
Chemical etching of pH-sensitive aggregation-induced emission-active gold nanoclusters for ultra-sensitive detection of cysteine†
Jianxing
Wang
a,
Xiangfang
Lin
a,
Lei
Su
 *ab,
Junfa
Yin
c,
Tong
Shu
a and
Xueji
Zhang
*a
*ab,
Junfa
Yin
c,
Tong
Shu
a and
Xueji
Zhang
*a
aBeijing Advanced Innovation Center for Materials Genome Engineering, Research Center for Bioengineering and Sensing Technology, School of Chemistry and Biological Engineering, University of Science and Technology Beijing, Beijing 100083, P. R. China. E-mail: sulei@ustb.edu.cn; zhangxueji@ustb.edu.cn
bBeijing Advanced Innovation Center for Food Nutrition and Human Health, Beijing Technology and Business University, Beijing 100048, P. R. China
cState Key Laboratory of Environmental Chemistry and Ecotoxicology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, P.R. China
First published on 27th November 2018
Abstract
This study reports the utilization of thiol-induced chemical etching of aggregation-induced emission (AIE)-active Au nanoclusters (NCs) for the facile, sensitive, and selective detection of cysteine. The AIE-active Au NCs were formed in an acidic solution containing excess Au(I)–thiolate complexes. At an acidic pH (2.0), the emission of these Au NCs was enhanced by cysteine at a concentratioin below 1 mM. However, the emission was quenched by cysteine at a high concentration, e.g., 500 mM, via the thiol-induced etching of gold, although the process occurred very slowly. Interestingly, in the absence of cysteine, increasing the solution pH enhanced the emission, while the presence of cysteine remarkably accelerated the etching-induced quenching process. The complete quenching of the emission by excess cysteine at pH 2.0 and the enhancement of the emission by the increasing pH in the absence of cysteine indicated that aurophilicity might not be involved in the AIE of the Au NCs prepared using glutathione (GSH) both as the reducing and protecting reagent. On the other hand, the etching process involved the penetration of cysteine molecules through the Au(I)–thiolate complexes, which could assemble or disassemble around the embedded Au NCs in response to the solution pH to get access to the innermost Au(0) cores. Therefore, a facile, sensitive, and selective method for the detection of cysteine was established. This method exhibited an extremely wide linear range as wide as nine orders of magnitude above the cysteine concentration, including two linear regions of the relative emission intensity of the Au NCs versus the logarithm of cysteine concentration, from 10 pM to 150 μM (correlation coefficient, 0.99851) and from 150 μM to 2 mM (correlation coefficient, 0.99866). An ultra-low limit of detection of 6.3 pM (S/N = 3) was also achieved. The developed method showed superior selectivity for cysteine relative to the 19 other natural amino acids and GSH. The method was applied for the analysis of human serum samples spiked with cysteine with satisfactory results. This study demonstrates the potential of the thiol-induced chemical etching approach as a powerful tool for studying luminescent metal NCs.
Introduction
Metal nanoclusters (NCs), typically comprising of a few to several hundreds of atoms, form a bridge between small organometallic complexes and large metal nanoparticles (NPs). Due to their ultra-small size, comparable to the Fermi wavelength of electrons, metal NCs often exhibit molecular properties such as HOMO–LUMO transitions, molecular chirality, and photoluminescence (PL).1,2 Despite the fact that their quantum yields (QYs) are often lower (e.g., <0.1%) than other luminescent materials, luminescent metal NCs have attracted extensive attention. So far, a full series of atomically precise luminescent metal NCs have been synthesized via the so-called “size-focusing” process based on thiol-induced chemical etching.3 These NCs have shown great potential in many optical fields, including biosensing, bioimaging, and solid-state lighting and display.2,4–6However, curiosity and interest in further chemical etching of metal NCs, especially Au NCs, have led to the development of a new research domain.7–15 The use of strong thiol etchants with Au NCs can lead to the dissolution of Au cores, which releases Au(I)–thiolate complexes and quenches their PL.16 The resultant Au(I)–thiolate complexes have been found to be useful for the preparation of novel luminescent nanomaterials,17–19 and the quenching of luminescent Au NCs by thiols has been used to develop sensors for detecting biothiols, such as glutathione (GSH),20 cysteamine,21 and lactate dehydrogenase.22
Recently, aggregation-induced emission (AIE)23 has presented an effective strategy to achieve luminescent Au NCs with higher QY in solution.24–28 The resultant AIE-type Au NCs have exhibited ultra-small metallic Au(0) cores enveloped by oligomeric Au(I)–thiolate complexes, which are capable of forming more complex and more rigid shell structures, thereby restraining intramolecular vibration- and rotation-induced internal non-radiative relaxation pathways.23,24 Currently, AIE-type Au NCs are receiving increasing attention and represent a growing field of scientific research and technological innovation.24–31
Herein, we report for the first time the cysteine-induced etching of AIE-type Au NCs and their application as ultra-sensitive cysteine sensors with an ultra-wide detection range, with concentrations extending from pM to mM. In this study, the AIE-type Au NCs were present in solution with excess Au(I)–thiolate complexes, which could assemble or disassemble around the Au NCs in response to the changes in the solution pH. Interestingly, we found that these Au(I)–thiolate assemblies surrounding the Au(0) core could serve as a pH-responsive “valve” to control the penetration of cysteine molecules through these Au(I)–thiolate network, and thus get access to and in further etching of the inner Au(0) core. Thus, a chemosensor based on AIE-type Au NCs was developed for the sensitive and selective detection of cysteine in human serum samples.
Experimental
Chemicals
HAuCl4·3H2O, glutathione (GSH), L-cysteine (Cys), ethylenediaminetetraacetic acid (EDTA), L-aspartic acid (Asp), L-alanine (Ala), L-asparagine monohydrate (Asn), L-arginine (Arg), L-glycine (Gly), L-glutamine (Gln), L-glutamic acid (Glu), L-histidine (His), L-isoleucine (Ile), L-leucine (Leu), L-lysine (Lys), L-methionine (Met), L-proline (Pro), L-phenylalanine (Phe), L-serine (Ser), L-tyrosine (Tyr), L-tryptophan (Trp), threonine (Thr), and L-valine (Val) were obtained from Sigma-Aldrich. All other chemicals were of analytical reagents grade and were purchased from Beijing Chemical Corporation (Beijing, China). Ultrapure water prepared by a Millipore-Q system (≥18 MΩ, Milli-Q, Millipore) was used throughout this study.Synthesis of the AIE-type GSH-protected Au NCs
The AIE-type Au NCs were synthesized according to our previous study.32 Briefly, an aqueous solution containing HAuCl4 (18 mM, 1 mL) and GSH (100 mM, 0.3 mL) was mixed with ultrapure water (8.7 mL) with gentle stirring at 25 °C, and then incubated at 70 °C for 24 h. Finally, the yellowish solution of the AIE-type Au NCs was obtained and stored at 4 °C. Prior to use, the Au NCs were not treated with any organic solvents.pH-Controlled cysteine-induced etching of the Au NCs
First, the as-synthesized Au NC solution and the cysteine solution were adjusted to the same pH value (2.0, 5.0, 8.0, or 11.0) using 1.0 M HCl or NaOH solution. Then, the Au NC and cysteine solutions were mixed together (v![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :v, 1:4, respectively) at 20 °C.
:v, 1:4, respectively) at 20 °C.
Sensitivity and selectivity of the Au NCs-based sensor for cysteine
First, all of the solutions used were adjusted to pH 11.0 using 1.0 M NaOH. To test the sensitivity of the sensor, the Au NC solution (200 μL) was incubated with cysteine solutions (800 μL, final concentrations 10 pM–2.0 mM) for 5 min at 20 °C. The resultant mixture was subjected to PL measurements. To test the selectivity of the sensor, GSH and other 19 non-cysteine natural amino acids (Ala, Arg, Asn, Asp, Gln, Glu, Gly, His, Ile, Leu, Lys, Met, Phe, Pro, Ser, Thr, Trp, Tyr, and Val) were used. Each solution of GSH or amino acid (800 μL, final concentration 1.0 mM) was incubated with the Au NC solution (200 μL) for 5 min at 20 °C, and then was subjected to PL measurement.Analysis of human serum
Blood samples from three healthy adult volunteers who had provided informed consent were collected by the school hospital of this university using Gel & clot activator tubes (Improve Medical Instruments Co. Ltd, Guangzhou, China). The serum samples were then prepared by centrifugation. The obtained serum samples were treated in an EDTA aqueous solution with a final EDTA concentration of 25 mM and an adjusted pH of 11.0 using 1.0 M NaOH before further use. A suitable volume of the pre-treated human serum was appropriately spiked with the standard cysteine solution to give three samples each with cysteine at final concentrations of 100 nM or 1 mM. These samples were used for analysis.Instrumentation
Fluorescence spectra were recorded on an F-4500 spectrometer (HITACHI). UV-vis absorption spectra were recorded on a Shimadzu UV-1800 spectrometer. X-ray photoelectron spectroscopy (XPS) was conducted with a VG Scientific (U.K.) X-ray photoelectron spectrometer (Model ESCALab220i-XL). Transmission electron microscopy (TEM) images were obtained using a JEOL JEM-2010 microscope.Results and discussion
Under UV light, the as-synthesized AIE-active Au NCs emitted intensely orange PL (Fig. 1A). In the PL spectrum, the Au NCs exhibited a main emission peak at 565 nm with a weak shoulder peak at 610 nm (Fig. 1B). Using fluorescein as a reference, the Au NCs were found to have a high QY of ∼6%. The PL lifetime measurements of the as-synthesized AIE-type Au NCs showed a microsecond-scale lifetime (1.22 μs, 61.91%) (Fig. 1C), indicating that the main components of the measured PL of the Au NCs originated from triplet states. The UV-vis absorption spectrum of the Au NCs shows a large energy gap (∼400 nm) (Fig. S1†), suggesting that the Au NCs had a small Au(0) core. XPS of the Au NCs shows that the Au 4f spectrum of the Au NCs could be deconvoluted into two peaks corresponding to the Au(I) and Au(0) components with binding energies of 84.3 eV and 83.7 eV, respectively (Fig. 1D). According to the XPS spectrum, Au(I) constitutes ∼75% of all Au atoms in the Au NC solution. The high Au(I) content in the Au NC solution indicated that besides the oligomeric Au(I)–thiolate complexes, forming the ligand shell of the Au NCs, there also existed excessive Au(I)–thiolate complexes in the Au NC solution, according to previous studies.24,32 These excess Au(I)–thiolate complexes were capable of assembly or aggregation, especially at acidic pH values,24,32 leading to the formation of membranous assemblies or crystalline polymeric networks. | ||
| Fig. 1 (A) Digital photo of the as-prepared AIE-active Au NCs under visible light (left) and 365 nm UV light (right), (B) PL (λex = 365 nm), (C) PL lifetime decay profiles, and (D) Au 4f XPS spectra of the as-prepared AIE-active Au NCs. | ||
The PL of the as-prepared AIE-type Au NCs was found to be sensitive to cysteine, a naturally sulfur-containing amino acid. The PL spectra were recorded after incubation of the as-prepared Au NC solution (pH 2.0) with cysteine at room temperature for 2 hours. As shown in Fig. S2,† the PL intensity of the Au NCs was dependent on the cysteine concentration. At pH 2.0 and lower cysteine concentrations (≤1.0 mM), the PL intensity of the as-prepared Au NCs increased with the increase in cysteine concentration, which indicated a cysteine-induced enhancement of the PL of the as-prepared AIE-type Au NCs. This enhancement could be associated with the adsorption of cysteine molecules onto the Au(0) cores, which decreased the surface defects,33 and further donating delocalized electrons of electron-rich atoms or cysteine groups to the Au(0) cores.34 At 1.0 mM cysteine concentration, the enhancement of the PL intensity of the Au NCs reached a maximum, producing an almost two-fold increase in the PL intensity. With further increase in the cysteine concentration, the enhancement effect weakened.
However, it is worth noting that when the incubation time of the Au NCs with cysteine was prolonged, it was found that although the enhanced PL intensity of the Au NCs induced by 1.0 mM cysteine was maintained for at least one week (Fig. S3†), the PL intensity of the Au NCs in the presence of 500 mM cysteine was completely quenched within 3 days (Fig. 2A). Moreover, the yellowish color of the Au NC solution gradually turned colorless (inset). The PL quenching in the presence of 500 mM cysteine was further examined by the UV-vis absorption and XPS spectra measurements, using 1 mM cysteine as a control. The time-dependent UV-vis absorption spectra of the Au NCs in the presence of 1 mM cysteine (Fig. S4†) show a minor increase in the absorbance below 500 nm within 2 hours, and the absorbance was subsequently maintained. However, significant changes could be seen in the UV-vis absorption spectra of the Au NCs in the presence of 500 mM cysteine. As shown in Fig. 2B, a small increase in absorbance over the whole wavelength range was observed within 2 hours. Subsequently, however, the absorbance below 500 nm decreased gradually, accompanied by the fading of the solution color. Further measurement of XPS spectra of the product solution (Fig. 3) revealed that the Au NCs incubated with 1 mM cysteine still exhibited two valent states, i.e., Au(I) at 84.3 eV and Au(0) at 83.7 eV. However, the Au NCs incubated with 500 mM cysteine only exhibited one oxidation state, Au(I) at 84.3 eV. This result reveals that the etching-induced oxidation caused decomposition of the Au(0) core by the high concentration cysteine, which is consistent with a previous report on the etching ability of cysteine towards gold.15
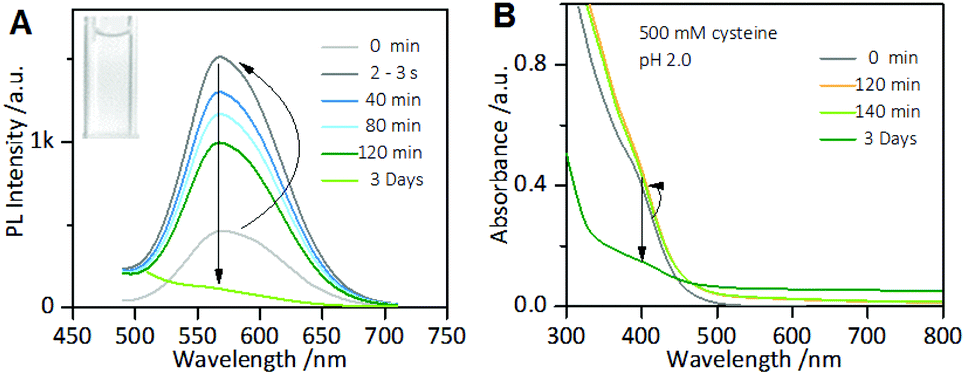 | ||
| Fig. 2 Time-dependent photoemission (A) and UV-vis absorption spectra (B) of the as-prepared AIE-active Au NC solution incubated with 500 mM cysteine at pH 2.0. Inset of (A): Digital photo of the AIE-active Au NC solution after adding 500 mM cysteine at pH 2.0 for 3 days under visible light. | ||
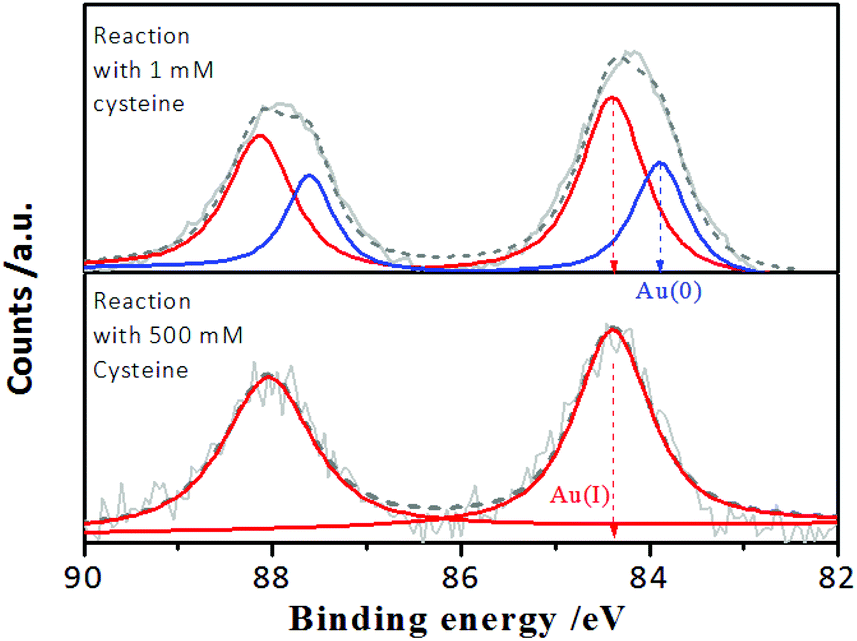 | ||
| Fig. 3 Au 4f XPS spectra of the AIE-active Au NCs after reaction for 3 days with 1 mM and 500 mM cysteine, respectively, at pH 2.0. | ||
The thiol-induced etching of nanoscale Au particles reportedly involved the direct interaction between thiol molecules and the outermost gold atoms of the Au(0) cores, leading to the thiols peeling off from the Au(0) cores via an Au–ligand complexation-driven oxidation dissolution pathway, which released the Au(I)–thiolate complexes into the solution.35,36 In the case of AIE-type Au NCs, the Au(0) cores were enveloped by the oligomeric Au(I)–thiolate complexes, which constitute the rigid ligand shell.24,32 Furthermore, the AIE-type Au NCs could be essentially embedded within the excess Au(I)–thiolate complex assemblies according to our recent study.32 Accordingly, the TEM image of the as-synthesized Au NCs (Fig. S5†) revealed that only the thick membranous morphology of the Au(I)–thiolate complex assemblies could be directly seen at pH 2.0. Therefore, the aforementioned observations of the cysteine-sensitive PL of the Au NCs clearly indicate that, on the one hand, cysteine molecules could penetrate the surrounding Au(I)–thiolate assemblies and the Au(I)–thiolate ligand shell and then access the innermost Au(0) cores. On the other hand, the full oxidation dissolution of the Au(0) cores by cysteine required a relatively high concentration of cysteine (e.g., 500 mM) and a long reaction time (e.g., several days), which reflected the relatively large resistance to cysteine penetration of the Au(I) assemblies and contact with the embedded Au(0) cores.
Interestingly, the cysteine-induced chemical etching of the Au NCs was found to be highly sensitive to the solution pH. Fig. 4 shows the effect of pH on the PL spectra of the Au NCs solution in the presence of cysteine. At elevated pH values, even 1 mM cysteine could quench the Au NCs PL. As shown in Fig. 4A, at pH 5.0, upon adding 1 mM cysteine, the PL intensity of the Au NCs increased. However, only 20 min later, the PL intensity began to decline slowly, and it reached nearly half of its maximum one week later. At pH 8.0, as shown in Fig. 4B, upon adding 1 mM cysteine, the PL intensity of the Au NCs first increased; however, only a few seconds later, the PL intensity began to decline and was quenched completely one week later. Furthermore, at pH 11.0, the quenching effect of cysteine on the PL of the Au NCs became more remarkable. As shown in Fig. 4C, upon adding 1 mM cysteine, the PL intensity of the Au NCs declined rapidly to nearly a quarter of its initial intensity within 40 min, and it was quenched completely 160 min later. Moreover, we observed that the yellowish color of the solution became colorless (inset). The resultant product solution was characterized with UV-vis absorption and XPS spectra, further confirming the cysteine-induced etching process. As revealed by UV-vis absorption spectra (Fig. 4D), an obvious decrease in the absorbance below 500 nm occurred after treating the Au NCs with 1 mM cysteine at pH 11.0. In the XPS spectra (Fig. S6†), only the Au(I) peak at 84.3 eV is present, corresponding to full oxidation of the Au(0) atoms. These results clearly show that increasing the pH could remarkably facilitate the etching-induced quenching of the luminescent Au NCs by cysteine. The solution pH is known to have the ability to control the assembly/disassembly of Au(I)–thiolate complexes through the protonation/deprotonation of pH-sensitive ligands.24,37–39 Increasing the solution pH could cause the decomposition of the assemblies of Au(I)–thiolate complexes in solution, which was observed using TEM and dynamic light scattering (DLS) techniques in our previous study.32 Therefore, the observed enhancement of the etching-induced quenching effect at higher pH values should enable increased cysteine penetration through the aforementioned Au(I)–thiolate surroundings to interact with the embedded Au(0) cores. In addition, increased pH is known to improve the thiol etching ability toward gold. Therefore, the enhanced etching-induced quenching effect at higher pH values should be a result of a synergistic effect of the reduced penetration resistance for cysteine molecules and the improved etching ability at higher pH values. The process of the etching-induced quenching of the AIE-active Au NCs by cysteine at higher pH values is illustrated in Scheme 1.
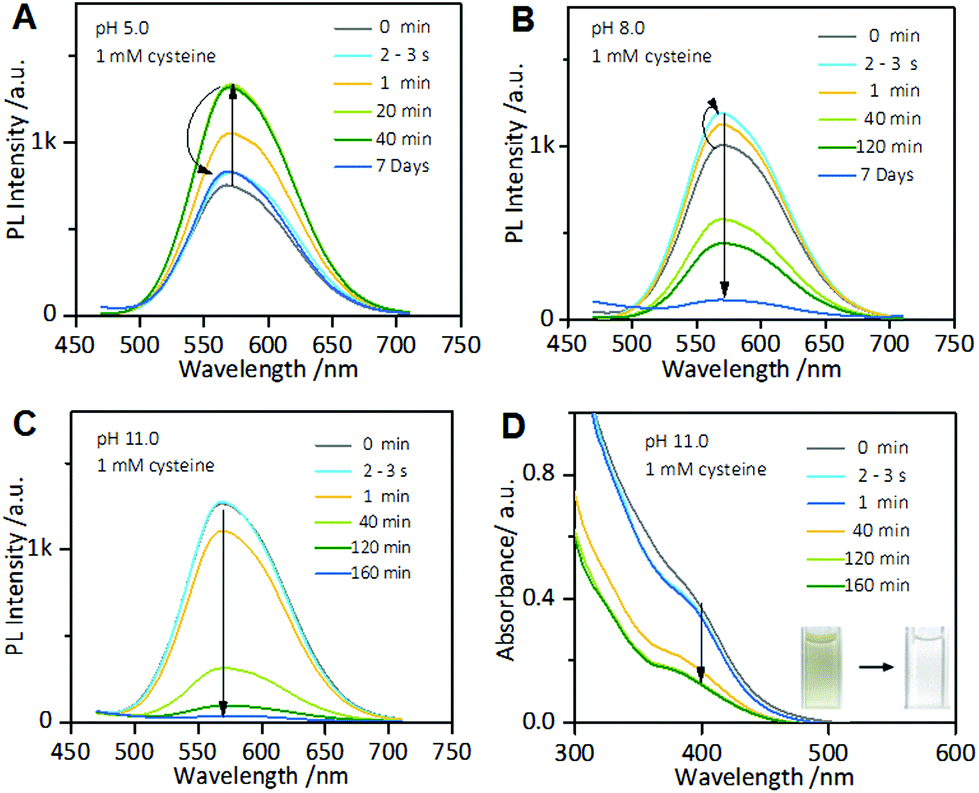 | ||
| Fig. 4 Time-dependent PL spectra of the AIE-active Au NC solution incubated with 1 mM cysteine at pH 5.0 (A), pH 8.0 (B), and pH 11.0 (C). (D) Time-dependent UV-vis absorption spectra of the AIE-active Au NC solution incubating with 1 mM cysteine at pH 11.0. Inset: Photographs of the solution before and after addition of 1 mM cysteine. | ||
 | ||
| Scheme 1 Schematic of the increase in the solution pH, prompting the permeation of the cysteine molecules through the Au(I)–thiolate complex surroundings to etch and quench the embedded Au NCs. (a → b) Increasing the pH prompted the disassembly of the Au(I)–thiolate complex network encapsulating the AIE-active Au NCs and enhanced the emission; (b → c) alkaline pH facilitated the penetration of cysteine through the Au(I)–thiolate surroundings thereby allowing access to the embedded Au NCs; (c → d) cysteine molecules etch the Au(0) cores, causing the decomposition of the AIE-active Au NC system and quenching of the emission. | ||
As an emerging luminescent Au NC system, the AIE-active thiolated-Au NCs should have a more rigid shell compared with their fluorescent counterparts with low QY, thereby restraining the intramolecular vibration- and rotation-induced internal nonradiative relaxation pathways (vide supra). Accordingly, their PL always contains predominantly microsecond lifetime components and are usually considered to be phosphorescence. However, the understanding of their complex surface/interfacial structures and the origin of their AIE is quite limited. For instance, it is not clear whether the rigid shell formed by the aggregation of the oligomeric Au(I)–thiolate complex ligands is maintained by aurophilic interactions or whether the AIE originates from aurophilic interactions, though it is well-known that aurophilicity of Au(I) can cause the rigidness of the Au(I) systems and microsecond-lifetime emission. In the present study, the etching-induced quenching of the PL of the AIE-active Au NCs could help to clarify this uncertainty. As mentioned above, the AIE-active Au NCs contain Au(0) cores and a rigid Au(I)–thiolate shell. Cysteine introduction could etch the Au(0) cores. If the rigid shell formed by the aggregation of the oligomeric Au(I)–thiolate complex ligands is maintained by aurophilic bonds between the Au(I) atoms of the neighbour Au(I)–thiolate complex ligands, and accordingly the AIE originates from aurophilic interactions, then the decomposition of Au(0) cores should not break the aurophilic bonds of neighbour Au(I)–thiolate complex ligands, which should continuously contribute to the emission of the system. However, it was observed that the addition of excess cysteine at pH 2.0 could etch the Au(0) cores (vide supra) and completely quench the emission of the system (Fig. 2A), indicating that aurophilic interactions might not have contributed to the formation of the rigid shell and the AIE of the Au NCs. Moreover, aurophilicity can be classified as a weak force, comparable to hydrogen bonding, and thus can be broken by the repulsive electrostatic forces.40 If aurophilic bonding between the neighbour Au(I)–thiolate complex ligands is responsible for the AIE of the AIE-active Au NCs, increasing the pH would break these aurophilic bonds and cause the quenching. On the contrary, the AIE was enhanced with the increasing pH, as shown in Fig. 5, which suggested the absence of aurophilic interactions. The lack of aurophilicity might be associated with the relatively large steric hindrance of the GSH molecule due to the charged Gly and Glu residues tethered to the Cys residue. The enhanced PL with the pH increase could be attributed to the fact that high pH conditions favor the electron transfer between gold and sulfur atoms.25
 | ||
| Fig. 5 PL spectra of AIE-active Au NC solution at different pH values in the absence of cysteine. | ||
Based on the above results, we next developed an AIE-type Au NCs-based chemosensor for highly sensitive and selective detection of cysteine. The cysteine concentration in healthy adult plasma is approximately 250 μM. The cysteine level in body fluids can serve as an indicator for some diseases.41–43 As mentioned above, at pH 11.0, cysteine molecules could rapidly quench the PL of the Au NCs. Therefore, pH 11.0 was used as the optimal pH value for testing the analytical performance of the sensor. As shown in Fig. 6A, the value of I/I0 decreased as the cysteine concentration increased, and two linear regions of the logarithm of the cysteine concentration are visible at 10 pM to 150 μM (correlation coefficient, 0.99851) and at 150 μM to 2 mM (correlation coefficient, 0.99866). The total linear range is as wide as 9 orders of magnitude, which is the widest linear range of all previous reported methods for the detection of cysteine. The limit of detection (LOD) at a signal to noise (S/N) of 3 was 6.3 pM, which is lower than that of all of metal NCs-based optical cysteine sensors reported so far (Table S1†). We also tested the specificity of the AIE-type Au NCs-based cysteine sensor by conducting control experiments using the remaining 19 natural amino acids and GSH to provide interference. The sensor showed superior selectivity for cysteine over the interferences, as shown in Fig. 6B. The developed AIE-type Au NCs-based cysteine sensor was also applied for the analysis of human serum samples without complicated derivation and separation procedures. A suitable volume of the blood serum samples pre-treated with 25 mM EDTA was appropriately spiked with the standard solution of cysteine and three samples at the same concentration of cysteine (100 nM or 1 mM). The results showed that the quantitative recoveries of the samples at 100 nM cysteine were between 95% and 109%, and the quantitative recoveries of the samples at 1 mM cysteine were between 97% and 107% (Table S2†). These results show that this sensor has the potential for facile and quantitative detection of free cysteine in human serum samples.
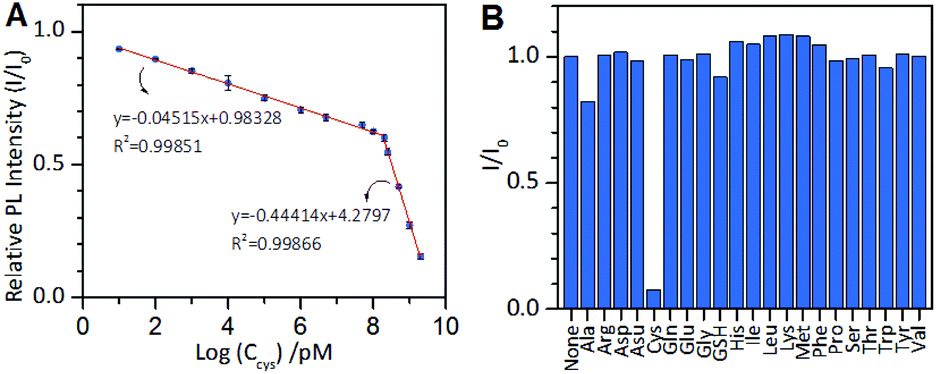 | ||
| Fig. 6 (A) Relative PL intensity (I/I0) of the AIE-active Au NCs vs. the logarithm of the cysteine concentration. The cysteine concentration ranges from 10 pM to 2 mM. The reaction time was 5 min (B) Specific responses of the AIE-active Au NCs to cysteine over a common interference of 1 mM. | ||
Conclusions
In the present study, we demonstrated that chemical etching of AIE-type Au NCs by cysteine can be applied to the facile, selective, and sensitive detection of cysteine. The AIE-type Au NCs were essentially in solution containing an excess of Au(I)–thiolate complexes. The Au(I)–thiolate complexes exhibited pH-sensitive permeability for cysteine, which affected the access of cysteine to the embedded Au NCs. Cysteine enhanced the emission of the Au NCs; however, at higher concentrations, cysteine etched the Au NCs. This etching caused PL quenching, which proceeded more quickly as the solution pH increased. The results indicate that the AIE of the Au NCs prepared with GSH as the reducing and protecting reagent might not involve aurophilicity. Furthermore, the AIE-type Au NCs-based cysteine sensor was constructed at an alkaline pH. The sensor exhibited an ultra-wide linear concentration range as wide as 9 orders of magnitude and an ultra-low LOD of 6.3 pM (S/N = 3). The sensor also showed superior selectivity for cysteine over the 19 other natural amino acids and GSH. With the developed sensor, human serum samples spiked with cysteine were analyzed with satisfactory results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the Beijing Advanced Innovation Center for Food Nutrition and Human Health, Beijing Technology and Business University (20181035), the National Natural Science Foundation of China (Grant No. 21727815), the Beijing Municipal Natural Science Foundation (2184107) and the Fundamental Research Funds for the Central Universities (FRF-BD-17-016A).Notes and references
- J. Zheng, C. Zhou, M. X. Yu and J. B. Liu, Nanoscale, 2012, 4, 4073–4083 RSC.
- R. C. Jin, C. J. Zeng, M. Zhou and Y. X. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- R. C. Jin, H. F. Qian, Z. K. Wu, Y. Zhu, M. Z. Zhu, A. Mohanty and N. Garg, J. Phys. Chem. Lett., 2010, 1, 2903–2910 CrossRef CAS.
- L. Shang, S. J. Dong and G. U. Nienhaus, Nano Today, 2011, 6, 401–418 CrossRef CAS.
- L. Y. Chen, C. W. Wang, Z. Yuan and H. T. Chang, Anal. Chem., 2015, 87, 216–229 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- Y. L. Liu, K. L. Ai, X. L. Cheng, L. H. Huo and L. H. Lu, Adv. Funct. Mater., 2010, 20, 951–956 CrossRef CAS.
- X. Yuan, Y. Q. Tay, X. Y. Dou, Z. T. Luo, D. T. Leong and J. P. Xie, Anal. Chem., 2013, 85, 1913–1919 CrossRef CAS PubMed.
- H. Kawasaki, K. Hamaguchi, I. Osaka and R. Arakawa, Adv. Funct. Mater., 2011, 21, 3508–3515 CrossRef CAS.
- R. P. Li, P. P. Xu, J. Fan, J. W. Di, Y. F. Tu and J. L. Yan, Anal. Chim. Acta, 2014, 827, 80–85 CrossRef CAS PubMed.
- Q. C. Mo, F. Liu, J. Gao, M. P. Zhao and N. Shao, Anal. Chim. Acta, 2018, 1003, 49–55 CrossRef CAS PubMed.
- W. W. Guo, J. P. Yuan and E. Wang, Chem. Commun., 2012, 48, 3076–3078 RSC.
- C. Zhang, X. Sun, J. Li and Y. N. Liu, Nanoscale, 2013, 5, 6261–6264 RSC.
- Y.-T. Tseng, Z. Yuan, Y.-Y. Yang, C.-C. Huang and H.-T. Chang, RSC Adv., 2014, 4, 33629–33635 RSC.
- Y. Chen, W. Y. Li, Y. Wang, X. D. Yang, J. Chen, Y. N. Jiang, C. Yu and Q. Lin, J. Mater. Chem. C, 2014, 2, 4080–4085 RSC.
- T. A. Dreier and C. J. Ackerson, Angew. Chem., Int. Ed., 2015, 54, 9249–9252 CrossRef CAS PubMed.
- T.-H. Chen, C.-C. Nieh, Y.-C. Shih, C.-Y. Ke and W.-L. Tseng, RSC Adv., 2015, 5, 45158–45164 RSC.
- T. Shu, L. Su, J. X. Wang, X. Lu, F. Liang, C. Z. Li and X. J. Zhang, Anal. Chem., 2016, 88, 6071–6077 CrossRef CAS PubMed.
- X. Lu, T. Wang, T. Shu, X. H. Qu, X. J. Zhang, F. Liang and L. Su, J. Mater. Chem. C, 2016, 4, 11482–11487 RSC.
- H. Jiang, X. Q. Su, Y. Y. Zhang, J. Y. Zhou, D. J. Fang and X. M. Wang, Anal. Chem., 2016, 88, 4766–4771 CrossRef CAS PubMed.
- T. Shu, L. Su, J. X. Wang, C. Z. Li and X. J. Zhang, Biosens. Bioelectron., 2015, 66, 155–161 CrossRef CAS.
- J. Liu, H. W. Li and Y. Q. Wu, RSC Adv., 2017, 7, 13438–13443 RSC.
- J. Mei, N. L. Leung, R. T. Kwok, J. W. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- Z. T. Luo, X. Yuan, Y. Yu, Q. B. Zhang, D. T. Leong, J. Y. Lee and J. P. Xie, J. Am. Chem. Soc., 2012, 134, 16662–16670 CrossRef CAS PubMed.
- A. Yahia-Ammar, D. Sierra, F. Merola, N. Hildebrandt and X. Le Guevel, ACS Nano, 2016, 10, 2591–2599 CrossRef CAS.
- B. Kuppan and U. Maitra, Nanoscale, 2017, 9, 15494–15504 RSC.
- F. F. Cao, E. G. Ju, C. Q. Liu, W. Li, Y. Zhang, K. Dong, Z. Liu, J. S. Ren and X. G. Qu, Nanoscale, 2017, 9, 4128–4134 RSC.
- Y. Xie, Y. Xianyu, N. X. Wang, Z. Y. Yan, Y. Liu, K. Zhu, N. S. Hatzakis and X. Y. Jiang, Adv. Funct. Mater., 2018, 28, 1702026–1702032 CrossRef.
- K. Pyo, V. D. Thanthirige, K. Kwak, P. Pandurangan, G. Ramakrishna and D. Lee, J. Am. Chem. Soc., 2015, 137, 8244–8250 CrossRef CAS.
- Y. H. Guo, X. Y. Tong, L. Y. Ji, Z. L. Wang, H. Y. Wang, J. M. Hu and R. J. Pei, Chem. Commun., 2015, 51, 596–598 RSC.
- H. H. Deng, X. Q. Shi, F. F. Wang, H. P. Peng, A. L. Liu, X. H. Xia and W. Chen, Chem. Mater., 2017, 29, 1362–1369 CrossRef CAS.
- J. X. Wang, N. Goswami, T. Shu, L. Su and X. J. Zhang, Mater. Chem. Front., 2018, 2, 923–928 RSC.
- M. L. Cui, J. M. Liu, X. X. Wang, L. P. Lin, L. Jiao, L. H. Zhang, Z. Y. Zheng and S. Q. Lin, Analyst, 2012, 137, 5346–5351 RSC.
- Z. K. Wu and R. C. Jin, Nano Lett., 2010, 10, 2568–2573 CrossRef CAS.
- L. R. P. de Andrade Lima and D. Hodouin, Hydrometallurgy, 2005, 79, 121–137 CrossRef CAS.
- T. Shu, J. X. Wang, L. Su and X. J. Zhang, Anal. Chem., 2016, 88, 11193–11198 CrossRef CAS PubMed.
- Y. Shichibu, Y. Negishi, H. Tsunoyama, M. Kanehara, T. Teranishi and T. Tsukuda, Small, 2007, 3, 835–839 CrossRef CAS PubMed.
- I. Odriozola, I. Loinaz, J. Pomposo and H. J. Grande, J. Mater. Chem., 2007, 17, 4843–4845 RSC.
- H. Y. Chang, Y. T. Tseng, Z. Yuan, H. L. Chou, C. H. Chen, B. J. Hwang, M. C. Tsai, H. T. Chang and C. C. Huang, Phys. Chem. Chem. Phys., 2017, 19, 12085–12093 RSC.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2008, 37, 1931–1951 RSC.
- K. G. Reddie and K. S. Carroll, Curr. Opin. Chem. Biol., 2008, 12, 746–754 CrossRef CAS PubMed.
- X. R. Yang, J. Z. Wu, S. Q. Jing, M. J. Forster and L. J. Yan, Biophys. Rep., 2018, 4, 104–113 CrossRef.
- J. Sun, F. Yang, D. Zhao, C. X. Chen and X. R. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 6860–6866 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nr08526a |
| This journal is © The Royal Society of Chemistry 2019 |