
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTailoring adhesion of anionic surfaces using cationic PISA-latexes – towards tough nanocellulose materials in the wet state†
J.
Engström
 ab,
T.
Benselfelt
ab,
L.
Wågberg
ab,
F.
D'Agosto
c,
M.
Lansalot
c,
A.
Carlmark‡
*a and
E.
Malmström
*a
ab,
T.
Benselfelt
ab,
L.
Wågberg
ab,
F.
D'Agosto
c,
M.
Lansalot
c,
A.
Carlmark‡
*a and
E.
Malmström
*a
aKTH Royal Institute of Technology, School of Chemistry, Biotechnology and Health, Wallenberg Wood Science Center, SE-100 44, Stockholm, Sweden. E-mail: anna.carlmark@ri.se; mavem@kth.se
bDepartment of Fibre and Polymer Technology, Wallenberg Wood Science Center, SE-100 44, Stockholm, Sweden
cUniversité de Lyon, Univ Lyon 1, CPE Lyon, CNRS UMR 5265, C2P2 (Chemistry, Catalysis, Polymers & Processes), LCPP, 69616 Villeurbanne, France
First published on 15th January 2019
Abstract
Cationic latexes with Tgs ranging between −40 °C and 120 °C were synthesised using n-butyl acrylate (BA) and/or methyl methacrylate (MMA) as the core polymers. Reversible addition–fragmentation chain transfer (RAFT) combined with polymerisation-induced self-assembly (PISA) allowed for in situ chain-extension of a cationic macromolecular RAFT agent (macroRAFT) of poly(N-[3-(dimethylamino)propyl] methacrylamide) (PDMAPMA), used as stabiliser in so-called surfactant-free emulsion polymerisation. The resulting narrowly distributed nanosized latexes adsorbed readily onto silica surfaces and to model surfaces of cellulose nanofibrils, as demonstrated by quartz crystal microbalance with dissipation monitoring (QCM-D) measurements. Adsorption to anionic surfaces increased when increasing ionic strength to 10 mM, indicating the influence of the polyelectrolyte effect exerted by the corona. The polyelectrolyte corona affected the interactions in the wet state, the stability of the latex and re-dispersibility after drying. The QCM-D measurements showed that a lower Tg of the core results in a more strongly interacting adsorbed layer at the solid–liquid interface, despite a comparable adsorbed mass, indicating structural differences of the investigated latexes in the wet state. The two latexes with Tg below room temperature (i.e. PBATg-40 and P(BA-co-MMA)Tg3) exhibited film formation in the wet state, as shown by AFM colloidal probe measurements. It was observed that P(BA-co-MMA)Tg3 latex resulted in the largest pull-off force, above 200 m Nm−1 after 120 s in contact. The strongest wet adhesion was achieved with PDMAPMA-stabilized latexes soft enough to allow for interparticle diffusion of polymer chains, and stiff enough to create a strong adhesive joint. Fundamental understanding of interfacial properties of latexes and cellulose enables controlled and predictive strategies to produce strong and tough materials with high nanocellulose content, both in the wet and dry state.
Introduction
Cellulose is a natural choice for new materials research due to its abundance and promising material properties.1–5 Cellulosic materials often suffer from incompatibility and mixing issues with synthetic polymers, due to their inherent hydrophilic nature. Nanosized cellulose materials, nanocrystals (CNCs) and nanofibrils (CNFs), have attracted significant interest as sustainable materials for bio-nanocomposite applications,6,7 functional materials in aero- or hydrogels,8,9 and strong and stiff films.5 In several strategies, CNFs or CNCs are combined with synthetic polymer matrices to increase stiffness and strength, as well as the content of renewables in the final material.10 In order to increase the compatibility, methods such as grafting-to or grafting-from of small molecules or polymers have been employed, as well as physisorption of either homopolymers or block copolymers.10–15 Irrespective of the aim being functionalisation in dispersion or compatibilisation for composites, the understanding of wet and dry state nanoscale interactions between a polymer and cellulose is crucial, and an extended fundamental knowledge is desirable.Polymers, including polyelectrolytes, can be designed to interact with celluloses of low charge density (such as filter paper and cotton) or high charge density (oxidised cellulose, carrying negatively charged groups such as carboxylate or sulphate ester); the former requiring strong affinity through non-ionic interactions whereas the latter is due to the entropy gain when counter-ions are released upon adsorption.16–20 There are still unresolved questions regarding nanoscale interactions between polyelectrolytes and surfaces, such as wet state interactions, the drying mechanisms and the influence of the surface properties. Different high resolution and in situ characterisation techniques can provide interesting, valuable insights into some of the remaining questions. Two examples are colloidal probe atomic force microscopy (AFM) and quartz crystal microbalance with dissipation monitoring (QCM-D). AFM can able to image the surface but also to measure adhesion between two surfaces in the wet state, whereas the adsorption kinetics and adsorbed amount onto different substrates can be quantified in detail with the aid of the QCM-D.21–25
Given the increasing expectations of greener chemistry, it is attractive to use water-borne chemistry to produce hydrophobic polymers for direct mixing with nanocellulose dispersions. Inspired by the well-established industrial and academic production of latexes, emulsion polymerization26–28 has received intensified interest lately. The adsorption of commercially available cationic latexes of polystyrene and a poly(styrene–butadiene) copolymer onto cellulose substrates has also already been studied.26,29–31 However, these earlier studies involve latexes stabilised by a low molecular weight surfactant, such as sodium dodecyl sulphate (SDS), or non-covalently bonded polymeric stabilisers.26,31,32 With the rapid development of controlled polymerisations a range of novel amphiphilic polymers are now available and applicable as stabilisers for colloidal dispersions.
One example is the use of reversible addition–fragmentation chain transfer (RAFT) polymerisation33–38 together with polymerisation-induced self-assembly (PISA)39–43 that creates a platform to produce tailored polymers and block copolymers as latexes.
PISA and macroRAFT agents allow easy access to latexes with adjustable particle sizes and low polydispersities and are hypothesised to result in better film properties due to the absence of stabiliser migration.44,45 If the chosen macroRAFT is a polyelectrolyte, the stabilized PISA-inspired latex system is anticipated to result in a larger adsorption to oppositely charged surfaces compared to conventional physisorbed polymeric- or small molecule (such as SDS) stabilized latexes.
Previously in our group, we have shown examples of cellulose modification using PISA-inspired latex systems, comprising a cationic poly(2-dimethyl aminoethyl methacrylate) PDMAEMA-based corona.21,22 Despite the interesting adsorption properties of PDMAEMA,46–49 one of the observed challenges is the hydrolysis of the monomer in aqueous media, even at neutral pH, resulting in methacrylic acid-residues along the polymer backbone.22 Anionic residues in the backbone give rise to lower cationic charge, thus resulting in lower adsorption to anionic surfaces. To circumvent this challenge, the hydrolytically more stable acrylamide analogue, N-[3-(dimethylamino)propyl] methacrylamide (DMAPMA), and its resulting polymer, PDMAPMA, was utilized in this work.50 Structurally similar polyacrylamides, such as commercial C-PAM, are well-known retention aids and dry strength additives in papermaking and have been used in many studies in conjunction with cellulose.51 PDMAPMA has been extensively investigated by McCormick et al. for biomedical applications and successful RAFT was demonstrated in buffer at pH below 7.52–54 It was hypothesised that PDMAPMA macroRAFT can be successfully used to stabilize latexes and at the same time provide a strong affinity to cellulose surfaces due to the higher cationic charge density, as compared to the previously reported PDMAEMA-stabilized latexes.21 To the best of our knowledge, this is the first investigation using PDMAPMA macroRAFT as a stabiliser for the synthesis of surfactant-free latexes and their applicability for cellulose modification.
In the present contribution, we focused on interfacial interactions and structural change upon adsorption to surfaces at the nanoscale. Fundamental questions, such as if the latex particle can deform and/or coalesce upon adsorption in aqueous media (wet state) and how physico-chemical properties of the latex can affect this process will be addressed. PDMAPMA-stabilised latexes with three different core polymers and Tg, PMMATg120, P(BA-co-MMA)Tg3 and PBATg-40, were synthesized. Adsorption measurements were performed in situ using QCM-D (on silica or CNF surfaces) and ex situ on cellulose filter papers, silica, CNF or glass substrates, to investigate the importance of surface roughness and charge density. The wet adhesion was assessed using AFM colloidal probe measurements. The results indeed show that latex nanoparticles constitute a promising toolbox to tailor nanoscale interactions in the wet or dry state.
Experimental section
Materials
N-[3-(Dimethylamino)propyl]methacrylamide (DMAPMA, Aldrich, 98%), methyl methacrylate (MMA, 99%), n-butyl acrylate (BA, 99%) 2,2′-azobis(2-methylpropionamidine) dihydrochloride (AIBA, 97%), 1,3,5-trioxane (Aldrich, ≥99%), potassium chloride (Merck), cetyltrimethylammonium chloride (CTAC) (25 wt% water solution) were all purchased from Sigma Aldrich and used as received, the monomers were activated passing through aluminum oxide columns. Hydrochloric acid (HCl, VWR Prolabo, 35 wt%, technical grade) and sodium chloride (VWR) were used as received. Poly(ethylene imine) (PEI, Mn = 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) was purchased from Acros. The water used was either deionised or Milli-Q water. RAFT agent 4-cyano-4-thiothiopropylsulfanyl pentanoic acid (CTPPA) was synthesised according to literature procedure.55–57 Munktell filter paper grade 3 was used for adsorption on filter paper (Ahlstrom, Munktell). The QCM-D crystals used were AT-cut crystals (5 MHz resonance frequency) with an active surface of sputtered silica (50 nm thickness) supplied by Q-sense AB. Silica wafers were supplied from Addison Engineering Inc. (San Jose, CA, US, single-side polished). Microscopy slides (Diameter 30 mm, Menzel Gläser, Thermo Scientific) were used for contact angle measurements. The surfaces were cleaned with sodium dodecyl sulphate solution (2 wt% in water) for 12 h and left in a sonication bath for 10 min followed by the careful rinsing sequence Milli-Q water, acetone and Milli-Q water prior to adsorption of dispersions. A cellulose nanofibril (CNF) gel (600 μeq g−1) was kindly received from RISE Bioeconomy (Formerly Innventia AB, Sweden).58 The gel with a solids content around 2 wt% was treated with one passage through a Microfluidizer (Microfluidizer M-110EH, MicrofluidicsCorp) using 1650 bar, 200 and 100 μm chambers in series. CNF dispersions were diluted to 0.2 wt% and sonicated (Sonication probe VCX Vibra-Cell, Sonics and Materials Inc.) for 15 min at 30% amplitude in an ice bath and centrifuged for 60 min at 4800 rpm (Beckman Coulter Avanti J-E) centrifuge to remove any agglomerates. The supernatant, consisting of free CNFs, was used for further analysis.
000 g mol−1) was purchased from Acros. The water used was either deionised or Milli-Q water. RAFT agent 4-cyano-4-thiothiopropylsulfanyl pentanoic acid (CTPPA) was synthesised according to literature procedure.55–57 Munktell filter paper grade 3 was used for adsorption on filter paper (Ahlstrom, Munktell). The QCM-D crystals used were AT-cut crystals (5 MHz resonance frequency) with an active surface of sputtered silica (50 nm thickness) supplied by Q-sense AB. Silica wafers were supplied from Addison Engineering Inc. (San Jose, CA, US, single-side polished). Microscopy slides (Diameter 30 mm, Menzel Gläser, Thermo Scientific) were used for contact angle measurements. The surfaces were cleaned with sodium dodecyl sulphate solution (2 wt% in water) for 12 h and left in a sonication bath for 10 min followed by the careful rinsing sequence Milli-Q water, acetone and Milli-Q water prior to adsorption of dispersions. A cellulose nanofibril (CNF) gel (600 μeq g−1) was kindly received from RISE Bioeconomy (Formerly Innventia AB, Sweden).58 The gel with a solids content around 2 wt% was treated with one passage through a Microfluidizer (Microfluidizer M-110EH, MicrofluidicsCorp) using 1650 bar, 200 and 100 μm chambers in series. CNF dispersions were diluted to 0.2 wt% and sonicated (Sonication probe VCX Vibra-Cell, Sonics and Materials Inc.) for 15 min at 30% amplitude in an ice bath and centrifuged for 60 min at 4800 rpm (Beckman Coulter Avanti J-E) centrifuge to remove any agglomerates. The supernatant, consisting of free CNFs, was used for further analysis.
Instrumentation and methods
More information and details about conventional instrumentation and methods used for this study can be found in ESI; NMR, Maldi-ToF MS, Gravimetric analysis (conversion during latex synthesis), SEC, DSC, PET (polyelectrolyte titration) and FTIR-spectroscopy.The number of particles per unit volume of the aqueous phase (Np) was calculated using the diameter obtained from DLS (DH, nm) according to the equation below:
 | (1) |
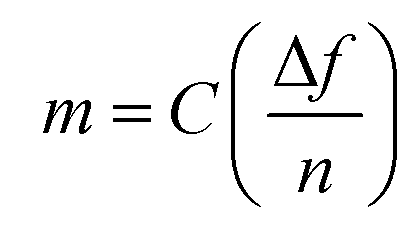 | (2) |
The QCM-D also detects the energy dissipation in the adsorbed layer which is related to the viscoelastic properties of the adsorbed layer. A thin, rigid film results in a low change in dissipation while a water-rich and viscoelastic film result in a larger change in dissipation. The dissipation factor, D, is defined as:
 | (3) |
300g (20000 rpm) with plastic containers at 5 °C cooling. Deionised water was added to the latexes before centrifugation to ease the separation of supernatant and the centrifuged pellet. The supernatant was collected separately and the pellet was re-dispersed using shaker table overnight.
000 rpm for 0.1 g L−1 concentrations and 13000 rpm for 0.2 wt%, respectively, for 20 min. The disintegration was performed with regular glass flasks.
RAFT polymerisation of DMAPMA in water using CTPPA
The synthesis of the macroRAFT agent PDMAPMA was performed in water, acidified to pH 6 using concentrated hydrochloric acid (HCl). In a typical experiment, RAFT agent (CTPPA) (0.4 g, 1.4 mmol) was added to a 50 mL round bottom flask. De-stabilised DMAPMA (6.15 g, 36.1 mmol) monomer was added to the flask and the RAFT agent was allowed to dissolve in monomer before addition of deionised water (22.08 g), targeting a monomer concentration of 1.63 M and a degree of polymerisation of 25. The reaction mixture was left stirring for 10 min in an ice-water bath. Addition of HCl was conducted carefully while monitoring the pH to reach pH = 6. The radical initiator AIBA (78.3 mg, 0.29 mmol and concentration ratio CTPPA:AIBA of 5:1) was added subsequently together with trioxane (0.272 g, added as an internal reference for NMR). The reaction mixture was degassed for 30 min under argon, and the polymerisation was then started by immersing the flask in an oil bath pre-heated to 70 °C. Aliquots were taken for 1H-NMR analysis every 20 min and full conversion was reached after 60 min (ESI Fig. S2†). The polymer was precipitated from water into ice-cold acetone 3 times, dried with a rotavapor and finally dried in a vacuum oven for 24 h at 50 °C. The precipitated polymer was analyzed with 1H-NMR in D2O, SEC in DMSO and in water (0.3 M sodium acetate/acetic acid buffer), MALDI-ToF and glass transition temperature was determined using DSC, ESI Fig. S1 and S2.† The final polymer was also analyzed for adsorption on QCM-D silica crystals at 0.1 g L−1 (ESI Fig. S3†). The established calibration curve for determining unknown concentrations of PDMAPMA in the latex water phase was performed as described in the NMR method section (ESI Table S4 and Fig. S6†).
Synthesis of PISA-inspired and conventional latexes
All latexes using PDMAPMA macroRAFT agent were synthesised targeting DP 400 for the hydrophobic core (ESI Table S1†). In a typical experiment, macroRAFT PDMAPMA (100 mg, 0.022 mmol) was dissolved in deionised water for 10 min in a round bottom flask under magnetic stirring. For poly(methyl methacrylate) (PMMA) latex, MMA (0.9 g, 8.90 mmol) was added dropwise to the flask, targeting a DP of 400 and final dry content of 15.3 wt%. The flask was kept in an ice water bath and radical initiator AIBA (0.74 mg, 217 μL of a 3.4 g L−1 aqueous solution) was added during stirring. The mixture was degassed for 30 min under argon and subsequently immersed in an oil bath pre-heated to 70 °C. All reactions were left to proceed for 120 min. Samples were regularly collected during the polymerisation, and conversion was determined by gravimetric analysis by drying the samples in an oven at 100 °C overnight. PBA latex was prepared from the same protocol using butyl acrylate (BA) instead of MMA. The copolymer of BA and MMA was synthesised from a 60/40 BA/MMA mixture (molar ratio). The resulting latexes were abbreviated PMMATg120, PBATg-40 and P(BA-co-MMA)Tg3, respectively. In all cases, the latexes were stored in the fridge and analyzed by cryo-TEM and DLS, while freeze-dried samples were analyzed by 1H-NMR in CDCl3, SEC in DMF and DSC (Table 1, Fig. 1, and ESI Tables S1–S2 and Fig. S4†). Two reference latexes were synthesised using AIBA as initiator and MMA as the monomer; one without PDMAPMA or any surfactant (abbreviated PMMAAIBA), and the second one using a commercial cationic surfactant (CTAC) (latex abbreviated PMMACTAC). Both latexes were produced using the protocol described above (ESI for more details and Tables S1 and S3†). | ||
| Fig. 1 Cryo-TEM images for (a) PMMATg120 latex (b) P(BA-co-MMA)Tg3 latex (c) PBATg-40 latex. | ||
| Latex (dry wt%)a |
M
nb [g mol−1] |
M
nc [g mol−1] |
Đ N | PdId |
D
Hd [nm] |
D
TEMe [nm] |
T
gf [°C] |
Chargeg [μeq g−1] |
|---|---|---|---|---|---|---|---|---|
| a Targeted dry content in brackets assuming 100% monomer conversion. b Estimation of molar mass from experimental determined conversions (86%, 73% and 79% for PMMA, P(BA-co-MMA) and PBA, respectively) and added macroRAFT agent. c Molar mass from DMF-SEC using PMMA standards. d Polydispersity index (PdI) and hydrodynamic diameter (DH) (Z-average) from DLS measurements in 10 mM KCl prior to work-up. e Average diameter of 10 particles in cryo-TEM. f T g from DSC. g Charge density measurement in Milli-Q water using PET. | ||||||||
| PMMATg120 (15.3%) | 39000 |
260000 |
1.5 | 0.01 | 215 | 180 ± 10 | 120 | 54 (±4) |
| P(BA-co-MMA)Tg3 (17.3%) | 37500 |
217000 |
1.6 | 0.01 | 124 | 130 ± 15 | 3 | 140 (±2) |
| PBATg-40 (15.3%) | 42000 |
240000 |
1.5 | 0.03 | 76 | 70 ± 20 | −40 | 130 (±4) |
Work-up protocol for removal of free PDMAPMA-chains
To remove any remaining PDMAPMA homopolymer chains from the water phase after latex formation, the crude reaction mixture was purified by successive centrifugation and re-dispersion steps. All latexes were diluted to half the reaction concentration with deionised water prior to centrifugation. Centrifugation (1 h at 48300g and 20000 rpm at 5 °C) was used to separate the latex nanoparticles from the water phase, sedimenting in the bottom of the plastic centrifuge tubes. The supernatant was freeze-dried before adding a known amount of D2O for analysis by 1H-NMR, observing the intensity of methylene protons at 2.8 ppm and 3.1 ppm (ESI Table S4†). To quantify the residual amount of PDMAPMA homopolymer, a calibration curve was constructed; using known concentrations of PDMAPMA using integral values from protons at 3.1 ppm (2.8 ppm gave same correlation) (ESI Fig. S6†). The pellet was re-dispersed by stirring on shaking table to reach similar to starting dry content (between 15 and 20 wt%), followed by characterization with DLS (ESI Tables S2 and S3†) and SEC in THF or DMF, Table 1. This sequence was up to 3 times to ensure efficient removal of any free polymer. The final latex fraction after 3 rounds of centrifugations was used for all analysis if nothing else is stated. Re-dispersion of the latex at for example 3 g L−1 in D2O did not give sufficient signal in 1H-NMR, hence no contribution in the results of calculating free soluble PDMAPMA. Results from centrifugation cycles can be seen from DLS, 1H-NMR data, QCM-D and SEM imaging in ESI Tables S2–S4 and Fig. S5–S8.†
Adsorption of latexes onto silica surface with the aid of QCM-D
The adsorption of latexes onto reference silica surfaces was monitored by QCM-D using a flow of 0.15 mL min−1. The latexes were introduced as dispersions at a concentration of 0.1 g L−1, diluted in either Milli-Q water or 10 mM NaCl, and adsorbed until saturation, followed by rinsing with Milli-Q water. After the adsorption experiment, the crystals were removed from the chamber and left to dry under in a Petri dish with an added wet tissue to supply humidity during drying, hence decreasing the rate of water evaporation to prevent the development of the “coffee ring” effect on the crystals. The effect of drying conditions was investigated by SEM (ESI Fig. S9 and S10†).Adsorption of latexes on a CNF layer formed in situ with the aid of QCM-D
The adsorption of latexes onto CNF surfaces was monitored by QCM-D at a flow rate of 0.15 mL min−1. The CNF surfaces were formed in situ by a two-step process. In the first step, an anchoring layer of poly(ethylene imine) (PEI) at 0.1 g L−1 (set to pH 10 with 10 mM NaOH in 10 mM NaCl) was adsorbed until saturation, followed by rinsing with Milli-Q water. In the second step, a layer of a CNF dispersion (0.1 g L−1) was adsorbed at pH 7, followed by rinsing with Milli-Q water in accordance with previous procedures.46,60 The latex (0.1 g L−1) was thereafter introduced until saturation adsorption, followed by rinsing with Milli-Q water. Reference crystals with only PEI and CNF layers were analysed without the adsorption of latexes in previous research.21 The crystals were subsequently analyzed by SEM for particle surface properties. QCM-D data for the in situ formed CNF layers are shown in Fig. 6 and CAM images are displayed in ESI Fig. S11 and Table S5.†Adsorption of latexes on a CNF layer formed ex situ on silica wafers
The adsorption of latexes onto silica wafer surfaces was also performed ex situ by simply saturating the surface with each component. In analogy to the QCM-D layer build up in situ, the CNF surfaces were formed by a two-step process. The first step was the introduction of an anchoring layer of PEI at 0.1 g L−1 (set to pH 10 with 10 mM NaOH) and 10 mM NaCl for 10 min, followed by rinsing step with Milli-Q. In the second step, a layer of a CNF dispersion was adsorbed at neutral pH 7, at a concentration 0.1 g L−1, and was left to adsorb for 20 min, followed by rinsing with Milli-Q water, in accordance to previous procedures.46,60 After rinsing with Milli-Q water, the latexes were adsorbed to the CNF coated wafer at a concentration of 0.5 g L−1 for 30 min, followed by a rinsing step with Milli-Q. A reference wafer with only the pre-formed PEI and CNF layer was also produced with the same procedure. The crystals were analyzed by CAM and SEM for particle surface properties. CAM measurements and SEM images of the ex situ layers are shown in ESI Fig. S12–S15 and ESI Table S6.†Adsorption of latexes onto filter papers
The latexes were also adsorbed onto Munktell cellulose filter paper grade 3 (2 × 3 cm2) using the following procedure: the latex was diluted to a concentration of 0.1 g L−1 in 10 mM NaCl (Milli-Q water) or in pure Milli-Q water in a 10 mL vial after which a filter paper was immersed in the dispersion. The vial was left on a shaker table for 24 h to ensure complete adsorption. The paper was thoroughly rinsed with Milli-Q water and left in Milli-Q water for 24 h on a shaking table where after the paper was rinsed again with Milli-Q water, left to dry at room temperature overnight and stored in a conditioned room at 23 °C and 50% RH for 24 h prior to analysis. Some of the samples were annealed in an oven at 160 °C for 3 or 12 h. The modified papers were characterized using SEM (Fig. 8), CAM and FTIR (ESI Table S8 and Fig. S17†).Measuring wet adhesion between silica surfaces treated with latexes using the AFM colloidal probe technique
The wet mechanical properties of the latex particles were investigated with the AFM colloidal probe technique using a MultiMode IIIa AFM (Veeco Instruments Inc. Santa Barbara, CA) with a PicoForce extension. Borosilicate microspheres with the radius 4.7–5.2 μm (Duke Standards, Thermo Scientific) were attached to tipless rectangular cantilevers (CLFC-NOCAL, Bruker) using a melting glue (Epikote 1001, Shell Chemical Co.) and a manual micromanipulator (HS 6 Manuell, Marzhauser Wetzlar GmbH & Co. KG). Each AFM probe had two cantilevers with spring constants of <1.4 N m−1 and >9.5 N m−1, respectively which were determined using AFM Tune IT version 2.7 (Force IT, Sweden). The different cantilevers were used to investigate how the applied load and the deflection sensitivity affected the adhesive properties. The latex particles (1 wt% in 10 mM NaCl solution) were adsorbed onto oxidised silicon wafers and the silica probe in situ for 1 h. The force-curves were measured in Milli-Q with or without 120 s surface delay and were further analysed using the software AFM force IT version 2.6 (Force IT, Sweden). Typical force measurements are shown in Fig. 9.Results and discussion
To lower the risk of monomer hydrolysis during the aqueous polymerisation we used DMAPMA as a monomer for the macroRAFT synthesis, compared to the previously published DMAEMA-based macroRAFT.21,22 PDMAPMA macroRAFT was then utilized for the chain-extension of hydrophobic monomers in surfactant-free emulsion polymerisation. In the previous study with PDMAEMA, the Tg was chosen to either 30 °C or 120 °C (nBMA or MMA), and here we tailored the Tg of the core in the range of −40 °C to more than 120 °C (BA, MMA and mixtures of them both). In this work, the synthesis and characterization of produced latexes have been studied, as well as the adsorption of the latexes to surfaces and the adhesion of adsorbed latex layers in the wet state using AFM colloidal probe force measurements.RAFT polymerisation of DMAPMA in water using CTPPA
The monomer conversion during RAFT polymerisation of DMAPMA was monitored by 1H-NMR (ESI Fig. S1 and S2c†). The isolated macroRAFT PDMAPMA, target degree of polymerisation (DP) of 25, was analyzed by SEC using DMSO or water as eluent (ESI Fig. S2a†) and MALDI-ToF MS (ESI Fig. S2b†). There was a good agreement between the Mn value obtained from MALDI-ToF MS (4700 g mol−1) and the value calculated based on the targeted molar mass and the conversion obtained by 1H-NMR (4530 g mol−1). However, the Mn values obtained by SEC performed in DMSO or water were significantly lower, i.e., 1700 g mol−1 (ĐN = 1.28) and 2400 g mol−1 (ĐN = 1.13), respectively. The justification for this behaviour may be the use of inapproriate standards (PEG and Pullulan, respectively). To further characterize the livingness of the resulting chains and to demonstrate the absence of hydrolysis of the RAFT moiety (pH set at 6 using the addition of HCl) the detector traces for the UV and RI signals were overlaid and showed that most of the chains comprised a trithiocarbonate chain end (as supported by Fig. S2a in ESI†). The control of the polymerisation was further verified by determining the average DP by 1H-NMR analysis of the obtained polymer, using the integrals for the hydrogens on either one of the two methylene peaks in the RAFT moiety close to the carboxyl group, around 2.2–2.3 ppm or 2.65–2.7 ppm, and relating this to the methylene group in the PDMAPMA repeating unit (–CH2–, either at 2.9 ppm or 3.1 ppm). DP was found to be in reasonable agreement with the targeted value of 25 and the MALDI generated DP value of 27 (ESI Fig. S1,† NMR).As expected, DMAPMA was more hydrolytically stable compared to previously used DMAEMA21,22 and no hydrolysis (formation of methacrylic acid and N,N-dimethylpropane-1,3-diamine) was observed from 1H-NMR (D2O) analysis during synthesis. Hence, positively charged PDMAPMA with a higher charge density control compared to PDMAEMA can be produced. The charge density of the PDMAPMA measured by PET at pH 7 was 5.6 meq g−1, (compared to 4.2 meq g−1 at pH 7 for PDMAEMA).22 Differential scanning calorimetry (DSC) was used to assess PDMAPMA's glass transition temperature, Tg, and it was found to be 144 °C, in reasonable agreement with the previously reported data (137 °C).61
Synthesis of PISA-inspired PMMA, P(BA-co-MMA), PBA latexes and conventional PMMA latexes
Surfactant-free emulsion polymerisation of MMA, BA or a mixture of both, mediated by PDMAPMA macroRAFT, resulted in stable latexes, Table 1, denoted with their core polymer and Tg, such as PMMATg120. A high monomer conversion (ca. 80%) was reached in all cases (ESI Fig. S4†), and as expected, the latexes were positively charged as determined by DLS (zeta potential: +30 mV) and PET. The particle diameter measured by DLS (DH) significantly changed with monomer composition: 215 nm for PMMA latex, 124 nm for P(BA-co-MMA) latex and 76 nm for PBA latex (Table 1). These values are in relatively good agreement with the particle diameter determined by statistical analysis of cryo-TEM images (Table 1 and Fig. 1). The contamination appearing as small particles in cryo-TEM images is interpreted as ice, Fig. 1b. When using the PISA strategy for the synthesis of block copolymer based nanoparticles,42 the resulting nano-objects usually show small sizes correlated with a controlled evolution of molar masses and low dispersity values (ĐN < 1.3). However, the Mn values from DMF-SEC were in all cases higher than expected, between 217000 and 260000 g mol−1 (targeted DP∼400, ca. 40000 g mol−1), even if the ĐN values remained relatively low and around 1.6 (Table 1 and ESI Fig. S4†).
SEC analyses performed in this study are based on the conventional calibration method using homopolymer standards and thus give relative molar mass values. Nevertheless, the large discrepancies between the theoretically calculated molar masses and those determined by SEC suggest that fewer PDMAPMA chains than anticipated were active in the chain extension step. Besides, dispersity values were relatively large for the final block copolymers (>1.5). Indeed, residual PDMAPMA chains were found in the final synthesised latexes, as shown by the small peak in the lower molar mass regions around 5000 g mol−1 in DMF-SEC analysis of a dried sample of PBA latex (ESI Fig. S5†). In order to remove PDMAPMA homopolymer chains, an efficient work-up protocol based on successive centrifugations was developed (ESI Table S4 and Fig. S6–S8†). The work-up did not affect the sizes of the latex particles but was found to increase the dispersion stability by increasing zeta potential (ESI, Table S2†). The work-up protocol was applied to all latexes unless otherwise stated. The NMR analysis of the supernatant from centrifugation showed an incorporation efficiency of the macroRAFT in the crude latexes of ca. 70–80% independent of the monomer type (BA or MMA) (ESI Table S4†). This affects the final molar mass of the PMMA-, P(BA-co-MMA)- and PBA-block with factors 1.3–1.4 (57000–80000 g mol−1), which is still much lower than the values obtained by SEC. One explanation to the deviating SEC results could be the amphiphilic nature of the copolymer, giving rise to overestimated values from the applied calibration curve. Comparing these data sets to previous studies using PDMAEMA-based macroRAFT agents,21,22 it can also be explained by the cationic charge of the macroRAFT (both PDMAEMA and PDMAPMA), which seems to disturb the addition–fragmentation steps in water due to electrostatic repulsion between the polymer chains. The subsequent self-assembly process would thus also be disturbed, explaining why the molar masses were higher than targeted and the presence of residual PDMAPMA chains in water. As this study aimed at producing narrowly size-distributed nano-objects by PISA, a full mechanistic understanding of this system was not further investigated, but the considerations mentioned above are worth to be pointed out.
In order to evaluate the specific use of PDMAPMA in macroRAFT-mediated emulsion polymerisation and how it affects the characteristics of the final latexes, two reference latexes without PDMAPMA were produced for the PMMA system (ESI Tables S1 and S3†). For latex PMMAAIBA, only AIBA and MMA were used in water. For latex PMMACTAC a cationic surfactant, CTAC, was also used. As expected, the particle size was significantly smaller with surfactant than without (60 nm compared to 300 nm). For both latexes, no control over the MMA polymerisation (ĐN 2.5–3.0 in DMF SEC) was observed, as expected, while fairly narrow particle size distributions (PdI < 0.07) were obtained. However, the PDMAPMA decorated latexes re-dispersed readily after drying, which was not possible with the reference PMMA particles. The use of PDMAPMA as macroRAFT results in the formation of very stable latexes (high zeta potential and latex stable during centrifugation cycles), shown by PMMATg120 latex being able to re-disperse in water, even after drying at room temperature (ESI Fig. S7†). This property emanates from the covalent anchoring of the PDMAPMA chains to the particle surface via the expected chain extension.
T g's for the latexes show a good correlation with previously reported values for homopolymers PMMA (120 °C)62,63 and PBA (−40 °C),63 and the measured Tg of the copolymer P(BA-co-MMA) (3 °C) was in good agreement with Tg-values calculated using the Fox equation.64
Adsorption dynamics and properties on silica surfaces
The adsorption and interactions of the different PDMAPMA-stabilised latexes with silica surfaces were studied in situ with QCM-D and ex situ. First, a solution containing only PDMAPMA macroRAFT chains was analysed with QCM-D, resulting in a shift of 2 Hz and 0.3 mg m−2 according to the Sauerbrey model (eqn (2)), ESI Fig. S3.† QCM-D was then used to investigate the adsorption dynamics and the adsorbed amount of the corresponding PDMAPMA-stabilised latexes, and to establish adsorption data for silica surfaces, before the colloidal probe measurements. Latexes having PDMAPMA in the corona exhibit significant adsorption onto silica surfaces, as shown in Fig. 2. However, it is noteworthy that it is crucial to remove the main part of the homopolymer of PDMAPMA since they otherwise hinder the latex adsorption, ESI Fig. S8† (QCM-D analysis after work-up protocol). | ||
| Fig. 2 QCM-D results for change in (a) frequency (left) and (b) dissipation (right), adsorption of the different latexes (0.1 g L−1) on silica surfaces: PMMATg120 (light blue), P(BA-co-MMA)Tg3 (pink) and PBATg-40 (purple). Latex dispersed in either Milli-Q (solid lines) or in 10 mM NaCl as supporting electrolyte (dashed lines). Inset (i) shows the adsorption kinetics during the first minutes (0–5 minutes) of the adsorption measurements in Milli-Q water. | ||
The three major differences between the latexes (PMMATg120, P(BA-co-MMA)Tg3 and PBATg-40) are the hydrodynamic volume, their charge density and the Tg of the core polymer. Similarly to the first PDMAEMA-stabilised systems,21,22 the reported PDMAPMA-stabilised latexes also show diffusion controlled kinetics of adsorption affected by the size, where smaller size gives faster adsorption, inset in Fig. 2i. From the adsorption patterns and difference between the latexes shown by QCM-D, Tg and size seem to contribute the most, the influence of charge density being less pronounced since no linear trend can be observed between the three latexes. To further investigate the polyelectrolyte properties of the PDMAPMA corona, salt (10 mM NaCl) was added resulting in increased adsorption above 70 Hz for all three latexes, see dashed lines in Fig. 2, in agreement with theories of polyelectrolyte adsorption.65,66 The corresponding masses show that the adsorbed amounts, including immobilized liquid, were 12.9 mg m−2 for PMMATg120, 18.6 mg m−2 for P(BA-co-MMA)Tg3 and 20.3 mg m−2 for PBATg-40 latex. Using QCM-D, it is also possible to monitor the change in the dissipation of the layer (eqn (3)) during adsorption, as shown in Fig. 2b. The dissipation of the latex layer was lowest for the PBATg-40 latex and highest for PMMATg120 latex, correlating the layer change in dissipation with the Tg of the core. The PBATg-40 and P(BA-co-MMA)Tg3 latexes give more stiff layer than PMMATg120, thus corroborating with previous studies for softer PBMA latex (Tg 30 °C) compared with PMMA latex (Tg 128 °C).21 The layers of adsorbed PBATg-40 latex and P(BA-co-MMA)Tg3 latex give rise to comparable frequency shift using salt, but PBATg-40 results in lower dissipation. The stiffer layer could be explained by the structural and viscoelastic difference between the different latexes, correlating to the Tg of the core. Possibly, the PBATg-40 can adsorb flatter on the surface turning the spherical shape into a more ellipsoidal shape. This would effectively expose more polyelectrolyte chains to the surface, neutralizing more surface charges and releasing counter-ions, see the proposed schematic difference in Fig. 3, thus giving rise to a stiffer layer onto the crystal, as observed in QCM-D data.
 | ||
| Fig. 3 SEM images of QCM-D crystals covered with latexes (0.1 g L−1, Milli-Q); (a) PMMATg120, (b) P(BA-co-MMA)Tg3 and (d) PBATg-40, (c) shows a schematic illustration of particles attached to the silica surface in wet state, prior to drying, adding also scaling estimate of Debye length in Milli-Q water and. | ||
There is a large frequency shifts when rinsing with Milli-Q water, as observed around 50 min. It is probably related to desorption of latexes or change in the properties of the adsorbed layer, i.e., conformational change, of the adsorbed layer. The results clearly show that the adsorption of latex is higher in the presence of salt and that it is possible to form monolayers for all latexes as observed by SEM, Fig. 3 and 4.
 | ||
| Fig. 4 SEM images of QCM-D crystals covered with latexes (0.1 g L−1, 10 mM NaCl); (a) PMMATg120, (b) P(BA-co-MMA)Tg3, (c) PMMATg120 (annealed at 140 °C for 3 h) (d) PBATg-40. The insets in (a) and (c) are for the same surfaces but at higher magnification 25k and 50k, respectively and scale bar show 150 nm. | ||
SEM was used to analyse the surfaces after adsorption of latexes both in situ during QCM-D measurements and ex situ on silica wafers. The surface coverage, Fig. 3, of adsorbed latexes from Milli-Q water is relatively low compared to the theoretical surface coverage of charged spheres which is around 50%.67 The SEM images also show accordingly with QCM-D that the different latexes adsorb to a different extent. Drying treatment and the Tg of the core affect the nanostructures formed on silica, resulting in deformation and larger sizes of the spherical objects for P(BA-co-MMA)Tg3 and PBATg-40 latexes when dried, compared with DLS and cryo-TEM in the wet state. The above phenomenon was also shown by Granier et al. using high-resolution imaging AFM for latex particles on inorganic surfaces.68
The surfaces were annealed at 140 °C for 3 h to investigate how particle morphology and film formation change upon temperature. No apparent changes were observed for the two low Tg latexes, but some visual difference appears before and after heat treatment for the PMMATg120 latex, Fig. 4a and c, comparing the contrast of the latex spheres (see inset in Fig. 4a and c). However, clearly no coalescence and film forming occur. PDMAPMA-stabilised latexes, visualized when using PMMA in the core, are shown to be more structurally stable upon drying and annealing, no coalescence, compared with the PDMAEMA-stabilised latexes in previous studies.21,22 The Tg of 144 °C for the macroRAFT PDMAPMA compared to 18 °C for PDMAEMA69 is one contributing factor to the particle stability. This corroborates previously discussed re-dispersibility of PMMATg120 after drying in room temperature (ESI Fig. S7†), a property not seen for PDMAEMA-stabilised. However, the thermal stability of the PDMAPMA corona is not enough to prevent the low Tg latexes from the film forming or deforming upon drying already at room temperature, which is shown by the observed clusters Fig. 4b and d.
Adsorption dynamics and properties of latexes on CNF surfaces
With the objective to utilize the latexes for modification of cellulose fibres/nanofibrils for subsequent applications in nanocomposites, their adsorption onto cellulose nanofibril (CNF) surfaces was also investigated using QCM-D. Some important parameters to consider when comparing the adsorption of latexes onto different surfaces, such as silica or CNFs, are; roughness, charge density, sensitivity towards salt addition and pH. A model surface of CNF has a charge density of around 600 μeq g−1 and should be considered as a more swollen three-dimensional layer compared to the smoother and more two-dimensional silica surface, even though it is well established that silica surfaces also have gel-like properties.70 Since the adsorption onto silica was found to be more pronounced in 10 mM NaCl, the same conditions were used for latex adsorption onto CNF surfaces. The results are summarised in Fig. 5 and it was observed that the normalised frequency shift (Fig. 5a) is significantly larger for CNF than for silica. The inset in Fig. 5a shows similar adsorption kinetics as for the silica surfaces, which means that the initial adsorption dynamics are controlled by particle diffusion. For the CNF-surfaces, all latexes show a normalised change in frequency above 200 Hz, compared with silica surfaces which all had a shift that did not exceed 125 Hz, most probably due to a higher surface charge density of the CNF layer.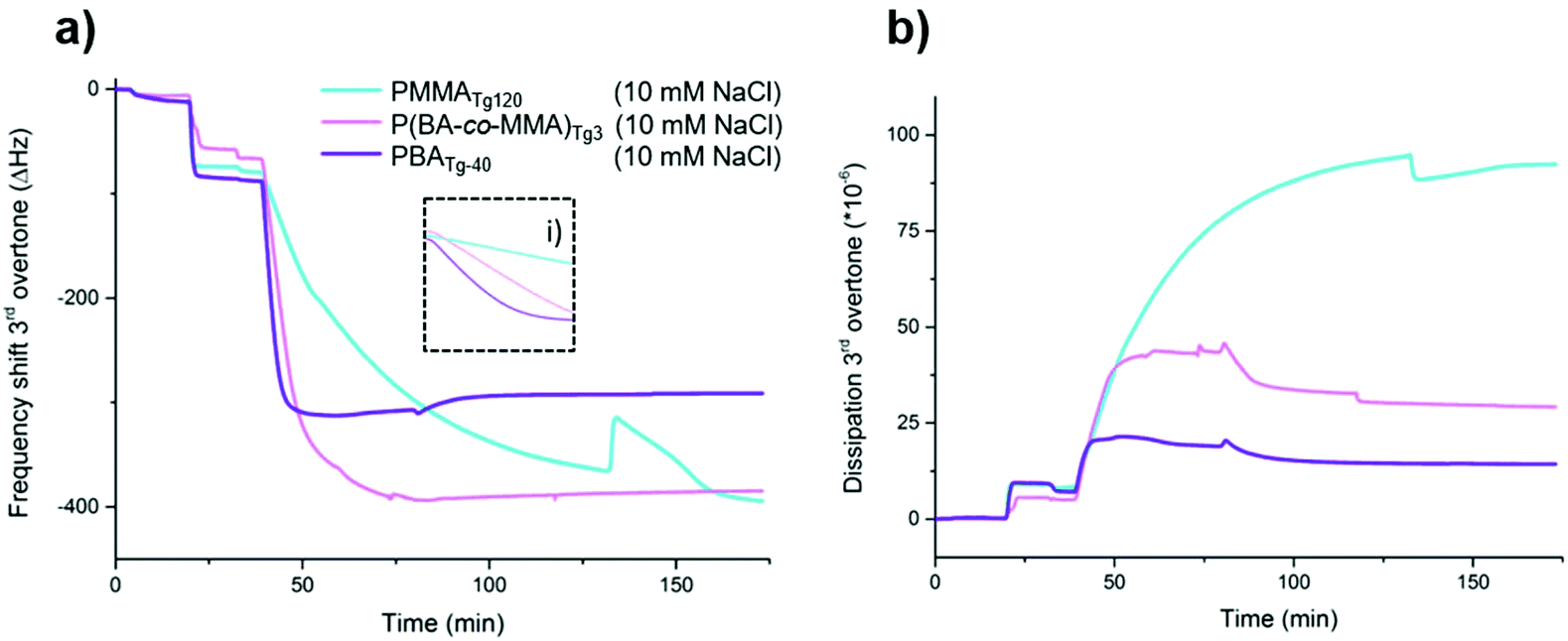 | ||
| Fig. 5 A summary of the QCM-D results as a change in (a) frequency (left) and (b) dissipation (right), adsorption of the different latexes (0.1 g L−1, 10 mM NaCl) on CNF surfaces; PMMATg120 (teal), P(BA-co-MMA)Tg3 (pink) and PBATg-40 (purple). Inset (i) shows the adsorption kinetics (ca. 40–50 min). | ||
Another reason for the larger adsorption onto CNF compared with silica could be the higher surface roughness of the swollen CNF layer, resulting in a larger available surface area. This latter effect is shown by the different adsorption patterns of the latex on the CNF surfaces, Fig. 6, comparing for example, the patterns created for PBATg-40 on CNF surfaces in Fig. 6c and silica surfaces in Fig. 6d. This is especially apparent for the two latexes with the lower Tg. The larger surface coverage for the latexes on the CNF surfaces corroborates with the larger frequency shift, Fig. 5, for the CNF surfaces, especially for the lower Tg latexes.
 | ||
| Fig. 6 SEM images of QCM-D crystals covered with latexes (0.1 g L−1, 10 mM NaCl); (a) PMMATg120, (b) P(BA-co-MMA)Tg3, (c) PBATg-40 adsorbed on CNF surfaces and (d) PBATg-40 adsorbed on silica surface for comparison. | ||
The surfaces of the CNF and the formed latex layers on the QCM-D crystals were analysed with CAM and resulted in the following contact angles (CA) for Milli-Q water on the surfaces; 44° for PMMATg120, 93° for P(BA-co-MMA)Tg3 and 85° for PBATg-40 respectively, before heat treatment. The reason why the PMMATg120 surface showed as low contact angle as 44° is probably due to the PMMATg120 latex maintaining its spherical shape with no hydrophobic core polymer being exposed to the water droplet, without heat treatment.
Adsorption of the latexes was also performed ex situ on silica wafers for comparison. The CAM analysis showed similar values as in situ QCM-D crystals before annealing, ESI Fig. S11 and Table S5.† A complete coalescence was detected after extensive annealing at 160 °C for 10 h, resulting in a more hydrophobic surface even for PMMATg120 layer (CA of 62° which is closer to the reported reference value for PMMA films of 68 °C (ref. 71)). To achieve accurate reference CA values for the latexes, they were also cast onto clean microscopy slides, without annealing, and analysed with CAM, as shown in ESI Fig. S16 and Table S7.† The results show that the P(BA-co-MMA)Tg3 and the PBATg-40 had contact angles of 92° and 99° respectively, whereas the PMMATg120 latex shows a CA close to 0° and more spreading of the water droplet than on the reference glass, which is in agreement with the Wentzel-equation for spreading of water droplets on surfaces showing contact angles below 90°.72
Adsorption of latexes onto filter papers
Filter papers were used to investigate the adsorption of the latexes onto a cellulose surface with a low charge density and large roughness. As for the previously reported cationic PDMAEMA-stabilised latexes,21 PDMAPMA-stabilised latex adsorbs readily on the rough fibre surfaces of the filter paper. Latex dispersed in 10 mM NaCl resulted in a higher surface coverage same as for the model surfaces (CNF and Silica), shown by the SEM images in Fig. 7 and FTIR analysis (ESI Fig. S17†). Similar to the adsorption of the latex onto CNF and silica substrates, the filter papers modified with PBATg-40 and P(BA-co-MMA)Tg3 latexes were hydrophobic (i.e. CA higher than 90°) after drying at room temperature, while the paper modified with PMMATg120 was hydrophilic prior to annealing, Fig. 8(i)–(iii) and ESI Table S8.† | ||
| Fig. 7 SEM images of filter papers covered with latexes (0.1 g L−1, 10 mM NaCl); (a) PMMATg120, (b) P(BA-co-MMA)Tg3, (c) water CA measurements prior to annealing (i) PMMATg120 (ii) P(BA-co-MMA)Tg3 and (iii) PBATg-40. Inset (iv) filter paper with PMMATg120 after annealing at 140 °C for 3 h and (d) PBATg-40. | ||
 | ||
| Fig. 8 SEM images of filter papers covered with latexes (0.1 g L−1, 10 mM NaCl); (a) PMMATg120 after annealing at 160 °C for 10 h, (b) PMMACTAC reference latex at room temperature without heat treatment. The CAs are measured after (i) annealing for 10 h at 160 °C and (ii) annealing for 3 h at 140 °C. | ||
The CAs of about 112° for P(BA-co-MMA)Tg3 latex and 136° for PBATg-40 latex, indicate that the particles coalesce to form films on the surface of the fibres. The filter papers are microscopically rough, why the resulting CAs can be exaggerated, both due to the roughness per se but also due to the existence of what is termed re-entrant surfaces.73 The CA's produced on the smoother microscopy glass (ESI Table S7†), also without any heat treatment, is in relatively good agreement with the paper surfaces, Fig. 7. Annealing for 3 h at 140 °C was insufficient for PMMATg120 to form a film on the fibres, why no change in contact angle was observed for the two surfaces shown in Fig. 7(i) and (iv). This was not the case for reference PMMACTAC, resulting in a hydrophobic filter paper with CA >90° already after annealing for 3 h at 140 °C (Fig. 8 and ESI Table S8†) and further highlights the effect of the stabilizing PDMAPMA-corona of the PMMATg120 latex. Not until the annealing was performed at 160 °C for 10 h, the PMMATg120 finally resulted in CAs above 90° as a consequence of coalescing particles, Fig. 8 and ESI Table S8.†
Chevalier et al. have reported a similar, increased structural stability against thermal treatment for grafted P(BA-co-styrene) latex having a polymeric corona.74 The surfactant-stabilised PMMACTAC latex also resulted in lower surface coverage on the filter paper, compared to the PDMAPMA-stabilised latexes, as observed in Fig. 8(b) together with a lower relative intensity for the carbonyl peak in PMMA at 1730 cm−1 as assessed by FTIR, ESI Fig. S17.†
Measuring wet adhesion between silica surfaces treated with latexes using the AFM colloidal probe technique
Atomic force microscopy (AFM) colloidal probe was used to investigate how the different Tg's (−40, 3 and 120 °C) of the core polymers in the PDMAPMA-stabilized latexes affect the wet state adhesion between treated silica surfaces. AFM images of the coated dried silica surfaces were recorded to image the coverage of silica surface (Fig. 9g, h and i) and were found to be in good agreement with the SEM images, where the PMMATg120 remains spherical on the surface while the P(BA-co-MMA)Tg3 particles collapse slightly and the PBATg-40 particles coalesce into a rather smooth film. When using the AFM colloidal probe technique, it is possible to alter the stiffness of the cantilever to investigate the effect from different pressures forcing the latex coated surfaces into contact. One can also alter the time of contact between the surface and the probe, results relating to chain mobility and relaxation of the polymeric components in the latex coatings (similar to the macro-scale use of a hot-press). Force curves from the colloidal probe measurements were acquired at up to a hundred different positions, and representative approach and retraction curves are shown in Fig. 9(a–f). More than one curve is displayed when there were significantly different behaviours at different positions on the surface. The approach curves (black) for the PMMATg120, Fig. 9a and d, show a step-wise increase in the repulsive force, which suggests that the probe move around adsorbed particles on the wet surface due to their inherent stiffness and weaker interaction with the silica as a result of the low contact area for each particle, Fig. 9d. For the P(BA-co-MMA)Tg3 latex, Fig. 9b and e, the approach curve is smooth and gradually becomes more repulsive when the particles are compressed to form a film between the probe and the silica surface. The PBATg-40 latex shows the most interesting approach behaviour in Fig. 9c and f where the probe jumps into contact from a distance of 100–150 nm. This suggests that it is highly favourable for surfaces coated with this latex to form a contact zone in order to avoid an interface against water. The observation further corroborates that these latexes form hydrophobic surfaces (CAM) and form stiff layers (QCM-D), Fig. 2, 5 and ESI Fig. S11–S16.†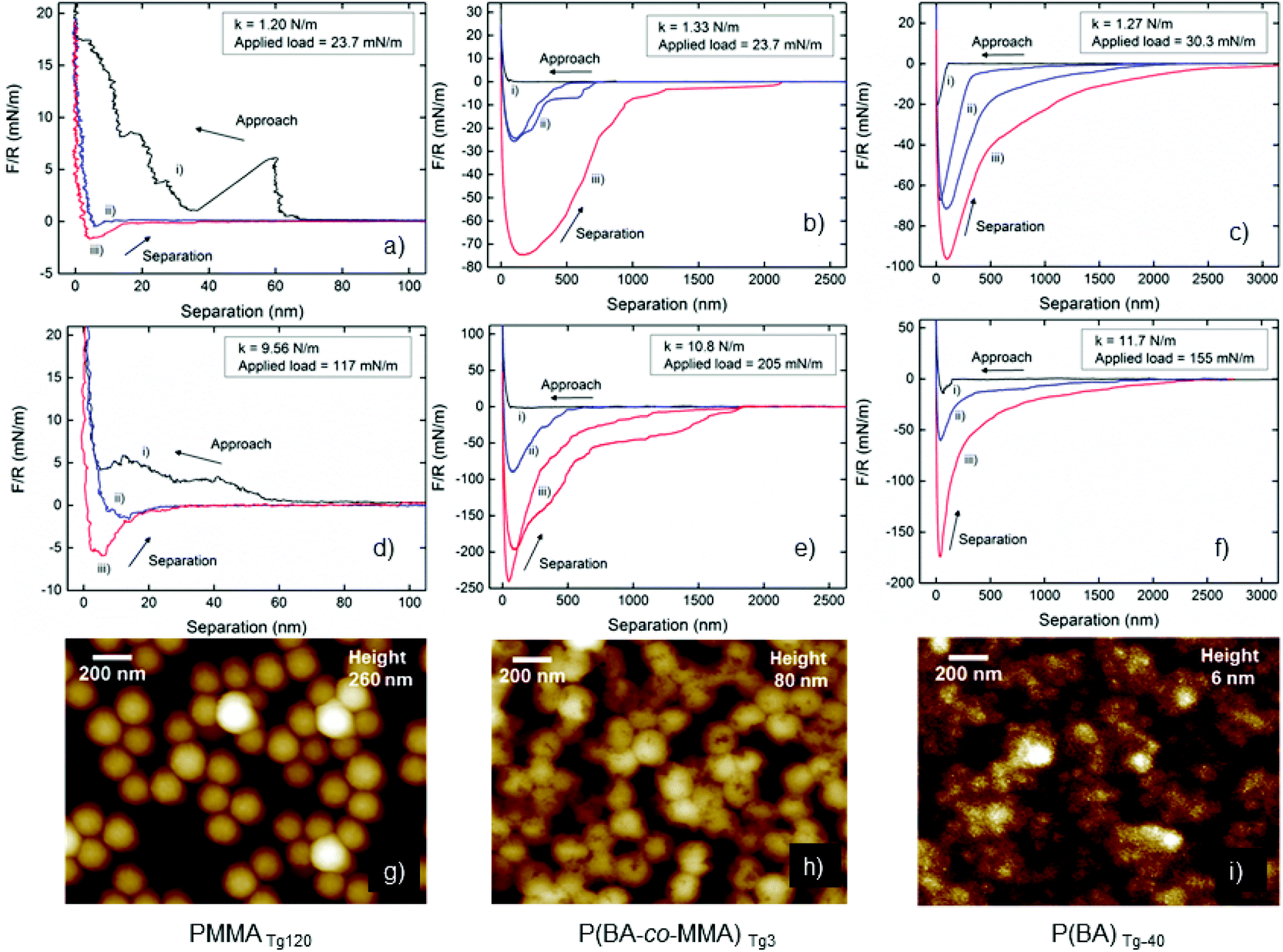 | ||
| Fig. 9 AFM colloidal probe wet-adhesion measurements between a latex-coated silicon probe and a latex-coated silica wafer. The bottom row shows AFM images after drying in room temperature. The force curves from the measurements (a–f) shows (i) an approach curve (ii) separation curves without surface delay and (iii) separation curve after 120 seconds surface delay. First row shows measurements for softer spring constants (<1.4 N m−1) and second row for stiffer spring constants (>9.5 N m−1). | ||
The separation curves for the stiffest latex PMMATg120 indicate very little interaction during separation demonstrating that essentially no coalescence occurs during contact between the probe and the silica wafer. The possible reason may be due to the repulsive properties of the cationic PDMAPMA-corona in the wet state but is most likely due to the experienced rigidity of the PMMATg120, shown when comparing to the other latexes with lower Tg. The Tg of the core polymer can be lowered by water, previously shown to change as much as 20 °C when saturated (1.92 g H2O per 100 g PMMA),75 however, this effect was clearly not sufficient for coalescence. Lowering of the Tg of the core, to below room temperature, largely increases the interaction between the coated probe and the coated silica surface, i.e., the two latex layers. The P(BA-co-MMA)Tg3 latex showed both longer interaction distances, above 2 μm, and force values above 200 mN m−1, with 120 s surface delay time, Fig. 9b and d. The force curves from the stiffer cantilever indicate a significant pressure sensitivity. It is also important to highlight the step-wise detachment where the force remains constant over hundreds of nm until sufficient force is generated to pull-off the probe from the surface. We suggest that coalesced particles and particle clusters at the edge of the contact zone are responsible for the step-wise separation when they release “one by one” as the distance between the probe and the surface increases. Similar behaviour was described for interacting polymer chains of cationic PDMAEMA in a PDMS matrix, analysed with an anionic probe, giving rise to step-wise separation.25 Lowering the Tg of the core further, the PBATg-40 reached separation distances of almost 3 μm and force values above 150 mN m−1 for the stiffest cantilever with 120 s surface delay time. For the softer cantilever, there was no significant difference between no surface delay and 120 s delay, which indicates that the polymers in the core can re-arrange rapidly due to high chain mobility to form strong adhesive joints in the wet state. PBATg-40 with a diameter of 76 nm showed the ability to form a film that retains adhesive contact up to 3 μm of separation, which can be translated to around 3800% elongation-at-break. The P(BA-co-MMA)Tg3 latex shows similar properties, and it appears that a Tg closer to room temperature provides suitable viscoelasticity to form impressive adhesive strength in the wet state. The considerable mobility of the chains for PBATg-40 will not result in strong adhesive contact, comparing to the higher Tg latex, (BA-co-MMA)Tg3, which show larger extent of non-reversible chain relaxation and strong adhesive contact, corroborating results from adhesion measurements performed on thin PBMA surfaces at different temperatures above and below Tg of PBMA.76
The average pull-off force (peak value) and the work of adhesion (area under the graph) can be calculated from the AFM colloidal probe measurements, Fig. 10. It can be observed that the pull-off force and work of adhesion increased after 120 s delay time for both the P(BA-co-MMA)Tg3 and the PBATg-40 latex. The resulting larger pull-off force, especially for the stiffer cantilevers, shows that the polymer chains in the particles require sufficient time to inter-diffuse between adjacent particles and disentangle at a low enough rate to give high adhesive interactions. If the polymers are too mobile, they will inter-diffuse but disentangle too quickly and thus result in a weak adhesive interaction, as observed when comparing P(BA-co-MMA)Tg3 latex to the PBATg-40 latex in Fig. 10.76
 | ||
| Fig. 10 AFM colloidal probe wet-adhesion measurements calculated as (a) pull-off force and (b) work of adhesion. Measurements correspond to latex-coated silicon probes and latex-coated wafers (1 h, 10 g L−1, 10 mM NaCl). Softer spring constants (<1.4 N m−1) and stiffer spring constants (>9.5 N m−1) are used. Error bars showing 95% confidence intervals. | ||
The low work of adhesion observed for the PMMATg120 latex is in accordance with the previous discussion on the PMMA rigidity and absence of film formation for this type of latex at around room temperature due to the small chain mobility.
Previously investigated polymeric systems such as multilayers of hyaluronic acid and poly(allylamine hydrochloride) (PAH) obtained by a layer-by-layer technique resulted in pull-off forces up to 15 mN m−1,77 without delay times. In this study, a monolayer of P(BA-co-MMA)Tg3 latex results in a nearly 6 times larger pull-off force, approaching 100 mN m−1. Another reported layer-by-layer assembly, using tri-block copolymer micelles, reach values as high as 180 mN m−1 without delay times (compared to 100 mN m−1 for (P(BA-co-MMA)Tg3 latex). However, the work of adhesion reported herein is almost one magnitude higher for the (P(BA-co-MMA)Tg3 latex than for the tri-block copolymer system, above 0.1 pJ without delay.78 Fantner et al. showed that the work of adhesion of collagen in bone was around 0.005 pJ (ref. 79) again, significantly lower than the herein reported values of 0.1 pJ for both P(BA-co-MMA)Tg3 and PBATg-40.
The strongest wet adhesion is achieved with a strongly-bonding cationic corona to adhere to the supporting surfaces and a “soft enough” core polymer, such as P(BA-co-MMA)Tg3 latex, that allows for inter-particle diffusion of chains, due to coalescence, in water and at the same time stiff enough to show strong mechanical properties at room temperature. When the Tg in the core is lower, PBATg-40 latex, the strength of the formed film is not sufficient to withstand as much pull-off force before failure.
Conclusions
A polyelectrolyte PDMAPMA macroRAFT was efficiently used in surfactant-free emulsion polymerisation for the formation of latexes with particle core of tailored Tg's (−40, 3 and 120 °C). These latexes adsorb readily onto both silica and CNF surfaces, as shown by QCM-D measurements, and to cellulose filter papers. The two lowest Tg latexes give rise to hydrophobic surfaces already after drying at room temperature while the high Tg latex results in a hydrophilic surface. The surface interactions between latex and treated surfaces show evident benefits from having a polyelectrolyte corona. Increasing salt concentration resulted in increased adsorbed mass and coverage of both silica and CNF surfaces. This was demonstrated with QCM-D and SEM, and also shown on cellulose filter paper using FTIR and SEM analyses. The PMMATg120 latex maintains its spherical shape until extensive annealing at 160 °C for more than 3 h whereas the lower Tg latexes deform and film form at room temperature. The effect of using a polyelectrolyte corona (PDMAPMA) and particle cores of different Tg was investigated by colloidal probe measurements in AFM. The P(BA-co-MMA)Tg3 latex showed very strong wet adhesion, due to an optimal combination of coalescence under pressure and polymer entanglement and disentanglement. The promising wet state interactions are assigned to the tailored combination of using cellulose-interacting polyelectrolyte PDMAPMA with the core polymer of Tg of around 3 °C. The strong adsorption to cellulose and the wet adhesion properties establish these PISA-latexes as very promising tools for the production of strong and tough materials with high nanocellulose content, easily formed by mixing of water dispersions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the Knut and Alice Wallenberg Foundation through the Wallenberg Wood Science Centre (WWSC) for financial support. Pierre-Yves Dugas (C2P2) is greatly acknowledged for his help with cryo-TEM imaging. Emma Ingo is greatly acknowledged for her hard work in the lab helping with centrifugation work-up of the latexes.Notes and references
- D. Klemm, F. Kramer, S. Moritz, T. Lindstrom, M. Ankerfors, D. Gray and A. Dorris, Angew. Chem., Int. Ed., 2011, 50, 5438–5466 CrossRef CAS PubMed.
- D. Klemm, B. Heublein, H. P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- I. Siró, D. Plackett, M. Hedenqvist, M. Ankerfors and T. Lindström, J. Appl. Polym. Sci., 2011, 119, 2652–2660 CrossRef.
- T. Saito, R. Kuramae, J. Wohlert, L. A. Berglund and A. Isogai, Biomacromolecules, 2012, 14, 248–253 CrossRef PubMed.
- M. Henriksson, L. A. Berglund, P. Isaksson, T. Lindström and T. Nishino, Biomacromolecules, 2008, 9, 1579–1585 CrossRef CAS PubMed.
- J.-M. Raquez, Y. Habibi, M. Murariu and P. Dubois, Prog. Polym. Sci., 2013, 38, 1504–1542 CrossRef CAS.
- K.-Y. Lee, Y. Aitomäki, L. A. Berglund, K. Oksman and A. Bismarck, Compos. Sci. Technol., 2014, 105, 15–27 CrossRef CAS.
- F. L. Hatton, J. Engstrom, J. Forsling, E. Malmstrom and A. Carlmark, RSC Adv., 2017, 7, 14947–14958 RSC.
- K. J. De France, T. Hoare and E. D. Cranston, Chem. Mater., 2017, 29, 4609–4631 CrossRef CAS.
- Y. Habibi, Chem. Soc. Rev., 2014, 43, 1519–1542 RSC.
- A. Boujemaoui, C. Cobo Sanchez, J. Engström, C. Bruce, L. Fogelström, A. Carlmark and E. Malmström, ACS Appl. Mater. Interfaces, 2017, 9(40), 35305–35318 CrossRef CAS PubMed.
- N. Lin and A. Dufresne, Macromolecules, 2013, 46, 5570–5583 CrossRef CAS.
- F. L. Hatton, E. Malmström and A. Carlmark, Eur. Polym. J., 2015, 65, 325–339 CrossRef CAS.
- A. Carlmark and E. Malmström, J. Am. Chem. Soc., 2002, 124, 900–901 CrossRef CAS.
- D. Roy, M. Semsarilar, J. T. Guthrie and S. Perrier, Chem. Soc. Rev., 2009, 38, 2046–2064 RSC.
- C. B. Bucur, Z. Sui and J. B. Schlenoff, J. Am. Chem. Soc., 2006, 128, 13690–13691 CrossRef CAS PubMed.
- M. R. Talingting, Y. Ma, C. Simmons and S. E. Webber, Langmuir, 2000, 16, 862–865 CrossRef CAS.
- L. Winter, L. Wågberg, L. Ödberg and T. Lindström, J. Colloid Interface Sci., 1986, 111, 537–543 CrossRef CAS.
- L. Wågberg, Nord. Pulp Pap. Res. J., 2000, 15, 12 CrossRef.
- L. Wågberg and R. Hägglund, Langmuir, 2001, 17, 1096–1103 CrossRef.
- J. Engstrom, F. L. Hatton, L. Wagberg, F. D'Agosto, M. Lansalot, E. Malmstrom and A. Carlmark, Polym. Chem., 2017, 8, 1061–1073 RSC.
- L. Carlsson, A. Fall, I. Chaduc, L. Wagberg, B. Charleux, E. Malmstrom, F. D'Agosto, M. Lansalot and A. Carlmark, Polym. Chem., 2014, 5, 6076–6086 RSC.
- I. R. Quevedo, A. L. J. Olsson and N. Tufenkji, Environ. Sci. Technol., 2013, 47, 2212–2220 CrossRef CAS PubMed.
- I. Popa, G. Gillies, G. Papastavrou and M. Borkovec, J. Phys. Chem. B, 2010, 114, 3170–3177 CrossRef CAS PubMed.
- A. Beaussart, T. C. Ngo, S. Derclaye, R. Kalinova, R. Mincheva, P. Dubois, P. Leclère and Y. F. Dufrêne, Nanoscale, 2014, 6, 565–571 RSC.
- B. Alince, P. Arnoldova and R. Frolik, J. Appl. Polym. Sci., 2000, 76, 1677–1682 CrossRef CAS.
- J. L. Keddie, Mater. Sci. Eng., R, 1997, 21, 101–170 CrossRef.
- S. C. Thickett and R. G. Gilbert, Polymer, 2007, 48, 6965–6991 CrossRef CAS.
- V. Favier, G. R. Canova, J. Y. Cavaillé, H. Chanzy, A. Dufresne and C. Gauthier, Polym. Adv. Technol., 1995, 6, 351–355 CrossRef CAS.
- E. L. Vinay Kumar, P. Salminen, D. Bousfield and M. Toivakka, Nord. Pulp Pap. Res. J., 2016, 31, 7 Search PubMed.
- R. H. Pelton, J. Polym. Sci., Part A: Polym. Chem., 1988, 26, 9–18 CrossRef CAS.
- B. Alince, M. Inoue and A. A. Robertson, J. Appl. Polym. Sci., 1976, 20, 2209–2219 CrossRef CAS.
- C. J. Ferguson, R. J. Hughes, D. Nguyen, B. T. T. Pham, R. G. Gilbert, A. K. Serelis, C. H. Such and B. S. Hawkett, Macromolecules, 2005, 38, 2191–2204 CrossRef CAS.
- M. Manguian, M. Save and B. Charleux, Macromol. Rapid Commun., 2006, 27, 399–404 CrossRef CAS.
- C. J. Ferguson, R. J. Hughes, B. T. T. Pham, B. S. Hawkett, R. G. Gilbert, A. K. Serelis and C. H. Such, Macromolecules, 2002, 35, 9243–9245 CrossRef CAS.
- R. T. A. Mayadunne, E. Rizzardo, J. Chiefari, Y. K. Chong, G. Moad and S. H. Thang, Macromolecules, 1999, 32, 6977–6980 CrossRef CAS.
- R. T. A. Mayadunne, E. Rizzardo, J. Chiefari, J. Krstina, G. Moad, A. Postma and S. H. Thang, Macromolecules, 2000, 33, 243–245 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- P. B. Zetterlund, S. C. Thickett, S. Perrier, E. Bourgeat-Lami and M. Lansalot, Chem. Rev., 2015, 115, 9745–9800 CrossRef CAS PubMed.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- M. Chenal, L. Bouteiller and J. Rieger, Polym. Chem., 2013, 4, 752–762 RSC.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- M. Lansalot, J. Rieger and F. D'Agosto, in Macromolecular Self-assembly, John Wiley & Sons, Inc., 2016, pp. 33–82, DOI:10.1002/9781118887813.ch2.
- J. Lesage de la Haye, I. Martin-Fabiani, M. Schulz, J. L. Keddie, F. D'Agosto and M. Lansalot, Macromolecules, 2017, 50, 9315–9328 CrossRef CAS.
- I. Martín-Fabiani, J. Lesage de la Haye, M. Schulz, Y. Liu, M. Lee, B. Duffy, F. D'Agosto, M. Lansalot and J. L. Keddie, ACS Appl. Mater. Interfaces, 2018, 10, 11221–11232 CrossRef PubMed.
- E. Larsson, C. C. Sanchez, C. Porsch, E. Karabulut, L. Wågberg and A. Carlmark, Eur. Polym. J., 2013, 49, 2689–2696 CrossRef CAS.
- S. Utsel, C. Bruce, T. Pettersson, L. Fogelström, A. Carlmark, E. Malmström and L. Wågberg, ACS Appl. Mater. Interfaces, 2012, 4, 6796–6807 CrossRef CAS PubMed.
- C. Bruce, I. Javakhishvili, L. Fogelstrom, A. Carlmark, S. Hvilsted and E. Malmstrom, RSC Adv., 2014, 4, 25809–25818 RSC.
- M. Vuoriluoto, H. Orelma, L.-S. Johansson, B. Zhu, M. Poutanen, A. Walther, J. Laine and O. J. Rojas, J. Phys. Chem. B, 2015, 119, 15275–15286 CrossRef CAS PubMed.
- P. van de Wetering, N. J. Zuidam, M. J. van Steenbergen, O. A. G. J. van der Houwen, W. J. M. Underberg and W. E. Hennink, Macromolecules, 1998, 31, 8063–8068 CrossRef CAS.
- T. Lindström, L. Wågberg and T. Larsson, On the nature of joint strength in paper - A review of dry and wet strength resins used in paper manufacturing, P. C. a. P. Mechanics, STFI Packforsk, Innventia.com, 2005 Search PubMed.
- Y. A. Vasilieva, D. B. Thomas, C. W. Scales and C. L. McCormick, Macromolecules, 2004, 37, 2728–2737 CrossRef CAS.
- L. C. Paslay, B. A. Abel, T. D. Brown, V. Koul, V. Choudhary, C. L. McCormick and S. E. Morgan, Biomacromolecules, 2012, 13, 2472–2482 CrossRef CAS PubMed.
- A. W. York, S. E. Kirkland and C. L. McCormick, Adv. Drug Delivery Rev., 2008, 60, 1018–1036 CrossRef CAS PubMed.
- T. Boursier, I. Chaduc, J. Rieger, F. D'Agosto, M. Lansalot and B. Charleux, Polym. Chem., 2011, 2, 355–362 RSC.
- G. Bouhadir, N. Legrand, B. Quiclet-Sire and S. Z. Zard, Tetrahedron Lett., 1999, 40, 277–280 CrossRef CAS.
- S. H. Thang, Y. K. Chong, R. T. A. Mayadunne, G. Moad and E. Rizzardo, Tetrahedron Lett., 1999, 40, 2435–2438 CrossRef CAS.
- L. Wågberg, G. Decher, M. Norgren, T. Lindström, M. Ankerfors and K. Axnäs, Langmuir, 2008, 24, 784–795 CrossRef PubMed.
- G. Sauerbrey, Z. Phys., 1959, 155, 206–222 CrossRef CAS.
- C. Aulin, I. Varga, P. M. Claesson, L. Wågberg and T. Lindström, Langmuir, 2008, 24, 2509–2518 CrossRef CAS PubMed.
- A. Das, S. Ghosh and A. R. Ray, Polymer, 2011, 52, 3800–3810 CrossRef CAS.
- C. B. Roth, A. Pound, S. W. Kamp, C. A. Murray and J. R. Dutcher, Eur. Phys. J. E: Soft Matter Biol. Phys., 2006, 20, 441–448 CrossRef CAS PubMed.
- A. I. Buzin, M. Pyda, P. Costanzo, K. Matyjaszewski and B. Wunderlich, Polymer, 2002, 43, 5563–5569 CrossRef CAS.
- T. G. Fox, Bull. Am. Phys. Soc., 1956, 1, 123 CAS.
- T. Saarinen, M. Österberg and J. Laine, J. Dispersion Sci. Technol., 2009, 30, 969–979 CrossRef CAS.
- L.-E. Enarsson and L. Wågberg, Langmuir, 2008, 24, 7329–7337 CrossRef CAS PubMed.
- Z. Adamczyk and P. Warszyński, Adv. Colloid Interface Sci., 1996, 63, 41–149 CrossRef CAS.
- V. Granier and A. Sartre, Langmuir, 1995, 11, 2179–2186 CrossRef CAS.
- S. Pal, S. Ghosh Roy and P. De, Polym. Chem., 2014, 5, 1275–1284 RSC.
- G. Vigil, Z. Xu, S. Steinberg and J. Israelachvili, J. Colloid Interface Sci., 1994, 165, 367–385 CrossRef CAS.
- Y. Ma, X. Cao, X. Feng, Y. Ma and H. Zou, Polymer, 2007, 48, 7455–7460 CrossRef CAS.
- C. Dorrer and J. Rühe, Soft Matter, 2009, 5, 51–61 RSC.
- A. Tuteja, W. Choi, G. H. McKinley, R. E. Cohen and M. F. Rubner, MRS Bull., 2008, 33, 752–758 CrossRef CAS.
- Y. Chevalier, C. Pichot, C. Graillat, M. Joanicot, K. Wong, J. Maquet, P. Lindner and B. Cabane, Colloid Polym. Sci., 1992, 270, 806–821 CrossRef CAS.
- L. S. A. Smith and V. Schmitz, Polymer, 1988, 29, 1871–1878 CrossRef CAS.
- G. Luengo, J. Pan, M. Heuberger and J. N. Israelachvili, Langmuir, 1998, 14, 3873–3881 CrossRef CAS.
- T. Pettersson, S. A. Pendergraph, S. Utsel, A. Marais, E. Gustafsson and L. Wågberg, Biomacromolecules, 2014, 15, 4420–4428 CrossRef CAS PubMed.
- A. Trager, S. A. Pendergraph, T. Pettersson, T. Halthur, T. Nylander, A. Carlmark and L. Wagberg, Nanoscale, 2016, 8, 18204–18211 RSC.
- G. E. Fantner, T. Hassenkam, J. H. Kindt, J. C. Weaver, H. Birkedal, L. Pechenik, J. A. Cutroni, G. A. G. Cidade, G. D. Stucky, D. E. Morse and P. K. Hansma, Nat. Mater., 2005, 4, 612–616 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nr08057g |
| ‡ Presently at RISE Research Institutes of Sweden AB, Drottning Kristinas Väg 61, 114 28, Stockholm Sweden. |
| This journal is © The Royal Society of Chemistry 2019 |