
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBio-interfactants as double-sided tapes for graphene oxide†
Felipe
Macul Perez
 ab,
Yendry Regina
Corrales Ureña
c,
Klaus
Rischka
*a,
Welchy
Leite Cavalcanti
a,
Paul-Ludwig Michael
Noeske
a,
Arta Anushirwan
Safari
b,
Gang
Wei
b and
Lucio
Colombi Ciacchi
b
ab,
Yendry Regina
Corrales Ureña
c,
Klaus
Rischka
*a,
Welchy
Leite Cavalcanti
a,
Paul-Ludwig Michael
Noeske
a,
Arta Anushirwan
Safari
b,
Gang
Wei
b and
Lucio
Colombi Ciacchi
b
aAdhesive Bonding Technology and Surfaces, Fraunhofer Institute for Manufacturing Technology and Advanced Materials IFAM, Wiener Straße 12, 28359 Bremen, Germany. E-mail: klaus.rischka@ifam.fraunhofer.de
bUniversity of Bremen, Hybrid Materials Interfaces Group, Faculty of Production Engineering, Center for Environmental Research and Sustainable Technology (UFT), and MAPEX Center for Materials and Processes, D-28359 Bremen, Germany
cUniversity of Fribourg, Adolphe Merkle Institute, Chemin des Verdiers 4, 1700 Fribourg, Switzerland
First published on 6th December 2018
Abstract
We present a versatile and highly substrate-independent approach for preparing multisandwich layers based on thermally reduced Graphene Oxide (rGO) which gets strongly attached by bio-interfactants using a layer-by-layer (LBL) aqueous dipping and rinsing process. The process allows for the deposition of homogeneous ultra-thin films (∼5.5 nm) in distinct surface topographies, thicknesses and compositions by varying the bio-interfactant layer(s). The layers formed on quartz or other semi conductive material are electrically conductive, flexible, and transparent. The here-developed approach could be applied for the fabrication of wearables, sensors, and antistatic transparent films.
Introduction
While much research on graphene is focused on achieving its natural outstanding properties (mechanical, electrical and optical), it remains a challenge to strongly attach graphene on diverse substrates in a simple way in order to explore applications of a two-dimensional ultra-thin continuous electrically conductive net.1Most of the processes that synthesize graphene or graphene-based layers are not scalable and versatile enough for cost-effective applications. The CVD process yields graphene with high electrical mobility and pure crystallinity but is limited regarding the number of possible substrates, and the transferring of the graphene layers requires several further process steps.2 This results in a strong limitation of the size of the final coated area. Moreover, CVD-deposited graphene inherently lacks functional groups based on sp3 sites to easily anchor or react chemically onto substrates,3–5 limiting its application where physical interactions with the environment are needed, for example in nanosensors, or in biological and nanomechanical devices.6
An alternative which resolves some of these challenges is to use graphene oxide (GO) in order to produce graphene-like films with properties akin to those of pristine graphene7 and topographies similar to the ones of thin graphene films. It is possible to reduce GO by several processes; from the most practical, which is heating to at least ∼175 °C,8,9 to less mild and less practical procedures, like chemical or electrochemical reductions,10–13 ultrahigh temperature annealing in vacuum up to 2400 °C,14 high hydrostatic pressure in super-critical liquids,15 microwave,16 UV irradiation17 or a combination of these.18,19 All of these processes result in a reduced, graphene-like form of GO (rGO). In this way, the natural sp2 aromatic rings that make graphene flakes electrically conductive20 can partially be restored and the essential remaining process challenge will be safeguarding the overlap of neighbouring sheets within the film.
Facing these needs is facilitated by a significant degree of freedom in process design and allows compromises between the attained properties and the complexity of the process. In general, the assembly of GO in form of nanometric coatings provides a more versatile way of achieving the desired coating architecture and the required substrate attachment prior to the transformation towards graphene.
Because of the growing interest in the topic, many new approaches have been employed to fabricate GO films on substrates, namely: dip coating,21 spray-coating,22 Langmuir–Blodgett film formation,18 spin-coating,19,23 vacuum-filtration,24 electrophoretic deposition25 and layer-by-layer (LBL) formation.10,13,17,26–28
The LBL route to produce thin films with graphene-based components allowed innovative materials to emerge, due to the possibility of tailoring the films’ electrical properties,27,29,30 achieving sub-nm thickness control,31 gas selectivity,32 water membranes purification capability33 and fast humidity sensing.28
Here we present a new and original LBL method that produces films of molecular bio-interfactants and GO structured by alternated dipping processes. We took advantage of common adsorption behaviour of biomolecules towards solids34 and GO.35 Such adsorption allows for example the attachment of eukaryotic or microbial cells to solid surfaces.36 In this work we use the denomination ‘bio-interfactant’ for the bio-molecules that serve as a platform for further material attachment, as introduced by Corrales et al.37 and explored further by Stamboroski et al.,38 as adhesive biolayers.
The adaptable nature of the selected bio-interfactants allows an essentially physical coupling of GO during the immobilisation on several distinct substrates, such as fused quartz, borosilicate glass, and polyimide (PI). The bio-interfactants used in this study were strategically selected in view of their versatile adsorption under diverse conditions39–47 and their potential key interactions with GO.35 They play an important role in the dynamics of thermal reduction of GO by allowing chemical selectivity during decomposition and anchoring of the rGO, and resulting in a peculiar rippled topography, following the classification used by Deng et al.48
The goal of this work is to explore the advantages of using bio-interfactants to attach graphene oxide in a coplanar arrangement on surfaces and investigate the processes that allow this nanoassembly configuration before and after the thermal reduction towards graphene.
Results and discussion
Immobilisation of GO and assembly GO/bio-interfactant
In order to test the versatility of the process, three substrates were selected for coating with GO: quartz, borosilicate and polyimide. With quartz glass we conducted analytical tests and endurance tests. Two types of ultrathin coatings were applied on the substrates, as summarized in Fig. 1. Either we used a process in which GO is deposited without the assistance of bio-interfactants and with lateral overlapping (similar to other works,5,21,49Fig. 1a) or a process in which GO and bio-interfactant were used in successive dipping processes (Fig. 1b). | ||
| Fig. 1 Overview of the processes used in this study. (a) Sequence of processes for coating an arbitrary substrate with rGO, by directly dipping in GO dispersion (step 2) followed by a thermal treatment process (step 3). (b) Process routes explored in this research, using a two-step process. In this strategy, step 1 and step 2 are iterative processes that can be repeated (first dipping in a liquid bio-interfactant formulation and later dipping in a GO dispersion), afterwards the material can be thermally treated (step 3). Anchor symbols represent adhesion quality of these coatings (characterized later in this report). | ||
The second approach is a new strategy that guarantees a robust sheet arrangement both in vertical and lateral direction, and reliably achieves electrically conductive films (as opposed to the first process, as discussed further and in Fig. S1, ESI†). The assembly is based on the LBL formation intercalating dipping processes in a bio-interfactant mixture and later on in a commercially available GO dispersion.
The following sample notation is applied in this paper:
1. “Substrate” is referred to a flat material where the coating is deposited on. We used quartz, borosilicate, and polyimide as substrates.
2. (GO)n is a coating made of GO where n is the number of combined dipping and rinsing processes in the GO dispersion, i.e. the number of step 2 repetitions (Fig. 1).
3. (GO/LAC)n and (GO/BSA)n are referred to coatings based on bilayered repeating units where a bio-interfactant (LAC which refers to a mixture of laccase and maltodextrin, or BSA which means Bovine Serum Albumin, respectively) is intercalated with GO in dipping processes for n times (always beginning with the bio-interfactant and ending with GO).50
The adsorption of bio-interfactants (step 1) on quartz was confirmed with two methods; ultraviolet-visible light transmittance spectroscopy (UV-Vis) (Fig. 2b and c) and with X-ray photoelectron spectroscopy (XPS) (Fig. S2, ESI†). The XPS atomic analysis indicated the addition of more nitrogen species after step 1, corresponding to the introduction of the nitrogen-rich bio-interfactants. The changes in N![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C ratios for adsorbed LAC and BSA reported here were also observed elsewhere.45,47 The UV-Vis characterisation of a quartz substrate after step 1 also detected changes in the substrate property by the reduction of light transmittance for both bio-interfactants, mainly in the UV waves (Fig. 2b and c), what is associated with the absorbance of light from these biomolecules.
:C ratios for adsorbed LAC and BSA reported here were also observed elsewhere.45,47 The UV-Vis characterisation of a quartz substrate after step 1 also detected changes in the substrate property by the reduction of light transmittance for both bio-interfactants, mainly in the UV waves (Fig. 2b and c), what is associated with the absorbance of light from these biomolecules.
 | ||
| Fig. 2 Light absorbance spectra of the three coating systems. Intermediate steps of the process were compared to each individual substrate; (a) GO1, (b) (GO/LAC)1 and (c) (GO/BSA)1. Full lines depict coating freshly produced. Dotted lines represent the absorbance of the samples after the thermal treatment. In (b) and (c) the continuous green lines represent the stage after bio-interfactants adsorption. Inset charts in pictures a to c indicate the contribution attributed only to GO, two guidelines are plotted as references, as the main charts a–c at 200 and 235 nm. (d) Light transmittance (wavelength = 550 nm) for ten levels of depositions (n = 1 to 10) and for the three different coating systems in two states, freshly prepared on top ((GO)n, (GO/LAC)n and (GO/BSA)n) and the same samples thermally reduced on the bottom ((rGO)n, (rGO/LAC)n and (rGO/BSA)n). For each condition and each coating an exponential curve was fitted, the line slope is indicated in the legend. (e) Sheet resistance of the thermally reduced coating systems varying from 1 to 10 layers of deposition and the corresponding light transmittance (same samples as (d) bottom). The model fitting the curves is a relationship derived from sheet resistance and transmittance equations for thin films50 without taking percolation effects into account. | ||
Step 2 (GO dip coating), was performed on bare quartz and also on quartz functionalized after step 1. It was observed that deposition of GO directly on the non-functionalised quartz (process in Fig. 1a) often produced samples with high distinction of coverage, as detailed in Fig. S1, ESI.† For this reason, samples presenting average light absorbance (such as in the example in Fig. 2a) were used in this study. The shape of the light absorbance spectrum of GO is depicted in insets of Fig. 2a–c. The contribution attributed to GO has a different profile for each of the three initial conditions, i.e., for GO adsorbed on quartz, LAC or BSA. Three absorbance peaks can be identified in the range of 190 to 350 nm, at 200, 235 and 300 nm. The two latter peaks, 235 and 300 nm, are GO features due to π–π* transitions of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds and to n to π* of the carbonyl groups respectively.51,52 The absorbance peak at 200 nm is manifested in different degrees for each surface; high and sharp on quartz substrate, low on LAC and the lowest for BSA.
C bonds and to n to π* of the carbonyl groups respectively.51,52 The absorbance peak at 200 nm is manifested in different degrees for each surface; high and sharp on quartz substrate, low on LAC and the lowest for BSA.
This peak was further investigated (Fig. S3, ESI†) by separating the GO mixture with a centrifuge and comparing the upper fraction (upGO) with the normal mixture. Both fractions were applied on a quartz substrate prepared with BSA, resulting in the coatings (upGO/BSA)1 and (GO/BSA)1. These coatings were evaluated with XPS and UV-Vis and revealed that the peak at ∼200 nm is associated with components present in the upper fraction of the mixture (Fig. S3, ESI†).
The effect of repeating the coating steps 1 and 2 several times was assessed following the light transmittance at 550 nm and compared to a control experiment without the use of step 1 (i.e., not assisted by bio-interfactants). The results (Fig. 2d) depict differences when the dipping process (and control experiment) was performed up to ten times. The control experiment demonstrates that sequential dipping on GO reduces slightly the light transmittance. On the other hand, when using bio-interfactants the light transmittance is reduced significantly with further process repetitions. The change of transmittance by process repetitions can be characterized by an exponential attenuation coefficient (α) following the Lambert–Beer law.53 This exponential decay is fitted in Fig. 2d for each coating and the resulting coefficient of determination R2 adjusted (R2adj.). (GO)n shows a low α coefficient and low R2adj., meaning that the model may not be suitable to describe the process in the absence of bio-interfactants. When LAC and BSA are used as bio-interfactants, α is −1.3% and −3.9%, respectively, suggesting that BSA is responsible for a greater reduction of transmittance. In both cases the data are well fitted by the exponential model, meaning that in each process repetition similar block assemblies are formed, with BSA allowing three times more light attenuation than LAC. Higher opacity is also observed when the bio-interfactants are used on other substrates (borosilicate and PI) (Fig. S4, ESI†).
Finally, step 3 is a low temperature (250 °C) thermal treatment in inert atmosphere (N2) to achieve electrical conductivity of the GO sheets and their interconnected 2D network. Under this circumstance, heat allows chemical reactions to take place, reducing the GO towards rGO. The initiation can be catalysed by trapped water (for example from the GO film54 and/or bio-interfactants55) and propagates via several possible paths, such as decarboxylation, attack on hydroxyls and/or ring-opening epoxides. The reaction propagation and its eventual termination depend on the type of chemical groups initially present on GO.54 The reaction starts by release of O2 and other O-containing species, which consumes the carbon backbone of GO.9 These reactions develop a percolation path between restored sp2 clusters,20 resulting in a permanent acquisition of electric conductivity. The temperature of 250 °C was chosen in view of the fact that such temperature characterizes the first event of thermal reduction of GO,56 where substantial removal of oxygen and preponderance of sp2 carbon bonds takes place.9 The effects of step 3 on the bio-interfactants were evaluated with UV-Vis on quartz (Fig. S5, ESI†). The results indicate that the bio-interfactants remain in some (modified) form on the substrate. Other studies also indicate a relatively large stability of immobilized laccase57,58 and BSA59,60 under higher temperatures.
The absorbance spectra of the coatings change after thermal treatment in three ways; (i) there is an increase in absorbance at 550 nm for all coatings and all levels of deposition (1–10) (evident as decrease of transmittance in Fig. 2d) (ii) the characteristic peak around 230 nm shifts towards 270 nm, consistent with increased π–π* transitions of CC bonds, and lastly, (iii) the removal of carbonyl bonds causes the disappearance of the broad peak at 300 nm due to loss of n–π* transitions.61–65
High resolution of C 1s XPS spectra of (GO)1 and the thermally reduced coatings (Fig. S6, ESI†) indicate prevalence of graphene–carbon hybridisation (sp2) induced by thermal reduction. The C 1s signal contributions associated with oxygen species are significantly lower in the reduced forms and in comparison to the aromatic component CC/C–C (∼285 eV), both indicating removal of O species and restoration of the sp2 network. Reduced samples with bio-interfactants also cause the presence of pronounced peak related to CC/C–C bonds in the XPS spectra. After thermal reduction, another obvious result is the gain of electrical conductivity (Fig. 2e). It was observed that for any given number of layered systems (n = 1 to 10), the sheet resistance is lowest for (rGO/BSA)n, then (rGO/LAC)n, followed by (rGO)n. Within each coating, higher process repetitions n also result in lower sheet resistance. Since higher conductivity (lower sheet resistance) is related to having more rGO,18 and the relative amount of GO can be compared with the light transmittance,17,66–69 it can be stated that the bio-interfactants do prime the substrate to adsorb more GO, ruling out that the lower transmissivity is caused by the bio-interfactants themselves. The experimental data in Fig. 2e were fitted with a model organically derived for bulk thin films,50 in which light transmittance and sheet resistance are related by the relationship T = e(k/Rs). In this equation, T is transmittance, Rs is the sheet resistance, and k is a characteristic constant of the material.
The results show that for (rGO/BSA)n and (rGO/LAC)n the resistance decreases exponentially with the amount of light absorbance, as it would be expected for bulk films (higher R2-adj). In the case of (rGO)n, instead, the model does not satisfactorily fit well the data. Indeed, in the absence of bio-interfactants the resistance of deposited rGO flakes is expected to be limited by the percolation pathways in the film, which thus behaves differently from an infinitely percolating bulk.
Effects of bio-interfactants on the layer structure
It is well documented that upon chemical reduction GO develops corrugations.18,70,71 This is attributed to the formation of new bonds at the epoxide rings in GO.72 The effect is more pronounced for thicker films18 and also for weak coupling with the SiO2 substrate, leading to delamination.71 Our result after thermal treatment of coating with one level of deposition (n = 1) (Fig. 3d–f) shows tunnel-like corrugations in the absence of biointerfactants, observed also elsewhere.18 In contrast, samples with bio-interfactants developed ripple-like48 shaped corrugations, which are not present in the initial microstructure of the fresh coatings (Fig. 3b and c). It has been proposed that ripples can be formed by many effects, for example (i) mismatch of substrate thermal expansion, (ii) trapped solvent and (iii) strong anchorage on substrate.48 These effects can cause stress on the film and thus rippling.73 Gas trapping due to GO reduction was observed elsewhere20 but only when over seven layers of GO constitute the coating. | ||
| Fig. 3 (a–c) Microstructure of the three investigated coatings (GO)1, (GO/LAC)1 and (GO/BSA)1 respectively, scanned with AFM after having been freshly prepared. (d–f) AFM image of thermally reduced coatings at 250 °C line order as in previous column. (g–i) Selected AFM phase images of (GO)1 and (GO/LAC)1 and (GO/BSA)1 respectively reduced at 180 °C; on (GO)1 and (GO/LAC)1 it was observed a high contrast of regions (illustrated in images (g) and (h)), which were linked to quartz substrate (hard phase), such contrasting phases were not observed in coatings with BSA, illustrated by (i). Regarding (GO/BSA)1, no AFM image was found to have the distinct 2-phase characteristic upon inspection of total 23 positions on five individual samples which included the four investigated temperatures, indicating high coverage of substrate independent of temperature of reduction when using BSA as bio-interfactant. | ||
AFM imaging in tapping mode delivered phase contrast images (Fig. 3g–i) in the three coatings with n = 1. The AFM phase changes are sensitive to the mechanical stiffness of the outermost material, the contrast in the images being indicative of phases that are harder and softer. These phases can be clearly related to either the quartz surface or the reduced GO flakes. Upon AFM scanning of at least 63 μm2 on each coating, (GO)1 and (GO/LAC)1 presented 6% and 11%, respectively, of high contrasting dark areas (quartz), exemplarily illustrated in Fig. 3g and h. No such contrasting regions were observed by evaluating 90 μm2 of (GO/BSA)1 coatings. The presence of gaps on coatings derived only from GO was also observed elsewhere.5,49 The AFM phase contrast supports the hypothesis that BSA leads to a higher coverage, translating into higher electrical conductivity as discussed above (Fig. 2d and e).
Taking into consideration the “double-sided tape” role of the bio-interfactants on the LBL formation, it can be assumed that specific amino acid residues of the bio-interfactants play a key role in anchoring GO flakes on the substrate. Amino acids affinity scales for interaction with quartz and GO have already been established.74,75 Arginine, lysine and histidine are the amino acid residues that display the strongest adsorption forces, both for GO and quartz. It was found that both bio-interfactants have around 10% of their solvent-accessible surface area covered with these residues. However, these amino acids show distinct distributions on the bio-interfactants (Fig. S7, ESI†); on BSA all the three of these anchoring amino acids are well scattered around the molecule, whereas on laccase they are more concentrated on one side of the molecule.
Molecular dynamics was used to examine in detail the differences that ensue from the structures of the two bio-interfactants. Our modelling results of laccase adsorption (Fig. S8, ESI†) indicate that its arrangement on the SiO2 (akin to step 1) is driven by the dipole direction and initial position. In six distinct simulations with different initial orientations, laccase either rotated or remained in the initial configuration, in order to orient its dipole moment perpendicular to the negatively charged silica surface. In contrast, BSA adsorption tends to be driven also by hydrophobic residues, leaving the majority of hydrophilic groups facing outwards and yet having a strong adsorption towards silica.76
Considering these facts and that GO is negatively charged at our experiment pH, it is expected that laccase would be less prone to bind to GO than BSA, in which the arginine, lysine and histidine anchoring sites are more uniformly distributed over all the protein's surface.77,78 Moreover, hydrophobic BSA/GO interactions may provide an additional driving force towards surface binding.79
The topography of the thermally reduced coatings, in particular the distinct rippling details, can be further examined in profiles of height and phase changes in selected areas of the AFM images (Fig. 4). Shallow peak-to-valley heights of <0.5 nm were found on (rGO)1 and (rGO/LAC)1, in contrast to higher peak-to-valley heights on (rGO/BSA)1 (>0.5 nm), as sampled in Fig. 4g–i and manifested in the corresponding roughness values Rq (Fig. 4a–c). The AFM phase image was used to measure the lateral spacing of these peaks on the flat regions of the coatings (exemplified in Fig. 4g–i). The amount of peaks per unit of area were 824, 918 and 492 peaks per μm2 on (rGO)1, (rGO/LAC)1 and (rGO/BSA)1, respectively. The choice of bio-interfactants prior to the thermal reduction thus clearly leads to larger or smaller ripples as compared to the unassisted process.
 | ||
| Fig. 4 Height and phase images obtained by AFM. Sequence of columns (left to right) correspond to (rGO)1, (rGO/LAC)1 and (rGO/BSA)1 on fused quartz glass substrates, respectively. a–c, AFM height image of the three coating systems reduced at 250 °C. Rq values are in nm and are measured on the flat regions of the scanned area. Images scaled to relative height values from 0 to 3 nm. d–f AFM phase image of the same locations from images a–c. Rq values are in degrees and referent to flat regions of the scanned area. (g–i) Height and phase values extracted from the marked region (white line) on pictures a–f. | ||
XPS was used in order to analyse the coating thickness throughout different temperatures. At the same time, with XPS the elemental compositions (C, O and N) could be extracted and compared. The atomic composition of these materials, listed in (Fig. 5a), was calculated excluding impurities from the substrate (detailed in Fig. S9, ESI†). The quantity of the bio-interfactants in the coatings could be estimated by using the atomic nitrogen surface concentration [N] of the samples and interpolating between the residual fraction on GO and the expected [N] values for these molecules (following the models 1GYC for laccase and 3V03 for BSA, both available at Protein Data Bank). By interpolating the measured [N] with the nitrogen concentration from the bio-interfactants and from the GO, it is expected that the coatings (GO/LAC)1 and (GO/BSA)1 carry about 2.6% and 13.5%, respectively, of atoms from the bio-interfactants.
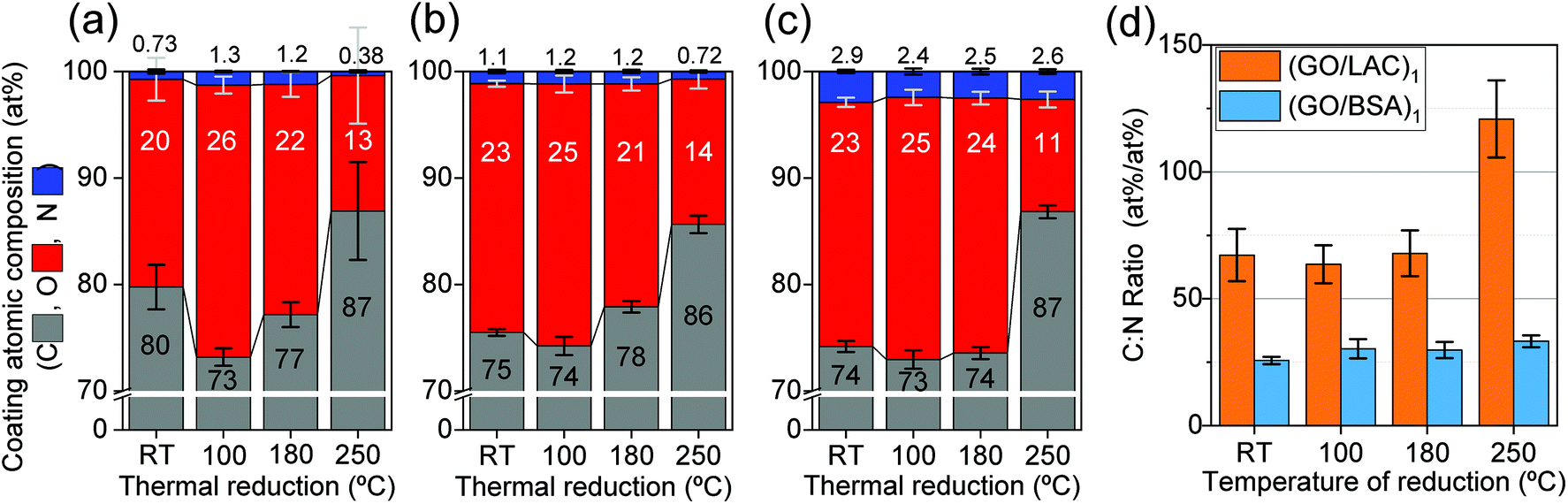 | ||
| Fig. 5 (a–c) XPS elemental analysis of layer composition (substrate content is subtracted) for four conditions of thermal reduction for (a) (GO)1, (b) (GO/LAC)1 and (c) (GO/BSA)1. The relative composition shows only the additional C, N and O elements added to the quartz substrate. (GO)1 was found to contain 0.75% atomic of N, which is attributed to be residual nitrogen expected to be found from Hummers process64,65 (d) atomic concentration ratio [C]:[N] in the coatings (from (a–b)) with bio-interfactants (GO/LAC)1 and (GO/BSA)1 for all conditioning temperatures. | ||
The evolution of the chemical stoichiometry with the applied temperature of reduction was used as an indication of the anchoring effect of the bio-interfactants. The [C]:[N] ratio indicates approximately the GO:bio-interfactant ratio (assuming that most carbon originates from GO and most nitrogen originates from the bio-interfactants). No significant change in the ratio was found up to 180 °C for both bio-interfactant-assisted coatings. However, at 250 °C (GO/LAC)1 loses significantly more nitrogen than carbon, whereas (GO/BSA)1 keeps the proportion close to a steady ∼30:1 ([C]:[N]) (Fig. 5d).
Moreover, the evolution of the [C]:[O] ratio also indicates a distinctive behaviour between the two coatings. Fig. 6b depicts that oxygen is preferentially released to a greater degree on (rGO/BSA)1 than on (rGO/LAC)1 for the highest temperature of reduction.
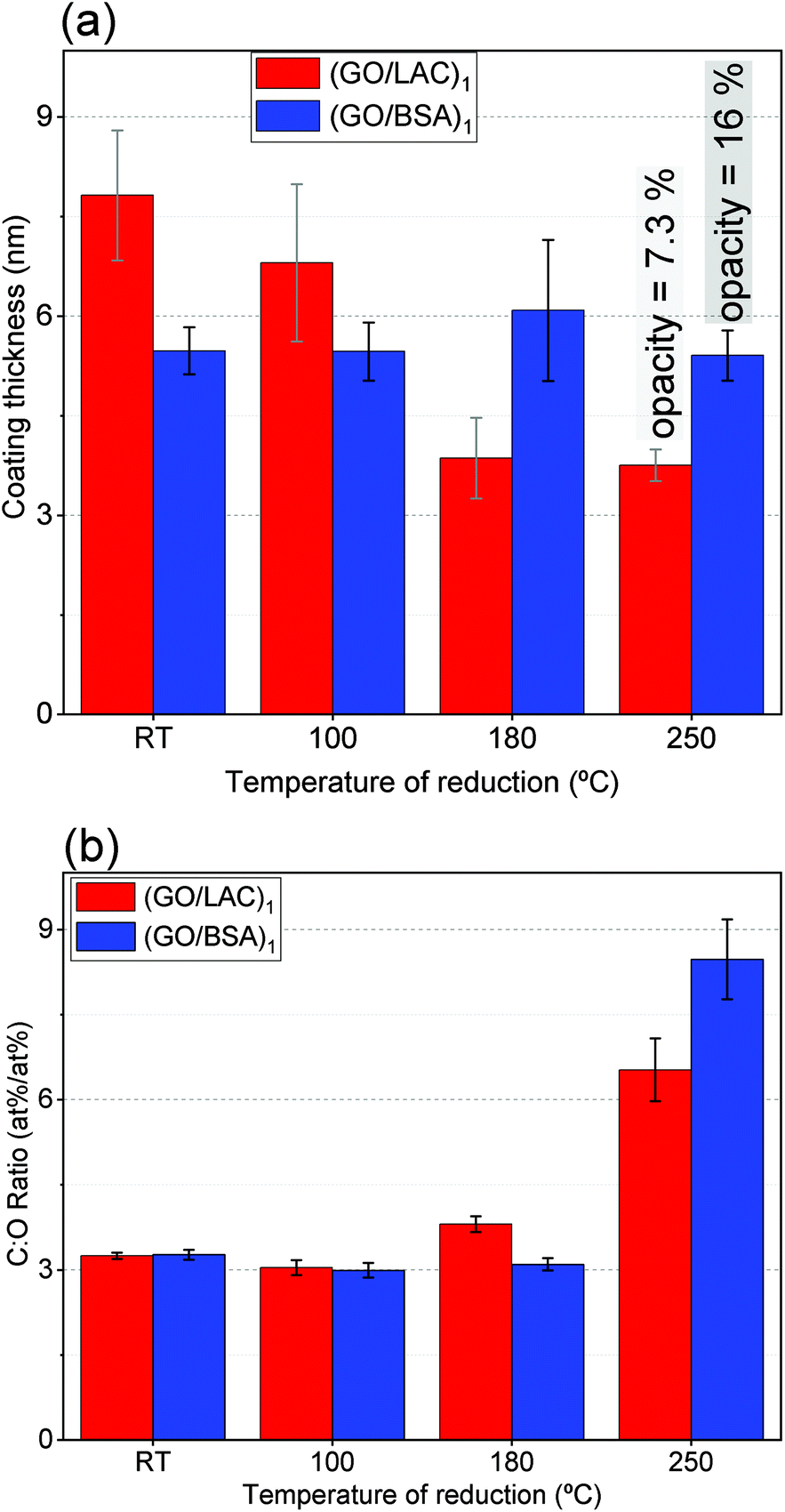 | ||
| Fig. 6 (a) Coating thickness of films with bio-interfactants and the relationship with higher temperatures of thermal reduction. (b) Atomic concentration ratio [C]:[O] of bio-interfactant assisted coatings throughout temperatures of reduction. | ||
As for the layer thickness of these coatings (Fig. 6a), calculated using XPS signal attenuation of the substrate, distinct behaviours were observed for each material. The thickness of BSA-assisted coatings remains in the range between 5 and 6 nm at all four temperatures. (GO/LAC)1 shows gradually lower thickness up to 180 °C and no further reduction up to 250 °C. There are limitations, however, on this calculation for (GO/LAC)1 since it was shown with AFM (Fig. 3h) that the coatings assisted by laccase cannot be assumed to be uniformly covered. For that reason AFM was also employed to verify the thickness of coatings with both bio-interfactants. Fig. S10, ESI† shows AFM areas where the coating thickness could be measured. Such measurements resulted in thicknesses of 1.80 nm and 5.75 nm for (rGO/LAC)1 and (rGO/BSA)1, respectively, versus the thicknesses calculated with XPS of 3.75 nm and 5.50 nm. The AFM measurement of (rGO/BSA)1 has good agreement with the thickness calculated with XPS. However, in the case of (rGO/LAC)1 the AFM measured a thickness 2 nm thinner than the one calculated with XPS.
This incongruence might be associated with the size of the sampled area with AFM, being 100000 times smaller than that area sensitive to XPS. The latter takes into account areas with thicker parts of the film (large area homogeneity especially of LAC-based coatings are depicted in Fig. S11, ESI†).
In summary, the thermal treatment of (GO/LAC)1 allows a great proportion of the nitrogen-containing species to be reduced and the coating thickness becomes thinner. On the other hand, BSA favours higher release of oxygen (comparing to LAC) and the carbon is kept in a similar proportion of nitrogen, while showing constant coating thickness. As discussed previously, two routes of oxygen release are possible during the thermal reduction at 250 °C; one that releases O2 and another that consumes the carbon backbone of GO (releasing CO and CO2).9 These aspects corroborate to the hypothesis that BSA provides stronger structural stability and facilitates a route of thermal reduction that consumes less carbon from the GO backbone. The selection of the bio-interfactants controls fixation of carbon from the GO backbone and change the dynamics of O release. Such selectivity leads to higher or lower [C]:[O] ratio, which is also directly associated with higher electrical conductivity within a rGO flake.20
Lastly, the thickness information from XPS can be compared to the light transmittance in order to propose a geometric model for the coatings. The opacity of each coating at 550 nm (complementary to its transmittance) is related to the number of graphene sheets, each layer increasing the opacity by 2.3%.68 Since the contribution from the adsorbed BSA is roughly 0.4% (Fig. S12, ESI†) it is expected that the coating contains around seven layers of GO among the adsorbed BSA. In the case of (rGO/LAC)n, we evaluated coatings resulting from depositing several layers with XPS (due to the limitations on the coating coverage, as discussed from Fig. 3h). As detailed in Fig. S13, ESI,† we observed a linear increase of 4 nm for each n number of depositions, and the proportion of bio-interfactant to GO also remains stable for different layers of deposition. This incremental increase by 4 nm per layer can be compared to the increase in opacity of 7% from Fig. 2d. These numbers, when compared, indicate that each process deposition gathers approximately three layers of GO for every preceding deposition of LAC. A plausible model for these coatings is that the adsorbed bio-interfactants have two ways of interacting with GO; in the case of (GO/LAC)n, adsorbed laccase anchors the available GO molecules as stacks of three flakes, on average. In the case of (GO/BSA)n, twice as much of GO is stacked on the coating because some BSA molecules might re-adsorb on GO during step 2, allowing even more pronounced assembly of the coating.
Adhesion
The capability of a coating to be strongly adhered to a substrate is key for applications where there is direct interaction with the medium, for example in wearables, sensors that have to endure exposure to tribological loadings or contact with liquids. In real-world applications, graphene needs to be supported (and kept) on a substrate in order to perform its function.1In order to test and compare the robustness of the thermally reduced coatings studied here, they were exposed to ultra-sound bath in deionized (DI) water in order to induce coating destabilization or destruction by cavitation. The corresponding decay of electrical conductivity was measured during the time of the experiment as a form to assess the durability of the coatings (Fig. 7a). Additionally, a commercially available transferred CVD graphene on quartz (Graphenea) was also exposed to this test for comparison. The values are relative to the first measurement at 0 minutes (when each relative conductivity is 100%). After only 7 minutes inside the ultrasound bath, there was no more detectable electrical conductivity on the commercial transferred CVD graphene film. Our own three other coatings derived from GO showed a slower decay in electrical conductivity. The ones prepared on previously adsorbed bio-interfactants, in particular with BSA, kept a high relative conductivity for longer time.
 | ||
| Fig. 7 (a) Evolution of relative electrical conductivity decay when samples were exposed to ultrasonic water bath. The samples (rGO/BSA)1, (rGO/LAC)1 and (rGO)1 were reduced at 250 °C. (b) Change of electrical conductivity of coatings after 180° peeling test with conventional tape; further peelings on the same sample were conducted with new parts of the tape. Numerical values in the (b) indicate the peeling resistance energy measured in the first peeling. | ||
A peeling test (Fig. 7b) with conventional pressure-sensitive tape was carried out on the three coatings to determine the changes of the film proprieties. The force from the tape peeling also was measured and resulted in values in the same range for the three coatings (Fig. 7b). However, the reduction in electrical conductivity was more dramatic for the (rGO)1 film. Further peelings had the same effect, and BSA in this case also acted as a component key for higher resistance. Light transmittance (at 550 nm) was measured before and after the sequence of ten peelings; the three coatings suffered minor changes in light transmittance (<3%).
In a recent study,5 it was observed that the peeling work (from nano-scratch techniques) of a GO film on glass decrease dramatically (from ∼350 J m−2 to 38 J m−2) after thermal reduction due to the removal of oxygen-containing groups, which were associated to be the main contributor to the van der Waals forces with the substrate. In parallel to these findings, our macroscale peeling tests indicated that the average energy of ∼300 J m−2 is enough to cause severe disruption in the conductivity of the rGO film. However, when assisted by the bio-interfactants, the electrical conductivity was sustained to near original values though the ten peelings.
Both enduring tests supports the model of strong anchoring when using bio-interfactants, especially BSA, as observed with other techniques. The destruction induced by both techniques indicates higher durability both at the nanoscale and at the macroscale when bio-interfactants are employed.
Conclusions
In summary, we have developed a method for attaching GO on multiple substrates with the use of bio-interfactants. The bio-interfactants showed distinct levels of GO adsorption and allowed further LBL intercalation assembly with GO. Proteic bio-interfactants can be selected according to their specific amino acid distributions in order to control the anchoring of GO and define the coating composition after a mild thermal reduction. In addition, the method allowed for the production of ultra-thin (5.5 nm) continuous coatings that are strongly bound on the substrate, sustaining both resistance to peeling and several hours inside an ultrasonic water bath.In view of wide interest and availability of the here-employed materials on the market, this new process may be suitable for scalable production of nanomaterials that require simultaneously a combination of the following properties: strong adhesion, transparency, antistaticity and extreme dimensions constrain.
Experimental section
Materials
Graphene oxide (GO) 4 mg ml−1 from Graphena was diluted with deionised water (<0.1 μS cm−1 form arium® pro VF, Sartorius) to 1 mg ml−1 (reaching pH ∼2.6). Bio-interfactants: LAC (laccase from Trametes versicolor, Sigma-Aldrich product number 38429, PDB code 1GYC), which in this commercial form has a concentration of ∼6%w (Fig. S5, ESI†) or BSA (bovine serum albumin, Sigma-Aldrich product number A6003, PDB code 3V03) were diluted (0.2 g L−1) in sodium acetate buffer solution. The acetate buffer solution (0.2 M at pH 4.75) was prepared with acetic acid and sodium acetate (reagent grade Sigma-Aldrich).Substrates: Quartz glass was acquired from GVB, Germany, afterwards cut, the quartz was cleaned using Piranha solution protocol. Briefly (Caution: piranha solution should be handled with great care.) 1:5 ratio (H2O2 (30%):H2SO4 (98%)) was prepared by pouring 75 mL of H2SO4 in a clean and dry beaker with a PTFE magnetic stirring bar. The beaker was placed in an ice-bath glassware (bath temperature kept at 0 °C). Then 15 mL of H2O2 (30%) was added slowly into the beaker (caution for highly exothermic reaction). The solution was mixed by aid of the stirring bar. When a homogeneous phase is achieved, the solution is ready to be used. As the magnetic stirrer continues to mix, quartz was added in the solution where it remained for 10 minutes. After that, the quartz substrates were placed in a beaker with DI water for 5 minutes. Next, the substrates were rinsed sequentially in two beakers with DI water and finally dried with nitrogen gun. XPS confirmed the clean state of the substrates (Fig. S5, ESI†). Borosilicate glass hydrolytic class 1 was acquired from Brand GmbH (product number 470-45). Polyimide film was purchased form Airtech (Termalimide film).
Fabrication of layer-by-layer (LBL) films
Step 1 (Fig. 1a) was performed by dipping the target substrate in a buffered dispersion of LAC or BSA for 1 h, followed by the rinsing of in DI water vigorously for about 5 seconds. Step 2 (Fig. 1a) was conducted by placing substrates in GO dispersion (1 mg ml−1) for 15 min, followed by rinsing with DI water (5 seconds vigorously). The samples were then dried with nitrogen gun until no apparent water drops were on the substrate. The back side of the samples were cleaned by wiping with delicate wipe towel in order to make sure that the layer was only formed on the top surface. For further layers depositions, steps 1 and step 2 were repeated (in the case of assembly with bio-interfactants). Films with multiple GO dipping (unassisted by bio-interfactants) were prepared performing consecutive repetitions of step 2.Thermal reduction, step 3, was conducted in a chamber furnace with feed of nitrogen. The cycle began with 90 min of nitrogen purge at room temperature, and then heating to target temperature at 10 K min−1 rate (target temperature either 100 °C, 180 °C or 250 °C). The temperature was kept at the target temperature for 10 minutes and finally let cool to room temperature (by turning off heat element).
Characterisations
AFM images were obtained with NanoWizard 3 NanoScience (JPK Instruments AG, Germany) in tapping mode. Samples characterized on Fig. 3a–f are 6 individual samples; the last three are the same samples from Fig. 4. Samples in Fig. 3g–i are selected from a group exposed to several temperatures of reduction and were selected to illustrate when two phases were clearly observed.X-ray photoelectron spectroscopy was measured with a Kratos Ultra Facility using the following parameters: base pressure of 4 × 10−8 Pa, sample neutralization with <5 eV electrons. The element composition of the LBL systems is calculated by the total measured atomic concentration of the sample (which may also comprise the substrate) minus the proportion of species contained on the substrate, using Si as a reference for proportion (it is assumed that GO does not contain Si). The background was corrected with Shirley method and the curve fitting used GL(30). The layer thickness was calculated using signal attenuation form the quartz substrate. It is assumed an exponential attenuation of the photoelectron Si 2p signal (which is attributed to the quartz), following the Beer–Lambert equation:80
| I = I0exp(−d/λa,b) |
81 for Si 2p electrons which are characteristic for the substrate. Samples on Fig. 5 and 6 are physically individual (i.e., one sample for each condition of temperature).
Light transmittance and absorbance were measured with Specord 210 plus, Analytic Jena AG, Germany. Samples measured with this technique were measured individually before dip coating in GO or bio-interfactants formulations in order to use a precise reference. Electrical conductivity was measured with multimeter 15XL, Wavetek (sensitive up to 2 GOhm). Sheet resistance was calculated form the measured resistance and adjusted to the rectangular ratio of the films (Fig. S14, ESI†). Universal mechanical testing machine (TA·XTplus 5 kg load cell) was used to peel the adhesive tape (tesafilm® transparent) with the angle of 180° at 5 mm s−1. The pressure applied on the films beforehand had been 2.5 kg cm−2 for 20 seconds. Molecules were graphically represented using Jmol or PyMol software (the PDB code used for BSA molecule was 3V03 and 1GYC for laccase from Trametes versicolor). Endurance in ultrasonic bath was measured on three samples (one for each condition studied in this report) and also compared to a commercial quartz coated with CVD graphene (Graphenea, Spain). The electrical conductivity was measured outside of the bath to avoid influence from the water, after measurement the samples were returned to the bath. SEM characterisations indicating the lateral homogeneity of several (rGO/BSA)n1 samples done with Helios 600 machine at 5 kV (Fig. S11, ESI†).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work benefitted from support from the CNPq program Science without Boarders programme (project 205247/2014-8) and the Swiss National Science Foundation through the National Center of Competence in Research Bio-Inspired Materials. Also, acknowledgement goes to Tobias Urbaniak for supporting with some UV-Vis measurements. AAS and LCC acknowledge funding via the DFG Research Training Network GRK 2247. We like to thank Dr Karsten Thiel for SEM images.References
- G. Zhao, X. Li, M. Huang, Z. Zhen, Y. Zhong, Q. Chen, X. Zhao, Y. He, R. Hu, T. Yang, R. Zhang, C. Li, J. Kong, J.-B. Xu, R. S. Ruoff and H. Zhu, Chem. Soc. Rev., 2017, 46, 4417–4449 RSC.
- M. Chen, R. C. Haddon, R. Yan and E. Bekyarova, Mater. Horiz., 2017, 4, 1054–1063 RSC.
- Z. Liu, J. Z. Liu, Y. Cheng, Z. Li, L. Wang and Q. Zheng, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 1–5 Search PubMed.
- V. Georgakilas, J. N. Tiwari, K. C. Kemp, J. A. Perman, A. B. Bourlinos, K. S. Kim and R. Zboril, Chem. Rev., 2016, 116, 5464–5519 CrossRef CAS PubMed.
- R. Kumar, R. M. Kumar, D. Lahiri and I. Lahiri, Surf. Coat. Technol., 2017, 309, 931–937 CrossRef CAS.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS PubMed.
- A. C. Ferrari, F. Bonaccorso, V. Fal, K. S. Novoselov, S. Roche, P. Bøggild, S. Borini, F. H. L. Koppens, V. Palermo, N. Pugno, J. A. Garrido, R. Sordan, A. Bianco, L. Ballerini, M. Prato, E. Lidorikis, J. Kivioja, C. Marinelli, T. Ryhänen, A. Morpurgo, J. N. Coleman, V. Nicolosi, L. Colombo, A. Fert, M. Garcia-Hernandez, A. Bachtold, G. F. Schneider, F. Guinea, C. Dekker, M. Barbone, Z. Sun, C. Galiotis, A. N. Grigorenko, G. Konstantatos, A. Kis, M. Katsnelson, L. Vandersypen, A. Loiseau, V. Morandi, D. Neumaier, E. Treossi, V. Pellegrini, M. Polini, A. Tredicucci, G. M. Williams, B. Hee Hong, J.-H. Ahn, J. Min Kim, H. Zirath, B. J. van Wees, H. van der Zant, L. Occhipinti, A. Di Matteo, I. A. Kinloch, T. Seyller, E. Quesnel, X. Feng, K. Teo, N. Rupesinghe, P. Hakonen, S. R. T. Neil, Q. Tannock, T. Löfwander and J. Kinaret, Nanoscale, 2015, 7, 4587–5062 RSC.
- I. Jung, D. A. Dikin, R. D. Piner and R. S. Ruoff, Nano Lett., 2008, 8, 4283–4287 CrossRef CAS PubMed.
- R. Larciprete, S. Fabris, T. Sun, P. Lacovig, A. Baraldi and S. Lizzit, J. Am. Chem. Soc., 2011, 133, 17315–17321 CrossRef CAS PubMed.
- N. A. Kotov, I. Dékány and J. H. Fendler, Adv. Mater., 1996, 8, 637–641 CrossRef CAS.
- M. Z. B. Dzukarnain, T. Takami, H. Imai and T. Ogino, Thin Solid Films, 2016, 615, 247–255 CrossRef CAS.
- S. Pei, J. Zhao, J. Du, W. Ren and H. M. Cheng, Carbon, 2010, 48, 4466–4474 CrossRef CAS.
- J.-W. Jeon, S. R. Kwon and J. L. Lutkenhaus, J. Mater. Chem. A, 2015, 3, 3757–3767 RSC.
- S. Barg, F. M. Perez, N. Ni, P. do Vale Pereira, R. C. Maher, E. Garcia-Tuñon, S. Eslava, S. Agnoli, C. Mattevi and E. Saiz, Nat. Commun., 2014, 5, 4328 CrossRef CAS PubMed.
- C. Y. Kong, W.-L. Song, M. J. Meziani, K. N. Tackett, L. Cao, A. J. Farr, A. Anderson and Y.-P. Sun, J. Supercrit. Fluids, 2012, 61, 206–211 CrossRef CAS.
- W. Chen, L. Yan and P. R. Bangal, Carbon, 2010, 48, 1146–1152 CrossRef CAS.
- K. K. Manga, Y. Zhou, Y. Yan and K. P. Loh, Adv. Funct. Mater., 2009, 19, 3638–3643 CrossRef CAS.
- H. A. Becerril, J. Mao, Z. Liu, R. M. Stoltenberg, Z. Bao and Y. Chen, ACS Nano, 2008, 2, 463–470 CrossRef CAS PubMed.
- J. Wu, H. A. Becerril, Z. Bao, Z. Liu, Y. Chen and P. Peumans, Appl. Phys. Lett., 2008, 92, 263302 CrossRef.
- C. Mattevi, G. Eda, S. Agnoli, S. Miller, K. A. Mkhoyan, O. Celik, D. Mastrogiovanni, G. Granozzi, E. Garfunkel and M. Chhowalla, Adv. Funct. Mater., 2009, 19, 2577–2583 CrossRef CAS.
- X. Wang, L. Zhi and K. Müllen, Nano Lett., 2008, 8, 323–327 CrossRef CAS PubMed.
- I. K. Moon, J. Il Kim, H. Lee, K. Hur, W. C. Kim and H. Lee, Sci. Rep., 2013, 3, 1112 CrossRef.
- S. Watcharotone, D. A. Diking Sasha Stankovich, R. Pinery, I. Jung, G. H. B. Dommett, G. Evmenenko, S. E. Wu, S. F. Chen, C. P. Liu, S. T. Nguyen and R. S. Ruoff, Nano Lett., 2007, 7, 1888–1892 CrossRef CAS PubMed.
- G. Eda, Y. Y. Lin, S. Miller, C. W. Chen, W. F. Su and M. Chhowalla, Appl. Phys. Lett., 2008, 92, 233305 CrossRef.
- M. Diba, D. W. H. Fam, A. R. Boccaccini and M. S. P. Shaffer, Prog. Mater. Sci., 2016, 82, 83–117 CrossRef CAS.
- F. Shao, C. Xu, W. Ji, H. Dong, Q. Sun, L. Yu and L. Dong, Desalination, 2017, 423, 21–29 CrossRef CAS.
- D. Wang and X. Wang, Langmuir, 2011, 27, 2007–2013 CrossRef CAS PubMed.
- D. Zhang, J. Tong and B. Xia, Sens. Actuators, B, 2014, 197, 66–72 CrossRef CAS.
- T. Cassagneau, F. Gurin and J. H. Fendler, Langmuir, 2000, 7318–7324 CrossRef CAS.
- Z. Gao, W. Yang, J. Wang, H. Yan, Y. Yao, J. Ma, B. Wang, M. Zhang and L. Liu, Electrochim. Acta, 2013, 91, 185–194 CrossRef CAS.
- D. W. Lee, T.-K. Hong, D. Kang, J. Lee, M. Heo, J. Y. Kim, B.-S. Kim and H. S. Shin, J. Mater. Chem., 2011, 21, 3438–3442 RSC.
- Y. H. Yang, L. Bolling, M. A. Priolo and J. C. Grunlan, Adv. Mater., 2013, 25, 503–508 CrossRef CAS PubMed.
- W. Choi, J. Choi, J. Bang and J. Lee, ACS Appl. Mater. Interfaces, 2013, 5, 12510–12519 CrossRef CAS PubMed.
- M. Rabe, D. Verdes and S. Seeger, Adv. Colloid Interface Sci., 2011, 162, 87–106 CrossRef CAS PubMed.
- D. Li, W. Zhang, X. Yu, Z. Wang, Z. Su and G. Wei, Nanoscale, 2016, 8, 19491–19509 RSC.
- B. Carpentier and O. Cerf, J. Appl. Bacteriol., 1993, 75, 499–511 CrossRef CAS.
- Y. R. Corrales Ureña, W. L. Cavalcanti, M. Soltau, K. Villalobos, K. Rischka, P.-L. M. Noeske, K. Brune and S. Dieckhoff, Appl. Adhes. Sci., 2017, 5, 10 CrossRef.
- S. Stamboroski, P. N. Stachera, Y. R. Corrales Ureña, G. H. Hrycyna, W. I. Taborda Ribas Neto, W. K. de Azambuja, D. Salz, J. Ihde, P.-L. M. Noeske and W. L. Cavalcanti, Appl. Adhes. Sci., 2016, 4, 6 CrossRef.
- L. Betancor, G. R. Johnson and H. R. Luckarift, ChemCatChem, 2013, 5, 46–60 CrossRef CAS.
- R. Fogel and J. L. Limson, Enzyme Microb. Technol., 2011, 49, 146–152 CrossRef CAS PubMed.
- S. Rodríguez Couto and J. L. Toca Herrera, Biotechnol. Adv., 2006, 24, 500–513 CrossRef PubMed.
- M. Hakamada, M. Takahashi and M. Mabuchi, Mater. Lett., 2012, 66, 4–6 CrossRef CAS.
- B. Jachimska, M. Wasilewska and Z. Adamczyk, Langmuir, 2008, 24, 6867–6872 CrossRef PubMed.
- B. Jachimska, K. Tokarczyk, M. Łapczyńska, A. Puciul-Malinowska and S. Zapotoczny, Colloids Surf., A, 2016, 489, 163–172 CrossRef CAS.
- Y. R. Corrales Ureña, L. Gaetjen, M. V. Nascimento, P. N. L. Filho, W. L. Cavalcanti, P.-L. M. Noeske and K. Rischka, Appl. Adhes. Sci., 2016, 4, 1–11 CrossRef.
- Y. R. Corrales Ureña, P. N. Lisboa-Filho, M. Szardenings, L. Gätjen, P.-L. M. Noeske and K. Rischka, Appl. Surf. Sci., 2016, 385, 216–224 CrossRef.
- A. Ithurbide, I. Frateur, A. Galtayries and P. Marcus, Electrochim. Acta, 2007, 53, 1336–1345 CrossRef CAS.
- S. Deng and V. Berry, Mater. Today, 2016, 19, 197–212 CrossRef CAS.
- L. J. Cote, F. Kim and J. Huang, J. Am. Chem. Soc., 2009, 131, 1043–1049 CrossRef CAS PubMed.
- N. N. Rosli, M. A. Ibrahim, N. Ahmad Ludin, M. A. Mat Teridi and K. Sopian, Renewable Sustainable Energy Rev., 2019, 99, 83–99 CrossRef CAS.
- S. N. Kazi, A. Badarudin, M. N. M. Zubir, H. N. Ming, M. Misran, E. Sadeghinezhad, M. Mehrali and N. I. Syuhada, Nanoscale Res. Lett., 2015, 10, 212 CrossRef PubMed.
- J. I. Paredes, S. Villar-Rodil, P. Solís-Fernández, A. Martínez-Alonso and J. M. D. Tascón, Langmuir, 2009, 25, 5957–5968 CrossRef CAS PubMed.
- G. Haacke, J. Appl. Phys., 1976, 47, 4086–4089 CrossRef CAS.
- M. Acik, G. Lee, C. Mattevi, A. Pirkle, R. M. Wallace, M. Chhowalla, K. Cho and Y. Chabal, J. Phys. Chem. C, 2011, 115, 19761–19781 CrossRef CAS.
- J. García de la Torre, Biophys. Chem., 2001, 93, 159–170 CrossRef.
- H.-K. Jeong, Y. P. Lee, M. H. Jin, E. S. Kim, J. J. Bae and Y. H. Lee, Chem. Phys. Lett., 2009, 470, 255–258 CrossRef CAS.
- L. F. Bautista, G. Morales and R. Sanz, Chemosphere, 2015, 136, 273–280 CrossRef CAS PubMed.
- J. Tully, R. Yendluri and Y. Lvov, Biomacromolecules, 2016, 17, 615–621 CrossRef CAS PubMed.
- P. Singh, H. Singh, V. Castro-Aceituno, S. Ahn, Y. J. Kim and D. C. Yang, RSC Adv., 2017, 7, 15397–15407 RSC.
- A. Gebregeorgis, C. Bhan, O. Wilson and D. Raghavan, J. Colloid Interface Sci., 2013, 389, 31–41 CrossRef CAS PubMed.
- D. Li, M. B. Müller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- K. Liu, J. J. Zhang, F. F. Cheng, T. T. Zheng, C. Wang and J. J. Zhu, J. Mater. Chem., 2011, 21, 12034–12040 RSC.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- Q. Bin Zheng, M. M. Gudarzi, S. J. Wang, Y. Geng, Z. Li and J.-K. Kim, Carbon, 2011, 49, 2905–2916 CrossRef.
- S. You, S. M. Luzan, T. Szabó and A. V. Talyzin, Carbon, 2013, 52, 171–180 CrossRef CAS.
- X. Li, Y. Zhu, W. Cai, M. Borysiak, B. Han, D. Chen, R. D. Piner, L. Colombo and R. S. Ruoff, Nano Lett., 2009, 9, 4359–4363 CrossRef CAS PubMed.
- H. Choi, Y. Lim, M. Park, S. Lee, Y. Kang, M. S. Kim, J. Kim and M. Jeon, J. Mater. Chem. C, 2015, 3, 1463–1467 RSC.
- R. R. Nair, P. Blake, A. N. Grigorenko, K. S. Novoselov, T. J. Booth, T. Stauber, N. M. R. Peres and A. K. Geim, Science, 2008, 320, 1308–1308 CrossRef CAS PubMed.
- Y. Wang, S. W. Tong, X. F. Xu, B. Özyilmaz and K. P. Loh, Adv. Mater., 2011, 23, 1514–1518 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- J. T. Robinson, M. Zalalutdinov, J. W. Baldwin, E. S. Snow, Z. Wei, P. Sheehan and B. H. Houston, Nano Lett., 2008, 8, 3441–3445 CrossRef CAS PubMed.
- H. C. Schniepp, J. L. Li, M. J. McAllister, H. Sai, M. Herrera-Alonson, D. H. Adamson, R. K. Prud'homme, R. Car, D. A. Seville and I. A. Aksay, J. Phys. Chem. B, 2006, 110, 8535–8539 CrossRef CAS.
- W. Bao, F. Miao, Z. Chen, H. Zhang, W. Jang, C. Dames and C. N. Lau, Nat. Nanotechnol., 2009, 4, 562–566 CrossRef CAS.
- A. Rimola, M. Sodupe and P. Ugliengo, J. Phys. Chem. C, 2009, 113, 5741–5750 CrossRef CAS.
- D. Stauffer, N. Dragneva, W. B. Floriano, R. C. Mawhinney, G. Fanchini, S. French and O. Rubel, J. Chem. Phys., 2014, 141, 044705 CrossRef CAS PubMed.
- K. Kubiak-Ossowska, K. Tokarczyk, B. Jachimska and P. A. Mulheran, J. Phys. Chem. B, 2017, 121, 3975–3986 CrossRef CAS PubMed.
- L. Pérez-Fuentes, C. Drummond, J. Faraudo and D. Bastos-González, Materials, 2017, 10, 893 CrossRef PubMed.
- T. B. Rouf and J. L. Kokini, J. Mater. Sci., 2016, 51, 9915–9945 CrossRef CAS.
- H. Zhang, Z. Zhu, Y. Wang, Z. Fei and J. Cao, Appl. Surf. Sci., 2018, 427, 1019–1029 CAS.
- J. F. Watts and J. Wolstenholme, An Introduction to Surface Analysis by XPS and AES, John Wiley & Sons, Ltd, Chichester, UK, 2003 Search PubMed.
- X. Wallart, C. Henry de Villeneuve and P. Allongue, J. Am. Chem. Soc., 2005, 127, 7871–7878 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S14 on XPS spectra and fitting of coatings, SEM, notes on process reliability, molecular dynamics, of the bio-interfactants, molecular visualisation, sample design, and UV-Vis spectra. See DOI: 10.1039/c8nr08607a |
| This journal is © The Royal Society of Chemistry 2019 |