
Superior oxygen electrocatalysts derived from predesigned covalent organic polymers for zinc–air flow batteries†
Jianing
Guo‡
,
Tingting
Li‡
,
Qiuli
Wang
,
Ningyuan
Zhang
,
Yuanhui
Cheng
and
Zhonghua
Xiang
 *
*
Beijing Advanced Innovation Centre for Soft Matter Science and Engineering, State Key Laboratory of Organic-Inorganic Composites, College of Chemical Engineering, College of Energy, Beijing University of Chemical Technology, Beijing, 100029, PR China. E-mail: xiangzh@mail.buct.edu.cn
First published on 15th November 2018
Abstract
Covalent organic polymers (COPs) as emerging porous materials with ultrahigh hydrothermal stability and well-defined and adjustable architectures have aroused great interest in the electrochemical field. Here, we reported a rational design approach for the preparation of a bifunctional electrocatalyst with the assistance of a predesigned bimetallic covalent organic polymer. With the predesigned nitrogen position and structural features of COP materials, the obtained CCOPTDP–FeNi–SiO2 catalyst affords a remarkable bifunctional performance with a positive half-wave potential (0.89 V vs. reversible hydrogen electrode: RHE, superior to the benchmark Pt/C) for ORR activity, and a low overpotential (0.31 V better than the benchmark IrO2) at 10 mA cm−2 for OER activity in alkaline solution. The potential gap between E1/2 and Ej=10 reaches 0.650 V, in line with that observed in the current state-of-the-art bifunctional oxygen electrode materials. Moreover, a homemade rechargeable Zn–air flow battery using the CCOPTDP–FeNi–SiO2 catalyst as an air cathode exhibits an almost twofold power density (112.8 vs. 64.8 mW cm−2) and a lower charge–discharge voltage gap, compared with a commercialized noble Pt/C + IrO2/C-driven Zn–air flow battery. More importantly, the CCOPTDP–FeNi–SiO2-driven battery maintains a better cycling stability compared to a noble metal-driven battery without performance decay. Accordingly, this work will open up new ways for fabricating practical oxygen electrodes for, but not limited to, metal air based battery applications.
Introduction
Rechargeable zinc–air flow batteries are a promising power technology for the future large-scale energy storage and release of electrical energy fields, owing to their high theoretical specific energy, low cost, safety, and economic viability.1–3 However, like most metal–air batteries, one of the main challenges for zinc–air flow batteries that needs to be overcome is the sluggish rates of the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER).4–6 Therefore, it is highly desired to develop an ideal bifunctional catalyst to make the obtained zinc–air flow batteries have excellent ORR/OER catalytic performances.7–9Recently, metallic (e.g., Fe, Co, and Ni) alloy catalysts have attracted intensive attention as low-cost and efficient oxygen electrocatalysts,10 which can exhibit superior activity in comparison to their individual entities.11,12 Among the various bimetallic alloys, FeNi alloy exhibits a great application prospect because iron or nickel shows high intrinsic activity for either the ORR or the OER,13,14 providing additional synergistic properties during the electrocatalysis and enhancing the electrocatalytic performance.15 Furthermore, it is believed that combining alloy nanoparticles and heteroatom doped carbon materials is a logical strategy to enhance the catalytic activity of the alloy, through increasing the number of active sites and providing efficient electron conduction channels.16,17 Wu et al. reported a new class of highly active and durable FeCoNi alloy particles attached onto nitrogen-doped graphene tube bifunctional electrocatalysts, which exhibited good stability across a wide potential window in alkaline media.18 Yang et al. synthesized a hybrid catalyst with NiCo alloy nanoparticles decorated on N-doped carbon nanofibers by a facile electrospinning method.19 However, most existing methods for synthesizing alloy-decorated carbon materials involve the mixing of the sources of transition metals, nitrogen, and carbon, followed by thermal treatment.15,20 In these processes, the metal precursors and N source are often randomly distributed or mixed on the carbon supports, which is difficult to control and form ordered electrocatalytically active sites. If realized, however, the order control of metallic active sites should provide us with a powerful means to tailor the structure–property relationship for the creation of a high-performance bifunctional electrocatalyst.
Recently, covalent organic polymers (COPs) as a new exciting type of porous material with a high specific surface area, excellent chemical stability and porosity have aroused great interest because of their enormous potential to catalyze the oxygen reduction reaction.21–23 Particularly, COPs not only possess a well-defined and precisely controllable capacity, such as robust tailoring of heteroatom incorporation and location of active sites, but also provide carbon and nitrogen atoms in the ligands, along with the flexibility to dope active transition metals into the frameworks.24,25 Our previous studies have designed and synthesized a large class of COPs with precisely controlled locations of N atoms and hole sizes as efficient metal-free electrocatalysts for ORR.26 Therefore, COPs are known to be good precursors for carbon materials with the possibilities and potential as highly efficient energy electrocatalysts for clean and renewable energy technologies.27,28
Herein, we report an in situ one-step method to prepare an efficient and durable bifunctional electrocatalyst, i.e., a metallic FeNi alloy encased in nitrogen-doped carbon based on predesigned covalent organic polymers towards both the ORR and OER. This novel FeNi carbon-based catalyst was synthesized via pyrolysis of Fe/Ni co-doped nitrogen-rich COPs with the assistance of silica (SiO2), which could effectively prevent FeNi alloy nanoparticle (NP) aggregation during the carbonization process. Meanwhile, FeNi alloy NPs are formed and embedded into the porous graphitic carbon layers during the pyrolysis process, preventing the corrosion of FeNi alloy by the harsh environment and promoting the stability of the catalyst.29 The obtained bifunctional electrocatalyst, named CCOPTDP–FeNi–SiO2, displays an outstanding bifunctional performance and stability for ORR (half-wave potential of 0.89 V vs. RHE) and OER (overpotential of 0.31 V at 10 mA cm−2) under alkaline conditions, and is even superior to the commercial Pt/C and IrO2 catalysts, respectively. A zinc–air flow battery constructed using the CCOPTDP–FeNi–SiO2 catalyst as an air-cathode exhibits a high power density of 112.8 mW cm−2 and a low voltage gap between charge and discharge.
Experimental
Bifunctional catalyst synthesis
In a glovebox, bis(1,5-cyclooctadiene)nickel(0) ([Ni(cod)2], 2.000 g), 2,2′-bipyridyl (1.138 g) and 1,5-cyclooctadiene (cod, 0.889 mL) were added to DMF (115 mL). Then the mixture was stirred at 80 °C for 30 min until completely dissolved. Then, meso-tetra (p-bromophenyl) porphine (0.433 g) and 3,8-dibromophenanthroline (0.315 g) were subsequently added to the resulting solution. Thereafter, the reaction vessel was heated at 85 °C for 10 h. After cooling to room temperature, concentrated HCl was added to the deep purple suspension. After filtration, the residual COPTDP was washed with CHCl3, THF, and deionized water, respectively, and dried in a vacuum oven at 80 °C overnight.COPTDP (0.100 g), FeCl3 (0.100 g) and NiCl2 (0.026 g) were added to DMF (10 mL) in a dried reaction bottle, respectively, and then put in an ultrasonic environment. After the ultrasound treatment, the mixture was stirred and refluxed at 120 °C for 12 h, and then cooled to room temperature. The resulting precipitate COPTDP–FeNi was then washed with ethanol and dried in a vacuum oven at 80 °C for 12 h. COPTDP–FeNi (0.116 g) was mixed with tetraethyl orthosilicate (TEOS, 0.29 mL) in a mortar, followed by mixing with formic acid (0.29 mL).30 The COPTDP–FeNi–SiO2 composite was dried in a 60 °C oven overnight.
The COPTDP–FeNi–SiO2 composite was loaded into a tube furnace and heated under an Ar atmosphere. The COPTDP–FeNi–SiO2 powder was heated to 900 °C and maintained at that temperature for 3 h. The resulting CCOPTDP–FeNi–SiO2 composite was mixed with 10% aqueous HF solution and stirred for 12 h to etch the silica, followed by filtering and washing with deionized water several times. Then, the product was dried in a vacuum oven at 80 °C for 12 h. Besides, CCOPTDP–FeNi and CCOPTDP were synthesized by direct pyrolysis of COPTDP–FeNi and COPTDP at 900 °C, respectively.
Physical characterization
The morphologies and structures of catalysts were analysed on an S4700 SEM instrument and a high-resolution transmission electron microscope (HRTEM, 2100F). The crystalline structures of catalysts were recorded on a D/MAX 2000 X-ray diffractometer using a Cu Kα line (λ = 1.54178 Å) as the incident beam with the 2θ scan from 5° to 90° at a step of 0.02°. X-ray photoelectron spectroscopy (XPS) analysis was carried out using an ESCALAB 250 operated at 150 W and 200 eV with monochromated Al Kα radiation. Solid-state NMR spectra were obtained on a Bruker AV300 spectrometer operating at 75.5 MHz for 13C NMR. BET data were obtained on an ASAP 2460.Electrochemical measurements
All electrochemical measurements were carried out at room temperature in a three electrode cell connected to an electrochemical analyzer (CHI 760E). A Pt wire and a saturated calomel electrode (SCE) were used as counter and reference electrodes, respectively. A rotating Pt-ring-disk electrode (RRDE, 5.61 mm diameter, PINE Instrument Inc.) was polished with alumina slurry (CH Instrument Inc.) in turn and subsequently rinsed with deionized water and dried in air. 5 mg of the as-prepared samples or Pt/C catalyst or IrO2/C catalyst were ultrasonically dispersed in 1 mL of anhydrous ethanol and 50 μL of Nafion solution (0.5 wt%) for about 30 min to form a homogeneous ink. Then, 20 μL of the as-prepared catalyst ink was added dropwise on a RRDE giving a loading of 0.386 mg cm−2 and drying at room temperature, while the Pt/C or IrO2/C catalyst electrode was loaded with 0.193 mg cm−2. The potential, measured against a SCE electrode, was converted to the potential vs. RHE according to ERHE = ESCE + 0.059PH + 0.24.Electrochemically active surface area (ECSA) measurements
The electrochemical double layer capacitance (Cdl) of the resulting catalysts was determined to estimate the ECSA of the catalysts within a potential window without faradaic response. Typically, a series of CV curves were obtained at various scan rates (25, 50, 75, 100, 150 and 200 mV s−1) within the potential range of 0.95–1.15 V (versus RHE) and 1.15–1.35 V (versus RHE) for ORR and OER in 1 M KOH, respectively. Thus, the gravimetric double-layer capacitance Cdl (F g−1) at a given scan rate (v) and mass of catalyst deposited on the electrode (m) can be related to Icapacitive as shown below.
The ECSA was calculated as follows
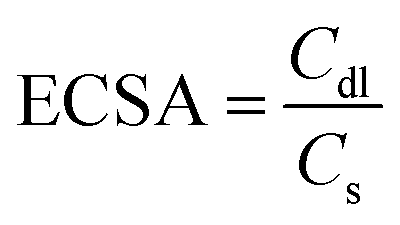
Zinc–air flow battery test
Zn plates (5 cm × 6 cm) were used as the anode. Bifunctional air electrodes were prepared by loading 3 mg CCOPTDP–FeNi–SiO2 hybrid ink on carbon paper plates (1 cm × 1 cm). Air cathodes were also used as the gas diffusion layer and the current collector in assembled batteries. The void between two electrodes was filled with an electrolyte containing 8 M KOH and 0.5 M ZnO to assemble one Zn–air flow battery. Reference bifunctional air electrodes were prepared by loading commercial Pt/C and IrO2 catalysts (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by weight ratio of Pt/Ir) at a similar mass loading (3 mg cm−2) on carbon paper plates. Zn–air flow battery performance was tested on a battery testing station (LAND).
:1 by weight ratio of Pt/Ir) at a similar mass loading (3 mg cm−2) on carbon paper plates. Zn–air flow battery performance was tested on a battery testing station (LAND).
Results and discussion
The synthetic strategy of the CCOPTDP–FeNi–SiO2 catalyst is schematically illustrated in Fig. 1. Briefly, COPTDP was synthesized with meso-tetra (p-bromophenyl) porphine and 3,8-dibromophenanthroline as monomers by the polymerization reaction. Then, Fe/Ni co-doped COPTDP composites were carbonized with the assistance of SiO2, converting into porous carbon material decorated FeNi alloy NPs. The solid state 13C CP/MAS NMR spectra of the as-synthesized COPTDP display four obvious characteristic peaks at 111.9, 126.3, 130.5 and 140.0 cm−1 attributed to the monomers (Fig. 2a), indicating that the skeletons of monomers are efficiently preserved in the COPTDP structure. The detailed structural characteristics of the CCOPTDP–FeNi–SiO2 hybrid were firstly investigated by X-ray diffraction (XRD). The relatively sharp peak at 26° corresponds to the (002) plane of graphitic carbon, while the others at 43.5°, 50.7°, and 74.7° are attributed to the diffraction from the (111), (200), and (220) planes of FeNi alloy (JCPDS 47-1417), respectively (Fig. 2b).15 X-ray photoelectron spectroscopy (XPS) measurements were conducted to investigate the surface chemical composition of the CCOPTDP–FeNi–SiO2 catalyst. The XPS full survey spectrum revealed the presence of C (86.9 at%), N (2.2 at%), and O (10.9 at%) (Fig. S1a†). There is almost no Fe and Ni content because Fe and Ni species may be encapsulated by porous graphitic carbon layers which cannot be detected.31 This result was confirmed from the following HRTEM result. The high-resolution N 1s spectrum (Fig. S1b†) can be deconvoluted into several peaks at 398.7, 400.1 and 400.8, which can be assigned to pyridinic N (29.2%), pyrrolic N (27.5%) and graphitic N (43.3%), respectively. The high content of pyridinic and graphitic N should be beneficial for ORR and OER.32,33 Nitrogen adsorption–desorption isotherms were obtained to investigate the Brunauer–Emmett–Teller (BET) surface area and pore structure of the CCOPTDP–FeNi–SiO2 catalyst (Fig. 2c). The N2 adsorption isotherm obtained for CCOPTDP–FeNi–SiO2 can be identified as a type-IV isotherm.34 The result indicates that CCOPTDP–FeNi–SiO2 has a big BET surface area of 435.5 m2 g−1. As displayed in the inset of Fig. 2c, the CCOPTDP–FeNi–SiO2 catalyst is mainly based on mesopores (most distributed at 3.8 and 23.7 nm) and macropores, which not only are more favorable for mass transport as compared to micropores that may be inaccessible to the electrolyte during the electrochemical reactions, but also provide enough active sites during the electrochemical reactions.35,36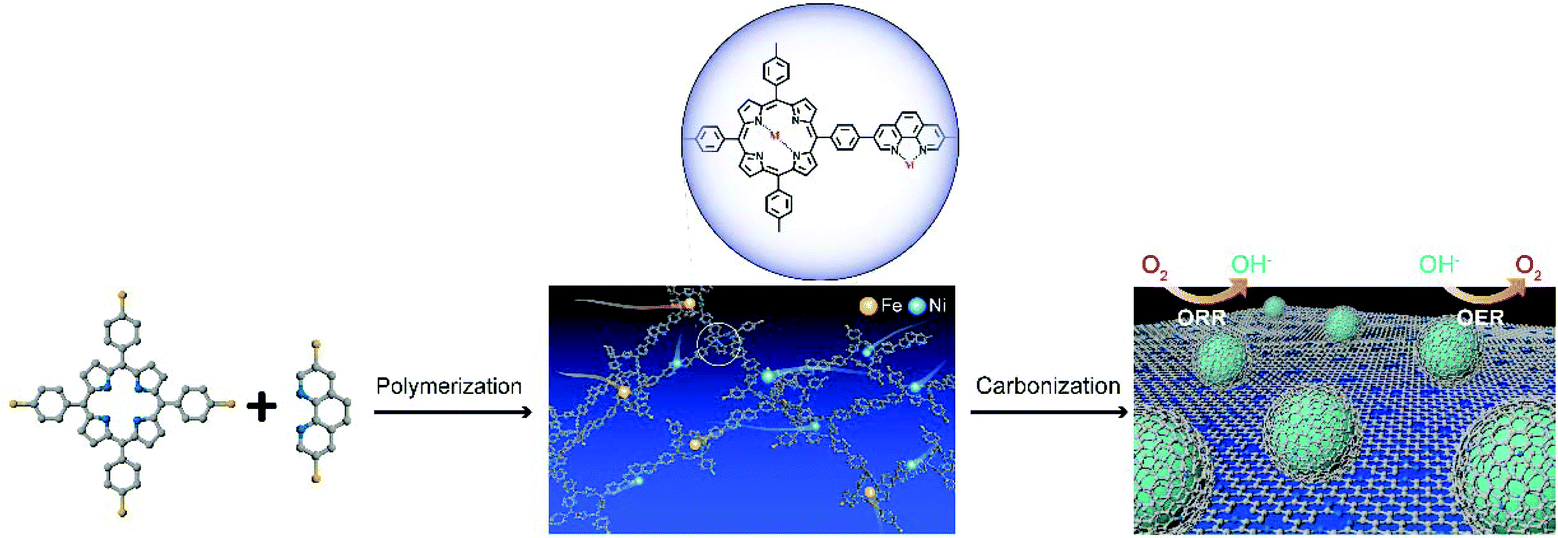 | ||
| Fig. 1 The schematic illustration of the synthesis of CCOPTDP–FeNi–SiO2. For clarity, gray, blue, yellow and purple spheres refer to carbon, nitrogen, iron and nickel atoms, respectively. | ||
 | ||
| Fig. 2 (a) 13C CP/MAS spectra of COPTDP and its peak assignments. (b) XRD patterns of CCOPTDP–FeNi–SiO2 and CCOPTDP–FeNi catalysts. (c) N2 isotherms of CCOPTDP–FeNi–SiO2. Inset: the pore size distribution curves of CCOPTDP–FeNi–SiO2. (d, e and f) The HRTEM images for CCOPTDP–FeNi–SiO2. (g) High-angle annular dark-field (HAADF)-STEM image of CCOPTDP–FeNi–SiO2 and the corresponding element maps of Fe and Ni for CCOPTDP–FeNi–SiO2. (h) The energy dispersive X-Ray (EDX) spectra of CCOPTDP–FeNi–SiO2. Inset: the detection part of the HAADF-STEM image. | ||
The morphology and microstructure were investigated by field scanning electron microscopy (SEM) and high-resolution transmission electron microscopy (HRTEM). The SEM image of the COPTDP presents a hierarchical structure, which can provide more active site exposure (Fig. S2a†). Fig. 2d presents the overview HRTEM image of CCOPTDP–FeNi–SiO2. As can be seen, CCOPTDP–FeNi–SiO2 reaches a high degree of carbonization, presenting a graphene-analogue layer structure. Moreover, FeNi alloy NPs are dispersed on the surface of the CCOPTDP support with an average size of ∼25 nm (Fig. S2b† and Fig. 2e), which is attributed to the SiO2 protective shell effectively preventing the reunion of the FeNi alloy NPs in carbonization. Consistent with the XRD results, the lattice fringes with spacings of 0.21 and 0.34 nm are attributed to the (111) lattice plane of FeNi alloy NPs and graphitic carbon layers, respectively (Fig. 2f). Furthermore, FeNi alloy NPs are embedded in about 12 porous graphitic carbon layers with a thickness of about 4 nm, which not only prevents FeNi NPs from direct contact with the electrolyte to improve catalyst stability, but also prevents inner FeNi alloy NPs from dissolution and agglomeration under harsh conditions.37 As discussed above, FeNi alloy NPs in CCOPTDP–FeNi–SiO2 were encased in the porous graphite shells, which is mostly in accordance with the XPS result (Fig. S1†). High-angle annular dark-field (HAADF)-STEM was performed to confirm the distribution of FeNi NPs in detail (Fig. 2g and Fig. S3†). It can be clearly seen that FeNi NPs of CCOPTDP–FeNi–SiO2 are uniformly distributed over CCOPTDP–FeNi–SiO2, while there is an obvious reunion and large particle size (∼60 nm) of FeNi NPs on CCOPTDP–FeNi (Fig. S2c and Fig. S4†), which also results in a high metal content and low carbon content of the CCOPTDP–FeNi catalyst (Tables S2 and S3†). The corresponding energy dispersive X-ray spectroscopy (EDS) map further verified the distribution of Fe, Ni, C, and N elements that match well with the HRTEM image of the CCOPTDP–FeNi–SiO2 (Fig. 2g and Fig. S3†). From the energy dispersive X-ray (EDX) spectroscopy results (Fig. 2h), it can be found that the encapsulated NPs are composed of homogeneously dispersed Fe and Ni elements, which supports the formation of metallic FeNi alloy.
To demonstrate the multifunctional electrochemical activities of the CCOPTDP–FeNi–SiO2 catalyst, the ORR performance was first investigated in O2-saturated 1 M KOH electrolyte. As a comparison, CCOPTDP–FeNi, CCOPTDP and Pt/C (20 wt% Pt) were also measured under the same conditions. The linear sweep voltammetric (LSV) curves in Fig. 3a confirm that the CCOPTDP–FeNi–SiO2 catalyst exhibits the best ORR activity with an onset potential of 0.990 V versus reversible hydrogen electrode (vs. RHE) and a half-wave potential (E1/2) of 0.890 V vs. RHE, among the ranks of the highest bifunctional performance of non-precious metal modified carbon-based catalysts (Table S1†). The measured E1/2 of CCOPTDP–FeNi–SiO2 is 16 mV more positive than that of Pt/C (0.874 V) and much surpasses that of CCOPTDP–FeNi (0.824 V) and CCOPTDP (0.806 V) (Fig. 3b). This may be attributed to the synergetic effect between the N-doped carbon material and alloy NPs improving catalytic activities. Meanwhile, the CCOPTDP–FeNi–SiO2 catalyst still shows the calculated kinetic current density (JK) value of 3.03 mA cm−2 equal to that of Pt/C (2.94 mA cm−2) at 0.87 V, which is much higher than those of CCOPTDP–FeNi (0.42 mA cm−2) and CCOPTDP (0.20 mA cm−2) catalysts (Fig. 3b, Fig. S5–S8†). The superior catalytic activity of CCOPTDP–FeNi–SiO2 toward the ORR is further verified from the Tafel plots obtained from the polarization curves. The CCOPTDP–FeNi–SiO2 catalyst has a Tafel slope of 46 mV dec−1, much lower than that of CCOPTDP–FeNi (54 mV dec−1), CCOPTDP (59 mV dec−1), and even Pt/C (57 mV dec−1), suggesting the favorable ORR kinetics in the CCOPTDP–FeNi–SiO2 electrocatalyst (Fig. 3c). This also indicates that the transfer of the first electron is probably the rate-determining step in the ORR catalyzed by CCOPTDP–FeNi–SiO2. The transferred number of electrons during the ORR measured by ring currents signaling efficient O2 activation on the CCOPTDP–FeNi–SiO2 catalyst presents a four-electron reduction path (Fig. S9†). The CCOPTDP–FeNi–SiO2 catalyst also shows excellent stability for ORR, as confirmed by cyclic voltammetry (CV) tests with cycling the potential between 0.7 V and 1 V vs. RHE at a scan rate of 100 mV s−1. Remarkably, there is almost no shift occurring over the onset potential and half-wave potential of LSV for CCOPTDP–FeNi–SiO2 after 5000 cycles (Fig. 3d), confirming the high stability of the prepared CCOPTDP–FeNi–SiO2.
 | ||
| Fig. 3 (a) ORR LSV curves of CCOPTDP–FeNi–SiO2, CCOPTDP–FeNi, CCOPTDP and Pt/C in O2-saturated 1 M KOH with a scan rate of 5 mV s−1 and a rotating speed of 1600 rpm. (b) The comparison of E1/2 and JK of different catalysts. (c) Corresponding Tafel plots derived from the ORR LSV curves of different catalysts. (d) ORR LSV curves of CCOPTDP–FeNi–SiO2 before and after 5000 potential cycles ranging from 0.65 V and 0.95 V (vs. RHE) in O2-saturated 1 M KOH. (e) OER LSV curves of CCOPTDP–FeNi–SiO2, CCOPTDP–FeNi, CCOPTDP and IrO2/C in O2-saturated 1 M KOH with a scan rate of 5 mV s−1 and a rotating speed of 1600 rpm. (f) Corresponding Tafel plots derived from the OER LSV curves of different catalysts. (g) Nyquist plots of the electrochemical impedance spectra of different catalysts. (h) Corresponding electrocatalytic surface area (ECSA) for ORR and OER of different catalysts. (i) The overall polarization curves of all samples in the whole ORR and OER region in 1 M KOH solution. | ||
In addition, the CCOPTDP–FeNi–SiO2 catalyst also exhibits remarkable activity and stability for the OER. The oxygen evolution activity of the CCOPTDP–FeNi–SiO2 catalyst was evaluated in O2-saturated 1 M KOH solution with IR-correction, and the corresponding LSV curves are shown in Fig. 3e. The CCOPTDP–FeNi–SiO2 catalyst reaches a current density of 10 mA cm−2 at an overpotential of 310 mV, which is smaller than that of IrO2 (315 mV), CCOPTDP–FeNi (330 mV) and CCOPTDP (470 mV). Meanwhile, the CCOPTDP–FeNi–SiO2 catalyst exhibits a comparable Tafel slope (57 mV dec−1) to that of the IrO2 catalyst (55 mV dec−1), but much lower than that of CCOPTDP–FeNi (60 mV dec−1) and CCOPTDP (107 mV dec−1), indicating an excellent OER kinetic process of this catalyst (Fig. 3f). Moreover, a continuous potential cycling test shows that there is no variation in the polarization curves, suggesting the high durability of CCOPTDP–FeNi–SiO2 in a long-term OER under 1 M KOH alkaline conditions (Fig. S10†). In addition, charge transport is also a crucial factor for the kinetics of the OER. Electrochemical impedance spectroscopy was performed to elucidate the superior OER activity of the CCOPTDP–FeNi–SiO2 catalyst. The smaller semicircle in the medium frequency region reveals the lower charge transfer resistance for the catalyst.38 As shown in Fig. 3g, the charge transfer resistance of the CCOPTDP–FeNi–SiO2 catalyst is obviously lower than that of CCOPTDP, suggesting a faster charge transfer process.
The apparent electrochemical surface area (ECSA) is more relevant to electrochemical activity; the ECSA of CCOPTDP–FeNi–SiO2, CCOPTDP–FeNi, and CCOPTDP is compared by obtaining cyclic voltammetry (CV) curves in 1.0 M KOH in the absence of O2 (Fig. S11 and S12†).22 The ECSAs of CCOPTDP–FeNi–SiO2 for ORR and OER are confirmed to be 126.3 m2 g−1 and 114.2 m2 g−1, respectively (Fig. 3h), higher than those of CCOPTDP (112.1 m2 g−1, 104.7 m2 g−1) and CCOPTDP–FeNi (46.2 m2 g−1, 49.4 m2 g−1). These results further confirm that CCOPTDP–FeNi–SiO2 has the best bifunctional electrocatalytic activity.39 The bifunctional oxygen electrode activity of the CCOPTDP–FeNi–SiO2 catalyst can be judged from the oxygen electrode activity parameter ΔE (ΔE = Ej=10, OER – E1/2, ORR) in 1 M KOH, which is an important metric to evaluate the bifunctional activity towards the ORR/OER.40 Generally, a smaller ΔE value indicates better bifunctional catalytic activity. Notably, the CCOPTDP–FeNi–SiO2 catalyst exhibits a low ΔE value of 0.650 V, much lower than that of CCOPTDP–FeNi (0.736 V) and CCOPTDP (0.794 V) (Fig. 3i), demonstrating that CCOPTDP–FeNi–SiO2 is an effective bifunctional electrocatalyst for ORR and OER.
Based on the excellent bifunctional catalytic performance of the CCOPTDP–FeNi–SiO2 catalyst, a zinc–air flow battery was constructed employing CCOPTDP–FeNi–SiO2 catalyst loaded carbon paper as the air cathode and Zn foil as the anode measured in 8.0 M KOH and 0.5 M ZnO electrolyte (Fig. 4a). For the control experiment, a mixed Pt/C + IrO2 (1:1 by weight) as the cathode was also tested as a reference. As shown in Fig. 4b, the three zinc–air flow batteries constructed with the CCOPTDP–FeNi–SiO2 catalyst as the air-cathode can easily power up a light-emitting diode (LED, 3.7 V) panel displaying “BUCT”, demonstrating its promising application in Zn–air flow batteries. Furthermore, the discharge maximum power density of the Zn–air flow battery with the CCOPTDP–FeNi–SiO2 electrode is 112.8 mW cm−2, which is higher than that of the Pt/C + IrO2/C air electrode (64.8 mW cm−2) under the same conditions (Fig. 4c). The corresponding discharge and charge polarization curves of CCOPTDP–FeNi–SiO2 and Pt/C + IrO2/C are presented in Fig. 4c. Compared with the Pt/C + IrO2/C-driven Zn–air flow battery, the CCOPTDP–FeNi–SiO2 cathode shows a lower charge–discharge voltage gap of 0.79 V, indicating a better round-trip efficiency. The stability of the CCOPTDP–FeNi–SiO2 cathode was first performed by battery charging and discharging cycles at a current density of 5 mA cm−2 (Fig. 4d). It is clearly seen that the CCOPTDP–FeNi–SiO2 catalyst has a better cycling performance without almost voltage gap change after 80 hours like Pt/C under the same conditions. Furthermore, the battery containing the CCOPTDP–FeNi–SiO2 cathode could withstand a high current density of 20 mA cm−2, maintaining a low voltage gap of 1.24 V (Fig. 4e). Impressively, the final potential difference of the battery containing the CCOPTDP–FeNi–SiO2 cathode performed at the current density of 10 mA cm−2 after 90 hours was almost the same as the initial potential (Fig. S13†). Based on the above results, the CCOPTDP–FeNi–SiO2 catalyst has the potential to replace the precious metal catalyst applied to the cathode catalyst of the zinc–air flow battery.
 | ||
| Fig. 4 (a) Schematic illustration of the rechargeable Zn–air flow battery. (b) Photographs of the Zn–air flow battery with an LED panel (3.7 V) powered by three-series Zn–air flow batteries in series with CCOPTDP–FeNi–SiO2 as the air-cathode. (c) Charge and discharge polarization curves and the corresponding power density plots of the primary zinc–air flow battery with the CCOPTDP–FeNi–SiO2 catalyst and Pt/C + IrO2/C as the air-cathodes, respectively. (d) Charge–discharge cycling performance of a rechargeable Zn–air flow battery based on CCOPTDP–FeNi–SiO2 and Pt/C + IrO2/C electrocatalysts at a current density of 5 mA cm−2. (e) Charge–discharge cycling curves of CCOPTDP–FeNi–SiO2 at different current densities of 5 mA cm−2, 10 mA cm−2, and 20 mA cm−2, respectively. | ||
Conclusions
In summary, a rational design approach is reported to develop CCOPTDP–FeNi–SiO2 as a novel and excellent OER/ORR bifunctional electrocatalyst. Attributed to the synergetic effects between the N-doped carbon material and alloy NPs, the as-obtained CCOPTDP–FeNi–SiO2 catalyst shows excellent ORR activity with a positive half-wave potential of 0.89 V versus RHE and superior OER activity with a low overpotential of 310 mV at 10 mA cm−2. The potential gap between E1/2 and Ej=10 reach as small as 0.650 V. Meanwhile, FeNi alloy NPs are embedded into the graphitic carbon layer during the pyrolysis process, promoting the stability of the catalyst. A rechargeable zinc–air battery constructed with a CCOPTDP–FeNi–SiO2 cathode showed a high performance with a small voltage gap between charge and discharge of 0.79 V (at 5 mA cm−2) and a good cycling life. Therefore, this work reveals a new pathway for designing and constructing multifunctional electrocatalysts for metal–air batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2017YFA0206500), the NSF of China (21676020; 51502012; 21620102007, 21606015), the Beijing Natural Science Foundation (17L20060, 2162032), the Young Elite Scientists Sponsorship Program by the CAST (2017QNRC001), the Start-up Fund for Talent Introduction of the Beijing University of Chemical Technology (buctrc201420; buctrc201714), the Talent Cultivation and Open Project (OIC-201801007) of the State Key Laboratory of Organic–Inorganic Composites, the Distinguished Scientist Program at BUCT (buctylkxj02) and the “111” project of China (B14004).Notes and references
- T. Wang, Z. Kou, S. Mu, J. Liu, D. He, I. S. Amiinu, W. Meng, K. Zhou, Z. Luo, S. Chaemchuen and F. Verpoort, Adv. Funct. Mater., 2018, 28, 1705048 CrossRef.
- S. Liu, M. Wang, X. Sun, N. Xu, J. Liu, Y. Wang, T. Qian and C. Yan, Adv. Mater., 2018, 30, 1870028 CrossRef.
- E. Davari and D. G. Ivey, Sustainable Energy Fuels, 2018, 2, 39–67 RSC.
- A. P. Tiwari, D. Kim, Y. Kim and H. Lee, Adv. Energy Mater., 2017, 7, 1602217 CrossRef.
- H. F. Wang, C. Tang, B. Wang, B. Q. Li and Q. Zhang, Adv. Mater., 2017, 29, 1702327 CrossRef PubMed.
- W. Niu, Z. Li, K. Marcus, L. Zhou, Y. Li, R. Ye, K. Liang and Y. Yang, Adv. Energy Mater., 2018, 8, 1701642 CrossRef.
- Q. Wang, L. Shang, R. Shi, X. Zhang, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Nano Energy, 2017, 40, 382–389 CrossRef CAS.
- Y. Zheng, Y. Jiao, Y. Zhu, Q. Cai, A. Vasileff, L. H. Li, Y. Han, Y. Chen and S. Z. Qiao, J. Am. Chem. Soc., 2017, 139, 3336–3339 CrossRef CAS PubMed.
- J. Yin, Y. Li, F. Lv, Q. Fan, Y. Q. Zhao, Q. Zhang, W. Wang, F. Cheng, P. Xi and S. Guo, ACS Nano, 2017, 11, 2275–2283 CrossRef CAS PubMed.
- S. K. Singh, V. Kashyap, N. Manna, S. N. Bhange, R. Soni, R. Boukherroub, S. Szunerits and S. Kurungot, ACS Catal., 2017, 7, 6700–6710 CrossRef CAS.
- X. F. Bai, C. C. Zhao, S. G. Li, Y. F. Song, R. P. Ge and Y. S. WeiWei, Angew. Chem., Int. Ed., 2017, 129, 12387–12391 CrossRef.
- X. Liu, M. Park, M. G. Kim, S. Gupta, G. Wu and J. Cho, Angew. Chem., Int. Ed., 2015, 54, 9654–9658 CrossRef CAS PubMed.
- L. Yang, X. Zeng, D. Wang and D. Cao, Energy Storage Mater., 2018, 12, 277–283 CrossRef.
- P. Li, X. Duan, Y. Kuang, Y. Li, G. Zhang, W. Liu and X. Sun, Adv. Energy Mater., 2018, 1703341 CrossRef.
- S. Ci, S. Mao, Y. Hou, S. Cui, H. Kim, R. Ren, Z. Wen and J. Chen, J. Mater. Chem. A, 2015, 3, 7986–7993 RSC.
- P. Cai, Y. Hong, S. Ci and Z. Wen, Nanoscale, 2016, 8, 20048–20055 RSC.
- P. Cai, S. Ci, E. Zhang, P. Shao, C. Cao and Z. Wen, Electrochim. Acta, 2016, 220, 354–362 CrossRef CAS.
- S. Gupta, L. Qiao, S. Zhao, H. Xu, Y. Lin, S. V. Devaguptapu, X. Wang, M. T. Swihart and G. Wu, Adv. Energy Mater., 2016, 6, 1601198 CrossRef.
- Y. Fu, H. Y. Yu, C. Jiang, T. H. Zhang, R. Zhan, X. Li, J. F. Li, J. H. Tian and R. Yang, Adv. Funct. Mater., 2018, 28, 1705094 CrossRef.
- J. C. Liang, K. Y. Zhou, G. Y. Chen, W. X. Zhang, J. A. Liu, W. Z. Zhang, Z. P. Zhang, W. Hou, M. Zhou, G. F. Liu and F. Niu, J. Solid State Electrochem., 2017, 21, 3315–3324 CrossRef CAS.
- P. Peng, Z. Zhou, J. Guo and Z. Xiang, ACS Energy Lett., 2017, 2, 1308–1314 CrossRef CAS.
- Y. Li, J. Guo, Y. Cheng, L. Dai and Z. Xiang, ACS Nano, 2017, 11, 8379–8386 CrossRef PubMed.
- Z. Xiang, D. Cao and L. Dai, Polym. Chem., 2015, 6, 1896–1911 RSC.
- Z. Xiang, Y. Xue, D. Cao, L. Huang, J.-F. Chen and L. Dai, Angew. Chem., Int. Ed., 2014, 53, 2433–2437 CrossRef CAS PubMed.
- Z. Xiang and D. Cao, J. Mater. Chem. A, 2013, 1, 2691–2718 RSC.
- Z. Xiang, D. Cao, L. Huang, J. Shui, M. Wang and L. Dai, Adv. Mater., 2014, 26, 3315–3320 CrossRef CAS PubMed.
- X. Zhuang, D. Gehrig, N. Forler, H. Liang, M. Wagner, M. R. Hansen, F. Laquai, F. Zhang and X. Feng, Adv. Mater., 2015, 27, 3789–3796 CrossRef CAS PubMed.
- Y. Su, Z. Yao, F. Zhang, H. Wang, Z. Mics, E. Canovas, M. Bonn, X. Zhuang and X. Feng, Adv. Funct. Mater., 2016, 26, 5893–5902 CrossRef CAS.
- X. Cui, P. Ren, D. Deng, J. Deng and X. Bao, Energy Environ. Sci., 2016, 9, 123–129 RSC.
- Y. J. Sa, D. J. Seo, J. Woo, J. T. Lim, J. Y. Cheon, S. Y. Yang, J. M. Lee, D. Kang, T. J. Shin, H. S. Shin, H. Y. Jeong, C. S. Kim, M. G. Kim, T. Y. Kim and S. H. Joo, J. Am. Chem. Soc., 2016, 138, 15046–15056 CrossRef CAS PubMed.
- Y. Lei, Q. Wang, Z. Chen, N. Wu, Y. Wang, B. Wang and Y. Wang, J. Mater. Chem. A, 2018, 6, 516–526 RSC.
- H. B. Yang, J. Miao, S. F. Hung, J. Z. Chen, H. B. Tao, X. Z. Wang, L. P. Zhang, R. Chen, J. J. Gao, H. M. Chen, L. Dai and B. Liu, Sci. Adv., 2016, 2, e1501122 CrossRef PubMed.
- J. Zhang and L. Dai, ACS Catal., 2015, 5, 7244–7253 CrossRef CAS.
- S. Gao, B. Fan, R. Feng, C. Ye, X. Wei, J. Liu and X. Bu, Nano Energy, 2017, 40, 462–470 CrossRef CAS.
- S. Cai, Z. Meng, H. Tang, Y. Wang and P. Tsiakaras, Appl. Catal., B, 2017, 217, 477–484 CrossRef CAS.
- Q. L. Zhu, W. Xia, L. R. Zheng, R. Zou, Z. Liu and Q. Xu, ACS Energy Lett., 2017, 2, 504–511 CrossRef CAS.
- S. Kumar, D. Kumar, B. Kishore, S. Ranganatha, N. Munichandraiah and N. S. Venkataramanan, Appl. Surf. Sci., 2017, 418, 79–86 CrossRef CAS.
- C. Tang, L. Zhong, B. Zhang, H. F. Wang and Q. Zhang, Adv. Mater., 2018, 30, 1705110 CrossRef PubMed.
- Y. Cheng, J. Guo, Y. Huang, Z. Liao and Z. Xiang, Nano Energy, 2017, 35, 115–120 CrossRef CAS.
- M. Kuang, Q. Wang, H. Ge, P. Han, Z. Gu, A. M. Al-Enizi and G. Zheng, ACS Energy Lett., 2017, 2, 2498–2505 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental setup including synthesis and characterization details. XPS spectra, SEM images, and LSV. See DOI: 10.1039/c8nr08330d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |