire composites
The morphology and microstructure of the ZnO NPs and PLLA fiber–ZnO NW composites were analyzed using scanning electron microscopy (SEM, Hitachi, SU 70, Japan) at an accelerating voltage of 15 kV or transmission electron microscope (TEM, JEM-2100F, JEOL, Japan) at an accelerating voltage of 200 kV. The samples for SEM and TEM experiments were prepared on conductive carbon tape and carbon-coated copper grid, respectively. The ZnO nanoparticles were diluted in absolute ethanol and then a drop of the sample solution was placed on the grid. For the PLLA microfibers, we carefully mounted a thin layer of microfiber on the grid and wetted it with ethanol to improve adhesion. All samples were dried in a vacuum oven at room temperature before the SEM and TEM observations. The crystal structure and crystallite size of the samples were determined by measuring powder X-ray diffraction (XRD, PANalytical, X'Pert ProMPD, U.K.) with Cu Kα radiation (λ = 1.5406 Å). Measurements were conducted in a θ–2θ geometry from 30° to 60° at 45 kV and 40 mA tube power.Mechanical fragmentation of PLLA–ZnO nanocomposites
In order to mechanically fragment the composites, 10 mg of PLLA or PLLA–ZnO membranes were dispersed in dimethyl sulfoxide (DMSO) for 30 min and serially passed through 10 times using a syringe with 18G, 23G, and 26G needles. The solution containing fragmented composites was then added to an 80% sucrose solution, and PLLA–ZnO nanocomposites were separated from PLLA fibers by density gradient centrifugation (3515g, 30 min). The composites obtained from the bottom layer were collected and further filtrated using a 70 μm cell strainer (Sigma Aldrich, USA). The final fragmented PLLA–ZnO nanocomposites were washed with phosphate-buffered saline (PBS) and used for further application. The size distribution of the fragmented complexes was manually measured under a microscope. The procedure for fragmentation and separation of PLLA–ZnO nanocomposites is summarized in Fig. S2.†Binding of ZBP peptide and ZBP–CEA antigen
Indicated amounts of ZnO-binding peptides (ZBP, amino acid sequence: RPHRKGGDARPHRKGGDARPHRKGGDA)1, labeled at their carboxyl terminals with fluorescein isothiocyanate (FITC, Peptron, Korea), were incubated with PLLA–ZnO nanocomposites for 1 h at 37 °C. ZnO nanowires coated with ZBP–FITC were washed 3 times with PBS, and then the relative binding of ZBP–FITC was assessed by measuring fluorescence intensity using Infinite®M200 PRO (Taken, Switzerland). For ZBP–CEA binding assay, 3× ZBP–CEA proteins purified as previously described1 were incubated with PLLA–ZnO nanocomposites for 1 h at room temperature. The bound proteins on the nanocomposites were collected by centrifugation and quantitated by Coomassie blue staining after SDS-PAGE.Cell viability and toxicity assay
A mouse dendritic cell line, DC2.4,17 was maintained in Roswell Park Memorial Institute medium (RPMI 1640) (Welgene, Korea), supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Welgene), 100 U ml−1 penicillin, and 100 μg ml−1 streptomycin (Gibco BRL, USA) at 37 °C in the presence of 5% CO2. The viability of DC2.4 cells treated with the indicated complexes was investigated up to 24 h by trypan blue staining and apoptosis assay. The cells were collected by trypsinization, and live cells were counted after trypan blue staining. Cellular apoptosis and necrosis were assessed by staining with annexin V-FITC (eBioscience, USA) and 7-aminoactinomycin D (7-AAD, Thermo Fisher Scientific, USA) using a flow cytometer (LSR II, BD Biosciences, USA) according to the manufacturer's instruction, as previously described.3Cytokine analysis
Cytokines secreted in culture supernatants were analyzed by cytometric bead array using a Mouse Inflammation Kit (BD Biosciences), according to the manufacturer's instructions. Culture media were changed before DC2.4 stimulation and the production of tumor necrosis factor (TNF)-α, IL-1β, IL-6, and IL-10 from stimulated DCs was measured 18 h after incubation.Flow cytometry analysis
PLLA and PLLA–ZnO nanocomposites were incubated with ZBP–FTIC at room temperature for 1 h and washed 3 times with PBS to remove unbound peptides. Subsequently, this complex was incubated with DC2.4 at 37 °C for the indicated times. The cellular uptake of ZBP–FITC was examined after harvesting the cells using a flow cytometer (LSR II, BD Biosciences). To analyze cellular activation, DC2.4 cells, cultured with PLLA and PLLA–ZnO nanocomposites, were washed with ice-cold FACS buffer (PBS containing 1% bovine serum albumin [BSA] and 1 mM EDTA) and blocked on ice for 30 min with ultra-block solution containing 10% rat sera, 10% hamster sera, 10% mouse sera (Sigma), and 10 μg ml−1 of 2.4G2 monoclonal antibody (BD Pharmingen, USA). DC maturation was examined after staining with FITC-conjugated anti-I-Ab (BD Pharmingen), PE-conjugated anti-CD40 (BD Pharmingen), APC-conjugated anti-CD80 (eBioscience), and PE/Cy7-conjugated anti-CD86 (BioLegend, USA) for 30 min on ice. The cells were analyzed using a flow cytometer.Lymphocytes collected from the spleens of immunized mice were cultured for 16 h in RPMI 1640 medium supplemented with 10% heat-inactivated FBS, 50 nM β-ME (Invitrogen, USA), 100 units per ml penicillin streptomycin, 2 mM L-glutamine (Welgene), and 20 μg ml−1 of purified CEA proteins in 24-well, flat-bottomed culture plates (5 × 106 cells per well). After incubation, GolgiPlug (BD Biosciences) was added for 6 h. Lymphocytes were washed three times with ice-cold FACS buffer. Cells were blocked with ultra-block solution for 30 min on ice and stained with BV-605-conjugated CD4 and Pe-cy7-conjugated CD8 antibodies (BD Pharmingen) for 30 min at 4 °C. After surface CD4 or CD8 staining, cells were washed 3 times with ice-cold FACS buffer and subjected to intracellular cytokine staining using the cytofix/cytoperm kit, according to the manufacturer`s instructions (BD Biosciences). Intracellular interferon (IFN)-γ) was stained using an APC-conjugated anti-IFN-γ antibody (BD Pharmingen) for 30 min at 4 °C. PE-conjugated anti-FoxP3 antibody (eBioscience) was also used to detect regulatory CD4 T cells. The stained cells were analyzed with a FACS Canto II flow cytometer (BD Biosciences). Flow cytometric data were analyzed by FlowJo software (version 8.8.6, FlowJo, USA).
Cytotoxic T lymphocyte (CTL) activity
Two weeks after the second vaccination, splenocytes were harvested from the immunized mice and CTL activity was measured using a crystal violet absorbance assay as previously described.18 CTLs were generated by stimulating the splenocytes with purified CEA antigen for 16 h followed by incubation with MC38/CEA cells, mouse adenocarcinoma cell line expressing human CEA antigen,1 as a target for 24 h. After washing, the target cells were stained with crystal violet (4 mg ml−1 in PBS) for 30 min at room temperature. The plate was then washed with PBS and the cells were lysed using methanol. Target cell survival was analyzed by measuring absorbance at 570 nm.Tumor growth
C57BL/6 mice (Orient Bio, Seoul, South Korea) were housed and maintained in the specific pathogen-free facility at Seoul National University (SNU) College of Medicine. Animal experiments were approved by the SNU IACUC (permission ID: SNU-090805-5) and performed in strict accordance with the recommendations in the National Guide Line for the care and use of laboratory animals. Tumor-bearing mice were humanely sacrificed when tumors exceeded 2 cm3 on the day of observation. Six-week-old C57BL/6 mice were injected subcutaneously at the right flank with MC38/CEA cells (1 × 105 cells per mouse). Seven days after tumor cell injection, the mice were immunized at the tail base with the indicated complex 3 times at weekly intervals. Five mice were used for each group. Tumors were measured by digital caliper (TESA SHOP-CAL, Willrich Precision, Switzerland). The tumor volume (mm3) was calculated using equation (A × B2)/2, where A is the long diameter and B is the short diameter. Tumor-bearing mice were humanely sacrificed with CO2 asphyxiation when tumors exceeded 2 cm in diameter on the day of observation and the surviving mice were monitored up to 30 d after tumor inoculation.Isolation of tumor infiltrating leukocytes
Tumor tissues were obtained aseptically from mice, cut into fragments, washed twice with PBS, and then digested with an enzyme cocktail (DNase and Collagenase IV, Sigma Aldrich) as described previously.19 The resulting cell suspension was filtered through nylon mesh screens to remove cell clumps, washed 3 times with PBS and resuspended in ice-cold FACS buffer. The tumor-derived cell mixtures were used for FACS analysis.Results and discussion
Characteristic features of synthesized PLLA fiber–ZnO NW composites are presented in Fig. 1. As shown in the scanning electron microscopy (SEM) images, ZnO NWs grew uniformly on the PLLA fibers. The average diameter of the PLLA fibers is 1.25 μm. After 2 h of hydrothermal reaction, the ZnO NW length increased to 1375 ± 247 nm and diameter of 122.5 ± 4 nm (Fig. S1D†). It has been shown that ZnO NWs can be grown as radial bundles from ZnO NPs dispersed in solution or coated on various substrates.3,20,21 However, it is interesting to note that we obtained a similar morphology of ZnO NWs as previous reports even with the seed layer of ZnO NPs inside the PLLA fibers (Fig. S1C†). The precursors dissolved in an aqueous solution as Zn2+ and OH− diffuse into PLLA fibers during hydrothermal reaction, and the ZnO crystals gradually nucleate and grow as radial NWs on the surface of ZnO NPs, potentially exposed on PLLA fibers (Fig. S1C†), serving as seeds. Furthermore, we confirmed that the ZnO NWs are synthesized evenly throughout the PLLA fiber membrane, which suggests that it is possible to scale up this methodology to mass production. We measured the XRD patterns to analyze the crystal structures of the composites. The diffraction peaks in Fig. 1B appear at 31.8°, 34.4°, 36.3°, 47.5°, 62.8°, and 67.7°, which can be assigned to (100), (002), (101), (102), and (110) planes of the ZnO NWs with hexagonal closed packed structure (PDF no. 36-1451), respectively.22 The diffraction peaks of ZnO NWs become sharper as ZnO NWs are grown from ZnO NPs. In order to obtain injectable forms of PLLA fiber–ZnO NW composites for in vivo testing, we fragmented and collected the PLLA fiber–ZnO NW composites by density gradient centrifugation after mechanical shearing in a DMSO solution using a syringe with 18G, 23G, and 26G needles (Fig. S2†). Fragmented composites ranged 1–70 μm in diameter, with an average of 15 μm.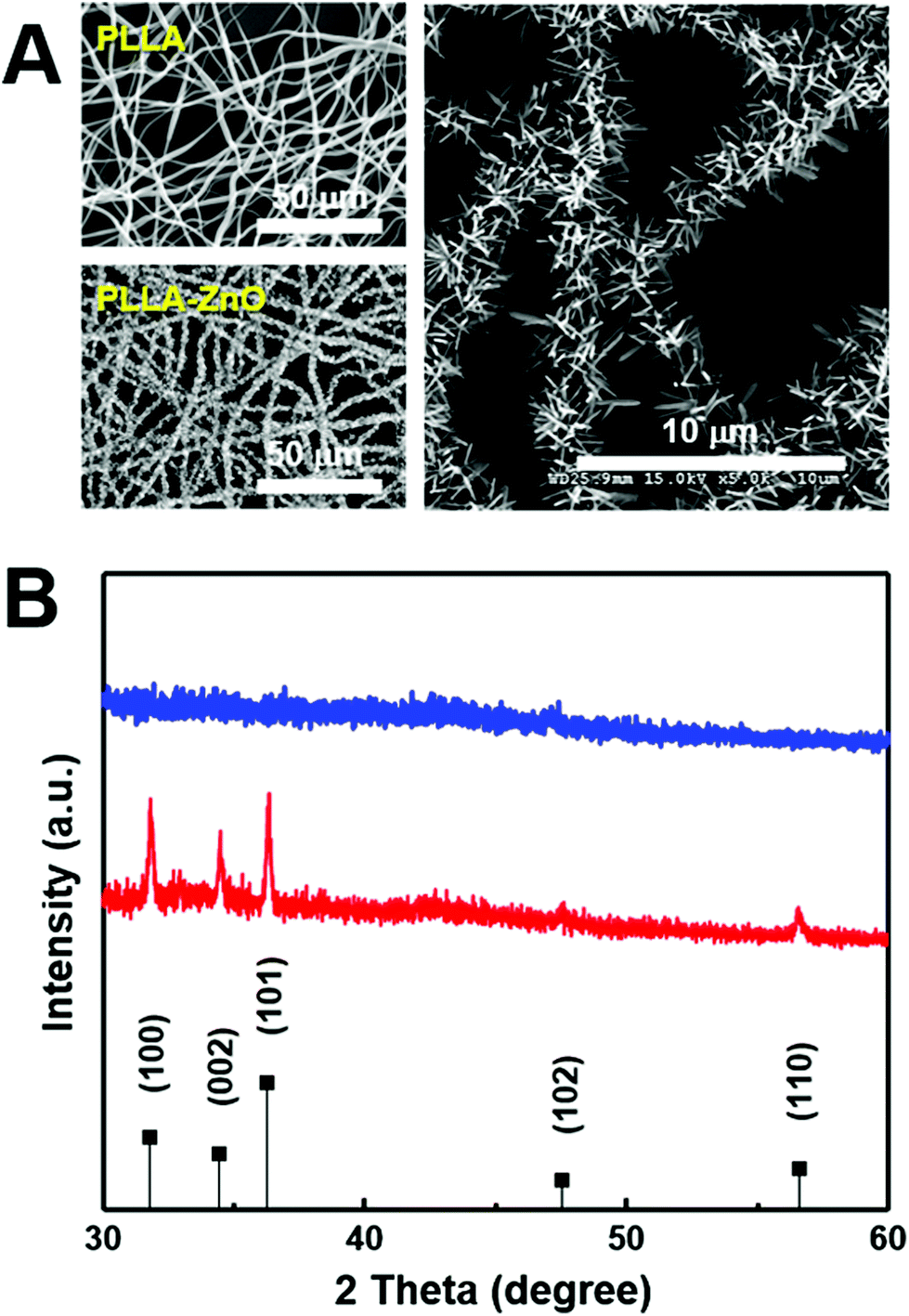 | ||
| Fig. 1 (A) SEM images of PLLA fibers (top left) and PLLA fiber–ZnO NW composites (left bottom and right at different magnifications). (B) XRD patterns of PLLA fiber (blue) and PLLA fiber–ZnO NW composite (red). ZnO peak positions are denoted in the bottom of the panel (B) by the black vertical line. | ||
To assess the cellular toxicity of PLLA fiber–ZnO NW composites, DC2.4 cells were treated with the nanocomposites, and live cell counts were compared with untreated control cells or PLLA fiber-treated cells (Fig. 2). Live cell counts gradually increased after 6 h of lag phase, indicating that DC2.4 cells can replicate in the presence of PLLA fiber–ZnO NW composites, albeit at a lower growth rate than that of untreated cells or PLLA fiber-treated cells (Fig. 2A). To assess for potential toxicity during the lag phase, we examined cellular necrosis and apoptosis (Fig. 2B and 2C). Apoptotic cell death gradually increased up to 12 h after incubation with PLLA fiber–ZnO NW composites, suggesting that cellular apoptosis is induced in a fraction of cells by PLLA fiber–ZnO NW composites, but then decreases to normal levels, comparable to that of untreated or PLLA fiber-treated cells 24 h after incubation. Even though the induction of apoptosis was also observed in cells treated with PLLA fibers, ZnO NW might be responsible for enhanced apoptotic cell death. Cells treated with PLLA fiber–ZnO NW composites showed a similar degree (4.1–5.3%) of early apoptosis when compared to those (4.1–5.5%) treated with PLLA fibers up to 24 h of incubation. However, late apoptotic cells, positive for both 7-aminoactinomycin D (7-AAD) and annexin V, peaked (∼25.5%) at 12 h after incubation with cells treated with PLLA fiber–ZnO NW composites. This value was significantly lower (4.7–14.5%) throughout the incubation period in cells treated with PLLA fibers. Interestingly, necrotic cells, as measured by membrane penetration of 7-AAD without annexin V staining, were barely detected (generally less than 2.0%) in all the experimental groups, suggesting that direct physical injury exerted by PLLA fibers or ZnO NWs might be negligible.
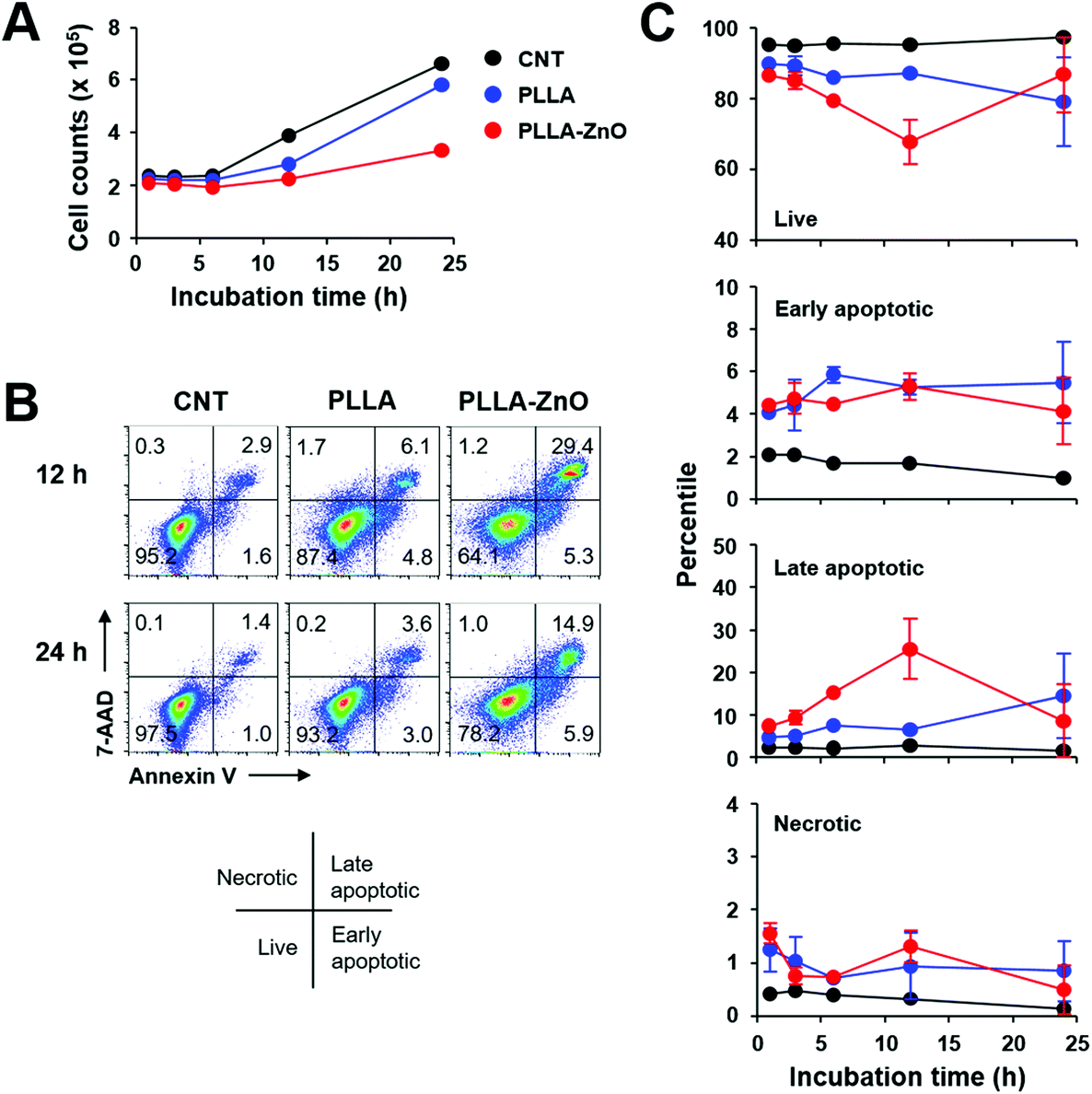 | ||
| Fig. 2 The effect of PLLA fiber–ZnO NW composites on cell viability. (A) DC2.4 cells were grown in the absence (CNT) or presence of 250 μg of PLLA fibers (PLLA) or PLLA fiber–ZnO NW composites (PLLA–ZnO), and their growth was monitored for up to 24 h. (B and C) Cellular apoptosis and necrosis were assessed by flow cytometry after annexin V and 7-AAD staining at the indicated times after cell culture. | ||
The kinetic responses of cellular apoptosis induced by PLLA fiber–ZnO NW composites were slightly different from those observed in our previous study using human embryonic kidney (HEK) 293 cells and ZnO NW arrays grown on glass coverslip.3 Early apoptotic phenotype peaked (∼40%) at 3 h and late apoptosis peaked (∼35%) at 6 h of incubation in HEK293 cells, grown on glass coverslips coated with ZnO NWs. This observation might be due to differences in cell type (phagocytic dendritic cell, DC2.4 vs. fibroblast, HEK293) or culture matrix (plastic culture plate vs. glass cover slip). Nevertheless, the degree of cellular apoptosis, but not necrosis, was significantly higher in both cell types during the initial stage of incubation, primarily due to the ZnO NWs. Since cellular apoptosis was rapidly induced during the early phase of incubation with ZnO NWs and gradually declined thereafter, cellular adaptation potentially helps to overcome apoptotic stress exerted by ZnO NWs, and cells can start to replicate even in the presence of ZnO NW composites, as consistently observed in our previous3 and current studies.
To utilize PLLA fiber–ZnO NW composites as an antigen delivery system, we used a ZnO-binding peptide (ZBP)1 and examined its binding capacity for nanocomposites (Fig. 3). The binding of ZBP–FITC to PLLA fiber–ZnO NW composites was assessed at titrated concentrations of ZBP–FITC after incubation for 1 h and then measured by fluorescence intensity and fluorescent microscopy (Fig. 3A and B). ZBP–FITC was saturated at ∼3.2 μM with 250 μg of PLLA fiber–ZnO NW composites. Although the peptide can bind nonspecifically to PLLA fibers and was saturated at the same concentration, the amount of bound peptides was approximately one third of that bound to PLLA fiber–ZnO NW composites, as measured by fluorescence intensity (Fig. 3B). Next, we used recombinant carcinoembryonic antigen (CEA) fused with 3× ZBP as a model antigen to investigate the applicability of the nanocomposites in cancer immunotherapy.1 ZBP–CEA consistently showed enhanced binding to PLLA fiber–ZnO NW composites when compared with PLLA fibers (Fig. 3C and D). Approximately 22 μg of ZBP–CEA protein showed saturated binding to 250 μg of PLLA fiber–ZnO NW composites, whereas only 15 μg of ZBP–CEA was needed to saturate the same amount of PLLA fibers when incubated with 80 μg of the antigen for 1 h. Non-specific adsorption of a protein, ovalbumin, has also been previously reported.23 Nevertheless, our results show that a tumor-associated protein antigen fused with ZBP could be more efficiently immobilized on PLLA fiber–ZnO NW composites than unmodified PLLA fibers, primarily via enhanced affinity to ZnO NW endowed by conjugated ZBP.1,2 In addition, the larger surface area of ZnO NWs may enhance the binding capacity of the nanocomposites over naked PLLA fibers.
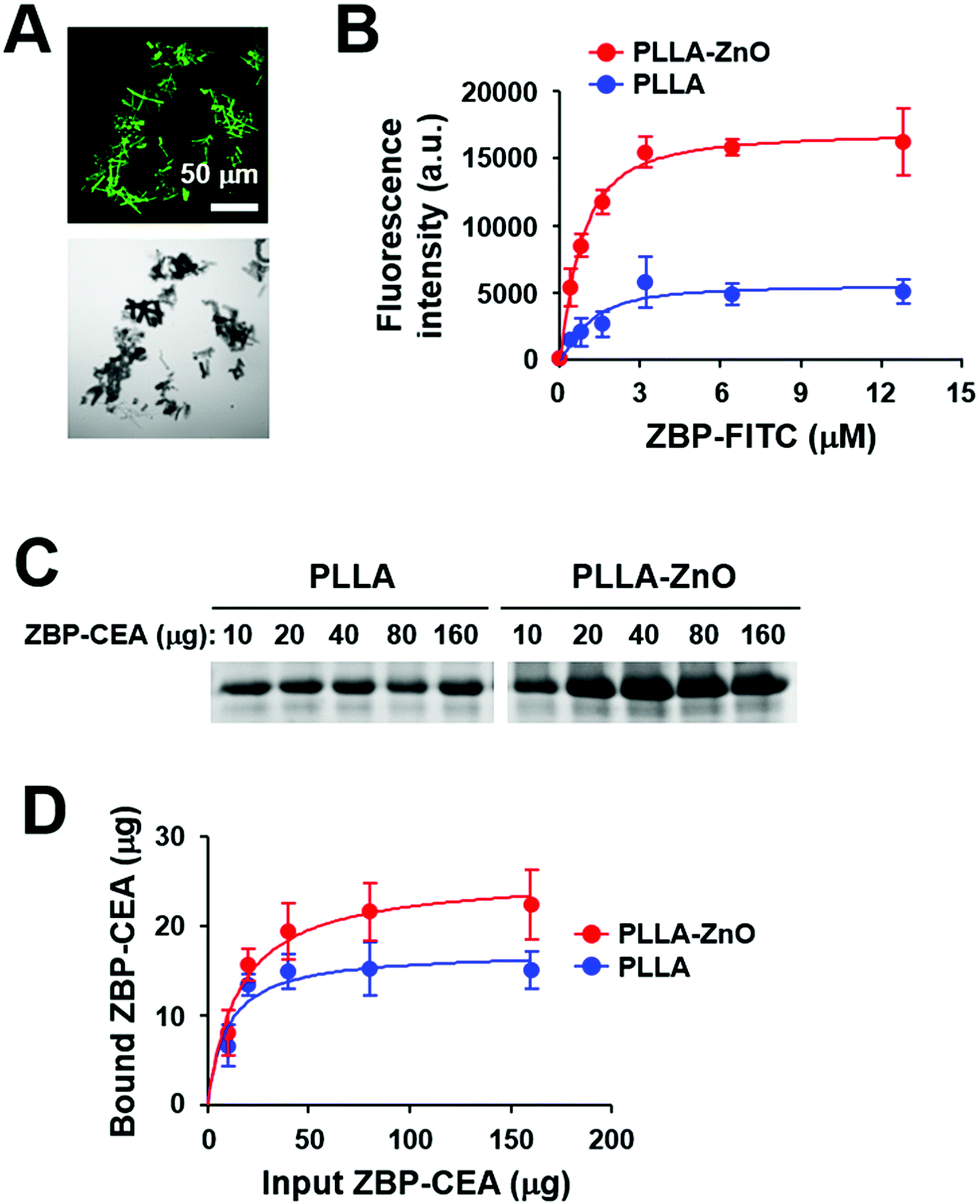 | ||
| Fig. 3 Immobilization of polypeptide on PLLA fiber–ZnO NW composites. (A) Microscopic images of fragmented PLLA fiber–ZnO NW composites coated with ZBP-labeled with FITC. Representative fluorescence (top) or differential interference contrast (bottom) images of the nanocomposite fragments are presented. (B) Indicated amounts of ZBP–FITC were incubated with the composites (250 μg) at room temperature for 1 h, and the relative binding of ZBP–FITC was assessed by measuring fluorescence intensity (a.u., arbitrary unit). The data are from three separate experiments. (C) Gel electrophoresis data showing the relative fraction of bound CEA antigen to PLLA fibers (PLLA) or PLLA fiber–ZnO NW composites (PLLA–ZnO). Bound CEA was resolved by sodium dodecyl sulphate polyacrylamide gel electrophoresis after incubation with the composites (250 μg) and the indicated amounts of CEA. (D) Bound CEA amounts from (C) were quantitated from three separate experiments and presented. | ||
Next, we assessed the efficacy of intracellular delivery of the associated antigen in dendritic cells by using the composites, since we previously observed efficient delivery of biomacromolecules, coated on the surface of radially grown ZnO NWs, into eukaryotic cells.3 As shown in Fig. 4A, most of the live cells (>90%) collected after 30 min of incubation at 37 °C on ZBP–FITC-coated PLLA fiber–ZnO NW composites were positive for FITC signal, and the fluorescence intensities were saturated thereafter. Only a fraction of DC2.4 cells (∼35%) incubated with an equal amount of PLLA fiber mixed with ZBP–FITC showed positive signals for FITC after 30 min of incubation, which rapidly declined to ∼10% thereafter. These results indicate that the peptide molecules coated on the nanowires are efficiently and rapidly delivered into antigen-presenting cells. The rapid saturation of the peptides also suggests that cells potentially uptake the peptide molecules coated on the tips of ZnO NWs right after cellular contact, as observed in our previous study.3
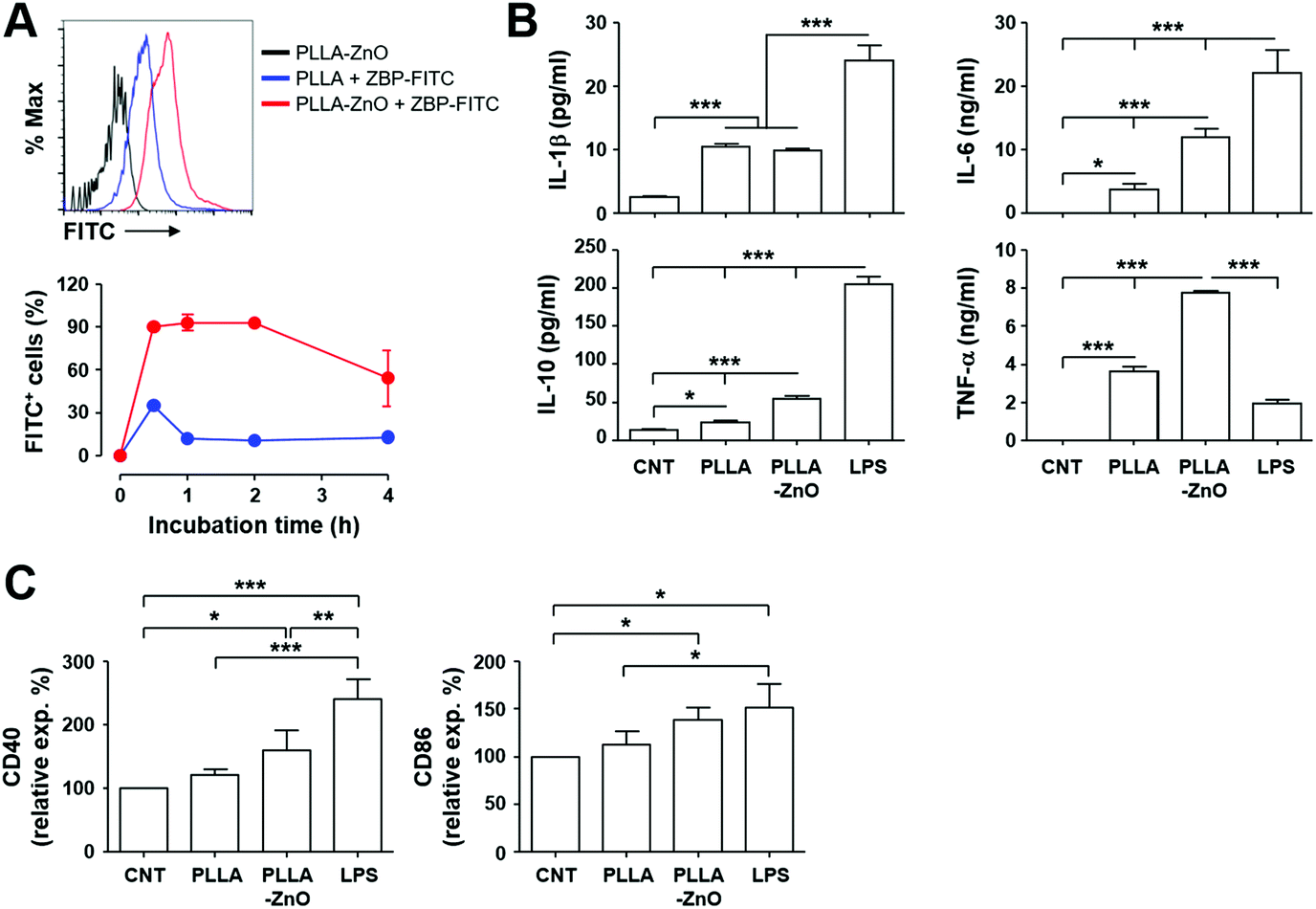 | ||
| Fig. 4 Intracellular uptake of peptides coated on PLLA fiber–ZnO NW composites and their stimulatory effect on dendritic cells. (A) DC2.4 cells were incubated with the indicated complexes coated with ZBP–FITC at 37 °C for the indicated times and analyzed by flow cytometry. Live cells were gated based on a scatter plot, and fluorescence intensities of cells were assessed. Representative flow cytometric data (top) and the kinetic changes of FITC-positive cellular fraction (bottom) are presented. (B) DC2.4 cells were incubated with the indicated complexes at 37 °C for 18 h and inflammatory cytokines were measured in the culture supernatants (n = 4). (C) Phenotypic changes of DC2.4 cells after stimulation with the indicated complexes were examined by staining for cellular surface markers. The relative surface expression levels of the indicated molecules in comparison with untreated cells (CNT) are presented (n = 4, see also Fig. S2† for MHC II and CD80). *, p < 0.05; **, p < 0.01; ***, p < 0.001. | ||
Since we and others consistently observed immunomodulatory and/or inflammatory effects of ZnO nanostructures on APCs, including macrophages and dendritic cells,2,7,10,24 we measured inflammatory cytokines secreted by DC2.4 cells after stimulation with PLLA fiber–ZnO NW composites. Treatment of the nanocomposites induced significantly higher levels of inflammatory cytokines, such as IL-1β, IL-6, IL-10, and TNF-α, when compare with untreated or PLLA fiber-treated cells. However, these cytokine responses, except TNF-α, were generally lower in cells stimulated with LPS as positive control (Fig. 4B). Nevertheless, all the cytokine levels, except IL-1β, were further elevated in cell culture supernatants from cells treated with the PLLA fiber–ZnO NW composites, when compared to those from PLLA fiber-treated cells, suggesting that ZnO NWs might be the primary stimulatory components in enhancing inflammatory responses in dendritic cells.
To further confirm cellular activation of dendritic cells, we examined activation markers after stimulation with the nanocomposites. The surface expression of the activation markers, CD40 and CD86, significantly increased after stimulation with PLLA fiber–ZnO NW composites when compared to that of untreated cells (Fig. 4C), whereas the expression of MHC II and CD80 only slightly increased without statistical significance (Fig. S3†). None of the activation markers were significantly elevated in cells treated with PLLA fibers, suggesting again that ZnO NWs might be the primary stimulants for cellular activation, even though to a lesser extent than stimulation with LPS. Taken together, these results clearly demonstrate that PLLA fiber–ZnO NW composites can stimulate antigen presenting phagocytes and induce inflammatory responses, largely owing to ZnO NW components.
Significant induction of inflammatory cytokines as well as activation of dendritic cells by PLLA fiber–ZnO NW composites prompted us to examine the potential applicability of the nanocomposites as immune-adjuvants for cancer immunotherapy. We directly immunized C57BL/6 mice with PLLA fiber–ZnO NW composites (1 mg per mouse) mixed with 100 μg of ZBP–CEA and analyzed CEA-specific cellular immunity. At 2 weeks after the second immunization, we examined T-cell responses in splenocytes by measuring their production of IFN-γ, a hallmark cytokine for cell-mediated immunity, in an antigen-dependent manner (Fig. 5A). The frequencies of IFN-γ-secreting CD4 or CD8 T cells in spleens of mice, immunized with the nanocomposites complexed with ZBP–CEA, significantly increased, when compared to those of non-immunized controls (CNT and PLLA–ZnO) or mice, immunized with PLLA fibers mixed with the same amount of ZBP–CEA antigens. It is notable that the presence of ZnO NWs in the complex significantly enhanced antigen-specific T cell responses by approximately 70–100%. Furthermore, substantial cytotoxic responses against CEA-expressing cancer cells (MC38/CEA) were induced in mice immunized with the nanocomposites complexed with ZBP–CEA, whereas no significant cytotoxicity was detected in the control groups (Fig. 5B). Even though splenocytes from mice immunized with PLLA fiber mixed with ZBP–CEA enhanced cytotoxic activity against cancer cells expressing CEA, those from mice immunized with the nanocomposites coated with the cancer antigen always showed slightly better CTL activity, but without statistical significance.
 | ||
| Fig. 5 (A) CEA-specific, IFN-γ-positive CD4 and CD8 T-cell responses of mice immunized with the indicated complexes. IFN-γ-positive T cells in splenocytes were detected in the presence (+) or absence (−) of CEA. Top panels: representative dot plots. Bottom panels: average percentiles of T cells from three independent experiments. (B) Cytotoxic activity of splenocytes from immunized mice. MC38 cells expressing CEA (MC38/CEA) were used as targets. (C) Tumor volume of mice (5 mice per group) injected with MC38/CEA cells. Mice were immunized three times with the indicated complexes at 7, 14, and 21 days after tumor cell injection. Mice bearing tumors over 2 cm3 at the day of observation were humanely sacrificed. Representative images of tumors collected from sacrificed mice and PLLA–ZnO/CEA group at day 30. **, p < 0.01; ***, p < 0.001. | ||
To further prove the functional significance of CEA-specific adaptive immunity for anti-cancer immunotherapy in vivo, mice inoculated with MC38/CEA cells were immunized 3 times at weekly intervals and tumor growth was monitored for a month. As shown in Fig. 5C, tumor growth was significantly suppressed in mice immunized with PLLA fiber–ZnO NW composites coated with ZBP–CEA and all of them survived, whereas other control treatments failed to protect the mice during the observation period and all the mice were sacrificed due to enlarged tumors. Since we observed that mice immunized with PLLA fibers, mixed with ZBP–CEA, also showed a significant increase in CEA-specific T cell responses but failed to suppress tumor growth, systemic immunosuppression and the infiltration of T cells in growing tumors were assessed by the proportion of T cells in spleens and tumors, respectively, at day 24 after tumor inoculation (Fig. 6). The systemic reduction of T cells as well as the gradual increase of regulatory T cells (TReg) in tumor-bearing hosts have been well established.25–27 In addition, immune contextures of growing tumors, especially tumor infiltrating CTLs, have been shown to correlate with clinical outcomes.15 In the spleens of tumor-bearing mice, we observed a drastic reduction of CD4 and CD8 T cells to generally less than 1 and 2% among whole splenocytes, respectively (Fig. 6A and Fig. S4†). In normal mice, CD4 and CD8 T cells generally represent 20 and 10% of splenocytes, respectively,25 indicating that all tumor-bearing mice suffered from systemic T cell suppression regardless of immunization. Nevertheless, mice immunized with PLLA fiber–ZnO NW composites, complexed with ZBP–CEA, retained significantly higher frequencies of CD4 T cells with lower TReg cell proportions in spleens than control groups. Additionally, this mice group showed significantly higher levels of tumor-infiltrating CD4 and CD8 T cells, despite similar levels of CD45+ immune cells and TReg cells in tumor tissues (Fig. 6B), suggesting a better immune contexture in the tumor microenvironment than in control groups.15 Taken together, these results suggest that inclusion of ZnO NWs complexed with a tumor antigen in a therapeutic cancer vaccine formulation, based on PLLA fiber matrix, can enhance antigen-specific T cell response and promote their infiltration into tumors, thereby, suppressing the tumor growth better than complexes without ZnO NWs.
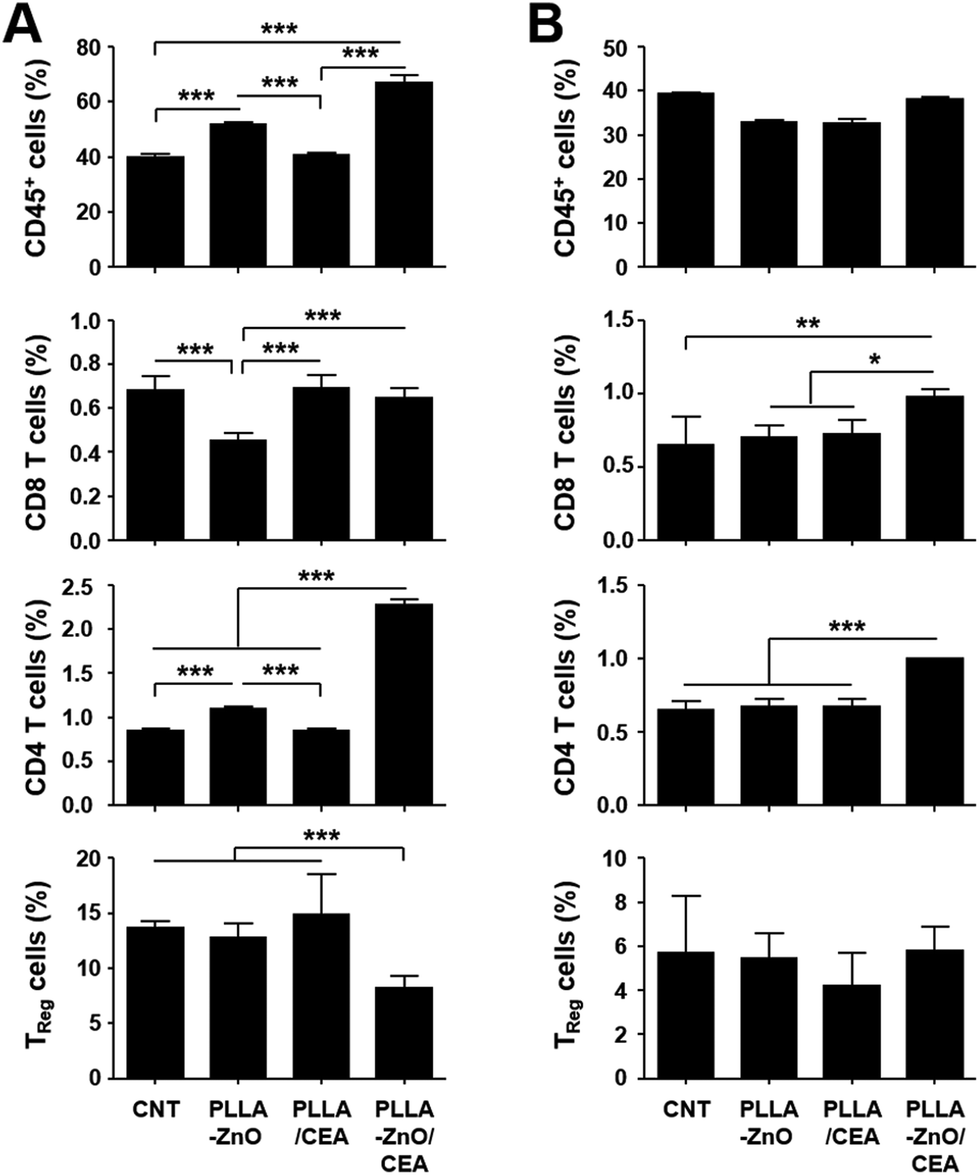 | ||
| Fig. 6 Relative fraction of hematopoietic immune cells (CD45+), CD4+, CD8+, and regulator T cells (TReg, CD4+/FoxP3+) were quantitated in spleens (A) or in tumor tissues (B) at day 24 after tumor inoculation in the indicated mice groups. See also Fig. S3† for representative flow cytometric data. CNT: non-immunized mice. *, p < 0.05; **, p < 0.01; ***, p < 0.001. | ||
The biocompatible PLLA-based formula has shown immense potential as a drug delivery system and as a scaffold for various biomedical applications.28 This organic material has been successfully applied to vaccine development for infectious diseases and cancers.29–33 However, recent studies have reported that a similar organic material, poly lactic-co-glycolic acid (PLGA), can also induce immune tolerance against the associated peptides for the treatment of autoimmune diseases, depending on particle size, shape, composition, and route of administration.34,35 It was shown that tolerogenic effects require PLGA microparticle uptake by macrophages expressing the scavenger receptor MARCO and are, in part, mediated by the activity of regulatory T cells, abortive T-cell activation and T-cell anergy.35 In addition, inert PLLA itself may have immune suppressive effects and/or promote tissue repair and foreign body reactions,36,37 enabling them to be used as scaffolds for tissue engineering. Therefore, considerable modification of the composition and structure of the inert organic material might be critical for its biomedical application as a vaccine carrier and/or adjuvant.33 The incorporation of immune-stimulating agents, such as Toll-like receptor (TLR) ligands, into the formulation is promising for cancer vaccine development to boost adaptive immunity.33 Here, we synthesized radially grown ZnO NWs on PLLA microfibers with a unique 3-dimensional structure and applied them as therapeutic cancer vaccines. Similar hybrid composites, comprised of ZnO nanostructures and a PLLA matrix, have been used for photocatalytic water purification,38 an anti-bacterial agents,39 and cellular differentiation of myoblasts.40 Nevertheless, our current study is the first application of the hybrid nanocomposite as a vaccine adjuvant. This inorganic–organic hybrid nanocomposite has mild cellular toxicity and can efficiently stimulate innate APCs and dendritic cells to express inflammatory cytokines and activation surface markers (Fig. 4). Induction of these innate immune responses may result from the direct recognition of ZNPs by Toll-like receptors (TLR4 or TLR6) or indirectly, by intracellular ROS generation resulting from disrupted cellular zinc homeostasis.6,9 Induction of in vivo inflammation by ZnO NWs was also previously reported.24
Conclusions
In this study, we demonstrated that PLLA fiber–ZnO NW hybrid nanocomposites can successfully induce tumor antigen-specific cellular immunity and significantly inhibit tumor growth in vivo, potentially, via efficient intracellular delivery of associated tumor antigen into APCs as well as stimulation of innate immunity. The multifunctional interaction of ZnO nanostructures with biological systems may also contribute to their usage as attractive anticancer agents.41 Even though the mechanistic correlation of ZnO NWs to systemic reduction of TReg and enhanced infiltration of T cells into tumor tissues needs to be further defined in a future study, our current results open a new avenue that extends the biomedical application of inorganic metal oxide-inert organic hybrid nanocomposites42 for cancer immunotherapy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Nano & Material Technology Development Program through the National Research Foundation of Korea (No. 2014M3A7B4052192). P. S., J. W. L., and N. Y. J. received a scholarship from the BK21-plus education program provided by the National Research Foundation of Korea.References
- N. H. Cho, T. C. Cheong, J. H. Min, J. H. Wu, S. J. Lee, D. Kim, J. S. Yang, S. Kim, Y. K. Kim and S. Y. Seong, Nat. Nanotechnol., 2011, 6, 675–682 CrossRef CAS PubMed.
- N. Y. Ha, H. M. Shin, P. Sharma, H. A. Cho, C. K. Min, H. I. Kim, N. T. Yen, J. S. Kang, I. S. Kim, M. S. Choi, Y. K. Kim and N. H. Cho, J. Nanobiotechnol., 2016, 14, 76 CrossRef PubMed.
- P. Sharma, H. A. Cho, J. W. Lee, W. S. Ham, B. C. Park, N. H. Cho and Y. K. Kim, Nanoscale, 2017, 9, 15371–15378 RSC.
- H. M. Xiong, Adv. Mater., 2013, 25, 5329–5335 CrossRef CAS PubMed.
- R. Roy, S. Kumar, A. K. Verma, A. Sharma, B. P. Chaudhari, A. Tripathi, M. Das and P. D. Dwivedi, Int. Immunol., 2014, 26, 159–172 CrossRef CAS PubMed.
- R. Roy, M. Das and P. D. Dwivedi, Mol. Immunol., 2015, 63, 184–192 CrossRef CAS PubMed.
- R. Roy, S. K. Singh, M. Das, A. Tripathi and P. D. Dwivedi, Immunology, 2014, 142, 453–464 CrossRef CAS PubMed.
- S. R. Saptarshi, B. N. Feltis, P. F. A. Wright and A. L. Lopata, J. Nanobiotechnol., 2015, 13, 6 CrossRef PubMed.
- S. R. Saptarshi, A. Duschl and A. L. Lopata, Nanomedicine, 2015, 10, 2075–2092 CrossRef CAS PubMed.
- J. Palomaki, P. Karisola, L. Pylkkanen, K. Savolainen and H. Alenius, Toxicology, 2010, 267, 125–131 CrossRef CAS PubMed.
- I. De Angelis, F. Barone, A. Zijno, L. Bizzarri, M. T. Russo, R. Pozzi, F. Franchini, G. Giudetti, C. Uboldi, J. Ponti, F. Rossi and B. De Berardis, Nanotoxicology, 2013, 7, 1361–1372 CrossRef CAS PubMed.
- J. S. Blum, P. A. Wearsch and P. Cresswell, Annu. Rev. Immunol., 2013, 31, 443–473 CrossRef CAS PubMed.
- M. Y. Bae, N. H. Cho and S. Y. Seong, Clin. Exp. Immunol., 2009, 157, 128–138 CrossRef CAS PubMed.
- H. Yang, N. H. Cho and S. Y. Seong, Clin. Exp. Immunol., 2009, 158, 174–185 CrossRef CAS PubMed.
- W. H. Fridman, F. Pages, C. Sautes-Fridman and J. Galon, Nat. Rev. Cancer, 2012, 12, 298–306 CrossRef CAS PubMed.
- A. Mildner and S. Jung, Immunity, 2014, 40, 642–656 CrossRef CAS PubMed.
- Z. H. Shen, G. Reznikoff, G. Dranoff and K. L. Rock, J. Immunol., 1997, 158, 2723–2730 CAS.
- T. C. Cheong, E. P. Shin, E. K. Kwon, J. H. Choi, K. K. Wang, P. Sharma, K. H. Choi, J. M. Lim, H. G. Kim, K. Oh, J. H. Jeon, I. So, I. G. Kim, M. S. Choi, Y. K. Kim, S. Y. Seong, Y. R. Kim and N. H. Cho, ACS Chem. Biol., 2015, 10, 757–765 CrossRef CAS PubMed.
- R. K. Pachynski, A. Scholz, J. Monnier, E. C. Butcher and B. A. Zabel, J. Visualized Exp., 2015 DOI:10.3791/52657.
- L. E. Greene, M. Law, J. Goldberger, F. Kim, J. C. Johnson, Y. F. Zhang, R. J. Saykally and P. D. Yang, Angew. Chem., Int. Ed., 2003, 42, 3031–3034 CrossRef CAS PubMed.
- C. Pacholski, A. Kornowski and H. Weller, Angew. Chem., Int. Ed., 2002, 41, 1188–1191 CrossRef CAS PubMed.
- S. Z. Deng, H. M. Fan, M. Wang, M. R. Zheng, J. B. Yi, R. Q. Wu, H. R. Tan, C. H. Sow, J. Ding, Y. P. Feng and K. P. Loh, ACS Nano, 2010, 4, 495–505 CrossRef CAS PubMed.
- J. Palacio, V. H. Orozco and B. L. Lopez, J. Braz. Chem. Soc., 2011, 22, 2304–2311 CAS.
- T. D. Zaveri, N. V. Dolgova, B. H. Chu, J. Lee, J. Wong, T. P. Lele, F. Ren and B. G. Keselowsky, Biomaterials, 2010, 31, 2999–3007 CrossRef CAS PubMed.
- S. Radoja, T. D. Rao, D. Hillman and A. B. Frey, J. Immunol., 2000, 164, 2619–2628 CrossRef CAS.
- J. Y. Liu, X. S. Zhang, Y. Ding, R. Q. Peng, X. Cheng, N. H. Zhang, J. C. Xia and Y. X. Zeng, J. Transl. Med., 2005, 3, 5 CrossRef PubMed.
- K. Oleinika, R. J. Nibbs, G. J. Graham and A. R. Fraser, Clin. Exp. Immunol., 2013, 171, 36–45 CrossRef CAS PubMed.
- P. Saini, M. Arora and M. Kumar, Adv. Drug Delivery Rev., 2016, 107, 47–59 CrossRef CAS PubMed.
- J. E. Eyles, V. W. Bramwell, E. D. Williamson and H. O. Alpar, Vaccine, 2001, 19, 4732–4742 CrossRef CAS PubMed.
- F. Coumes, C. Y. Huang, C. H. Huang, J. Coudane, D. Domurado, S. Li, V. Darcos and M. H. Huang, Biomacromolecules, 2015, 16, 3666–3673 CrossRef CAS PubMed.
- J. Wang, S. Li, Y. Han, J. Guan, S. Chung, C. Wang and D. Li, Front. Pharmacol., 2018, 9, 202 CrossRef PubMed.
- P. Di Bonito, L. Petrone, G. Casini, I. Francolini, M. G. Ammendolia, L. Accardi, A. Piozzi, L. D'Ilario and A. Martinelli, Int. J. Nanomed., 2015, 10, 3447–3458 CrossRef CAS PubMed.
- J. M. Silva, M. Videira, R. Gaspar, V. Preat and H. F. Florindo, J. Controlled Release, 2013, 168, 179–199 CrossRef CAS PubMed.
- Z. Hunter, D. P. McCarthy, W. T. Yap, C. T. Harp, D. R. Getts, L. D. Shea and S. D. Miller, ACS Nano, 2014, 8, 2148–2160 CrossRef CAS PubMed.
- D. R. Getts, A. J. Martin, D. P. McCarthy, R. L. Terry, Z. N. Hunter, W. T. Yap, M. T. Getts, M. Pleiss, X. Luo, N. J. King, L. D. Shea and S. D. Miller, Nat. Biotechnol., 2012, 30, 1217–1224 CrossRef CAS PubMed.
- S. Kadowaki, K. Sugimoto, Y. Tsurumaki, Y. Tabata, Y. Ikada, J. Fujita and K. J. Mori, Biomed. Pharmacother., 1993, 47, 385–391 CrossRef CAS.
- M. Santoro, S. R. Shah, J. L. Walker and A. G. Mikos, Adv. Drug Delivery Rev., 2016, 107, 206–212 CrossRef CAS PubMed.
- A. Sugunan, V. K. Guduru, A. Uheida, M. S. Toprak and M. Muhammed, J. Am. Ceram. Soc., 2010, 93, 3740–3744 CrossRef CAS.
- H. Rodriguez-Tobias, G. Morales, A. Ledezma, J. Romero and D. Grande, J. Mater. Sci., 2014, 49, 8373–8385 CrossRef CAS.
- S. Trujillo, E. Lizundia, J. L. Vilas and M. Salmeron-Sanchez, Colloids Surf., B, 2016, 144, 152–160 CrossRef CAS PubMed.
- G. Bisht and S. Rayamajhi, Nanobiomedicine, 2016, 3, 9 CrossRef PubMed.
- S. H. Mir, L. A. Nagahara, T. Thundat, P. Mokarian-Tabari, H. Furukawa and A. Khosla, J. Electrochem. Soc., 2018, 165, B3137–B3156 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary figures (Fig. S1–S4). See DOI: 10.1039/c8nr08704k |
| ‡ Theses authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |