
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanoassemblies of ultrasmall clusters with remarkable activity in carbon dioxide conversion into C1 fuels†
Avik
Halder‡
 a,
Joseph
Kioseoglou‡
b,
Bing
Yang
a,
Karthika Lakshmi
Kolipaka
a,
Soenke
Seifert
c,
Jan
Ilavsky
c,
Michael
Pellin
a,
Mukhles
Sowwan
d,
Panagiotis
Grammatikopoulos
*d and
Stefan
Vajda
*ae
a,
Joseph
Kioseoglou‡
b,
Bing
Yang
a,
Karthika Lakshmi
Kolipaka
a,
Soenke
Seifert
c,
Jan
Ilavsky
c,
Michael
Pellin
a,
Mukhles
Sowwan
d,
Panagiotis
Grammatikopoulos
*d and
Stefan
Vajda
*ae
aMaterials Science Division, Argonne National Laboratory, Argonne, Illinois 60439, USA. E-mail: vajda@anl.gov
bDepartment of Physics, Aristotle University of Thessaloniki, GR-54124 Thessaloniki, Greece
cX-ray Science Division, Argonne National Laboratory, 9700 South Cass Avenue, Argonne, Illinois 60439, USA
dNanoparticles by Design Unit, Okinawa Institute of Science and Technology Graduate University, 1919-1, Onna-Son, Okinawa 904-0495, Japan. E-mail: pgrammatikopoulos@oist.jp
eInstitute for Molecular Engineering, The University of Chicago, Chicago 60637, USA
First published on 20th February 2019
Abstract
Cu nanoassemblies formed transiently during reaction from size-selected subnanometer Cu4 clusters supported on amorphous OH-terminated alumina convert CO2 into methanol and hydrocarbons under near-atmospheric pressure at rates considerably higher than those of individually standing Cu4 clusters. An in situ characterization reveals that the clusters self-assemble into 2D nanoassemblies at higher temperatures which then disintegrate upon cooling down to room temperature. DFT calculations postulate a formation mechanism of these nanoassemblies by hydrogen-bond bridges between the clusters and H2O molecules, which keep the building blocks together while preventing their coalescence.
Efficient conversion of CO2 is a way to remove the pollutant gas from the atmosphere1,2 and for the synthesis of fuels, hydrocarbons, and industrially relevant chemicals such as carboxylic acids, esters, lactones, etc.3–5 Cu-Based catalysts have been widely used where one can control the metal–oxide or metal–carbide interfaces (forming the metal and substrates, respectively) to increase the catalyst efficiency.6 Generally, these processes are energy demanding and are operated at temperatures of about 200–300 °C or higher, and at a high pressure of about 50–100 atmospheres.7,8 Finding a CO2 conversion catalyst which will operate under environmentally benign conditions remains an active area of research. There is a very interesting recent study with the CeOx/Cu2O/Cu(111) catalyst where the ceria islands forming at the step edges of Cu2O showed pronounced activity for CO2 conversion at near atmospheric pressure and at a temperature of about 300 °C.6,9 As another example, subnanometer copper clusters on oxide surfaces have been found to be highly active as well, under low pressures.10,11
Cu clusters containing a precise number of atoms form an excellent category of materials for which the number of surface atoms is well defined and, thus, are well within the resolution capabilities of the DFT formulation.12 An addition or subtraction of even a single atom from the cluster can have a drastic effect on the catalytic performance of such ultrasmall particles.10 For example, Cu4 clusters deposited on hydroxylated alumina supports have shown high activity in CO2 conversion into methanol compared to copper clusters of other sizes.10 Key factors determining the performance of a cluster also include the oxidation state of the clusters, the charge transfer between the cluster and the substrate, and the binding energy between the cluster and reaction intermediates.9,10,13–16 A significant advancement toward enhanced catalytic performance would be by increasing the cluster coverage of surfaces, while maintaining the specific size of the active sites.17 Increasing the surface coverage of clusters while retaining their specific size is a challenging endeavor, as coalescence into large nanoparticles, which can often have a detrimental effect on performance, should be avoided.18 On the other hand, atomic clusters and nanoparticles can be assembled into nanostructures exhibiting new propensities and/or improved stability.19–24 This paper presents results from in situ X-ray characterization of the catalysts,24,25 showing that by changing the reaction environment, the transient assembly of subnanometer clusters into three-dimensional nanostructures can be influenced during the course of the reaction. Ab initio DFT calculations provide an insight into the cluster assembly/disassembly process.
The copper clusters were prepared in a magnetron sputtering/condensation source housed in a high-vacuum apparatus12,26 and size-selection occurred with atomic precision in a quadrupole mass selector coupled to the cluster source by an ion guide assembly.26 The 4-atom clusters were thereafter softly landed on the substrate with an impact energy lower than 1 eV per atom. This step ensures that the clusters do not undergo fragmentation upon landing.27,28 The substrates were prepared by atomic layer deposition (ALD) of alumina on top of a native oxide of a silicon wafer (SiO2/n-type, phosphorus-doped Si (100)), yielding an ∼3 monolayer (ML) thin hydroxylated amorphous alumina film. The clusters of a single size were deposited on the substrate at a surface coverage of 5% of a ML, containing a total of 1.11 × 1013 Cu4 clusters on the sample surface, at an estimated average cluster-to-cluster distance of 3 nm. Next, the catalysts were studied under two reaction gas mixtures/conditions: (i) Condition A: CO2 and H2 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio, fed to the reactor of 40 cm3 volume at a rate of 20 sccm under 1.1 atm pressure and 100 ppm oxygen present, and (ii) Condition B: CO2, H2, and He in a 1:3:1 ratio, supplied at a rate of 5 sccm with 1000 ppm oxygen present. The applied temperature ramp is shown in Fig. S1a.† The catalytic activity was monitored using a mass spectrometer and the turnover rate (TOR) was calculated as the total number of product molecules formed per total Cu atom per second.25
:3 ratio, fed to the reactor of 40 cm3 volume at a rate of 20 sccm under 1.1 atm pressure and 100 ppm oxygen present, and (ii) Condition B: CO2, H2, and He in a 1:3:1 ratio, supplied at a rate of 5 sccm with 1000 ppm oxygen present. The applied temperature ramp is shown in Fig. S1a.† The catalytic activity was monitored using a mass spectrometer and the turnover rate (TOR) was calculated as the total number of product molecules formed per total Cu atom per second.25
Under condition A, the TOR of methanol formation reached 0.004 molecules per total Cu atom per second, as shown in Fig. 1a, which compares very well with the performance of some of the most active catalysts reported in the literature.6,9,29 The activity peaked at 275 °C and then dropped at higher temperatures, when a rise in the methane signal was observed (Fig. 1b). We note that the drop in the methanol signal could in part be caused by the formation of CO at high temperatures through the reverse water gas shift (rWGS) reaction; however, under the applied reaction conditions it was not possible to clearly distinguish the formation of CO due to an overlapping signal from the fragments of CO2 present at high concentrations. The rise of methane and the CO signal at high temperatures was previously reported and explained by the higher activation energy barrier for the respective channels.11 Methane activity set on at 325 °C, and peaked at 375 °C with a TOR of 0.07 molecules per total Cu atom per second, at the highest reaction temperature applied.
 | ||
| Fig. 1 TOR of (a) methanol and (b) methane for the Cu4 cluster on the alumina support under condition A, respectively. TOR of methanol (c) and methane (d) produced under condition B, respectively. | ||
Under condition B the peaks in both methanol and methane formation were observed at 375 °C, with a TOR of 0.012 and 0.65 molecules per total Cu atom per second respectively, as shown in Fig. 1c and d. These TORs for methanol were higher by a factor of 3 and 10, respectively, than under condition A with lower oxygen traces (the resulting signals are compared in Fig. S1b and S1c†). We also note the apparent differences in activity observed at identical temperatures during the heat-up and cool-down cycle; these differences may have various triggers such as differences in the oxidation state or in the average size of the nanoassemblies, as will be discussed below. However, other causes cannot be excluded either, such as a potential continuously evolving internal structure within the assembly.
In situ XANES spectra obtained at the Cu K-edge (8.976 keV) under condition B indicate that copper started reducing with the rise in temperature, as shown in Fig. 2a. The oxidation state of copper in the clusters was estimated by fitting the XANES spectra with a linear combination of bulk Cu standards Cu, Cu2O, CuO, and Cu(OH)2 (the spectra of the standards are shown in Fig. S2† and the representative fit results are shown in Fig. S3†). The initial composition within the cluster ensemble at room temperature resembled Cu(OH)2, but changed during the temperature ramp to a mixture of Cu0, Cu+1, and Cu+2 with ∼50% contribution from Cu0 at high temperatures, as shown in Fig. 2b. From in situ GISAXS observations, the drop in the oxidation state correlates with the formation of nanoassemblies which grew in size with rising temperature. Horizontal cuts of GISAXS data during temperature ramping are shown in Fig. 2c, indicating horizontal cluster assembly (2D scattering images and schematics of the assembled nanoparticles are shown in Fig. S4,† whereas horizontal cuts collected at 275 °C and 375 °C are analyzed in Fig. S5 and S6,† respectively). Interestingly, no growth in the vertical direction is observed, as indicated by the vertical cuts in Fig. S7.†
 | ||
| Fig. 2 In situ characterization for the Cu4 cluster sample under condition B, respectively: (a) GIXANES spectra recorded at the Cu K-edge, (b) change in the composition of copper with temperature obtained from the linear combination fit of the spectra, and (c) horizontal cuts of GISAXS data during the heat up and cool down cycle during the temperature ramp shown in Fig. S1a.† (d) Overlaid plot of the change in the oxidation state of copper with the evolution of the particle size of the assembly. The dashed line drawn at 1 nm indicates the cut-off below which the used GISAXS geometry did not allow the resolution of the particle size. The estimated fitting error in the cluster composition obtained from XANES is between 5–10% (relative). | ||
To demonstrate the correlation between the oxidation state of copper and the particle size of the assembly, we overlaid the plots showing their concurrent evolution. As shown in Fig. 2d, above 275 °C the particle sizes increased, reaching a lateral diameter of 4.5 nm at 375 °C; at the same temperature, Cu attained an average oxidation state of 0.7. The nanoassemblies started falling apart as the temperature dropped below 225 °C. A simultaneous re-oxidation of copper with disassembly can also be observed, with a sharp rise in the oxidation state between 175 °C to 125 °C, as copper was oxidized to CuO.
Under condition A, copper was reduced at a lower temperature (275 °C) and the lowest average oxidation state attained was 0.6, as shown in Fig. S8† (superimposed with that under condition B, for comparison). Interestingly, under condition A no nanoassembly formation was observed, as indicated by GISAXS data presented in Fig. S9.† The results show that the Cu4 cluster based nanoassemblies have significantly higher activity relative to individual Cu4 clusters under the applied reaction conditions, underlining the effect of reaction conditions on the nature and morphology of the catalyst as well as its catalytic performance.
A similar correlation between the cluster size and the copper oxidation state was also observed for clusters of different sizes, i.e. Cu12, as shown in Fig. S10 and S11.† Neither Cu12 clusters nor their assemblies showed measurable activity towards methanol formation, and only very low activity towards methane formation was detected.
Theoretical investigation was performed to elucidate the underlying driving force for the stable nanoassembly formation where the “building blocks” can maintain their individual properties. Two primary experimental observations were especially taken into account: (i) GISAXS observations of larger-size clusters decomposing toward their original sizes indicate that their bonding is not strong. This fact implies nanocluster agglomeration, i.e. a collection of clusters held together by weak forces (such as van der Waals, electrostatic, capillary, etc.),30 rather than coalescence toward larger nanoparticles.31 (ii) Agglomeration enhancement by increased concentration of a highly electronegative atom such as oxygen points towards hydrogen bond formation as the mechanism providing the binding forces.
The VASP ab initio simulation package32 with Projector Augmented Wave (PAW) pseudopotentials33,34 was implemented to investigate the structure and interactions of the Cu-based clusters. The Perdew, Burke, and Ernzerhof (PBE) Generalized Gradient Approximation (GGA)35,36 is more accurate than the Local-Density Approximation (LDA) in the description of hydrogen bonded cases,37,38 for which the LDA severely overestimates the strength of the hydrogen bonds.39,40 Consequently, to consider the hydrogen bonded cases of CuO, the GGA+U approach was employed. The energy cut-off for the plane wave basis set was 520 eV. The Gaussian smearing method with a smearing width of 0.05 eV was selected for the filling of the electronic levels. A 6 × 6 × 4 Γ-centered k-point mesh for the primitive cell was used. The U–J parameter was set at 7,41 a value that produces lattice constants for the primitive CuO cell (a = 4.723 Å, b = 3.447 Å, c = 5.115 Å, beta = 99.658, u = 0.577) in agreement with the experimental values.42
We analyzed the interaction between pairs of Cu4O4 clusters relaxed into their energetically favorable configurations (Fig. S12†) taking into account all possible related parameters such as the relative orientation, the initial distance between them, etc. As expected, when chemical bonds are formed between the clusters, the total energy of the system is decreased significantly. As an example, in Fig. 3a we present the relaxed configuration of two Cu4O4 clusters, where the formation of chemical bonds between them is demonstrated, accompanied by a drop in energy by 5.00 eV.
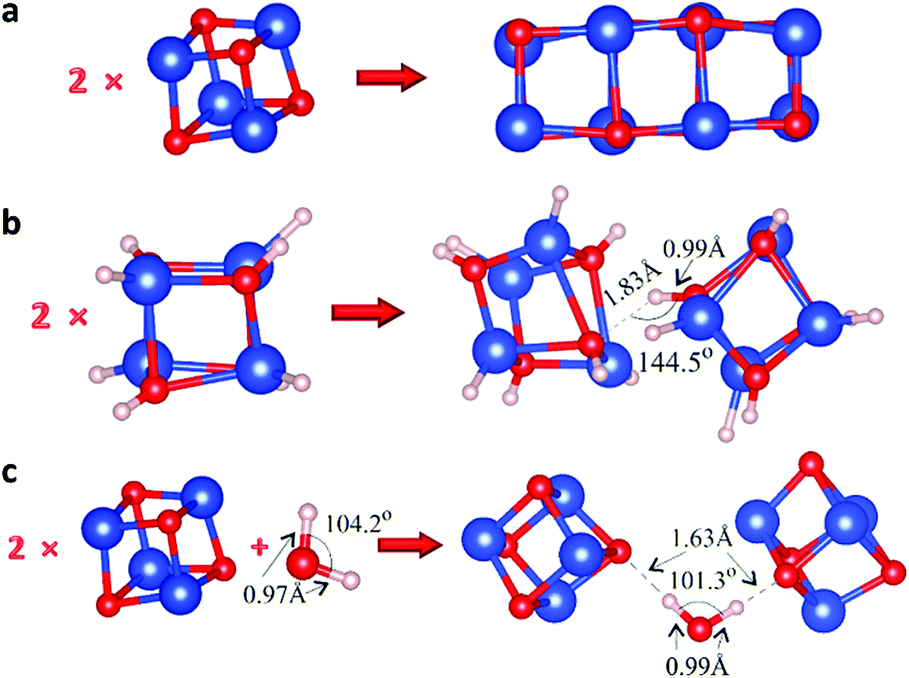 | ||
| Fig. 3 The initial components (on the left) and the energetically favorable relaxed atomistic configurations (on the right) of (a) two Cu4O4 clusters, where the formation of chemical bonds between them is demonstrated, (b) two Cu4O4H8 clusters with an isolated H⋯O hydrogen bond and no further interaction between the clusters, and (c) two Cu4O4 with a H2O molecule containing two H⋯O hydrogen bonds. | ||
Subsequently, to put our hydrogen bond hypothesis to the test, we saturated all the dangling bonds in our primary clusters with hydrogen to prevent any potential covalent bonding between them, and to enable only the formation of hydrogen bonds between them; more specifically, we saturated the Cu4O4 nanoclusters with eight hydrogen atoms (Fig. S13†). On the relaxed model on the left-hand side of Fig. 3b, the O–H bond length is ∼0.96 Å, while the Cu–O one is ∼1.43 Å. We investigated the interaction between pairs of Cu4O4H8 in order to analyze the possible formation of hydrogen bonds between the saturated hydrogen atoms and oxygen atoms. This was achieved by constructing and relaxing multiple configurations in order to identify H⋯O hydrogen bonds between the two Cu4O4H8 moieties (e.g. Fig. S14†). Indeed, in accurately constructed models like that of Fig. 3b we isolated H⋯O hydrogen bonds with no further interaction between the two Cu4O4H8 clusters. In these cases, the formation energy was found to be equal to 1.3 eV, which falls within the high end of the H⋯O hydrogen bond energy range.43
However, even though this mechanism can, in principle, predict the weak electrostatic bonding required for the loose agglomeration of the clusters, it cannot account for the counter-intuitive dissociation of the agglomerates upon cooling down; the reverse behavior would be expected instead, where the bonds break apart upon heating (rather than cooling) of the samples. Therefore, we also tested another potential mechanism, i.e. the formation of water molecule bridges providing adhesion between the clusters. In principle, it is a process similar to adding water to sand to make its grains stick together in order to build a sandcastle; note, however, that the analogy is only qualitative since in the case of sandcastles water forms capillary, not hydrogen-bond, bridges. In Fig. 3c, one such relaxed configuration is presented, where the two H⋯O hydrogen bonds are indicated. Initially, the H–O distances were ∼1 Å, which is close to the H–O chemical bond length, but after relaxation they reached 1.63 Å, indicating H⋯O hydrogen bonds. Moreover, the formation energy Ef = Etotal − 2 × ECu4O4 − EH2O equals ∼0.14 eV per bond, leading to the conclusion that these are very weak H⋯O hydrogen bonds. Also note that in this case there is no need for hydrogen saturation of the primary particles.
At elevated temperatures, water is expected to be produced, as seen in other Cu-based systems.4,29,44 In addition, water can also be formed from the reaction of O2 and H2. As a result, the formation of water on the clusters can lead to a local rise in H2O concentration, enough to act as the necessary adhesive. Naturally, upon heating, some of the water that holds the agglomerate together is expected to evaporate, but it can be replenished by H2O being formed as a reaction product, thus forming a dynamic equilibrium; no such dynamic behavior is expected of H2, which is a feed gas, and, as such, its concentration within the system does not change. Once the system starts cooling, water production decreases (or ceases altogether) while water molecules forming cluster bridges may still evaporate, thus offsetting the dynamic equilibrium and eventually resulting in the disintegration of the agglomerates. Therefore, the latter mechanism can account for all the observed experimental behavior of the system, whereas the former two may only potentially serve as auxiliary mechanisms.
Conclusions
The observed enhanced activity of Cu4 nanoassemblies, compared with individual clusters as reported in ref. 10 and 11, could be due to the higher oxidation state of the Cu clusters (an average oxidation state of 0.7 was determined) or the synergistic/proximity effect from the close lying clusters within the nanoassemblies.45,46 The assemblies fall apart gradually during cool-down with a simultaneous rise in the oxidation state of the particles. At high temperatures, due to the increased activity of the clusters, there would be a rise in the local concentration of H2O which plays an active role in keeping the clusters from coalescing. The stability of the individual clusters within the assemblies could be substantiated from the ab initio calculation in two possible ways: (i) weak H⋯O bond formation between adjacent clusters instead of metallic bond formation (which would lead to coalescence) and (ii) water molecules forming hydrogen bond bridges between clusters keeping them in proximity but separated. This is consistent with the fact that the local concentration of H2O drops during the cool-down ramp and the clusters gradually fall off, thus returning back to sub-nm size particles. The cluster nanoassemblies presumably reverse to their original size components, as the activity is found to be reversible on double ramp measurements.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work at Argonne was supported by the US Department of Energy, BES Materials Sciences under Contract DEAC02-06CH11357 with U Chicago Argonne, LLC, operator of Argonne National Laboratory. The work at the Advance Photon Source (beamline 12-ID-C) was supported by the US Department of Energy, Scientific User Facilities under Contract DEAC02-06CH11357 with U Chicago Argonne, LLC, operator of Argonne National Laboratory. The work performed at OIST was supported by the Okinawa Institute of Science and Technology Graduate University. The authors are also grateful to Mr Sakis Varkas for providing the back cover art photograph.Notes and references
- G. A. Olah, G. K. Prakash and A. Goeppert, J. Am. Chem. Soc., 2011, 133, 12881–12898 CrossRef CAS PubMed.
- T. R. Karl and K. E. Trenberth, Science, 2003, 302, 1719–1723 CrossRef CAS PubMed.
- A. G. George A. Olah and G. K. Surya Prakash, Beyond Oil and Gas: The Methanol Economy, 2nd, Updated and Enlarged Edition, Wiley-VCH, Weinheim, Germany, 2011 Search PubMed.
- A. Halder, M. Kilianová, B. Yang, E. C. Tyo, S. Seifert, R. Prucek, A. Panáček, P. Suchomel, O. Tomanec, D. J. Gosztola, D. Milde, H.-H. Wang, L. Kvítek, R. Zbořil and S. Vajda, Appl. Catal., B, 2018, 225, 128–138 CrossRef CAS.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- J. A. Rodriguez, P. Liu, D. J. Stacchiola, S. D. Senanayake, M. G. White and J. G. Chen, ACS Catal., 2015, 5, 6696–6706 CrossRef CAS.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B.-L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, Science, 2012, 336, 893–897 CrossRef CAS PubMed.
- C. Baltes, S. Vukojević and F. Schüth, J. Catal., 2008, 258, 334–344 CrossRef CAS.
- J. Graciani, K. Mudiyanselage, F. Xu, A. E. Baber, J. Evans, S. D. Senanayake, D. J. Stacchiola, P. Liu, J. Hrbek, J. F. Sanz and J. A. Rodriguez, Science, 2014, 345, 546–550 CrossRef CAS PubMed.
- B. Yang, C. Liu, A. Halder, E. C. Tyo, A. B. F. Martinson, S. Seifert, P. Zapol, L. A. Curtiss and S. Vajda, J. Phys. Chem. C, 2017, 121, 10406–10412 CrossRef CAS.
- C. Liu, B. Yang, E. Tyo, S. Seifert, J. DeBartolo, B. von Issendorff, P. Zapol, S. Vajda and L. A. Curtiss, J. Am. Chem. Soc., 2015, 137, 8676–8679 CrossRef CAS PubMed.
- E. C. Tyo and S. Vajda, Nat. Nanotechnol., 2015, 10, 577 CrossRef CAS PubMed.
- Z. Weng, Y. Wu, M. Wang, J. Jiang, K. Yang, S. Huo, X.-F. Wang, Q. Ma, G. W. Brudvig, V. S. Batista, Y. Liang, Z. Feng and H. Wang, Nat. Commun., 2018, 9, 415 CrossRef PubMed.
- E. L. Uzunova, N. Seriani and H. Mikosch, Phys. Chem. Chem. Phys., 2015, 17, 11088–11094 RSC.
- P. C. P. Caldas, J. M. R. Gallo, A. Lopez-Castillo, D. Zanchet and J. M. C. Bueno, ACS Catal., 2017, 7, 2419–2424 CrossRef CAS.
- B. Yoon, H. Häkkinen, U. Landman, A. S. Wörz, J.-M. Antonietti, S. Abbet, K. Judai and U. Heiz, Science, 2005, 307, 403–407 CrossRef CAS PubMed.
- M. Nesselberger, S. Kunz, F. F. Schweinberger, K. Schlögl, K. J. J. Mayrhofer, U. Heiz and M. Arenz, ECS Trans., 2013, 50, 1333–1338 CrossRef.
- T. W. Hansen, A. T. DeLaRiva, S. R. Challa and A. K. Datye, Acc. Chem. Res., 2013, 46, 1720–1730 CrossRef CAS PubMed.
- Z. Luo, A. W. Castleman and S. N. Khanna, Chem. Rev., 2016, 116, 14456–14492 CrossRef CAS PubMed.
- S. Lee, M. Di Vece, B. Lee, S. Seifert, R. E. Winans and S. Vajda, ChemCatChem, 2012, 4, 1632–1637 CrossRef CAS.
- S. Lee, M. Di Vece, B. Lee, S. Seifert, R. E. Winans and S. Vajda, Phys. Chem. Chem. Phys., 2012, 14, 9336–9342 RSC.
- B. Y. Xia, W. T. Ng, H. B. Wu, X. Wang and X. W. Lou, Angew. Chem., Int. Ed., 2012, 51, 7213–7216 CrossRef CAS PubMed.
- C. J. Fullerton and R. L. Jack, J. Chem. Phys., 2016, 145, 244505 CrossRef PubMed.
- Y. Lei, F. Mehmood, S. Lee, J. Greeley, B. Lee, S. Seifert, R. E. Winans, J. W. Elam, R. J. Meyer, P. C. Redfern, D. Teschner, R. Schlögl, M. J. Pellin, L. A. Curtiss and S. Vajda, Science, 2010, 328, 224–228 CrossRef CAS PubMed.
- S. Lee, B. Lee, S. Seifert, S. Vajda and R. E. Winans, Nucl. Instrum. Methods Phys. Res., Sect. A, 2011, 649, 200–203 CrossRef CAS.
- C. Yin, E. Tyo, K. Kuchta, B. V. Issendorff and S. Vajda, J. Chem. Phys., 2014, 140, 174201 CrossRef CAS PubMed.
- S. Duffe, N. Grönhagen, L. Patryarcha, B. Sieben, C. Yin, B. von Issendorff, M. Moseler and H. Hövel, Nat. Nanotechnol., 2010, 5, 335 CrossRef CAS PubMed.
- M. Di Vece, S. Palomba and R. E. Palmer, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 073407 CrossRef.
- J. Yoshihara, S. C. Parker, A. Schafer and C. T. Campbell, Catal. Lett., 1995, 31, 313–324 CrossRef CAS.
- B. Buesser and S. E. Pratsinis, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 103–127 CrossRef CAS PubMed.
- P. Grammatikopoulos, E. Toulkeridou, K. Nordlund and M. Sowwan, Model. Simul. Mater. Sci. Eng., 2015, 23, 015008 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396–1396 CrossRef CAS.
- A. Stroppa, D. Di Sante, S. Horiuchi, Y. Tokura, D. Vanderbilt and S. Picozzi, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 014101 CrossRef.
- J. Ireta, J. Neugebauer and M. Scheffler, J. Phys. Chem. A, 2004, 108, 5692–5698 CrossRef CAS.
- F. Sim, A. St. Amant, I. Papai and D. R. Salahub, J. Am. Chem. Soc., 1992, 114, 4391–4400 CrossRef CAS.
- K. Laasonen, F. Csajka and M. Parrinello, Chem. Phys. Lett., 1992, 194, 172–174 CrossRef CAS.
- M. Nolan and S. D. Elliott, Phys. Chem. Chem. Phys., 2006, 8, 5350–5358 RSC.
- S. Asbrink and L.-J. Norrby, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1970, 26, 8–15 CrossRef CAS.
- O. Markovitch and N. Agmon, J. Phys. Chem. A, 2007, 111, 2253–2256 CrossRef CAS PubMed.
- J. C. Matsubu, V. N. Yang and P. Christopher, J. Am. Chem. Soc., 2015, 137, 3076–3084 CrossRef CAS PubMed.
- W. Karim, C. Spreafico, A. Kleibert, J. Gobrecht, J. VandeVondele, Y. Ekinci and J. A. van Bokhoven, Nature, 2017, 541, 68 CrossRef CAS PubMed.
- M. Nesselberger, M. Roefzaad, R. Fayçal Hamou, P. Ulrich Biedermann, F. F. Schweinberger, S. Kunz, K. Schloegl, G. K. H. Wiberg, S. Ashton, U. Heiz, K. J. J. Mayrhofer and M. Arenz, Nat. Mater., 2013, 12, 919 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nr06664g |
| ‡ Equally contributing authors: AH and JK. |
| This journal is © The Royal Society of Chemistry 2019 |