
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly adjustable 3D nano-architectures and chemistries via assembled 1D biological templates†
Jacqueline F.
Ohmura
 ab,
F. John
Burpo
ab,
Chamille J.
Lescott
a,
Alan
Ransil
cb,
Youngmin
Yoon
d,
William C.
Records
ae and
Angela M.
Belcher
*abc
ab,
F. John
Burpo
ab,
Chamille J.
Lescott
a,
Alan
Ransil
cb,
Youngmin
Yoon
d,
William C.
Records
ae and
Angela M.
Belcher
*abc
aDepartments of Biological Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, 76-561, Cambridge, Massachusetts 02139, USA
bKoch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, 77 Massachusetts Avenue, 76-561, Cambridge, Massachusetts 02139, USA
cDepartments of Materials Science and Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, 76-561, Cambridge, Massachusetts 02139, USA
dDepartments of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, 76-561, Cambridge, Massachusetts 02139, USA
eDepartments of Chemical Engineering, and Massachusetts Institute of Technology, 77 Massachusetts Avenue, 76-561, Cambridge, Massachusetts 02139, USA
First published on 19th December 2018
Abstract
Porous metal nanofoams have made significant contributions to a diverse set of technologies from separation and filtration to aerospace. Nonetheless, finer control over nano and microscale features must be gained to reach the full potential of these materials in energy storage, catalytic, and sensing applications. As biologics naturally occur and assemble into nano and micro architectures, templating on assembled biological materials enables nanoscale architectural control without the limited chemical scope or specialized equipment inherent to alternative synthetic techniques. Here, we rationally assemble 1D biological templates into scalable, 3D structures to fabricate metal nanofoams with a variety of genetically programmable architectures and material chemistries. We demonstrate that nanofoam architecture can be modulated by manipulating viral assembly, specifically by editing the viral surface coat protein, as well as altering templating density. These architectures were retained over a broad range of compositions including monometallic and bi-metallic combinations of noble and transition metals of copper, nickel, cobalt, and gold. Phosphorous and boron incorporation was also explored. In addition to increasing the surface area over a factor of 50, as compared to the nanofoam's geometric footprint, this process also resulted in a decreased average crystal size and altered phase composition as compared to non-templated controls. Finally, templated hydrogels were deposited on the centimeter scale into an array of substrates as well as free standing foams, demonstrating the scalability and flexibility of this synthetic method towards device integration. As such, we anticipate that this method will provide a platform to better study the synergistic and de-coupled effects between nano-structure and composition for a variety of applications including energy storage, catalysis, and sensing.
Introduction
Porous metal frameworks, have made significant contributions to a diverse set of key technologies including aerospace,1 separation and filtration,2,3 energy,4,5 catalysis6–8 sensing,9,10 and health11 for the past century.12 This is due to several distinct properties of porous metal frameworks including good permeating selectivity, thermal/sound insulation, high surface area, low density, and excellent electrical conductivities.1,4,13,14While synthetic methods have been developed and commercialized for structural porous metals, finer control over nano and microscale features must be gained to reach the potential of 3D metal frameworks in energy, catalytic, and sensing applications.1,13,14 Methods including casting, foaming, and compaction sintering are the most mature approaches for porous metal framework synthesis. These methods enable expedited, low cost synthesis of bulk porous metal, yet exhibit poor structural control below the mm scale. While suitable for structural applications, the narrow range of synthetically controlled parameters as well as large pore sizes are lacking for the design and fabrication of metal frameworks for energy, catalytic, and sensing applications.
As a result, synthetic methods are being developed to enable such structural control. These methods fall mainly into two categories: phase removal and templated growth.1,13–16 Phase removal methods, such as milling, intrinsically require energy intensive post processing, expensive fabrication facilities, and can alter the surface chemistry of the material. Alternatively, resulting pore features in methods such as dealloying are limited in size and chemical scope due to the dependency on manipulations at material interfaces. Templated methods, on the other hand, allow for nano to microscale pore control without these constraints. While some material templates require expensive or energy intensive equipment,9,17 biological templates have the potential to eliminate these drawbacks. Under mild conditions of standard temperature and pressure, biological organisms can form templates in a wide range of complex shapes extending from the nano to mesoscale.18,19 Additionally, the tight genotype–phenotype link of most biological organisms enables tunable surface chemistries to control material nucleation and growth.19–24
To date, biological principles have been successfully utilized in materials synthesis. While bioinspired materials apply biological principles to traditional synthetic approaches,25–27 biotemplated materials take additional advantage of the unique properties of biological components incorporating them into synthesis protocols. Thus, bio-templating can leverage the chemical motifs of biology,18,27,28 and, in some cases, the unique structures of biology30–37 in the fabrication of high performing materials.
While technologies, such as protein engineering, have demonstrated the ability to create complex hierarchical structures with biotemplates, biotemplated structures do not typically demonstrate the scale required to produce materials for device integration nor the breadth of architectural and material options required for device development. With respect to scale, concentration of assembled structures and macroscopic dimensions of the final assembled materials often fall short of enabling even mm scale production.38–41 Thus, to optimize and integrate biotemplated materials into devices, biological components are typically templated as single nanowires in solvent suspensions, then processed into a desired form factor. Binder addition, lyophilization, or carbonization of filler materials are two examples of such processes.35,42–44 To achieve desired structures without significant post processing, micron level patterning such as 3D printing45,46 and fiber spinning47,48 have been utilized. While these patterning approaches have enabled the design of excellent structural materials, the feature sizes of these materials do not display the nanoscale control desired for 3D electrode design and do not fully realize the potential of biological components to assemble into 3D constructs without the aid of specialized machinery. Furthermore, the genetic component of biological templates to produce a range of architectures from the same templating process as well as versatility of a 3D biotemplate to extend across multiple, tunable material chemistries has yet to be fully leveraged. Thus, our interest is in the assembly of nano and microscale biotemplates for architectural control in scalable 3D metallic nanofoams.
With respect to biotemplate selection, the form factor of the M13 phage, an fd virion with a simple cylindrical structure of 930 nm × 6.5 nm, is an ideal building block for higher order 3D structures. This virion is composed of 2700 copies of the main p8 coat protein surrounding a single stranded DNA backbone. The ends of the M13 phage are capped with 5 copies of each of 4 minor coat proteins. The well characterized genotype–phenotype link between the genetic code and structure of the coat proteins is well documented enabling facile modifications to the surface of the M13 phage.49 These modifications have been demonstrated to endow M13 mutants with the ability to nucleate material growth as well as bind to specific ions or material surfaces.21,42 As such, the M13 was the main template utilized in this study.
In this study we demonstrate a scalable, biologically assisted synthetic method for the fabrication of nanoporous metal foams which exhibit control over multiple forms of nano to microscale architectural features. Additionally, we demonstrate that this synthetic method can be extended to fabricate metal foams with tunable chemistries across a wide range of catalytically relevant elements. While gluataraldehyde crosslinking of biological templates as well as metal deposition onto individual biological templates has been carried out in previous studies,29,45 we carry out a mechanistic study for the application of an architecturally adaptable porous metal nanofoam. Knowledge gleaned is then leveraged to show that protein engineering in tandem with other biologically controllable handles, such as variation in template density, can be utilized to dramatically alter the nano to micro architectures in biotemplated materials. We provide characterization of strut thickness, strut crossover density, and overall morphology of these materials via SEM, TEM, and image analysis to quantify and confirm structural control. We next establish that the architecture endowed from 3D whole organism assembled templates can be extended across a variety of material chemistries via electroless deposition including monometallic and bimetallic combinations of each nickel, copper, cobalt, and gold as well as phosphorous and boron doped nickel while retaining template structure. X-ray diffraction, EDS mapping, TEM, and SEM characterization is presented to confirm architecture retention as well as the core–shell nature and/or tuneability of each element in bimetallic nanofoams. Interestingly, our results not only revealed an increase in surface area, but also a decrease in average crystallite size in biotemplated metals. Finally, we demonstrate the scalability of M13 templated metallic nanofoams. In addition to cm scale free standing metallic foams, biotemplated foams were integrated into an array of substrates common to device driven applications. We believe this scalability in tandem with increased surface area and decrease in average crystallite size show great potential in the application of biotemplated metal foams toward integration and performance in energy and catalytic applications.
Experimental
Growth of bacteriophage
10 mL of bacterial culture (E. coli K12 ER2738) and 1 mL of phage infected bacterial culture were added to 1 L of LB in a 2 L Erlenmeyer flask. The flask was rotated at 225 rpm at 37 °C for 20 hours. The culture was then centrifuged at 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rcf for 10–20 minutes. Supernatant was mixed volumetrically with polyethylene glycol (PEG)–NaCl solution (200 g of PEG 8000 and 146 g of sodium chloride per liter of water) in a 5:1 ratio and was refrigerated at 4 °C for one to two days. After centrifugation at 11000 rcf for 30 minutes, supernatant was discarded, and the white phage pellet was resuspended in Millipore deionized water. The resuspended phage was transferred to 30 mL centrifuge tubes where a 5:1 ratio of supernatant to PEG–NaCl was added again, followed by a two hour incubation in ice and centrifugation at 11000 rcf for 30 minutes. The final phage pellet resulting from this step was resuspended in DI water and stored at 4 °C. Phage was also amplified utilizing a GE wave bioreactor system 20/50 EHT in a 10 L Cellbag (CB0020L10-31). 100 mL of both bacterial and phage infected bacterial cultures were used at the start of the amplification process. Additionally, 20 mM of MgCl2 was added to the LB media. Phage was amplified for 20 hours during which oxygen was set at maximum input (50%). Prior to purification, 30 mM of EDTA was added to the culture to prevent precipitation of MgCl2 and placed in a cold room overnight. Bacteria was filtered via 1.2 μm filter from Sartopure PP2 Maxicaps (5591303P1—SS) and 1 μm 10 in long GE Health care ULTA Capsule GF filter (KGF-A-0110FF) in series. Phage was then purified from LB media via water buffer exchange column utilizing a hollow fiber cartridge (56-4102-43) concentrating the volume of phage solution to 1 L. One round of PEG–NaCl purification as described previously was then carried out to further purify the phage, followed by a purification supplement.
000 rcf for 10–20 minutes. Supernatant was mixed volumetrically with polyethylene glycol (PEG)–NaCl solution (200 g of PEG 8000 and 146 g of sodium chloride per liter of water) in a 5:1 ratio and was refrigerated at 4 °C for one to two days. After centrifugation at 11000 rcf for 30 minutes, supernatant was discarded, and the white phage pellet was resuspended in Millipore deionized water. The resuspended phage was transferred to 30 mL centrifuge tubes where a 5:1 ratio of supernatant to PEG–NaCl was added again, followed by a two hour incubation in ice and centrifugation at 11000 rcf for 30 minutes. The final phage pellet resulting from this step was resuspended in DI water and stored at 4 °C. Phage was also amplified utilizing a GE wave bioreactor system 20/50 EHT in a 10 L Cellbag (CB0020L10-31). 100 mL of both bacterial and phage infected bacterial cultures were used at the start of the amplification process. Additionally, 20 mM of MgCl2 was added to the LB media. Phage was amplified for 20 hours during which oxygen was set at maximum input (50%). Prior to purification, 30 mM of EDTA was added to the culture to prevent precipitation of MgCl2 and placed in a cold room overnight. Bacteria was filtered via 1.2 μm filter from Sartopure PP2 Maxicaps (5591303P1—SS) and 1 μm 10 in long GE Health care ULTA Capsule GF filter (KGF-A-0110FF) in series. Phage was then purified from LB media via water buffer exchange column utilizing a hollow fiber cartridge (56-4102-43) concentrating the volume of phage solution to 1 L. One round of PEG–NaCl purification as described previously was then carried out to further purify the phage, followed by a purification supplement.
Supplemental purification
Amplified clones were further purified via addition of 4 mg ml−1 DNase (from a 2000× stock solution) along with standard 1× Dnase buffer (2.5 mM MgCl2, 10 mM Tris-HCl, 0.5 mM CaCl2, at pH 7.6@25 °C). Samples were heated to 37 °C for 1 hour before being placed at 4 °C for 48 h. Samples were dialyzed with a 100 kDa dialysis filter and dialyzed against 6 mM EDTA, 1 mM EDTA, and 0.25 mM EDTA in succession over the course of 48 hours. Representative AFM images of protein preparations before and after this additional purification can be found in the supplement, section 1. Phage concentration was then measured via UV-vis absorbance in the units of plaque forming units (pfu) per milileter.50Nanofoam fabrication
Phage templates were prepared via glutaraldehyde crosslinking. 10 μL of 2.5 × 1012 to 50 × 1013 pfu mL−1 of phage was deposited onto a substrate of choice typically a 1 cm × 1 cm silicon wafer, stainless steel mesh, strip of carbon paper, or silica filter. The support structure was then inverted onto the well of a standard 12 well plate filled with 0.2 mL 50% glutaraldehyde. In the case of silica, the support structure was first submerged into a solution of 5 × 1012 pfu mL−1 then removed and submerged into glutaraldehyde. After remaining in contact with glutaraldehyde for 20 minutes, the samples were rinsed in 100 mL of Millipore water and transferred to 4 L of DI water for 12–24 hours of dialysis. Templates were then sensitized with a minimum of 100 μL of 10 mM tetraamine palladium chloride per phage gel for 4–8 hours followed by overnight dialysis. Templates were then placed in metal specific electroless deposition solutions from 9–20 minutes for cobalt nanofoams, 20 minutes to 2 hours for nickel nanofoams, and 2.5 to 6 hours for copper nanofoams depending on desired nanowire thickness. Clones exhibiting glutamic acid rich sequences have previously demonstrated great cation affinity.51 As such, nanofoam fabrication was initially carried out with the clone EEAE to improve the adhesion of the platinum sensitizer to the virus coat. As this study is focused on scalability, we selected EEAE over the clones EEE or EEEE due to the stability of the EEAE insert in the M13 construct. Further investigation, however, demonstrated that glutamic acid enrichment was not required for successful sensitization and that, in the absence of divalent salt, nanofoam architectures remained consistent across M13 mutants containing sequences other than EEAE (Fig. 2 panels c and e, Fig. S2†). Nonetheless, the clone EEAE is also distinguished by high yield and minimally invasive growth in the laboratory. For this reason, unless further specified, all nanofoams presented in the text are synthesized with the M13 phage clone EEAE.Metal deposition solutions
Deposition solutions were prepared from 10× stock solutions of 1 M, pH 7 MOPS; 0.4 M EDTA; and 0.67 to 1 M dimethyl amine borane. Immediately before use, each deposition solution was prepared to the following specifications specific to the desired deposition metal. In each case, the solutions were made by adding the components in the following order: buffer, water, metal salt, and reducing agent. This order significantly increases stability of the deposition solutions. Copper ELD solution was composed of 0.032 M CuSO4, 0.040 M EDTA, 0.1 M MOPS (3-(N-morpholino)propanesulfonic acid), 0.067–0.1 M dimethylamine borane. Nickel ELD solution was composed of 0.032 M NiSO4, 0.064 M sodium D-lactate, 0.1 M MOPS, and 0.067–0.1 M reducing agent (dimethylamine borane or sodium hypophosphite). Cobalt ELD solution was composed of 0.032 M CoSO4, 0.064 M lactate, 0.1 M MOPS, 0.067 M dimethylamine borane.Addition of second metal
Au was added to preformed nickel or cobalt nanowires via immersion of templated nanonetworks into 0.01 M NaAuCl4·H2O, 0.32 M Na2SO3, 0.08 M Na2S2O3·5H2O at pH 9. This solution was pH balanced after the addition of the metal ions to avoid precipitation. Gold was deposited onto copper samples via galvanic displacement. Upon completion of copper templating, the nanonetwork was rinsed on a shaker for 20 minutes, then subsequently immersed in 0.5 mL of 10 mM gold chloride for up to 30 minutes (alternative gold concentrations are reported in supplement). Cu–Ni was formed via deposition of copper utilizing a solution from a previous electroless deposition study onto freshly templated and subsequently rinsed nickel nanonetworks.52 The solution contained 0.28 M NaH2PO2, 0.085 M NiSO4, 1.6 mM CuSO4, 0.3 M Na3C6H5O7, and 1 M NH4CH3CO2 balanced to pH 5. 3–10 mL of solution was utilized per sample. Deposition time ranged from 20 minutes to 2 hours. To achieve highest copper incorporation two successive 2 hour treatments of copper deposition were required.Electron microscopy
Samples comprised of nanowires or phage bound nanoparticles in water solution were imaged via transmission electron microscopy (TEM, FEI Technai G2 Spirit TWIN system and JEOL 2010). Nanofibers were prepared on carbon film on 400 mesh copper grids by dropping 10 μL of solution onto the grid for 10 minutes before wicking off the extra fluid. Grids were then washed 5–8 times by dropping 10 μL of liquid on the surface of the grid and wicking off the fluid immediately. Samples were imaged at 120 kV beam acceleration voltage.Samples composed of thin 3D nanowire network films were characterized via the SEI setting of a 6700 JEOL scanning electron microscope (SEM) at 10 kV. Image analysis was completed via ImageJ plugin Diameter J53 (Fig. 5). All samples were compared at the same magnification and SEM settings. Contrast was adjusted uniformly across all images reported in Fig. 2via ImageJ. Energy dispersive X-ray spectroscopy (EDS/EDX) was performed via a JEOL 5910 general purpose SEM with a Bruker EDX system at 15 kV. Samples were not coated with gold or similar surface treatment prior to imaging. Instead, samples were dried in a desiccator under vacuum or lyophilized prior to imaging. High resolution compositional mapping was performed via Zeiss Merlin high-resolution scanning electron microscope at 15 kV. Contrast was adjusted uniformly across all maps via Photoshop cc 2017. HRTEM and STEM/EDX were performed on a JEOL 2100 FEG transmission electron microscope operating at 200 kV. TEM samples were prepared by physically separating a phage-templated thin film from its substrate, resuspending in acetone, and sonicating in a water bath for two minutes at 480 W. 10 μL of the solution was dropped on carbon mesh on 400 mesh copper grids (Electron Microscopy Sciences) for 5 minutes before wicking off. The grids were then washed by dropping 10 μL of ultrapure water and wicking off the fluid after 1 minute.
X-ray diffraction
X-Ray Diffraction (XRD) was performed via a PANanalytical X'Pert Pro Multipurpose Diffractometer, a conventional 1.8 kW sealed X-ray tube source, using a Cu target (Kα = 1.5418 Å), and a vertical circle theta:theta goniometer with a radius of 240 mm. Data was obtained with 40 mA, 45 kV generator at 80° min−1 scan speeds with 2° of anti-scatter slit, 10 mm beam mask, and 10 mm of irradiated length of automatic mode and 0.04 rad of soller slit. XRD pattern analysis was performed via Highscore plus software “search and match” functionality. The reported XRD data was collected with by employing a multiple position sample changer coupled to a stage that rotates each sample about the surface normal direction at an angular velocity of 16 seconds per rotation. Each sample was prepared on a zero background plate that was inserted into a pre-defined sample holder that fit into the spinner stage. The height variation of the sample holders was addressed by taking measurements of multiple NIST 660b LaB6 standards to ensure the consistency of the XRD data collection. Data was ploted via Origin on a log scale y-axis. All samples were plotted on the same scale, reference patterns were included for clarity of peaks of interest and were scaled to the maximum peak intensity of the pattern of interest.
BET
To make large-scale samples for BET, 32.4 ml of M13 bacteriophage (EEAE clone) at a concentration of 125 × 1012 pfu ml−1 was poured into a 14.5 cm diameter glass dish. 3 ml of 50% Glutaraldehyde was added. The sample was covered and allowed to crosslink for about 21 hours, resulting in a gel. 3 DI baths of 90 ml each were added sequentially to wash glutaraldehyde out of the sample. Due to the thickness of the sample, baths were left for up to 10 hours each. 30 ml of 10 mM Pd(II) tetraamine chloride aqueous solution was added and the sample was allowed to sit overnight. 4 DI baths of 90 ml each were added to wash the sample. These baths were left for up to 2 hours each. 80 ml of Ni Electroless Solution was added, with initial solution temperature at 4 °C. Deposition was allowed to continue for 23 minutes under house vacuum. The resulting nanofoam was scraped from the bottom of the tube and flash frozen using liquid nitrogen, then lyophilized to produce a dry nanowire powder. This sample was placed in BET flask and heated in air to 180 °C for 2 hours, ±2 °C per minute in order to remove any volatile compounds (TGA analysis suggests that this process does not remove the M13 backbone, Fig. S14†). It was then degassed overnight on a Micromeritics ASAP 2020 Surface Area and Porosity Analyzer, with a 60 minutes evacuation time, 20 μm Hg vacuum setpoint, and a 150 °C holding temperature with a 30 minutes hold time. The mass was recorded after degassing and before measurement. The BET measurement was taken using nitrogen gas and liquid nitrogen as the coolant.Double layer capacitance
Double layer capacitance was utilized to determine the surface area of copper nanofoams. Capacitance of 3 bio-templated nanofoams was compared to that of a standard Cu electrode with surface area 0.9 cm2 and AFM measured roughness of 1.01. Conditions utilized during capacitance measurements included an electrolyte of Ar sparaged 1 M NaClO4 and an Hg/HgSO4:KCl reference electrode (0.658 V vs. NHE). Double layer capacitance was measured at 5–50 mV s−1 scan rate at −1.05 V, ±0.05 V. A −1.6 V pre-reduction step was also included to ensure that all measured surfaces were in the reduced oxidation state. After electrochemical measurements, nanofoam masses were determined via Agilent ICP-OES VDV 5100 performed on samples dissolved in 0.5 ml of aqua regia.Results and discussion
Biotemplated electroless deposition mechanism
Nanofoams were fabricated by depositing metal onto assembled M13 clones. To achieve this, M13 phage hydrogel templates were first prepared via glutaraldehyde facilitated crosslinking. Electroless deposition (ELD) was then utilized to deposit metal on the surface of the M13 hydrogels. As a brief introduction, ELD is typically a 4-step process. In the first step, referred to as activation, a template surface is chemically treated to bind a sensitizing agent. Next, during sensitization, the template surface is coated with an activating compound typically called an activator. During activation, and subsequent deposition, the activator is converted to a form which facilitates autocatalytic metal deposition onto the template surface.54,55 Adapting ELD to M13 phage templates, the surface charge of virion's major coat protein, p8, eliminated the need for an activation step. Instead, virus hydrogels were sensitized directly with the positively charged activator, tetraamine palladium chloride. Following sensitization, palladium decorated M13 hydrogels were exposed to the dimethylamine borane based ELD solution (Fig. 1A).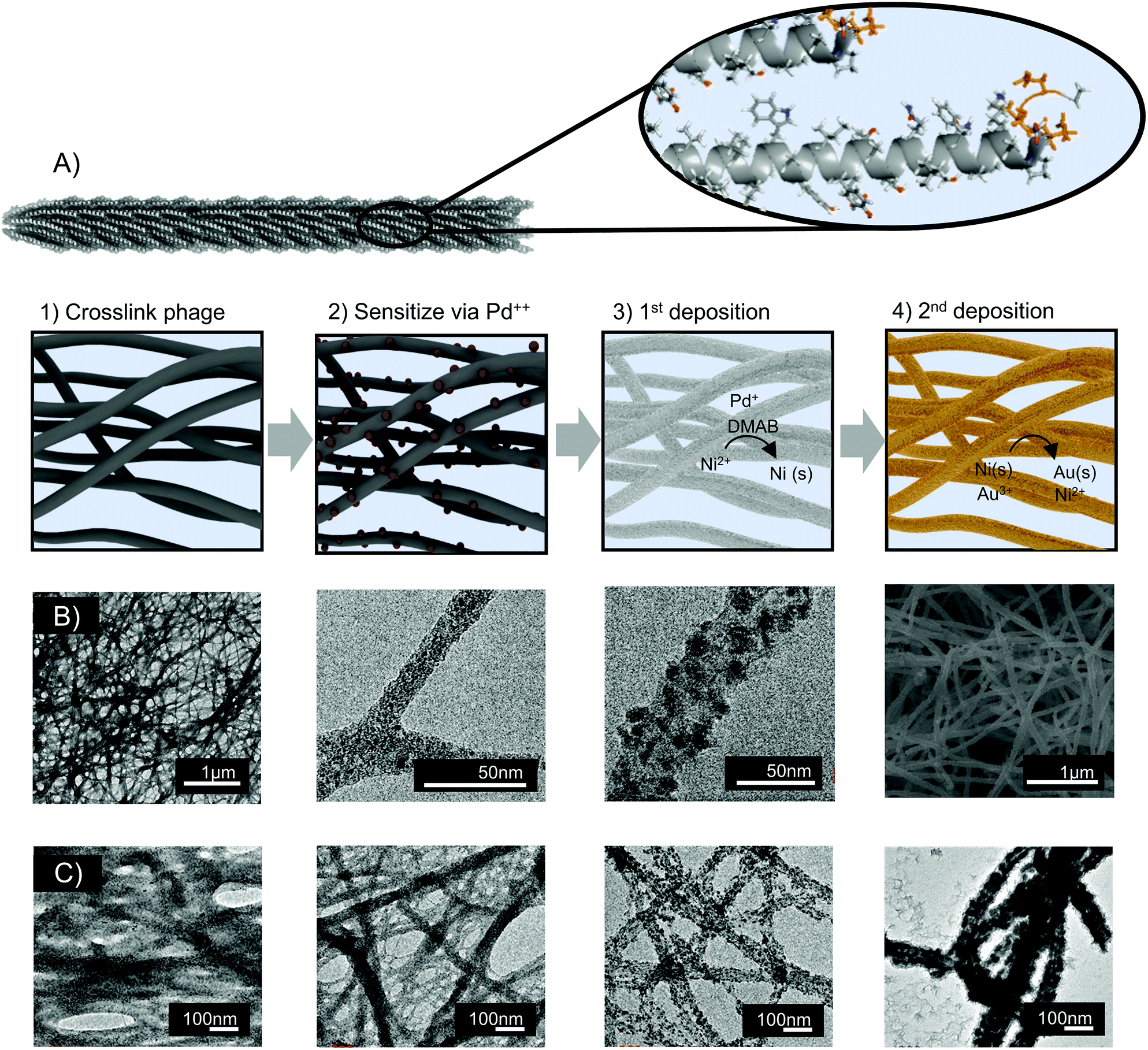 | ||
| Fig. 1 Synthesis of nanofoams utilizing M13 templates (A) rendering of M13, p8 surface protein with glutamic acid rich EEAE N-terminus insert, and synthesis scheme of biotemplated metal nanofoam synthesis (B) TEM micrographs depicting growth mechanism and crosslinked M13 virons, Pd binding, and nickel deposition after 30 seconds of deposition. SEM image of nickel deposition after 20 minutes of deposition (C) the pore formation mechanism is depicted left to right from bare M13 virons crosslinked with gluataldehyde, palladium sensitized phage gels, 30 seconds of deposition, to 18 minutes of deposition. | ||
Time-lapse TEM images were used to probe the mechanism of metal nanofoam growth. Before the introduction of metal ions, the virus hydrogel template was observed to be composed of interconnected fibers of virus bundles (Fig. 1B). During the introduction of palladium ions and the reducing agent, nanoparticles were nucleated onto the surface of these virus bundles. As electroless deposition progressed, the particles ripened and coalesced to form struts in an interconnected nanofoam with controlled final diameter thicknesses from 100–300 nm (Fig. 1B and S3†). Growth and coalescence of adjacent particles were consistent with a typical 2D ELD growth mechanism. In addition to this growth mechanism, the 3D M13 hydrogel templates are characterized by the bundling and coalescence of adjacent virus fibers (Fig. 1C). As a result, this bundling phenomenon is suspected to be the main mechanism by which sample porosity is derived. As the number of phage templates remains constant throughout the deposition process, the bundling of adjacent phage fibers would result in a decrease of observed nanofoam ‘struts’ and increase in fiber to fiber distances. This morphological shift is in fact observed during metal deposition (Fig. 1C).
Architectural design: the role of salts and p8 structure in template architecture
Fiber bundling, observed during metal deposition, was further manipulated to produce a range of nanofoam architectures. M13 phage can be engineered to display affinity towards specific metal ions based on coat protein charge and amino acid character. We leveraged this protein sequence based affinity to tune the degree of virus, and therefore fiber, bundling which resulted in diverse nanofoam architectures.As the clone displaying EEAE on the p8 protein is rich in negative charges, virus bundling can be induced through the addition of divalent cations (Fig. 2A, S3 and S4†). Switching between aggregated and non-aggregated states was observed during addition of calcium or magnesium to purified, highly concentrated EEAE virus solutions (Fig. S3 and S4†). Aggregation was reversible upon addition of a metal chelating agent, disodium EDTA to the solution, a change observed via a turbidity shift in the virus solution from clear to opaque white after salt addition (Fig. 2B). Extending this effect to modulate nanofoam architecture, nanofoams were synthesized from EEAE templates in the presence of Ca++ and Mg++ ions (“free salt”) and separately synthesized in the presence of chelated Ca++ and Mg++ ions (“complexed salt”). EEAE nanofoams synthesized in the “free salt” condition exhibited a very dense architecture (Fig. 2F), while EEAE nanofoams synthesized in the presence of “complexed salt” resulted in structures identical to that of salt free templates (Fig. 1C and 2E).
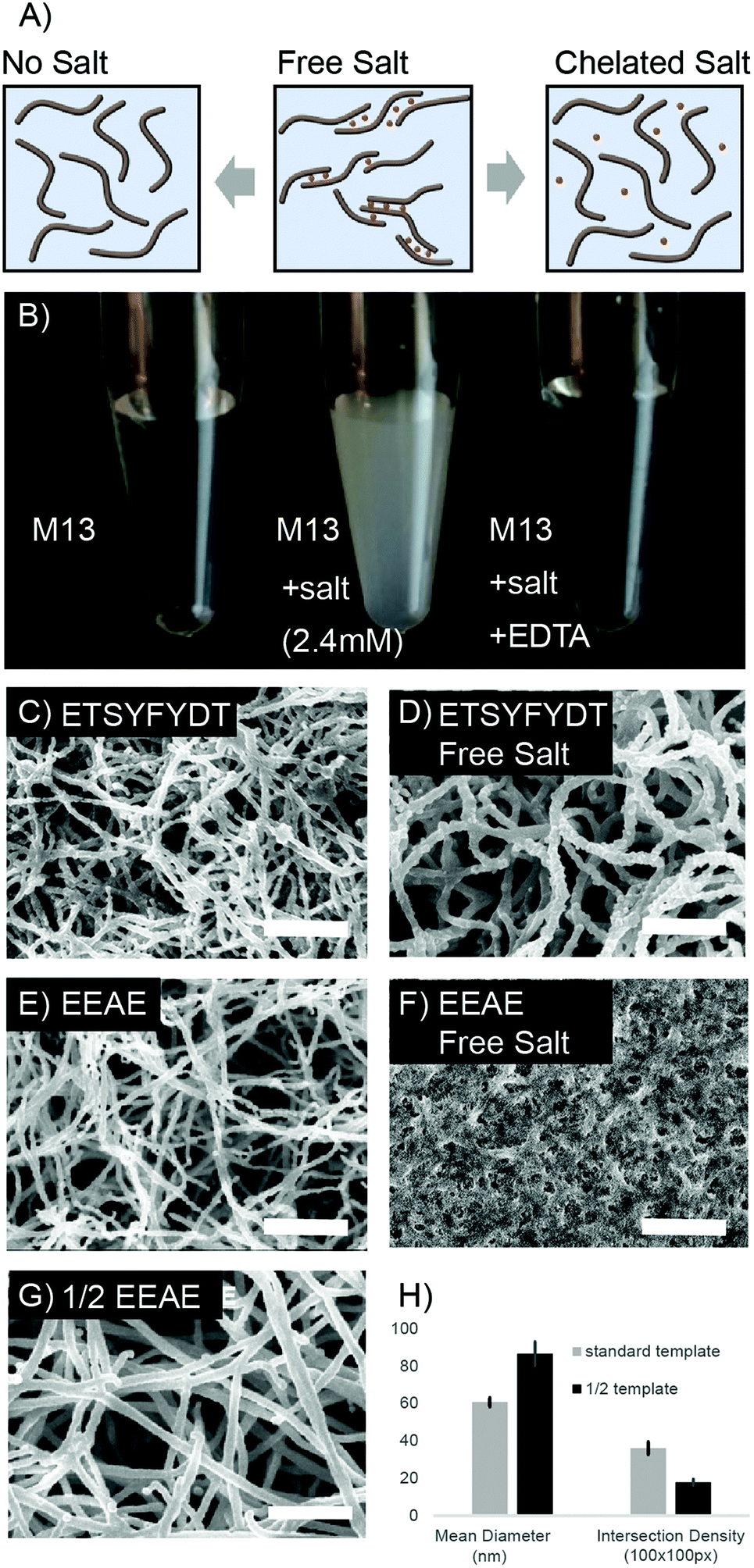 | ||
| Fig. 2 Architectural effects of via surface protein modifications, salt presence, and templating density (A) schematic of M13 coat protein – salt interactions (B) bench photograph of reversible salt aggregation in M13 virons. (C–F) SEM images of nanofoam architectures varied through surface coat modifications in tandem with the presence of free calcium (G) architectural effect of diminishing M13 template concentration to 50%. All other synthetic conditions match that of sample depicted in (E). All scale bars represent 1 micron (H) quantified architectural effects of decreasing M13 template density 50%. | ||
This phenomenon was further extended to an M13 mutant presenting an amino acid sequence less susceptible to salt induced aggregation, “ETSYFYDT”. Design of the ETSYFYDT clone is detailed in the supplement, section 2. While the “complexed salt” ETSYFYDT nanofoams (Fig. 2C) displayed morphologies very similar to that of the “complexed salt” EEAE nanofoams (Fig. 2E), the morphology of the “free salt” E-T-S-Y-D-T nanofoams (Fig. 2D) resulted in a structure differing from that of the “free salt” EEAE nanofoams. Instead, the “free salt” ETSYFYDT nanofoams were characterized by a lower density of thicker nanofoam struts. This could be suggestive of a moderate amount of virion bundling due to salt mediated virion–virion associations as compared to a greater amount of salt mediated virion–virion associations in that of the EEAE nanofoams. This behaviour would be consistent with M13 based aggregation observed in a side-by-side comparison of the EEAE and ETSYFYDT clones in the presence of Mg++ and Ca++ ions (S3 and S4†). As such, these results suggest that the manipulation of M13 clone–clone associations can result in unique architectural variants of bulk assembled M13 based nanofoams.
Architectural design: template density modifies intersection density
While yielding less dramatic architectural changes, the concentration of viruses utilized to fabricate the 3D template also dictated nanofoam structure. Nanofoams templated with concentrations of 20 × 1012 pfu ml−1 and 40 × 1012 pfu ml−1 were quantified in terms of average strut intersections per 100 square pixels and average fiber diameter (Fig. 2G and H). Nanofoams with a lower template concentration, displayed close to half of the intersection density of nanofoam struts as compared to controls with the standard template concentration exposed to ELD solution for the same time period. Additionally, strut diameters in the low concentration samples were larger than the higher concentration conditions. This effect was presumably due to the greater separation between neighboring struts, diminishing the effect of neighboring strut ion diffusion gradients. The variation in strut thickness, however, remains adjustable as strut thickness can be further tuned via exposure time to electroless deposition solution (Fig. S6†).Compositional range and control
A strength of this fabrication technique lies in the decoupling of architecture from chemical composition; as compared to methods such as dealloying,16 where architecture is dependent on composition. Instead, the chemical flexibility of this system was demonstrated over a range of catalytic and energy storage materials of interest including monometallic transition metals as well as bimetallic compositions. In each case, nanofoam architecture of the 3D assembled templates was retained.First, nanofoams of monometallic transition metals nickel, cobalt, and copper were synthesized. The nanofoam structure in each material system was retained and XRD indicated a predominantly amorphous and nano-crystalline structure indicated by peak broadening (Fig. S4†).
Transition metal phosphides and borides have gained increasing attention for their performance in reactions such as the hydrogen evolution reaction.56–60 Typical to metals deposited via electroless deposition, elements incorporated into the reducing agent, typically phosphorous or boron, can be incorporated into the final deposited metal.61 Biological templating demonstrated this phenomenon. Altering the concentration of reducing agent as well as selecting either a phosphorous or boron based reducing agent resulted in control over the final boron or phosphorous content in the nanofoams (Table 1). In each case overall structure was not altered.
| Reducing agent | Concentration | At% B | At% P |
|---|---|---|---|
| DMAB | 32 mM | 0.70 ± 0.04 | — |
| DMAB | 96 mM | 2.56 ± 0.34 | — |
| H2PO2− | 100 mM | — | 7.03 ± 0.15 |
Bimetallic and core–shell nanofoams
Bimetallic materials, particularly combinations of noble and transition metals, have displayed enhancements in applications such as gold–copper for carbon dioxide reduction (CDR),62 gold–metal oxides for heterogeneous oxidation,63 and core–shell morphologies for the oxygen reduction reaction.64 As a result, the synthesis of multi-metallic nanonetworks was explored via the addition of a noble metal, gold, to each of the monometallic transition metals as well as the ability to blend two transition metals onto one nanofoam. Table 2 reports multi-material systems explored as well as the range of mass percent integration of secondary material achieved in this study. Nanofoam architecture was retained in each case (Fig. 3, S8 and S11†). Further examinations of compositional control in these studies indicated the potential to tune final bimetallic ratios over the range depicted in Table 2 (Fig. S11†).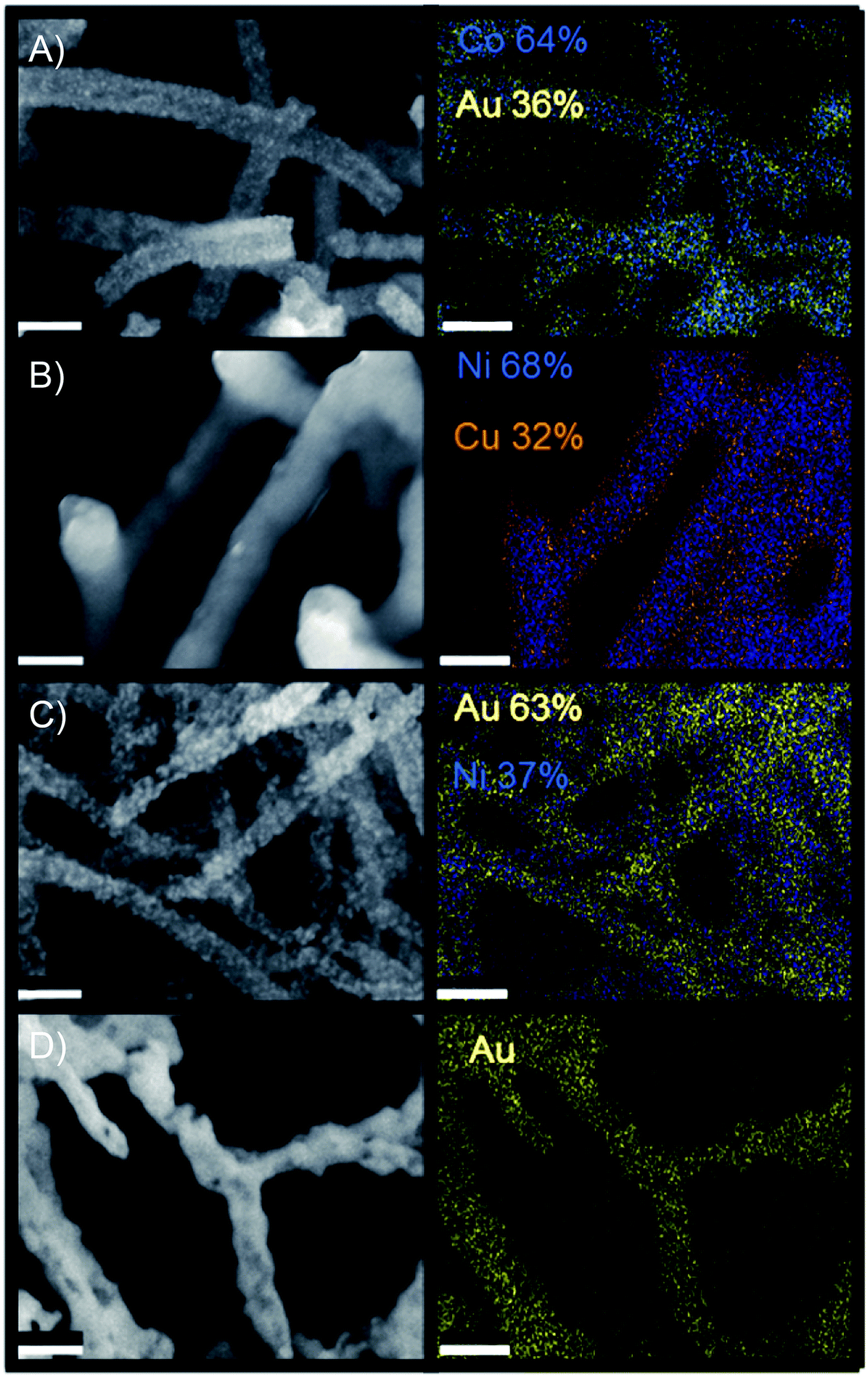 | ||
| Fig. 3 SEM of multimetallic nanofoam structures, scale bar depicts 100 nm (A) cobalt sample after gold deposition treatment (B) nickel sample after copper deposition treatment (C) nickel sample after gold deposition treatment (D) copper sample after gold treatment. | ||
| Metal 1 | Metal 2 | wt% incorporation metal 2 |
|---|---|---|
| Ni | Au | 0–60% |
| Cu | Au | 0–99% |
| Co | Au | 0–63% |
| Ni | Cu | 0–98% |
Of the bimetallic systems explored, Cu–Ni demonstrated the most distinct core shell type morphology. These networks were synthesized via successive electroless deposition of nickel onto the M13 biotemplate followed by copper deposition onto the resulting nickel structure. Elemental mapping (EDS) indicated enrichment of nickel within the nanowire cores (Fig. 3). Core–shell wires were also indicated via TEM analysis (Fig. S10†). Additionally, only separate patterns of nickel and copper were reported via XRD, yielding no evidence of intermetallic Cu–Ni alloys (Fig. S9†).
Au–Co, Au–Ni, and Au–Cu nanofoams were also synthesized. This was achieved via galvanic displacement of gold onto the transition metal templated structures. 3D morphology was retained after the gold treatment using sulfate–thiosulfate solutions to inhibit the uncontrolled formation of large gold nanoparticles in the Co–Au and Ni–Au structures (Fig. S8 and S11†). Unlike the Cu–Ni networks, compositional mapping via EDS (Fig. 4) did not clearly indicate a core shell structure, however, a gold enrichment at the nanowire surfaces was indicated via TEM line scan (Fig. S10†). XRD indicated the formation of gold crystals within the structure (Fig. S9†). This suggests that the structure is bimetallic with individual gold domains preferentially distributed throughout the nanowire surface as opposed to a distinct core shell structure. Additional notes on the comparison between the gold and copper deposited bimetallic materials is reported in supplement section 6.
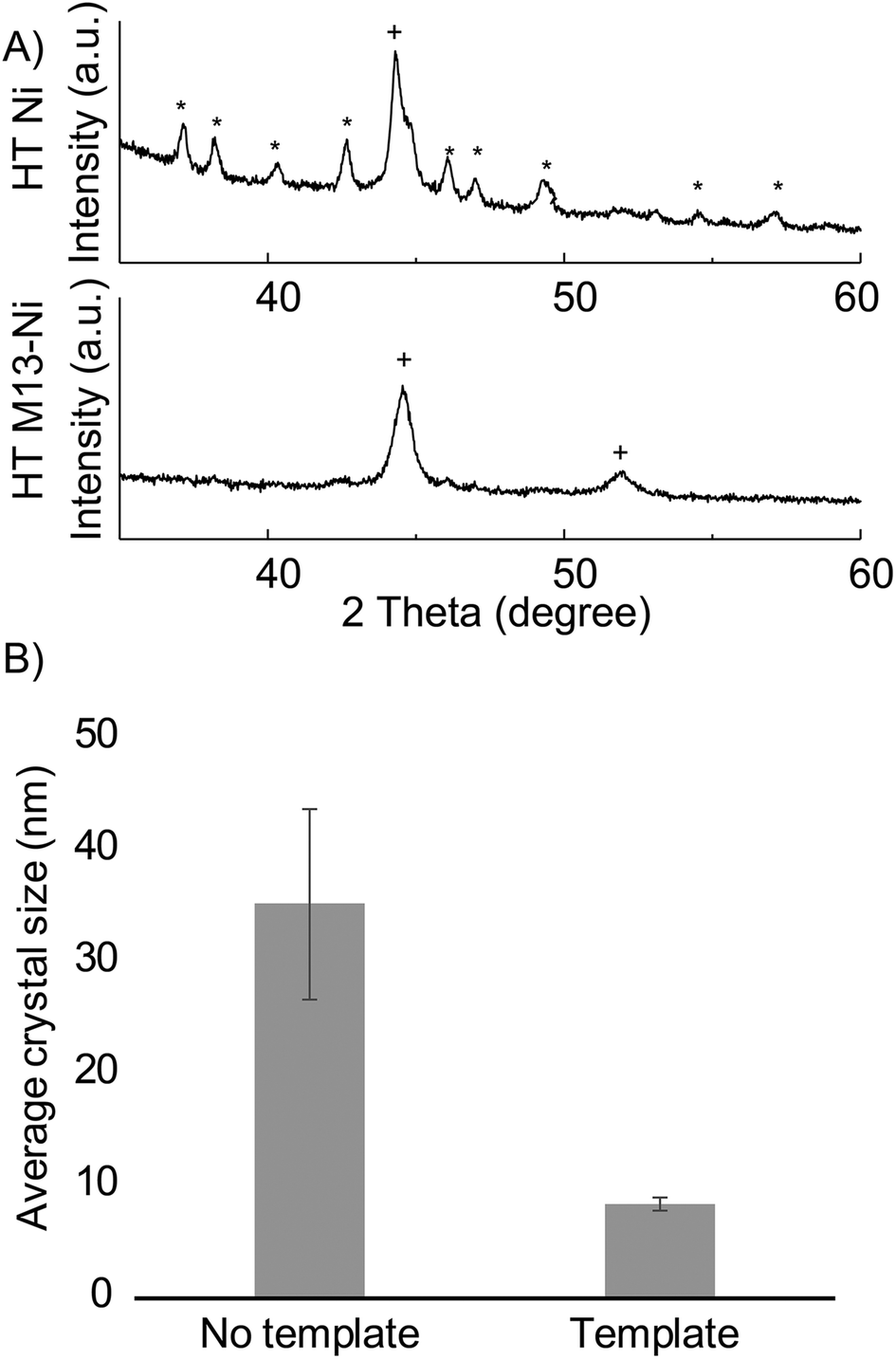 | ||
| Fig. 4 (A) XRD patterns of templated vs. non-templated electroless nickel each deposited for 20 minutes. Ni3B peaks are labeled with * according to 04-005-4901 reference pattern, Ni peaks are labeled with + according to 04-10-6148 (B) comparison of average crystal size in templated vs. non-templated electroless nickel post heat treatment associated crystal planes are detailed in the supplement. | ||
Material microstructure and properties are enhanced by templating process
In addition to retaining structure independent of deposited material, this synthetic method enables material modifications on a crystallographic level. Templated samples exhibited an altered phase composition and increased surface area as compared to non-templated samples. We anticipate that these modifications will be advantageous to nanofoam utilization as increased defect concentrations, such as grain boundaries, have been correlated to higher catalytic activity,65 and enhanced surface area is desirable for increased sensitivity and space economy in application utilization.During annealing at 300 °C, non-templated (control) nickel crystalized into, NiB3 and face centered cubic Ni. Templated nickel, however, exhibited significant reduction the formation of NiB3 – to the point of almost complete suppression (Fig. 4A, S12†). Additionally, Scherrer analysis revealed smaller crystal sizes in that of bio-templated nickel nanofoams as compared to non-templated samples (Fig. 4B and Table S1†). The observed microstructural material changes between templated and non-templated nickel may be the result of altered diffusion within the planar 2D nickel in the non-templated plane as compared to 1D diffusion within in the strut of the nanofoam architecture.
Geometric surface area is typically utilized as the representative surface area of electroless and electro deposited materials.39 This synthetic method increased surface area over a factor of 50 as compared to the geometric surface area of a 2D template of the same footprint. Specific surface area of nanofoams also exhibited significant gains compared to that of other templated 3D structures, and is in agreement with the theoretical surface area for smooth templated wires of comparable diameters (ESI section 7†). Copper nanofoams indicated 5.1 ± 1.1 m2 g−1via double layer capacitance and nickel nanofoams demonstrated 6.2 m2 g−1 measured via BET (Fig. S13–S15†) as compared to specific surface areas below 0.1 m2 g−1 for comparable, yet coarser, templated architectures.5,66
Suitability for device integration
The ability of bio-templated porous metal to adhere to a variety of surface chemistries as well as conform to unique shapes on the macro scale is paramount in the ability to utilize this synthetic method towards a plethora of applications including energy storage and catalysis. As a result, we demonstrated that biotemplated porous metals can be integrated into a variety of structures. While characterized foams were synthesized as thin films on a silicon wafer or FTO glass, the biotemplated porous metals were also successfully deposited into the pores of stainless steel meshes, into the pores of larger commercial metallic nanofoams, into carbon paper, and on glassy carbon electrodes (Fig. 5). Beyond support integration, the process is scalable to create free-standing cm scale nanofoam structures (Fig. 5E, S16 and S17†). | ||
| Fig. 5 Versatility of nanofoam synthesis SEM images of (A) free standing nickel nanofoam (B) and SEM of M13 nanofoam microstructure for comparison to nanofoams grown in (C) stainless steel commercial foam, (D) carbon paper, (E) a steel mesh, (F) and silica filter. | ||
Conductivity of nanofoams was measured via four point probe measurements (ESI, section 11†). Both nickel and copper nanofoam films demonstrated conductivities suited for electrode functionalities. Nanowires above 100 nm displayed resistance measurements ranging from 5.8 × 10−6 to 1.07 × 10−6 Ωm, similar to that of graphite parallel to the basal plane (Tables S2–S4†). The resistance values were about one order of magnitude greater than bulk copper and nickel. This is likely due to the decreased cross-sectional metal area as well as decreased crystallite size and therefore increased grain boundaries within the nanowires (Fig. 4).
Finally, M13 templated metal nanofoams were demonstrated as glycerol oxidation catalysts. The low cost of nickel in comparison to the expensive noble metals typically used in electrocatalysis provides an appealing option as a catalyst material for glycerol oxidation.67 To this end, the activity of phage derived nickel nanofoams exceeded that of non-templated controls (S 18). Furthermore, modifying M13 templating density, and therefore nanofoam strut crossover density, altered electrochemical behavior of the nanofoams. Each templating density displayed a different maximum current density in cyclic voltammetry. Modifications in M13 coat protein resulted in even more distinct alterations in electrochemical oxidation behavior (S 19). Though beyond the scope of this study, we anticipate further investigations of electrochemical behavior could elucidate the specific mechanism by which phage derived architectural properties result in these altered electro-catalytic activities. Nonetheless, the present data demonstrates that the controlled architectural diversity in metal nanofoams derived from biological templates can be utilized to alter electrocatalytic behavior.
Conclusions
We have demonstrated that biotemplating can be utilized to fabricate chemically and architecturally versatile 3D nanofoams in a scalable manner. We show that protein engineering can produce controlled architectural modifications in assembled biotemplated materials. As biological structures exist on an extremely wide range of length scales, these results also suggest that further control over structure can be achieved by hierarchically assembling bio-templates of desired, application specific feature sizes.We demonstrated the flexibility of our process over a wide range of material chemistries including both monometallic and two-component systems including combinations of boron, phosphorous, copper, nickel, cobalt, and gold. Two-component nanofoams were tunable over ranges as broad as 0–99 wt% secondary material and demonstrated morphologies of both core–shell and homogeneous structures. While previous studies have been limited to a few monometallic material chemistries,34,42,68 as well as exhibited altered architectures when altering material chemistry,42 our results suggest that this method could be utilized in screening applications where architecture retention across several material chemistries is required.
Furthermore, we show that in addition to over a 50-fold increase in surface area as compared to each nanofoams 2D geometric footprint, a significant decrease in average crystallite size was observed in resulting bio-templated nanofoams. Beyond integration of these biotemplated nanofoams into a variety of support structures, we have demonstrated that the process can be scaled to free standing nanofoams on the cm scale. Excellent conductivity measurements in tandem with M13 template specific electrocataytic behavior indicates that this synthetic method can be utilized for the development of novel electrodes.
In conclusion, this study demonstrates that biotemplated structures can fill a nanoscale design gap between that of conventional aerogels and commercial metal foams. The ability to alter metal nanofoam architecture and composition through methods such as protein engineering or the incorporation of biological structures at application relevant feature sizes, provides great potential to rapidly create and screen application specific electrode structures and chemistries. As such, we anticipate that this method will provide a platform to better study the synergistic and decoupled effects between nano-structure and composition for a variety of applications including energy storage, catalysis, and sensing.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding for this work was provided by the Institute for Collaborative Biotechnologies through Grant W911NF-09-0001 from the U.S. Army Research Office.This work made use of the Massachusetts Institute of Technology (MIT) Materials Research Science and Engineering Center (MRSEC) Shared Experimental Facilities supported by the National Science Foundation via Grant DMR-0819762. J. F. O. gratefully acknowledges the National Science Foundation Graduate Research Fellowship (NSFGRFP). We thank Dr Yong Zhang and Dr Patrick Boisvert at the Center for Materials Science and Engineering (CMSE) at MIT for their assistance with electron microscopy. We thank Dr Charlie Settens at the Center for Materials Science and Engineering (CMSE) for his assistance with X-ray diffraction. We also thank Dr Nir Pour for assistance with the BioLogic, Dr Briana Dunn for the AFM image in Fig. S1,† and Griffin Clausen for sharing his high throughput sequencing insights for the selection of the ETSYFYDT clone.
Notes and references
- B. C. Tappan, S. A. Steiner III and E. P. Luther, Angew. Chem., Int. Ed., 2010, 49(27), 4544–4565 CrossRef CAS PubMed.
- J. Liu, N. Canfield and W. Liu, Ind. Eng. Chem. Res., 2016, 55(13), 3823–3832 CrossRef CAS.
- W. Liu and N. Canfield, J. Membr. Sci., 2012, 409–410, 113–126 CrossRef CAS.
- S. Wang, Z. Ren, Y. Guo and P. X. Gao, CrystEngComm, 2016, 18(17), 2980–2993 RSC.
- C. Xu, B. M. Gallant, P. U. Wunderlich, T. Lohmann and J. R. Greer, ACS Nano, 2015, 9(6), 5876–5883 CrossRef CAS PubMed.
- Y. Yoon, A. S. Hall and Y. Surendranath, Angew. Chem., Int. Ed., 2016, 55(49), 15282–15286 CrossRef CAS PubMed.
- A. S. Hall, Y. Yoon, A. Wuttig and Y. Surendranath, J. Am. Chem. Soc., 2015, 137(47), 14834–14837 CrossRef CAS PubMed.
- A. B. Shigarov, V. V. Kireenkov, V. A. Kuzmin, N. A. Kuzin and V. A. Kirillov, Catal. Today, 2009, 144(3–4), 341–349 CrossRef CAS.
- K. O. Iwu, A. Lombardo, R. Sanz, S. Scirè and S. Mirabella, Sens. Actuators, B, 2016, 224, 764–771 CrossRef CAS.
- Y. J. Lee, C. Oh, J. Y. Park and Y. Kim, IEEE Trans. Nanotechnol., 2011, 10(6), 1298–1305 Search PubMed.
- D. W. Müller, A. M. Matz and N. Jost, spired, Biomimetic Nanobiomater., 2013, 2(2), 76–83 CrossRef.
- M. De Meller, Produit métallique pour l'obtention d'objets laminés, moulés ou autres, et procédés pour sa fabrication. FR 615147D, 1925.
- L. F. Dumée, L. He, B. Lin, F. M. Ailloux, J. B. Lemoine, L. Velleman, F. She, M. C. Duke, J. D. Orbell, G. Erskine, P. D. Hodgson, S. Gray and L. Kong, J. Mater. Chem. A, 2013, 1(48), 15185–15206 RSC.
- L. P. Lefebvre, J. Banhart and D. C. Dunand, Adv. Eng. Mater., 2008, 10(9), 775–787 CrossRef CAS.
- J. Jiang, Y. Li, J. Liu, X. Huang, C. Yuan and X. W. Lou, Adv. Mater., 2012, 24(38), 5166–5180 CrossRef CAS PubMed.
- J. Weissmüller, R. C. Newman, H. J. Jin, A. M. Hodge and J. W. Kysar, MRS Bull., 2009, 34(8), 577–586 CrossRef.
- J. Kibsgaard, A. Jackson and T. F. Jaramillo, Nano Energy, 2016, 29, 243–248 CrossRef CAS.
- M. M. Tomczak, D. D. Glawe, L. F. Drummy, C. G. Lawrence, M. O. Stone, C. C. Perry, D. J. Pochan, T. J. Deming and R. R. Naik, J. Am. Chem. Soc., 2005, 127(36), 12577–12582 CrossRef CAS.
- M. B. Dickerson, K. H. Sandhage and R. R. Naik, Chem. Rev., 2008, 108(11), 4935–4978 CrossRef CAS PubMed.
- R. Derda, S. K. Y. Tang, S. C. Li, S. Ng, W. Matochko and M. R. Jafari, Molecules, 2011, 1776–1803 CrossRef CAS PubMed.
- G. A. Weiss and S. S. Sidhu, J. Mol. Biol., 2000, 300(1), 213–219 CrossRef CAS PubMed.
- Z. Liu, J. Qiao, Z. Niu and Q. Wang, Chem. Soc. Rev., 2012, 41(18), 6178–6194 RSC.
- A. Hernandez-Garcia, D. J. Kraft, A. F. Janssen, P. H. Bomans, N. A. Sommerdijk, D. M. Thes-wessie, M. E. Favretto, R. Brock, F. A. De wolf, M. W. Werten and P. Van Der Schoot, Nat. Nanotechnol., 2014, 9(9), 698–702 CrossRef CAS PubMed.
- S. Borukhin, L. Bloch, T. Radlauer, A. H. Hill, A. N. Fitch and B. Pokroy, Adv. Funct. Mater., 2012, 22(20), 4216–4224 CrossRef CAS.
- T. F. Otero, J. G. Martinez and J. Arias-Pardilla, Electrochim. Acta, 2012, 84, 112–128 CrossRef CAS.
- Z. Chen, Y. Peng, F. Liu, Z. Le, J. Zhu, G. Shen, D. Zhang, M. Wen, S. Xiao, C. P. Liu and Y. Lu, Nano Lett., 2015, 15(10), 6802–6808 CrossRef CAS PubMed.
- S. Ling, Z. Qin, W. Huang, S. Cao, D. L. Kaplan and M. J. Buehler, Sci. Adv., 2017, 3(4), e1601939 CrossRef PubMed.
- L. Gu and D. J. Mooney, Nat. Publ. Gr., 2016, 16(1), 56 CAS.
- C. Remuñán-López and R. Bodmeier, J. Controlled Release, 1997, 44(2–3), 215–225 CrossRef.
- A. K. Wanekaya, W. Chen, N. V. Myung and A. Mulchandani, Electroanalysis, 2006, 18(6), 533–550 CrossRef CAS.
- C. T. Wirges, J. Timper, M. Fischler, A. S. Sologubenko, J. Mayer, U. Simon and T. Carell, Angew. Chem., Int. Ed., 2009, 48(1), 219–223 CrossRef CAS PubMed.
- E. A. Lewis, D. J. Lewis, A. A. Tedstone, G. Kime, S. Hammersley, P. Dawson, D. J. Binks, P. O'Brien and S. J. Haigh, Chem. Mater., 2016, 28(16), 5582–5586 CrossRef CAS.
- N. M. Dorval Courchesne, S. A. Steiner, V. J. Cantú, P. T. Hammond and A. M. Belcher, Chem. Mater., 2015, 27(15), 5361–5370 CrossRef CAS.
- R. Kirsch, M. Mertig, W. Pompe, R. Wahl, G. Sadowski, K. J. Böhm and E. Unger, Thin Solid Films, 1997, 305(1–2), 248–253 CrossRef CAS.
- P. Y. Chen, X. Dang, M. T. Klug, N. M. D. Courchesne, J. Qi, M. N. Hyder, A. M. Belcher and P. T. Hammond, Chem. Mater., 2015, 27(5), 1531–1540 CrossRef CAS.
- J. C. Zhou, C. M. Soto, M. S. Chen, M. A. Bruckman, M. H. Moore, E. Barry, B. R. Ratna, P. E. Pehrsson, B. R. Spies and T. S. Confer, J. Nanobiotechnol., 2012, 10(1), 18 CrossRef CAS PubMed.
- L. I. Vera-Robles, G. Van Tran Nhieu, C. Laberty-Robert, J. Livage and C. Sanchez, Adv. Eng. Mater., 2013, 15(10), 954–961 CAS.
- G. T. Hess, C. P. Guimaraes, E. Spooner, H. L. Ploegh and A. M. Belcher, ACS Synth. Biol., 2013, 2(9), 490–496 CrossRef CAS PubMed.
- A. R. Chandrasekaran and O. Levchenko, Chem. Mater., 2016, 28(16), 5569–5581 CrossRef CAS.
- D. Han, S. Pal, J. Nangreave, Z. Deng, Y. Liu and H. Yan, Science, 2011, 332(6027), 342–346 CrossRef CAS.
- Y. Ke, L. L. Ong, W. M. Shih and P. Yin, Science, 2012, 338(6111), 1177–1183 CrossRef CAS.
- D. Oh, J. Qi, B. Han, G. Zhang, T. J. Carney, J. Ohmura, Y. Zhang, Y. Shao-Horn and A. M. Belcher, Nano Lett., 2014, 14(8), 4837–4845 CrossRef CAS PubMed.
- K. Gerasopoulos, E. Pomerantseva, M. McCarthy, A. Brown, C. Wang, J. Culver and R. Ghodssi, ACS Nano, 2012, 6(7), 6422–6432 CrossRef CAS PubMed.
- S. M. Jung, J. Qi, D. Oh, A. Belcher and J. Kong, Adv. Funct. Mater., 2017, 27(4), 1603203 CrossRef.
- P. Y. Chen, M. N. Hyder, D. Mackanic, N. M. D. Courchesne, J. Qi, M. T. Klug, A. M. Belcher and P. T. Hammond, Adv. Mater., 2014, 26(30), 5101–5107 CrossRef CAS.
- J. Wang, M. Yang, Y. Zhu, L. Wang, A. P. Tomsia and C. Mao, Adv. Mater., 2014, 26(29), 4961–4966 CrossRef CAS PubMed.
- L. F. Deravi, N. R. Sinatra, C. O. Chantre, A. P. Nesmith, H. Yuan, S. K. Deravi, J. A. Goss, L. A. MacQueen, M. R. Badrossamy, G. M. Gonzalez, M. D. Phillips and K. K. Parker, Macromol. Mater. Eng., 2017, 302(3), 1–14 Search PubMed.
- M. Rother, M. G. Nussbaumer, K. Renggli and N. Bruns, Chem. Soc. Rev., 2016, 45(22), 6213–6249 RSC.
- J. W. Kehoe and B. K. Kay, Chem. Rev., 2005, 105(11), 4056–4072 CrossRef CAS PubMed.
- A. G. Sabelnikov, Anal. Biochem., 1972, 48(2), 629–632 CrossRef CAS PubMed.
- K. T. Nam, Science, 2006, 312(5775), 885–888 CrossRef CAS.
- R. Touir, H. Larhzil, M. Ebntouhami, M. Cherkaoui and E. Chassaing, J. Appl. Electrochem., 2006, 36(1), 69–75 CrossRef CAS.
- N. A. Hotaling, K. Bharti, H. Kriel and C. G. Simon, Biomaterials, 2015, 61, 327–338 CrossRef CAS PubMed.
- M. Paunovic, M. Schlesinger and D. D. Snyder, Fundamental Considerations, 2011 Search PubMed.
- F. A. Lowenheim and S. Senderoff, in Modern Electroplating, ed. M. Schlesinger and M. Paunovic, John Wiley & Sons, 1964, vol. 111 Search PubMed.
- P. Xiao, W. Chen and X. Wang, Adv. Energy Mater., 2015, 5(24), 1500985 CrossRef.
- M. Zeng, H. Wang, C. Zhao, J. Wei, K. Qi, W. Wang and X. Bai, ChemCatChem, 2016, 8(4), 708–712 CrossRef CAS.
- J. Masa, P. Weide, D. Peeters, I. Sinev, W. Xia, Z. Sun, C. Somsen, M. Muhler and W. Schuhmann, Adv. Energy Mater., 2016, 6(6), 1502313 CrossRef.
- Y. Shi, Y. Xu, S. Zhuo, J. Zhang and B. Zhang, ACS Appl. Mater. Interfaces, 2015, 7(4), 2376–2384 CrossRef CAS PubMed.
- E. J. Popczun, J. R. McKone, C. G. Read, A. J. Biacchi, A. M. Wiltrout, N. S. Lewis and R. E. Schaak, J. Am. Chem. Soc., 2013, 135(25), 9267–9270 CrossRef CAS PubMed.
- M. Schlesinger and M. Paunovic, in Modern Electroplating: Fifth Edition, ed. M. Schlesinger and M. Paunovic, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2011 Search PubMed.
- D. Kim, J. Resasco, Y. Yu, A. M. Asiri and P. Yang, Nat. Commun., 2014, 5, 4948 CrossRef CAS PubMed.
- Y. Wang, S. Van De Vyver, K. K. Sharma and Y. Román-Leshkov, Green Chem., 2014, 16(2), 719–726 RSC.
- Y. Kang, J. Snyder, M. Chi, D. Li, K. L. More, N. M. Markovic and V. R. Stamenkovic, Nano Lett., 2014, 14(11), 6361–6367 CrossRef CAS PubMed.
- X. Feng, K. Jiang, S. Fan and M. W. Kanan, ACS Cent. Sci., 2016, 2(3), 169–174 CrossRef CAS PubMed.
- H. C. Shin, J. Dong and M. Liu, Adv. Mater., 2003, 15(19), 1610–1614 CrossRef CAS.
- V. L. Oliveira, C. Morais, K. Servat, T. W. Napporn, G. Tremiliosi-Filho and K. B. Kokoh, J. Electroanal. Chem., 2013, 703, 56–62 CrossRef CAS.
- M. Young, W. Debbie, M. Uchida and T. Douglas, Annu. Rev. Phytopathol., 2008, 46(1), 361–384 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nr04864a |
| This journal is © The Royal Society of Chemistry 2019 |