
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRaman tweezers microspectroscopy of circa 100 nm extracellular vesicles†
Sergei G.
Kruglik
 *a,
Félix
Royo
b,
Jean-Michel
Guigner
c,
Laura
Palomo
bd,
Olivier
Seksek
e,
Pierre-Yves
Turpin
f,
Irène
Tatischeff
g and
Juan M.
Falcón-Pérez
bd
*a,
Félix
Royo
b,
Jean-Michel
Guigner
c,
Laura
Palomo
bd,
Olivier
Seksek
e,
Pierre-Yves
Turpin
f,
Irène
Tatischeff
g and
Juan M.
Falcón-Pérez
bd
aLaboratoire Jean Perrin, Sorbonne Université, CNRS UMR 8237, 4 place Jussieu, Paris, 75005, France. E-mail: sergei.kruglik@upmc.fr
bCIC bioGUNE, CIBERehd, Bizkaia Science and Technology Park, Derio, Bizkaia 48160, Spain
cIMPMC, Sorbonne Université, CNRS UMR 7590, IRD, MNHN, Paris, 75005, France
dIKERBASQUE, Basque Foundation for Science, Bilbao, Bizkaia 48013, Spain
eIMNC, Université Paris Sud, CNRS UMR 8165, Orsay, 91405, France
fUR6 Enzymologie de l'ARN, Sorbonne Université, Paris, 75005, France
gRevInterCell, 49T rue François Leroux, Orsay, 91400, France
First published on 8th January 2019
Abstract
The technique of Raman tweezers microspectroscopy (RTM) for the global biomolecular content characterization of a single extracellular vesicle (EV) or a small number of EVs or other nanoscale bioparticles in an aqueous dispersion in the difficult-to-access size range of near 100 nm is described in detail. The particularities and potential of RTM are demonstrated using the examples of DOPC liposomes, exosomes from human urine and rat hepatocytes, and a mixed sample of the transfection reagent FuGENE in diluted DNA solution. The approach of biomolecular component analysis for the estimation of the main biomolecular contributions (proteins, lipids, nucleic acids, carotenoids, etc.) is proposed and discussed. Direct Raman evidence for strong intra-sample biomolecular heterogeneity of individual optically trapped EVs, due to variable contributions from nucleic acids and carotenoids in some preparations, is reported. On the basis of the results obtained, we are making an attempt to convince the scientific community that RTM is a promising method of single-EV research; to our knowledge, it is the only technique available at the moment that provides unique information about the global biomolecular composition of a single vesicle or a small number of vesicles, thus being capable of unravelling the high diversity of EV subpopulations, which is one of the most significant urgent challenges to overcome. Possible RTM applications include, among others, searching for DNA biomarkers, cancer diagnosis, and discrimination between different subpopulations of EVs, lipid bodies, protein aggregates and viruses.
Introduction
The existence of a specific method of cellular communication based on the release of specialized membranous nano- and/or micrometre-sized vesicles has been discovered in recent years, and the number of related scientific publications is now increasing exponentially.1–9 The general name for such cell-derived bioparticles is extracellular vesicles (EVs).4 They are secreted from living cells and can be isolated from a variety of body fluids such as blood plasma, urine, saliva, and milk, among others. The population of EVs is largely heterogeneous; their size varies between 30 nm and 3 μm, and depending on the origin, function or size, they bear various names: exosomes, micro-vesicles, ectosomes/microparticles, apoptotic bodies, etc.4,8,9The chemical composition of EVs depends on the physiological state of the cells from which they originate.1,3–5 Progenitor cells secrete EVs containing growth and/or differentiation messages, whereas differentiated cells generate differentiation and senescence signals. Being released in an extracellular medium, EVs can interact with neighbouring cells and directly induce a signalling pathway or affect cellular phenotype via the transfer of new receptors or even genetic material. For example, EVs may carry messenger- and micro-RNAs, which can be functionally active in the recipient cells and/or regulate their gene expression.10 Other EVs can transmit immune properties to their target cells.11,12 Due to their innate ability to easily enter cells whose lipid membrane otherwise possesses strong defensive means against penetration, EVs are thought to be used as potent efficient “Trojan horses” for drug delivery within cells.13,14 Diseased cells are even more prone to secrete EVs that contain signatures of a number of specific illnesses, such as cancer, hepatic, cardiac and neurological disorders, AIDS, infections or allergic diseases.6,15–19 For example, the concentration and protein content of EVs might be useful for early detection of cancer.20,21 Therefore, EVs are also important interesting candidates that can be used for the evaluation of “healthy” and “ill” states, for diagnostic purposes.
In this work, we describe in detail the method of Raman tweezers microspectroscopy (RTM) for EV characterization, in the important – but difficult to access – nanoscale domain, with particles’ size ranging from approximately 50 nm to a few hundreds of nm.
The RTM technique combines optical trapping22–27 with Raman probing.28 The main idea consists of utilizing the same laser radiation for both optical trapping of the particles of interest and excitation of Raman scattering from the particle's constituent biomolecules. The historical perspective is briefly addressed: the effect of optical trapping was reported by Arthur Ashkin in 197022 for transparent micron-sized latex spheres. His pioneering studies on optical entrapment and manipulation of single cells,23 and viruses and bacteria24 were published in 1987. Useful tutorial reviews concerning optical trapping25 and micro-manipulation26 appeared in 2004 and 2008, respectively, whereas extending optical trapping to the nanoscale on the example of lipid vesicles in the 100 nm size range was reported in 2011.27
Raman characterization of optically trapped particles was performed for the first time for glass and quartz spheres in 1984.29 Application of the Raman technique to optically trapped biological particles in aqueous medium was reported almost a decade later, in 2002 for cellular organelles30 and entire cells,31 in 2003 for single unilamellar lipid vesicles,32 and in 2004 for Raman analysis of liposomal membrane composition33 and single bacterial spores.34 The above list of papers is non-exhaustive; numerous reviews35–40 cover in detail this field of research. Note that the general application of Raman spectroscopy for biomedical and clinical research has also been recently reviewed.41,42
It is noteworthy that the terms “Raman tweezers microspectroscopy” (RTM)33,43,44 and “laser tweezers Raman spectroscopy” (LTRS)31,38 describe essentially the same method in the context of our study. We chose the term RTM as it directly points to the fact that the same laser beam is used for both optical trapping and Raman probing, and since it was suggested in the very first papers43,44 utilizing this approach. LTRS can be considered as a more general term; it describes all possible situations, including multiple-beam experiments in which optical trapping is performed by one or many laser beams that are distinct from the Raman probing beam(s) spatially, spectrally, or both.38
We emphasize the critical importance of the size of bioparticles in RTM experiments. Raman measurements of micrometre and even sub-micrometre objects (organelles,30,45 cells,31,46 protein inclusions,47 phospholipid vesicles,32,48,49 triglyceride-rich lipoproteins,50etc.) might be considered almost routine at the current state of the experimental art. However, within the very important size range near 100 nm, the situation is different: because of the intrinsic weakness of non-resonant Raman scattering, the Raman spectrum of trapped nanosized bioparticles in an aqueous environment is dominated by the contribution from water, whereas the contribution from constituent biomolecules is indeed extremely weak, making the collection of the informative signal nontrivial.
The first Raman spectrum of EVs at the nanoscale was reported in 2009, in a paper devoted to drug vectorization.13 The first pilot study revealing the potential of RTM in EV characterization appeared in 2012, focusing on EVs from Dictyostelium discoideum and exosomes from human urine.51 The following study52 regarding single exosomes from eight different cell lines (both cancerous and non-cancerous) confirmed the ability of Raman spectroscopy to distinguish various exosome subpopulations through principal component analysis; it appeared in 2015. Since then, very few studies regarding Raman characterization of optically trapped EVs (or any other nanosized bioparticle) have been published.53–55
We note one very recent Raman study concerning EVs from mesenchymal stromal cells56 in which the enhancement of the informative signal was achieved by water evaporation and investigation of air-dried drops of an EV suspension. This approach, being interesting and promising per se, bears the disadvantage of losing information about individual bioparticles, such as any other analytical study of a bulk sample.
Another method to circumvent the problem of the weakness of the non-resonant Raman signal is to use surface-enhanced Raman spectroscopy (SERS).57 SERS-based characterization of biomolecules and nanosized bioparticles, in particular exosomes, has been literally exploding in the last few years.58–65 Two different approaches are typically employed.
In the first, “direct” approach,58,62–64 the intrinsic vibrational spectrum of the particle's constituent biomolecules is enhanced via the SERS effect providing molecular specificity. The main problem arises from the fact that the signal enhancement depends strongly on the distance between the molecule and the SERS substrate and vanishes at distances greater than a few nanometers.57 Moreover, Raman modes corresponding to molecular vibrations perpendicular to the surface are preferably enhanced.57 Consequently, an overall SERS vibrational spectrum is somewhat fragmented, distorted, and often difficult to interpret. Because of the short-range SERS enhancement, only those biomolecules that are attached to or incorporated into the particle's membrane can be detected and analysed.
According to the second, “indirect” approach,59–61,65 SERS nanotags functionalized with bio-recognition molecules, which specifically bind to a specific target, are used as quantitative reporters. This SERS strategy produces interesting results, is very sensitive and competes directly with fluorescent molecular probes. However, general information about the particle's constituent biomolecules is lost; only the amount of target molecules is assessed via the signal coming from SERS nanotags.
In summary, RTM of nanosized bioparticles provides some unique information inaccessible to other analytical methods: it combines single- (or very few) particle(s) selectivity, due to optical trapping, with label-free characterization of the particle global biomolecular content, owing to non-resonant Raman detection. Indeed, as a nanoparticle's size is smaller than the optical trap, the informative Raman signal originates from all constituent biomolecules, both attached to the membrane and within the particle's volume, thus providing unique interpretable vibrational fingerprints. The current work is devoted to a detailed analysis of the RTM technique in focusing on EVs within the ∼100 nm size range. More specifically, we investigated the temporal and concentration dependences of the optical trapping process, and depending on the problem under study, we can switch from single- to few- to many-vesicle trapping regimes. An experimental strategy that allows one to obtain non-resonance Raman spectra of good quality from a single EV or very few EVs has been proposed; this strategy directly circumvents the problem of a weak Raman signal without resorting to bulk dry samples or SERS signal enhancement.
On the basis of our results, we are making an attempt to convince the scientific community that RTM is a promising method of single-EV research: to our knowledge, it is the only technique available at the moment that provides unique information about the global biomolecular composition of individual vesicles, thus being capable of unravelling the high diversity of EV subpopulations, which is one of the most significant urgent challenges to overcome.
The results obtained can also be applied to other water-dispersed nanosized bioparticles, such as lipoproteins, protein aggregates and viruses.
Experimental section
Sample preparation
The prepared DOPC liposomes were kept in PBS solution and were found to be stable over time (at least 3 months without any change in the size distribution); no sign of aggregation has ever been observed, even upon sedimentation of the sample by centrifugation and resuspension in a bigger volume.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 30 min, and the supernatant was centrifuged again at 100000g for 75 min. The pellet was resuspended in PBS and again ultra-centrifuged at 100000g for 75 min. The final pellet was suspended in 150 μl of PBS and stored at −80 °C. All urine samples were obtained from the Basque Biobank for research (BIOEF, Basurto University hospital) upon informed consent and with evaluation and approval from the corresponding ethics committee (CEIC code OHEUN11-12 and OHEUN14-14).
000g (exo-free medium). Hepatocytes were treated with the hepatotoxic drug Acetaminophen when indicated. A dose of 10 mM Acetaminophen (APAP, Sigma-Aldrich) was added to the exo-free medium, and EVs were isolated as previously described.69 Briefly, the culture supernatant was centrifuged at 1500g for 10 min to remove the lifted cells and cellular debris. The resultant supernatant was filtered using 0.22 μm pore filters, followed by ultra-centrifugation at 10000g and 100000g for 30 min and 75 min, respectively. The pellet was resuspended in PBS, and ultra-centrifuged again at 100000g for 75 min. The final pellet of small EVs was resuspended in 150 μL of PBS, and then stored at −80 °C. EVs isolated from primary rat hepatocytes have been characterized by us elsewhere.67,70
000g for 30 min, and the supernatant was centrifuged again at 100000g for 75 min. The pellet was resuspended in PBS and again ultra-centrifuged at 100000g for 75 min. The final pellet was suspended in 150 μl of PBS and stored at −80 °C. All urine samples were obtained from the Basque Biobank for research (BIOEF, Basurto University hospital) upon informed consent and with evaluation and approval from the corresponding ethics committee (CEIC code OHEUN11-12 and OHEUN14-14).
000g (exo-free medium). Hepatocytes were treated with the hepatotoxic drug Acetaminophen when indicated. A dose of 10 mM Acetaminophen (APAP, Sigma-Aldrich) was added to the exo-free medium, and EVs were isolated as previously described.69 Briefly, the culture supernatant was centrifuged at 1500g for 10 min to remove the lifted cells and cellular debris. The resultant supernatant was filtered using 0.22 μm pore filters, followed by ultra-centrifugation at 10000g and 100000g for 30 min and 75 min, respectively. The pellet was resuspended in PBS, and ultra-centrifuged again at 100000g for 75 min. The final pellet of small EVs was resuspended in 150 μL of PBS, and then stored at −80 °C. EVs isolated from primary rat hepatocytes have been characterized by us elsewhere.67,70
Sample characterization
Raman signal was collected in a backscattering geometry, spectrally filtered off with a matched combination of beamsplitter and long-pass filter (Semrock RazorEdge “U”), dispersed by a 500 mm focal length spectrograph (Acton SpectraPro 2500i) with a 400 mm−1 grating optimized for 850 nm, and registered by a back-illuminated NIR CCD detector (Princeton Instruments SPEC-10 400BR/LN) cooled to 140° K using liquid nitrogen.
The Raman signal from nanosized bioparticles is extremely weak; that is why true confocal signal collection using a diffraction-limited pinhole is problematic, as it substantially reduces the throughput of the detection pathway. Instead, a “semi”-confocal configuration was implemented using a 50 μm slit with a height of 10 mm at the spectrometer entrance, together with a 75 mm achromatic focusing lens. In this case, the effective volume of Raman signal collection can be estimated as a cylinder with a diffraction-limited diameter of approximately 0.86 μm and an axial length of approximately 3.5 μm,72 giving Vcol ≈ 2 μm3 = 2 fl.
The spectral resolution of our RTM setup with a 50 μm spectrometer slit width is about 5 cm−1. Frequency calibration was performed using Raman lines of toluene with an absolute accuracy of ±2 cm−1 and a relative frequency position accuracy of ±1 cm−1. Spectra were acquired using the WinSpec software; further data treatment was performed with the IgorPro for Windows software package. The details of the raw Raman spectra treatment have been described elsewhere.51 In this work, for Raman kinetics measurements on the basis of spectral band areas, an additional step of automatic background correction using spline functions has been introduced.
000 kit (Agilent) and run in 2100 Bioanalyzer (Agilent). The electrophoretic profile and gel representation were obtained using 2100 Expert Software (Agilent).
Results and discussion
Size, structure and concentration of the studied bioparticles
Fig. 1 presents the NTA size distribution for the bioparticles from the samples studied, including DOPC liposomes of various sizes (panel A), exosomes from human urine (panel B), and exosomes from rat hepatocytes (panel C). Fig. 2 shows the Cryo-TEM images of the same samples; more images can be found in Fig. S1–S6 of the ESI.†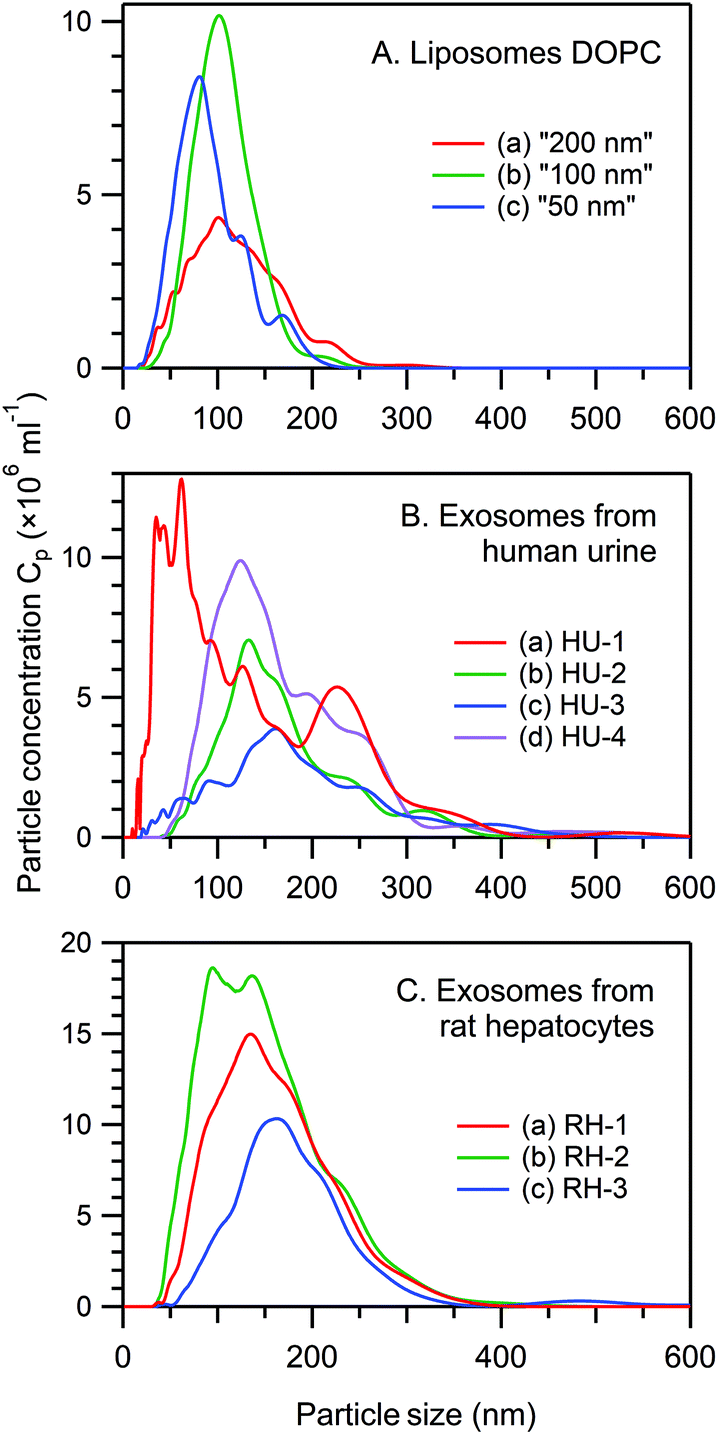 | ||
| Fig. 1 NTA data regarding the size distribution of bioparticles from the samples studied. Panel A, DOPC liposomes “200 nm” ((a), particle concentration Cp = 5.0 × 108 ml−1), “100 nm” ((b), Cp = 7.4 × 108 ml−1), and “50 nm” ((c), Cp = 6.4 × 108 ml−1). Panel B, exosomes from human urine, samples HU-1 ((a), Cp = 1.6 × 109 ml−1), HU-2 ((b), Cp = 8.3 × 108 ml−1), HU-3 ((c), Cp = 6.3 × 108 ml−1), and HU-4 ((d), Cp = 1.3 × 109 ml−1). Panel C, exosomes from rat hepatocytes, samples RH-1 ((a), Cp = 2.1 × 109 ml−1), RH-2 ((b), Cp = 2.8 × 109 ml−1), and RH-3 ((c), Cp = 1.3 × 109 ml−1). | ||
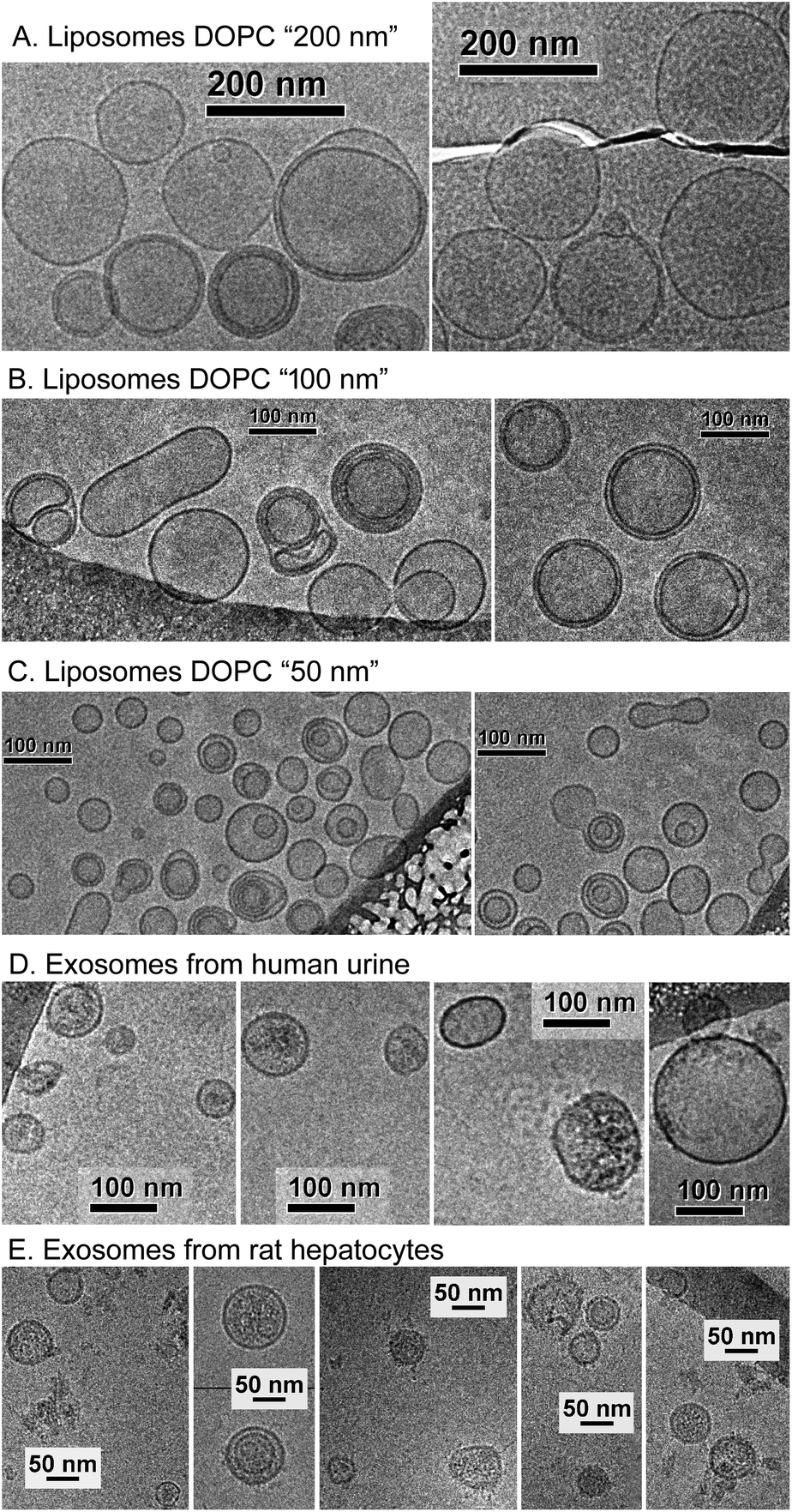 | ||
| Fig. 2 Cryo-TEM images of bioparticles from the samples studied: DOPC liposomes “200 nm” (panel A, Cp = 5.0 × 1013 ml−1), “100 nm” (panel B, Cp = 7.4 × 1013 ml−1), “50 nm” (panel C, Cp = 6.4 × 1013 ml−1), exosomes from human urine, sample HU-1 (panel D, Cp = 1.6 × 1011 ml−1), and exosomes from rat hepatocytes, sample RH-1 (panel E, Cp = 2.1 × 1011 ml−1). | ||
In general, liposomes “50 nm” and “100 nm” are statistically slightly larger, whereas liposomes “200 nm” are smaller than the respective membrane's pore size used for their fabrication. Multiple-compartment and multi-layer liposomes are rather widespread. For the EV samples, the size varies between 30 nm and 300 nm, with the majority of exosomes being in the range from 50–120 nm, although a few larger vesicles were also detected, as were multiple-compartment ones. Whereas liposomes are “empty” when they have only water inside (Fig. 2A–C and S1–S3†), the majority of exosomes are filled with biomaterials (Fig. 2D, E and S4–S6†). Occasionally for the rat hepatocytes (Fig. 2E and S6†) and the urine (Fig. S4 and S5†) samples, small round particles with sizes of approximately 20–40 nm, both with and without a lipid envelope, can be observed. Since contamination by water vapour is characterized by the smallest black “stones” with an irregular form, the appearance of these particles cannot be explained by water contamination. For the moment, their nature is unclear: they may represent protein aggregates or indeed be very small exosomes.
The particle concentration (Cp) in various experiments (NTA, Cryo-TEM, RTM) was calculated from the NTA profiles taking into account the corresponding dilution coefficients. In general, a properly set NTA experiment requires Cp in the range from 108–109 ml−1. A Cryo-TEM experiment requires much higher Cp (at least 1011 ml−1, better 1013–1014 ml−1), which is essentially the maximal concentration available after EV preparation. In RTM, a wide range of Cp (108–1014 ml−1) has been studied (see below).
Concept of the RTM experiment
Raman spectra in our RTM experiment are obtained in separate “experimental runs”. One run consists of 200 spectra acquired in series, each spectrum being accumulated for 3 s; thus, the total time for one measurement is 10 min. According to our experience, the accumulation time of 3 s is a good compromise between temporal resolution and Raman spectra quality. Since the process of particle trapping by light is stochastic and, in the case of poorly prepared samples, can lead to signal contamination by trapped debris or some unwanted material, it is critically important to store each recorded 3 s spectrum to perform an appropriate spectral sorting and averaging afterwards.It is noteworthy that, in general, any duration of a time window and any number of spectra recorded in one run can be set according to particular experimental requirements.
Fig. 3 explains the concept of our RTM experiment using the example of DOPC liposomes “100 nm” with a lipid concentration of 20 μg ml−1 in PBS. For this test experiment we have chosen liposomes instead of EVs for the following reasons. First, DOPC liposomes are chemically uniform, and their Raman spectra are always the same. Therefore, calculating the area under the whole spectrum, we can estimate the number of bioparticles in the optical trap with a better accuracy than on the basis of a single characteristic Raman band. Second, the procedure of liposome preparation by using calibrated 100 nm pores allows a narrower size distribution to be obtained (Fig. 1A), compared to the case of EVs (Fig. 1B and C). Third, we have chosen to study “empty” liposomes filled with water buffer, since these model bioparticles represent the lightest objects with the smallest amount of biomaterial at a given size. If one can optically trap them and record Raman spectra of good quality, then it would certainly be possible to study by RTM the similar-sized, heavier EVs possessing, in addition to the lipid membrane, other biomolecular contents (proteins, nucleic acids, etc.). Our experiment was inspired by the report of successful optical trapping of single lipid vesicles with sizes as small as 50 nm.27
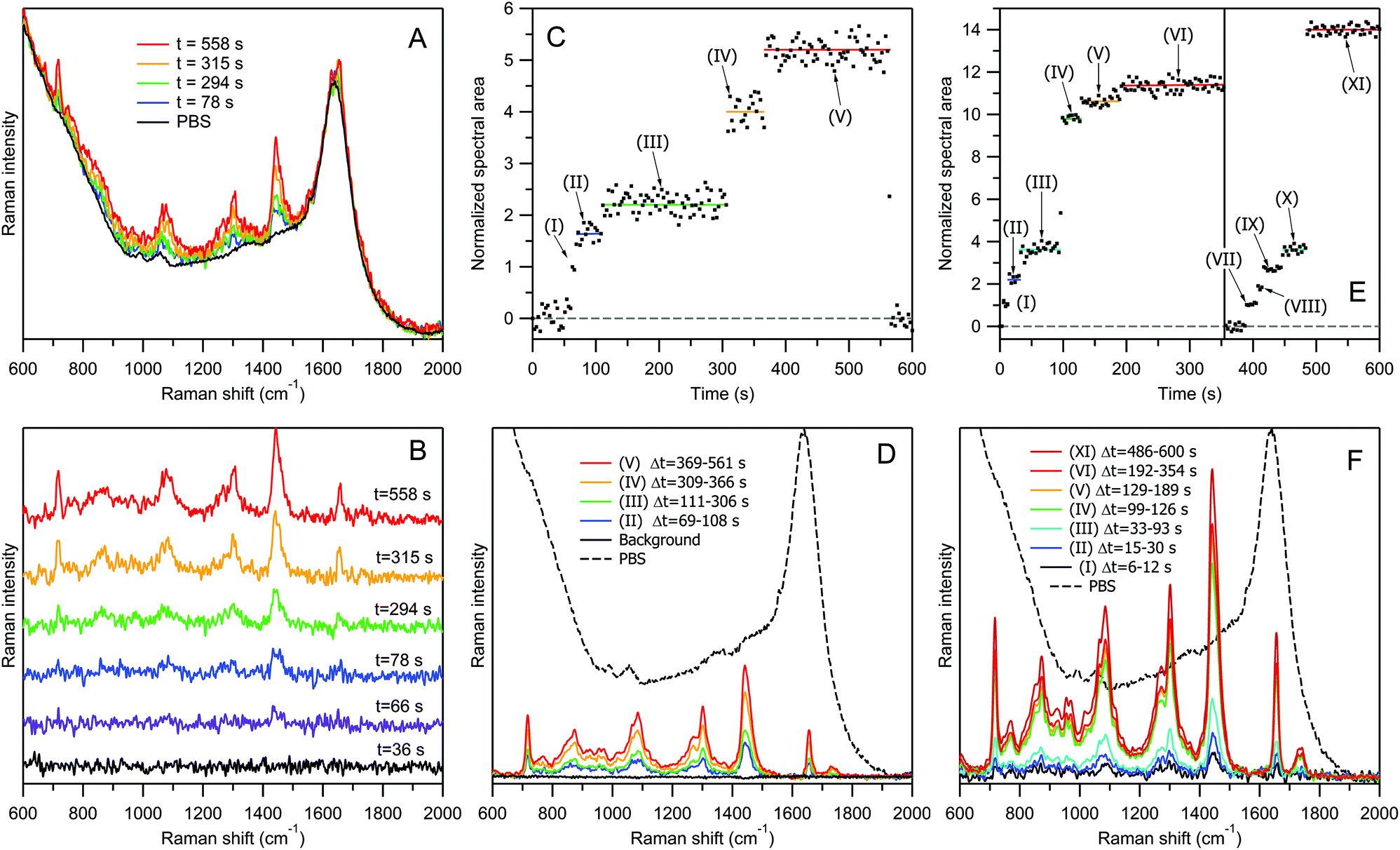 | ||
| Fig. 3 Explanation of the RTM experiment using DOPC liposomes “100 nm” for two particle concentrations: Cp = 1.5 × 1011 ml−1 (panels A–D) and Cp = 7.5 × 1011 ml−1 (panels E and F). Panel A, selected raw Raman spectra recorded at indicated time delays after the start of the experiment, together with an averaged spectrum of PBS. Panel B, selected treated Raman spectra after PBS contribution subtraction, for 6 representative time delays; the acquisition time was 3 s for each spectrum. Panel C, time dependence of the normalized intensity for all 200 treated Raman spectra. Panel D, averaged Raman spectra of different “particle sets” corresponding to different plateaus in panel C. Panels E and F, the same as in panels C and D, but for five-fold higher liposome concentrations. Black dashed curves in panels D and F represent the averaged Raman spectrum of PBS for intensity comparison. | ||
Fig. 3A shows the representative raw Raman spectra of DOPC liposomes “100 nm” recorded at four selected time delays after the start of the acquisition, along with an averaged spectrum of PBS. All 200 raw spectra recorded in this experimental run are shown in Fig. S7 of the ESI.† The treated Raman spectra obtained from the corresponding raw spectra by PBS contribution subtraction, at six representative time delays, are shown in Fig. 3B. Raman intensity normalization on the water band at 1640 cm−1 has always been performed during this subtraction step. Thus, the treated Raman spectra represent the net contribution from lipids, providing quantitative information about the trapped biomaterial since the number of water molecules in the optical trap can be reasonably assumed to be very large and constant. The first black curve in Fig. 3B (time delay t = 36 s) is flat, suggesting that there is no signal from lipids at this time point. For the second spectrum at t = 66 s, a very weak but well distinguishable Raman band appears at ∼1440 cm−1, corresponding to CH2 deformations of lipid chains. The spectrum becomes increasingly pronounced at t = 78 s (third blue curve) and t = 294 s (fourth green curve). Then, the Raman bands become approximately twice stronger for the fifth yellow spectrum (t = 315 s), and then increase again for the last red spectrum recorded at t = 558 s.
Fig. 3C shows the time dependence of the normalized Raman intensity (total area under the contour in 600–1800 cm−1 range) for all of the 200 treated spectra presented in Fig. S8 of the ESI.† The Raman signal recording starts with the beginning of sample irradiation, and during the first ∼60 s no liposome is trapped. It is worth noting that micromolar concentrations of lipids are not detectable by non-resonance Raman spectroscopy in a water dispersion without optical trapping. At t = 63–66 s, the first liposome is captured (time range (I) in panel C). Then, at t = 69 s, a second transition occurs, and within the time interval from t = 69 s to t = 108 s (II), a very weak but well distinguishable Raman signal appears, fluctuating around some average value indicated by the blue horizontal line.
Bendix and Oddershede measured the time series of the positions visited by an optically trapped lipid vesicle with a sampling frequency of 22 kHz (Fig. 1 of ref. 27). They found that the trapped vesicle rapidly oscillates with an amplitude of up to 200 nm around some equilibrium point, with the histogram of all visiting positions forming a Gaussian distribution and the positional power spectrum being fitted by a Lorentzian function with a corner frequency of 300 Hz.27 From these observations, we infer that the nanovesicle's trapping – namely, the establishment of its equilibrium position in the optical trap – occurs much faster than the sampling time of 3 s in our RTM experiment, and that its fast oscillatory movement within the trap is beyond our time resolution. What we observe instead is random averaged intensity fluctuation, influenced also by the subtraction of the strong Raman signal from water.
In Fig. 3C, at t = 111 s, the trapping of the third liposome occurs, with the plateau (III) extending up to t = 306 s (green line). The next transition occurs at t = 309 s, with an approximately twofold increase in intensity, indicating that the trapped liposome contains a larger amount of lipids than the previous particles. Then, at t = 369 s, the signal rises again, producing the fifth plateau up to t = 564 s.
Starting from this moment, the sample stage is moved back and forth for a few millimetres by a specially designed mechanism to destroy the optical trap. As a result, no new trapping event can occur until the end of the measurement. Raman spectra recorded at the beginning (t < 60 s) and end (t > 564 s) of the experimental run were used to calculate the averaged spectrum of PBS (black curves in Fig. 3A and D), which was later employed for normalization and subtraction purposes.
Each of the five transitions in Fig. 3C (at t = 63, 69, 111, 309, and 369 s) corresponds to an event of liposome trapping, and the difference in the intensity changes between different plateaus relates to the liposome size variability and to the possibility of trapping multiple-compartment vesicles.
Fig. 3D presents Raman spectra corresponding to different “particle sets” of Fig. 3C. A Raman spectrum of a “particle set” is calculated as an averaged spectrum of all time points within the same plateau corresponding to the same number of trapped particles. For example, the weakest blue curve (Fig. 3D(II)) was obtained by averaging 14 spectra within the time range from 69 s to 108 s, corresponding to the averaged Raman spectrum from two trapped liposomes. The strongest red curve (Fig. 3D(V)) is an average of 65 spectra within the time range Δt = 369–561 s, corresponding to ∼5 trapped liposomes.
The experiment was repeated several times, and the time traces varied from one run to the other, reflecting the stochastic nature of optical trapping. In general, a particular form of kinetics is determined by a complex interplay of many parameters: (i) particles’ characteristics (concentration, size and density), (ii) stochastic Brownian motion (temperature), (iii) optical trapping forces (including parameters such as the laser wavelength and power, objective magnification and numerical aperture), and (iv) a very minor gravitational force contribution.
Concentration, size and particle density dependence
In our sample containing DOPC liposomes “100 nm” with a concentration of 20 μg ml−1 in lipids, the number of particles per unit volume was measured by NTA to be Cp = 1.5 × 1011 ml−1. Thus, within the estimated volume of Raman signal collection of Vcol ≈ 2 fl, the average number of randomly moving liposomes, before trapping, is estimated to be N = Cp × Vcol ≈ 0.3. It is possible to perform RTM measurements at even lower concentrations; however the increasing waiting time for a trapping event will eventually set a practical limit of detectable concentration to be ∼109 ml−1 or N ≈ 0.002 for empty liposomes “100 nm” under our experimental conditions.Fig. 3E and F show the RTM results for a more concentrated suspension of liposomes. The five-fold concentration increase, from 20 μg ml−1 to 100 μg ml−1 in lipids (N ≈ 1.5), changes the temporal evolution of Raman intensity in Fig. 3E in terms of two main aspects. First, at the beginning of the measurements, the waiting time for the trapping of the first vesicle is considerably reduced, the trapping occurring sometimes almost immediately; this happened in the case of transition (I). Second, the attraction of a vesicle into the optical trap can occur either consecutively, one-by-one, as in the previous “low-concentration” case of Fig. 3C and during the transitions (I) → (II) and (II) → (III) in Fig. 3E, or simultaneously, when several vesicles are trapped within the same 3 s accumulation time interval, as during the transition (III) → (IV) at t = 96 s. Then, again, the transitions (IV) → (V) and (V) → (VI) are presumably due to a single-vesicle trapping. At plateau (VI), the Raman signal reaches saturation, as no further transition occurs.
The vertical line at t = 355 s in Fig. 3E denotes the moment at which the sample stage was briefly shaken, causing the loss of the optical trap; the trapping process subsequently restarts. The new trapping of the first liposome occurs at t = 390 s. Then, after a series of single-vesicle transitions (VII) → (VIII) → (IX) → (X), a new saturation plateau (XI) is reached again via an abrupt multi-vesicle transition (X) → (XI) at t = 486 s. This observation suggests that, with increasing particle concentration, the probability of simultaneous, i.e., within the 3 s accumulation time window, trapping of several particles into the optical trap also increases.
Fig. 3F presents the averaged Raman spectra corresponding to various particle sets of Fig. 3E.
Now, let us move from empty lipid vesicles to EVs filled with biomaterials, and examine the process of optical trapping and Raman scattering in an aqueous dispersion of exosomes from human urine, with a protein concentration of ∼10 μg ml−1 (Fig. 4A, B, S9 and S10†). Qualitatively, the temporal evolution of the Raman signal from exosomes (Fig. 4A) looks similar to that from liposomes (Fig. 3C and E), with both single-particle 0 → (I) → (II), (III) → (IV), 0 → (V) → (VI) → (VII) → (VIII), (IX) → (X) and multi-particle (II) → (III), (VIII) → (IX) transitions, and eventual signal saturation within the high-intensity plateaus (IV) and (X). Here again, at t = 303 s (vertical line), the sample stage was briefly shaken, causing the release of exosomes from the optical trap, and the restart of the trapping process.
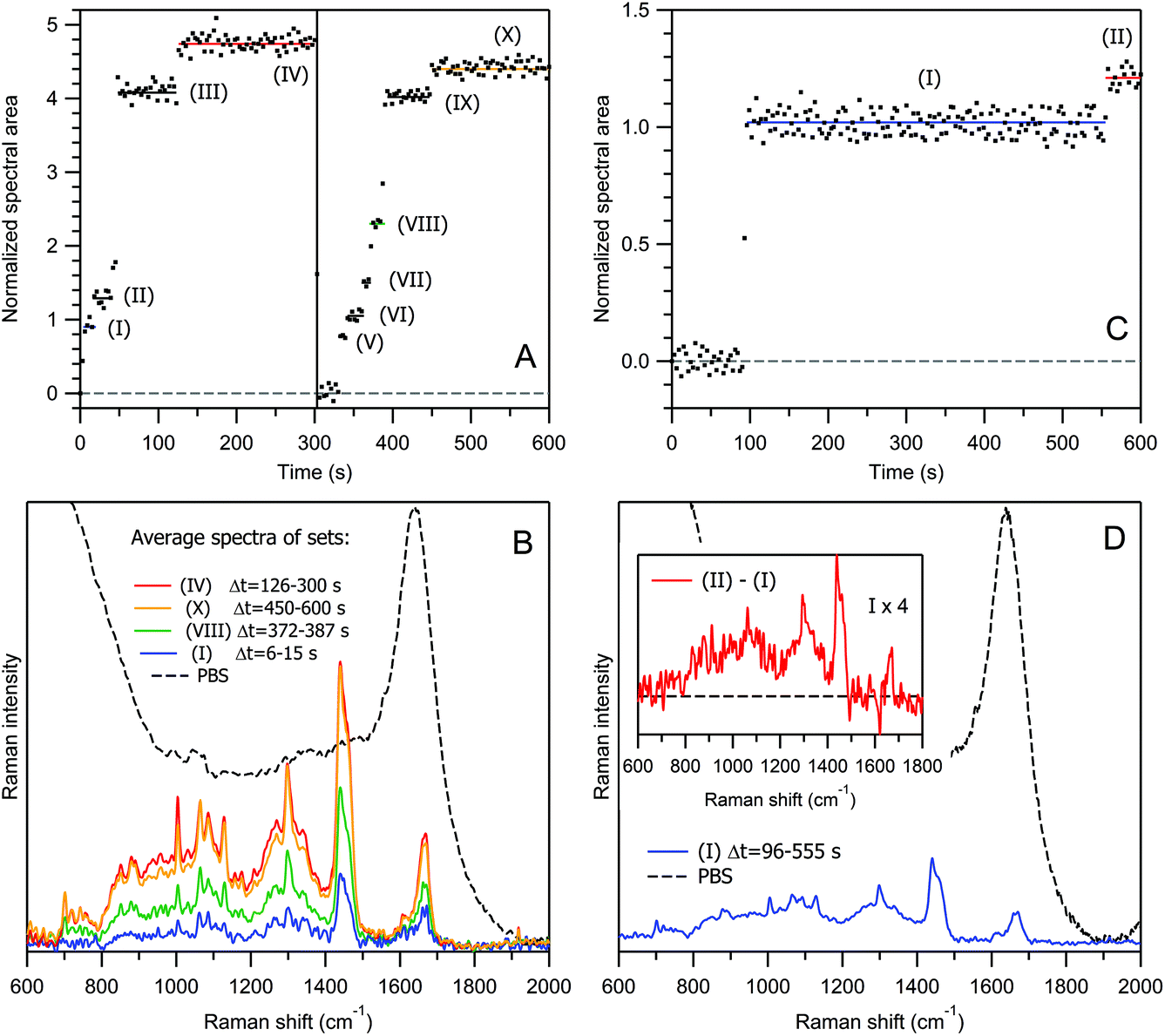 | ||
| Fig. 4 Normalized Raman intensity kinetics (panels A and C) and averaged Raman spectra of different particle sets (panels B and D) for exosomes from human urine from two different samples, HU-1 (Cp = 1.6 × 1010 ml−1, panels A and B) and HU-2 (Cp = 4.1 × 108 ml−1, panels C and D). Inset in panel D, Raman spectrum corresponding to the difference between species (I) and (II) in panel C; its intensity is multiplied by 4 to visually enhance the resulting spectral features. The black dashed curves in panels B and D represent the Raman spectrum of PBS for intensity comparison. | ||
However, there is also a significant difference. The number of exosomes per unit volume is Cp = 1.6 × 1010 ml−1 (N = 0.03), i.e. an ∼10 to ∼50-fold lower value than in the case of liposome samples studied above. Indeed, it is not surprising that denser particles of a similar size are trapped more efficiently.
For exosomes from human urine, the size distribution is usually more heterogeneous than in the case of DOPC liposomes. Moreover, exosomes are chemically heterogeneous, also, in contrast to liposomes. This can be observed well from Fig. 4B by comparing the averaged Raman spectra of two independent vesicle sets within the saturation plateaus, (IV) (red curve) versus (X) (yellow curve). The relative amount of proteins with respect to lipids is lower in the latter case as evidenced from the intensity ratio of the protein marker band at 1004 cm−1 (Phe) to the lipid triplet at 1060–1130 cm−1 and CH2 twisting of the lipid acyl chain at 1298 cm−1 (see also the corresponding positive and negative peaks in the Raman difference spectrum in Fig. S11 of the ESI†).
Because of these heterogeneities in size and chemical composition and possible multi-particle transitions at higher concentrations, the relationship between the Raman intensity kinetics and the number of trapped particles is more difficult to establish for exosomes than for liposomes. To verify that the particle sets (I) and (V) in Fig. 4A and the corresponding blue spectrum in Fig. 4B relate to single-exosome trapping, we have conducted an additional experiment at a ∼40-fold lower exosome concentration of Cp = 4.1 × 108 ml−1. The result is shown in Fig. 4C and D: the trapping of the first exosome occurs at t = 96 s, and then the Raman signal remains constant until t = 555 s; the normalized intensities and the averaged spectra corresponding to the first plateaus are quite similar for both concentrations. Moreover, this intensity level has been reached in a stepwise manner in less than two time points of 3 s. It is improbable that not one but two, or more than two, exosomes were trapped simultaneously in all three realizations, especially in the low-concentration case.
It is interesting to note that the trapping of an empty exosome consisting predominantly of lipids occurs during the transition (I) → (II) in Fig. 4C. This conclusion was made after the analysis of the Raman difference spectrum between the two states (inset in Fig. 4D), which does not exhibit protein marker bands from Phe, Tyr or Trp amino acid residues (see Table 1 and the discussion below for Raman spectra interpretation). This explains well the weak increase in the Raman intensity during the transition (I) → (II) with respect to the transition (0) → (I), corresponding to an exosome filled with proteins.
| Frequency | Biomolecule | Assignment |
|---|---|---|
| ν = Stretching mode, δ = deformation mode; A = adenine, C = cytosine, G = guanine, T = thymine, U = uracil; Trp = tryptophan, Tyr = tyrosine, Phe = phenylalanine. This band assignment is based on studies on proteins,47,85–90 lipids,32,33,46,91–93 “Raman signature of life”,94–98 nucleic acids,99–103 and carotenoids.104–106 | ||
| 1720–1750 | Lipids |
ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) in ester COOR O) in ester COOR |
| 1670–1690 | Nucleic acids |
ν(CO) in pyrimidines |
| 1640–1700 | Proteins | Amide I: ν(CO) |
| 1650–1670 | Lipids |
ν(CC) in acyl chain |
| 1619–1621 | Proteins | Trp (W1) |
| 1615–1617 | Proteins | Tyr (Y1) |
| 1605–1607 | Proteins | Phe (F1) |
| 1602–1604 | Lipids | Ergosterol |
| 1570–1580 | Nucleic acids | Purine A, G ring |
| 1550–1555 | Proteins | Trp (W3) |
| 1515–1540 | Carotenoids | Polyene ν(CC) |
| 1450–1490 | Nucleic acids | Purine A, G ring |
| 1435–1465 | Proteins | Backbone δ(CH2, CH3) |
| Lipids | δ(CH2, CH3) in acyl chain | |
| 1330–1380 | Nucleic acids | Pyrimidine and imidazole rings A/G stacking |
| 1339, 1361 | Proteins | Trp (W5, W4) |
| 1300–1350 | Proteins | Backbone δ(CαH), ν(Cα–C) |
| 1295–1305 | Lipids | δ(CH2) in acyl chain |
| 1260–1270 | Lipids |
δ(CH2) in acyl chain |
| 1230–1305 | Proteins | Amide III: ν(C–N) + δ(NH) |
| 1200–1260 | Nucleic acids | U, C ring; sugar puckering |
| 1207–1210 | Proteins | Phe (F3), Tyr (Y3) |
| 1175–1177 | Proteins | Tyr (Y4) |
| 1155–1160 | Carotenoids | Polyene ν(C–C) |
| 1050–1160 | Proteins | Backbone ν(Cα–N, Cα–C, C–N) |
| Lipids | ν(C–C) in acyl chain | |
| 1090–1100 | Nucleic acids | Phosphodioxy νs(PO2−) |
| 1032 | Proteins | Phe (F4) |
| 1012 | Proteins | Trp (W6) |
| 1004 | Proteins | Phe (F5) |
| 930–960 | Proteins | α-Helix backbone ν(C–Cα–N) |
| 878–880 | Proteins | Trp (W7) |
| 852–857 | Proteins | Tyr (Y5) |
| 828–837 | Proteins | Tyr (Y6) |
| 820–900 | Phospholipids | ν(O–C–C–N+), ν(C4–N+) |
| 810–836 | Nucleic acids | Phosphodiester νs(O–P–O) |
| 782–788 | Nucleic acids | Pyrimidine C, T, U ring |
| 758–759 | Proteins | Trp (W8) |
| 725–751 | Nucleic acids | A |
| 717 | Phospholipids | ν s(C–N+) |
| 700–704 | Lipids | Cholesterol |
| 668–683 | Nucleic acids | G |
| 643–645 | Proteins | Tyr (Y7) |
| 621–623 | Proteins | Phe (F6) |
The common feature of time traces in Fig. 3C, E and 4A, C is the formation of a saturation plateau when the Raman signal does not rise anymore, within one experimental run of 10 min. Generally, the exact number of trapped particles in such a plateau is rarely possible to determine, although a reasonable estimation can be made: in the experiments of Fig. 3 and 4, from 1 to approximately 14 particles have been trapped.
Fig. 5 presents the dependence of the normalized Raman signal within the highest-intensity saturation plateau versus particle concentration for empty DOPC liposomes of three different sizes, “50 nm” (a), “100 nm” (b), and “200 nm” (c), and for the exosomes from human urine (d). Because of the exosomes’ chemical heterogeneity, and for the sake of comparison with chemically homogeneous liposomes, the amplitude of the Raman band at 1440–1460 cm−1 due to δ(CH2, CH3) deformations of lipids and proteins, normalized to the water band at 1640 cm−1, has been used instead of the total spectral area.
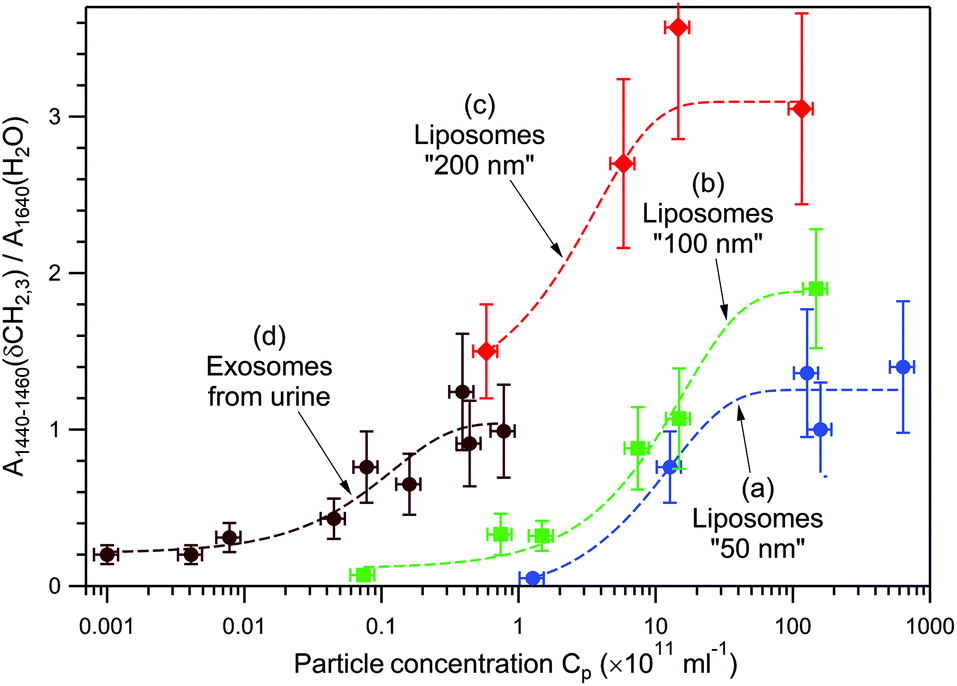 | ||
| Fig. 5 Normalized Raman signal of the highest-intensity saturation plateau, within one experimental run of 10 min, versus particle concentration, for DOPC liposomes “50 nm” (a), “100 nm” (b), “200 nm” (c), and urinal exosomes (d). Dashed curves through the data points were drawn to highlight the tendencies. | ||
The following three regions can be distinguished: (i) a low-intensity constant level at low concentrations, (ii) a logarithmic intensity rise at intermediate concentrations, and (iii) intensity saturation at high concentrations. The low-intensity constant level is explained by the trapping of only one vesicle independently of Cp. In this range, only the time delay between the optical trap formation and the trapping event depends on Cp, whereas the Raman intensity does not. The intermediate-concentration range is characterized by a near-linear dependence of the Raman intensity on the logarithm of Cp. This experimental finding can be rationalized by the following qualitative argument. A colloidal system's behaviour depends on its chemical potential, which in turn depends on the concentration of colloidal particles in the dispersion, more specifically on ln(Cp).73 When an optical trap is created in the dispersion by a strong laser field, the system is perturbed, and subsequent particle entrapping provides gradual equilibration of chemical potentials. The higher the chemical potential of a colloidal system, the larger the number of particles that can be trapped. Since the process is stochastic, the number of trapped particles and the time to reach the saturation plateau may vary from one measurement to another (compare plateaus VI and XI in Fig. 3E, and IV and X in Fig. 4A). The upper limit of the number of trapped particles (high-concentration region of intensity saturation in Fig. 5) depends on a complex interplay between optical forces, particles’ kinetic energy due to Brownian motion, and particle concentration, since the diffusion of particles is inherently concentration-dependent.73,74 In other words, once the effective “core” volume of an optical trap is fully occupied with nanoparticles, the chances to trap more particles substantially decrease. Such a behaviour has been routinely observed in our experiments; this also means that DOPC liposomes and EVs under study do not aggregate, since the filled optical trap does not serve as a nucleation centre for further spontaneous aggregation, at least within the studied concentration range, Cp < 1014 ml−1.
Concerning the “core” size of an optical trap, as a reference, the signal collection volume of ∼2 μm3 may accommodate up to 2400 spheres of 100 nm diameter, taking into account the sphere's random packing factor of 0.63.75 Therefore, since from one to tens of nanovesicles are usually trapped under our experimental conditions, we conclude that the optical trap's effective “core” volume is much smaller than the signal collection volume; taking into account the results of Bendix and Oddershede,27 the upper limit of the effective “core” volume is estimated to be on the order of 0.1 μm3.
Returning back to the single-exosome transition at low Cp in Fig. 4C(I), the average number of randomly moving EVs, before trapping, in such a “core” volume is N = 4.1 × 10−5; therefore, the probability of simultaneous trapping more than one exosome is indeed very low.
The upper value of exploitable Cp is limited by the following two physical effects. First, the increase in Cp causes a decrease in the sample transparency, and even in slightly opaque water dispersions, the strong Rayleigh wing/Mie scattering completely obscures the weak Raman signal, making Raman measurements impossible. Second, forced aggregation of nanovesicles in the optical trap may occur at very high Cp, giving rise to very strong Raman bands that dominate the spectrum and do not correspond to vesicle chemical composition, but rather to the structure of an aggregate (so-called morphology-dependent resonances35).
Data analysis of Fig. 5 allows the following main tendencies to be formulated. First, larger vesicles of the same chemical composition produce a stronger Raman signal at the same Cp and are easier to detect at smaller Cp. For example, at Cp = 1012 ml−1, the Raman signal from liposomes “200 nm” is approximately 3.3 and 4.9 times stronger than those from liposomes “100 nm” and “50 nm”, respectively. Second, heavier (filled with biomaterials) vesicles of approximately the same size can be detected at smaller Cp. For example, the curve (d) corresponding to exosomes from human urine shifts by a factor of ∼102 to lower concentrations with respect to the curve (b) corresponding to empty liposomes “100 nm”.
Taking this into account, we conclude that for heterogeneous mixtures of EVs, the RTM technique possesses a certain selectivity for large and heavy (filled) vesicles at the expense of small and light (empty) ones. Nevertheless, because of the stochastic nature of optical trapping, small and/or light vesicles can still be trapped sometimes, as in the case of transition (I) → (II) in Fig. 4C and D. In our opinion, a proper EV sorting, such as using the asymmetric flow field–flow fractionation (AF4) method,76–79 performed prior to RTM analysis, may provide additional information about the total EV population under study.
Strategy of RTM measurements
Our strategy of RTM measurements for a sample containing EVs of ∼100 nm size consists of the following main steps: (i) start exposition of the sample to the laser light; (ii) wait until vesicle trapping and saturation of the Raman signal occur; (iii) accumulate Raman spectra from one particle set during a certain period of time, usually for several minutes; and (iv) repeat this cycle many times to obtain reliable statistics. Note that during the event of multi-vesicle trapping, a bright diffusive spot appears at the centre of the optical trap in the objective's focal plane (Fig. S12†).If one wants to record single-vesicle spectra (like set (I) in Fig. 4C and D), it is necessary to choose Cp within the low-concentration region of the curve in Fig. 5 and, because of the extreme weakness of the Raman signal, to accumulate spectra for a longer time than in the case of multi-vesicle trapping to obtain an averaged spectrum of comparable quality.
Sabelnikov and Kempf have discussed the feasibility of using Raman tweezers in single-cell research;80 their analysis can be further extended to single-vesicle research. From Fig. 1 of ref. 80, one might estimate that at least 71 independent measurements are necessary for a statistically significant result if at least 15 observations of a significant difference between the studied and the control samples (x ≥ 15) are required, and if the probability of trapping a representative particle is p = 0.5. Even for x ≥ 5 and p = 0.9, at least 12 independent measurements are required. Taking into account that one RTM measurement of a single-vesicle set takes minutes, this approach is far from being “fast”,53 in contrast to what was claimed originally.51 As a possible compromise to accelerate RTM experiments, and at the same time to enhance the spectral quality, we propose to record the Raman spectra of multi-vesicle sets (like sets (IV) and (X) in Fig. 4A and B) combining the contributions from 5–10 trapped vesicles. Since each vesicle is attracted into the optical trap independently of the others, the averaged Raman spectrum of such a multi-vesicle set contains information about all of the trapped EVs, effectively performing some type of “pre-averaging” of individual spectral contributions.
No long-term photodegradation of the trapped particles has been observed under our experimental conditions, except for weak carotenoid photobleaching in some preparations, which is a well-known effect due to the tail of carotenoid absorption in the NIR region. The main damage due to the high power density of light in the optical trap might consist of sudden explosions of the trapped vesicles; the debris are then moved immediately away from the trap via Brownian motion. The larger the diameter of a vesicle, the more probable the explosion event, and under our experimental conditions, micrometre-sized vesicles were sometimes affected, whereas exosomes and even “200 nm” DOPC liposomes were found to be robust.
Another possible detrimental process is knocking out of the already trapped particle by an arriving one. Here, two different scenarios may occur: first, both particles escape from the trap; second, one particle is replaced by the other. In the first case, the informative Raman signal disappears (only the water contribution remains); this case cannot be distinguished from the above-discussed explosion event. In the second case, the Raman spectrum changes according to the difference in particles’ chemical composition; if the difference is small, the knocking-out event is difficult to notice. However, it should be noted that at usually employed exosome concentrations (Cp = 108–1011 ml−1), such an event of direct ballistic interaction between particles is expected to be very rare, as the average number of randomly moving exosomes, before trapping, is on the order of N = 10−5–10−2 within an effective “core” volume of 0.1 μm3. According to our experience, much more probable is the knocking-out effect due to large particles/debris/aggregates in a poorly prepared sample.
In any case, since we are recording Raman spectra every 3 s, all the data obtained prior to the detrimental event can be successfully analysed.
RTM diagnostic feasibility
The use of Raman spectroscopy in biomedical analysis for detecting directly, without exogenous markers, molecular changes in cells and tissues is well known.41,42,81–83 Here, we performed a model experiment targeting the RTM diagnostic potential with EVs as information messengers.Rat hepatocytes had been treated with the hepatotoxin Acetaminophen, and EVs secreted by both treated and untreated (control) cells were collected, isolated by differential ultracentrifugation, and examined by RTM. Acetaminophen is a widely used drug that can cause hepatic failure,84 and is considered as a good experimental model to unravel potential biomarkers of hepatotoxicity. Thus, Raman analysis of EVs secreted by hepatocytes with and without Acetaminophen exposure could provide specific signals for the evaluation of serum EVs and detect hepatic damage in a specific and non-invasive manner.
The aggregated results are presented in Fig. 6. Spectra 6(a) and 6(b) are averaged normalized Raman spectra obtained via two independent measurements of 5 and 7 exosome sets, respectively, collected from rat hepatocytes treated with the hepatotoxin. Their (1:1) difference is presented in Fig. 6(d), which is effectively zero within the accuracy of measurements. Spectrum 6(c) is the averaged spectrum of 6 independent exosome sets from the control sample. Changes in the chemical composition of exosomes from treated versus control cells are shown in the (1:1) difference spectrum 6(e), which contains prominent positive and negative features, due to the increase or decrease of the corresponding molecular component.
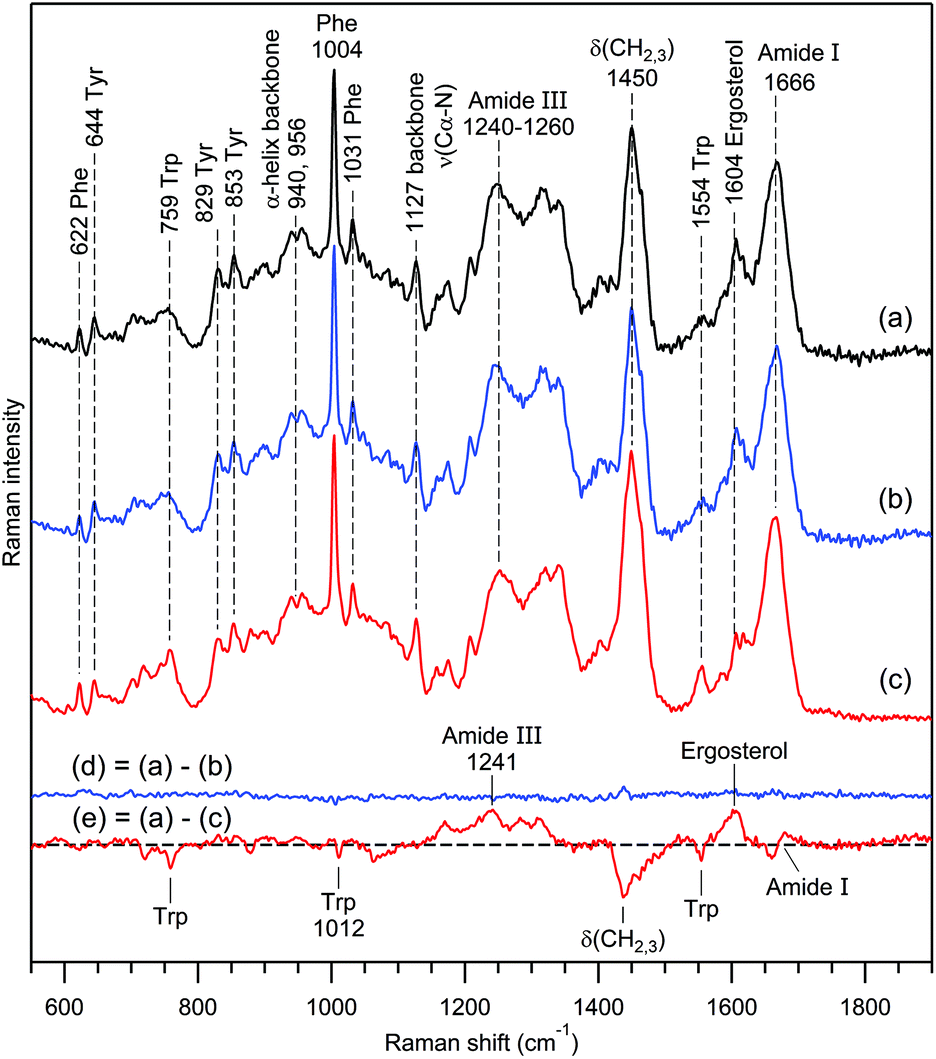 | ||
| Fig. 6 Study of RTM/EV diagnostic feasibility. Curve (a) is the normalized Raman spectrum of exosomes obtained from rat hepatocytes treated with the hepatotoxin Acetaminophen. Curve (b) is the same as (a) but for another independent measurement. Curve (c) is the normalized Raman spectrum of exosomes from the control non-treated sample. The water buffer contribution was subtracted, and the slowly changing background was corrected using cubic spline interpolation for all spectra. Curves (d) and (e) are the (1:1) difference (d) = (a)–(b) and (e) = (a)–(c), respectively. Raman bands assignment according to Table 1. | ||
The Raman band assignment for the biomolecular composition of EVs is presented in Table 1, and the most-characteristic bands are indicated by vertical dashed lines in Fig. 6.
In general, dominant contributions to non-resonance Raman spectra of EVs come from proteins, lipids, nucleic acids and carotenoids; other biomolecules usually provide weaker contributions. Changes in the protein composition in Fig. 6e are manifested by: (i) a decrease of the tryptophan content, (ii) an enhancement of the amide III band related to the protein's secondary structure, (iii) an intensity decrease of bending vibrations δ(CH2, CH3) of the protein's backbone and lipid acyl chain, and (iv) a shift of the amide I band to higher frequencies, revealing a redistribution between α-helical and β-strand structures.85,86
It is worth noting the enhancement of the Raman band around 1602–1604 cm−1 upon hepatotoxin action. This band was reported for the first time in studies of living yeast cells94,98 and dubbed “Raman signature of life”.94 Further studies45,95–97 clarified that its appearance can serve as an indicator of a specific mitochondrial metabolic activity; an assignment to ergosterol has been proposed.96,97
From a comparison of the Raman difference spectra 6(d) and 6(e), it is clear that the treatment of rat hepatocytes by the hepatotoxin Acetaminophen causes substantial changes in the biomolecular contents of the exosomes produced by these cells, in agreement with recent reports.107 Raman spectra of independent individual vesicle sets exhibit predominant contributions from proteins and lipids with rather small intra-sample chemical heterogeneity, which is smoothed out upon averaging of the sufficiently large number of trapped exosomes. A previous experiment on exosomes from human urine (Fig. 4B) revealed a similar tendency. In such cases, the RTM diagnostics using EVs is undoubtedly feasible. The straightforward approach is to apply one of the methods of multivariate statistical analysis, e.g., clustering algorithms, regression, classification and unmixing models.52,55,81–83
Intra-sample biomolecular heterogeneity of EVs
Nevertheless, the almost perfect reproducibility of the data in Fig. 6 (curve d) was surprising to us. Therefore, we performed numerous RTM experiments on exosomes from various rat hepatocyte preparations to acquire larger statistics. As a result, we indeed observed strong intra-sample chemical heterogeneity in some preparations.The result of one such experiment is presented in Fig. 7A. Raman spectra (a–c) correspond to 3 independent vesicle sets of the same sample containing exosomes from rat hepatocytes without any treatment. From the analysis of Raman markers (see below), we conclude that this sample contains very different types of exosomes. Vesicle set (a) is characterized by a major contribution from non-saturated lipids, low protein content and the absence of nucleic acids. The second set (b) reveals moderate–strong contributions from lipids and proteins, and low nucleic acid content, whereas the third set (c) is characterized by strong contributions from proteins and nucleic acids and low lipid content. It is interesting that the Raman spectra in Fig. 6c and 7A(c), originating from different non-treated rat hepatocyte samples, are somewhat similar, having, however, the following main distinctions: the spectrum in Fig. 7A(c) contains a major contribution from nucleic acids (bands at 725, 784, 1097 and 1576 cm−1), whereas the spectrum in Fig. 6c reveals strong Trp contribution (bands at 759 and 1554 cm−1). From the position and shape of the phosphodiester stretch at 810–840 cm−1 in the difference spectrum (Fig. 7A(d)), we infer that nucleic acids in question possess an A-type helical structure.102,103
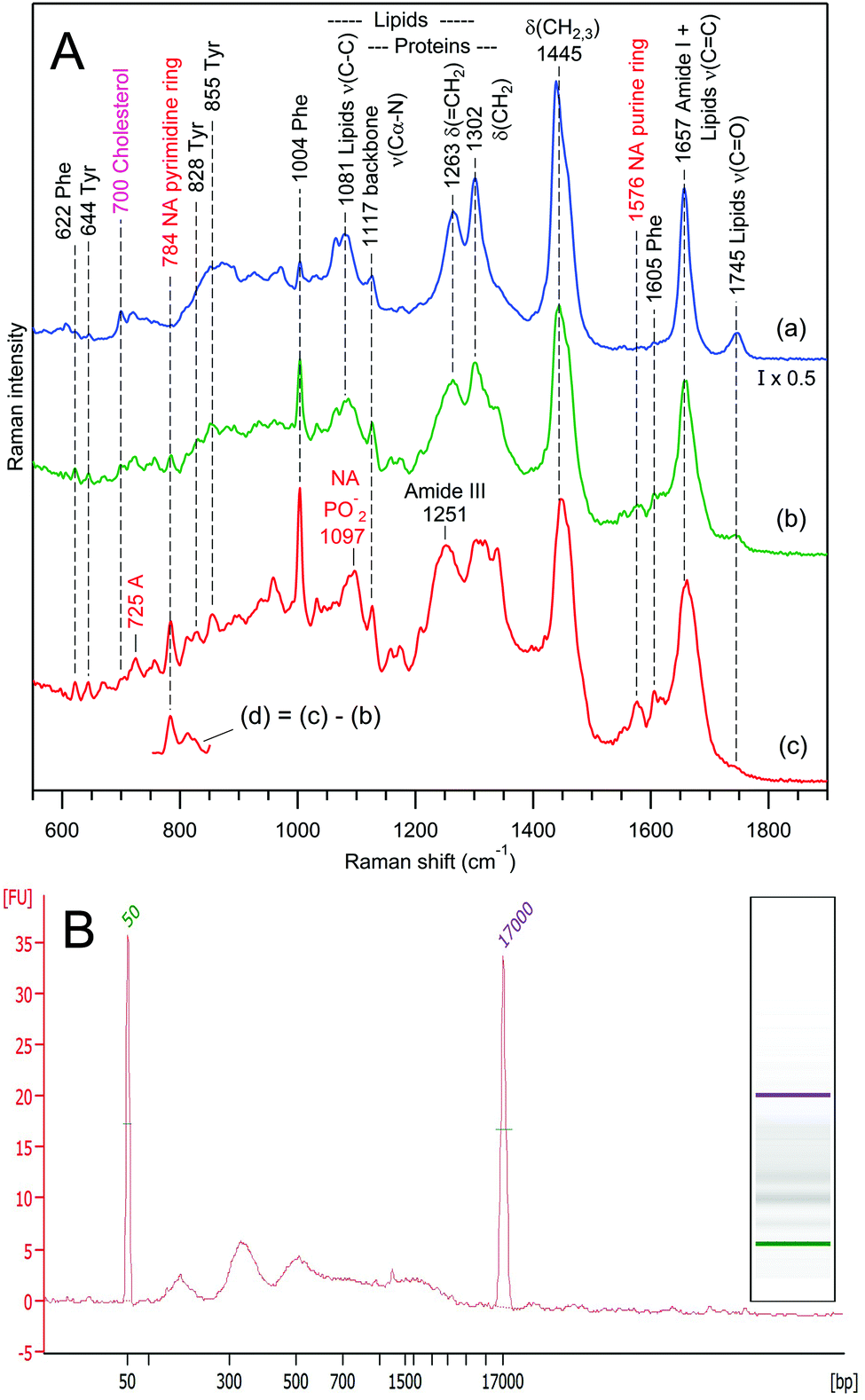 | ||
| Fig. 7 Panel A, Raman spectra of 3 independent vesicle sets (a–c) from the same sample containing exosomes from rat hepatocytes. The intensity of spectrum (a) was multiplied by 0.5 for convenience of visual comparison. The water buffer contribution was subtracted from all spectra. Curve (d) was obtained as the (1:1) difference (d) = (c)–(b). Raman bands assignment according to Table 1. Panel B, electrophoretic size profile (in base pairs) of isolated DNA generated by Bioanalyzer for the same sample. The digital gel recreation of the electrophoresis shows a scale pattern. | ||
To verify the presence of nucleic acids via a Raman-independent method, DNA isolation and Qubit™ quantification were performed for the same EV preparation. The Qubit™ quantification estimated a total of 0.37 ng of DNA per million particles. The electrophoretic profile reveals a characteristic pattern compatible with apoptotic DNA fragmentation, which supports the presence of DNA among EVs (Fig. 7B).
The effect is not limited to exosomes from rat hepatocytes. We have also observed chemically heterogeneous distributions of exosomes in preparations from human urine (Fig. 8) and from the mouse hepatic cell line MLP-29, also (both RTM and Bioanalyzer data, not shown).
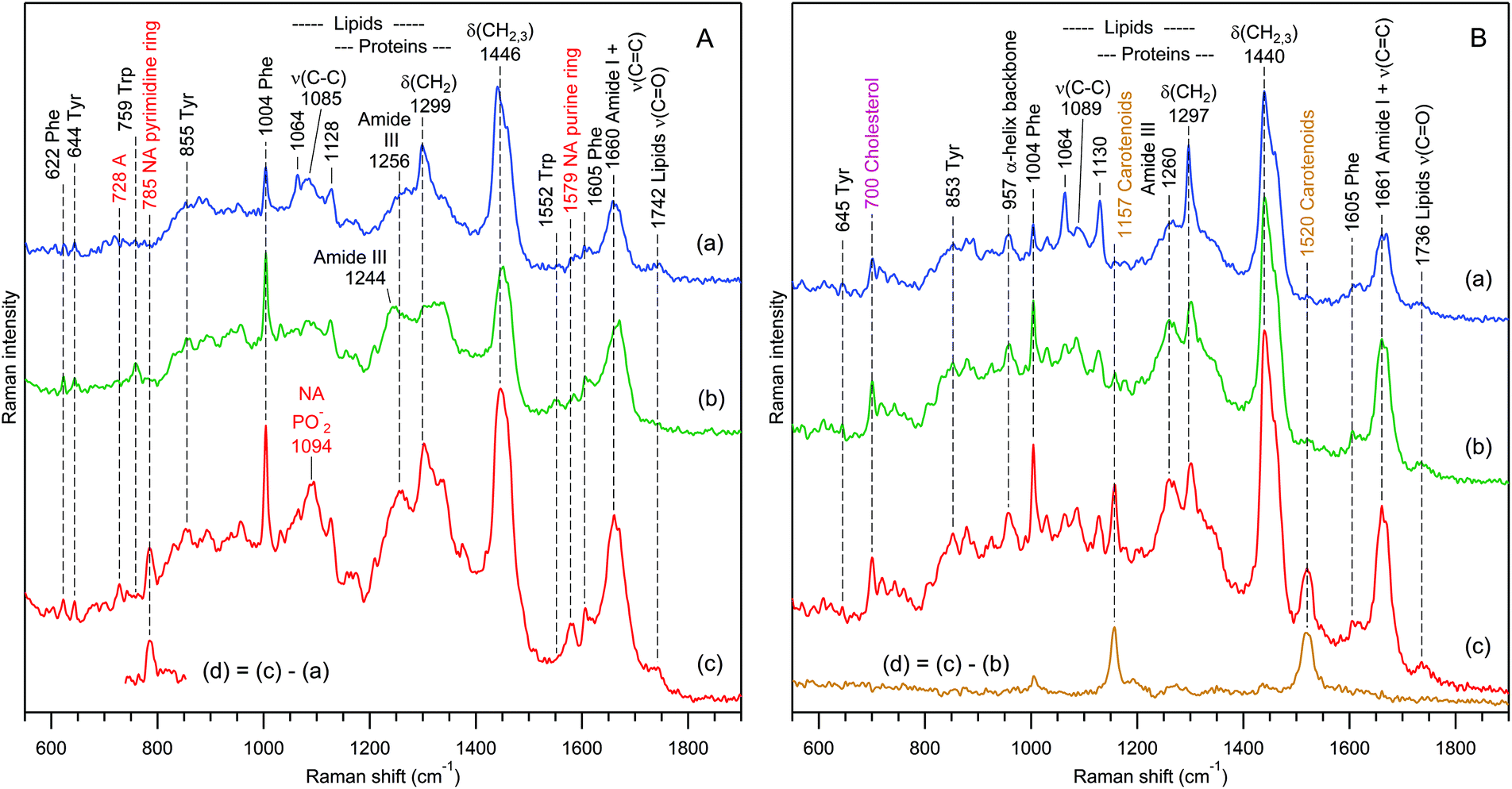 | ||
| Fig. 8 Raman spectra of 3 independent exosome sets (a–c) for samples prepared from the urine of 2 different donors (A and B). The difference spectra (d) reveal the presence of nucleic acids (A) and carotenoids (B) in sets (c). The water buffer contribution was subtracted. Raman bands assignment according to Table 1. | ||
Fig. 8 presents selected RTM results for exosomes from human urine of two different donors. Again, as in the case of rat hepatocytes, one can distinguish three families of EVs on the basis of Raman qualitative analysis. For the first donor's sample (Fig. 8A), there are EVs containing the following: (a) mainly lipids with a weak protein contribution, (b) mainly proteins with a very weak lipid contribution, and (c) strong contributions from proteins, lipids, and nucleic acids. The characteristic fragment of the Raman difference spectrum in Fig. 8A(d) suggests that nucleic acids possess predominantly a B-type helical structure.102
In contrast, the Raman spectra of the second donor's sample (Fig. 8B) contain strong contributions from cholesterol92,93 (characteristic Raman band at 700 cm−1) and carotenoids105 (characteristic Raman band at 1520 cm−1), and do not exhibit any signature of nucleic acids. The grouping is performed in the following manner: (a) predominantly saturated lipids with weak protein contribution, (b) strong contributions from proteins and lipids, and (c) like (b), but with an additional strong carotenoid contribution, which is clearly revealed by a characteristic105,106 Raman difference spectrum (d).
In the case of strong chemical heterogeneity (Fig. 7 and 8), the difference between any two averaged Raman spectra of the same sample is largely non-zero, and varies from one measurement to the other, in contrast to the case of weak intra-sample heterogeneity of Fig. 6. Unfortunately, at the moment, we are not able to establish unequivocally the origin of such strong intra-sample biomolecular variability of EVs. There is most likely more than one origin; the study is still in progress. In this paper, we would only like to attract attention to the effect. The mere fact of the presence of strong biomolecular heterogeneity in EVs might signify the occurrence of important processes within the cell source. For example, it has been shown in our previous work51 that Dictyostelium discoideum cells produce EVs of different compositions during the growth and starvation phases of their life cycle.
Whenever the intra-sample biomolecular heterogeneity of EVs is comparable to or stronger than the inter-sample one, the diagnostic capability of RTM using EVs may be compromised. So far, in a preliminary RTM study, we were not able to pass the blind test of distinguishing between urinal EVs from healthy donors and cancer patients.
Nevertheless, the Raman spectral heterogeneity observed for a given EV sample reflects its biological heterogeneity. In our opinion, the notion of a reference “normal” Raman spectrum of EVs from a healthy donor at the current state of EV preparation/separation art is rather simplified. Deciphering the biological EV heterogeneity108–112 at the level of a single EV or very few EVs is becoming one of the most significant challenges to overcome to understand and control the EV-mediated inter-cellular communication and to validate the appealing promises of EV liquid biopsy113,114 for early non-invasive diagnosis of many human diseases, including cancers. Although we are waiting for an efficient method of EV sorting into subpopulations with homogeneous size distributions, RTM already provides a unique possibility to test the EV biomolecular heterogeneity of a given sample at the single-EV level.
Biomolecular component analysis based on Raman markers
Reconstruction of the biomolecular composition of an EV from its Raman spectrum is a complicated mathematical problem. Even in the case of a single protein, the complete quantitative spectral interpretation is still in progress.86 The superposition of contributions from various proteins, lipids, nucleic acids and other biomolecules constituting an EV further complicates its spectra interpretation. Nevertheless, in some situations, rigorous mathematical treatment of the Raman spectra of a bio-sample based on biomolecular component analysis115 is possible, such as for plaques in human coronary atherosclerosis,116,117 lesions in human breast tissue,118,119 or cell nuclei.115,120 However, a similar quantitative treatment of the unknown biomolecular composition of an arbitrary EV seems difficult at the moment. Note that the widely used methods of multivariate statistical analysis,81–83 in particular principal component analysis,52,55 provide a robust separation of Raman data into clusters, but information about individual biomolecular composition is usually not obtained.Therefore, for a qualitative, and sometimes quantitative, estimation of global biomolecular content, we employ the method of biomolecular component analysis based on separate Raman markers. The idea is not new – the particularity lies in the choice121 of Raman markers. For example, two strong dominant Raman contours – within the range of 1435–1465 cm−1 due to backbone δ(CH2, CH3) deformations in proteins and lipids and within the range of 1640–1690 cm−1 due to overlapping contributions from ν(CC) and ν(CO) stretches in proteins, lipids and nucleic acids – were found to be non-optimal as they cannot provide a reliable separation of individual biomolecular contributions.
Table S1 of the ESI† presents our choice of markers that are essentially non-overlapping narrow and strong Raman bands characteristic of major EV constituting biomolecules. Typical Raman spectra of several nucleic acids, lipids, and proteins recorded by using our Raman setup are presented in Fig. S13–S15 of the ESI.† The most reliable quantitative estimation of concentration can be performed for nucleic acids and carotenoids because their marker bands, pyrimidine ring stretch at 782–788 cm−1 and polyene carbon double bond stretch at 1515–1540 cm−1, respectively, do not overlap with any other major Raman band and are sufficiently strong to dominate the background. Moreover, for nucleic acids, the type of helical structure can be estimated103 by using the position and band shape of the phosphodiester stretch at 810–840 cm−1.
For proteins and lipids, the situation is less straightforward, as their major Raman bands related to the backbone structure largely overlap. For lipids, the most characteristic region lies within 1050–1130 cm−1: saturated acyl chains are responsible for the strong narrow bands in this region (Fig. 4B, 8B(a) and S14†), whereas unsaturated chains with at least one CC double bond are characterized by a broad unresolved contour with a maximum at 1079–1088 cm−1 (Fig. 3, 7A and S14†). Another spectral region characteristic for lipids is 1720–1750 cm−1 which contains CO double-bond stretching of ester COOR. This band does not overlap with any other Raman band thus being specific for many membrane lipids; unfortunately, its weak intensity makes reliable concentration estimation difficult. At the same time, several particular lipids, such as cholesterol and ergosterol, possessing strong characteristic Raman bands at 700–704 and 1602–1604 cm−1, respectively, can be reliably detected and quantified.
Protein Raman spectra (Fig. S15†) are dominated by (i) backbone stretching and bending modes and (ii) contributions from 3 amino acid residues containing aromatic rings: Phe, Tyr and Trp. Since the backbone Raman bands of proteins and lipids largely overlap, we suggest using sharp narrow Raman bands of aromatic amino acids for a qualitative estimation of the protein content in the overall Raman spectrum of EVs. Although quantitative estimation of protein concentration by using this approach is difficult, we are nevertheless able to address another very important question, namely, whether the trapped bioparticles contain any protein contribution. The same question may be addressed for lipids, also, effectively separating EVs and aggregates of proteins.
Fig. 9 explains our biomolecular component analysis approach for the estimation of the local concentration of nucleic acids in interaction with bioparticles.
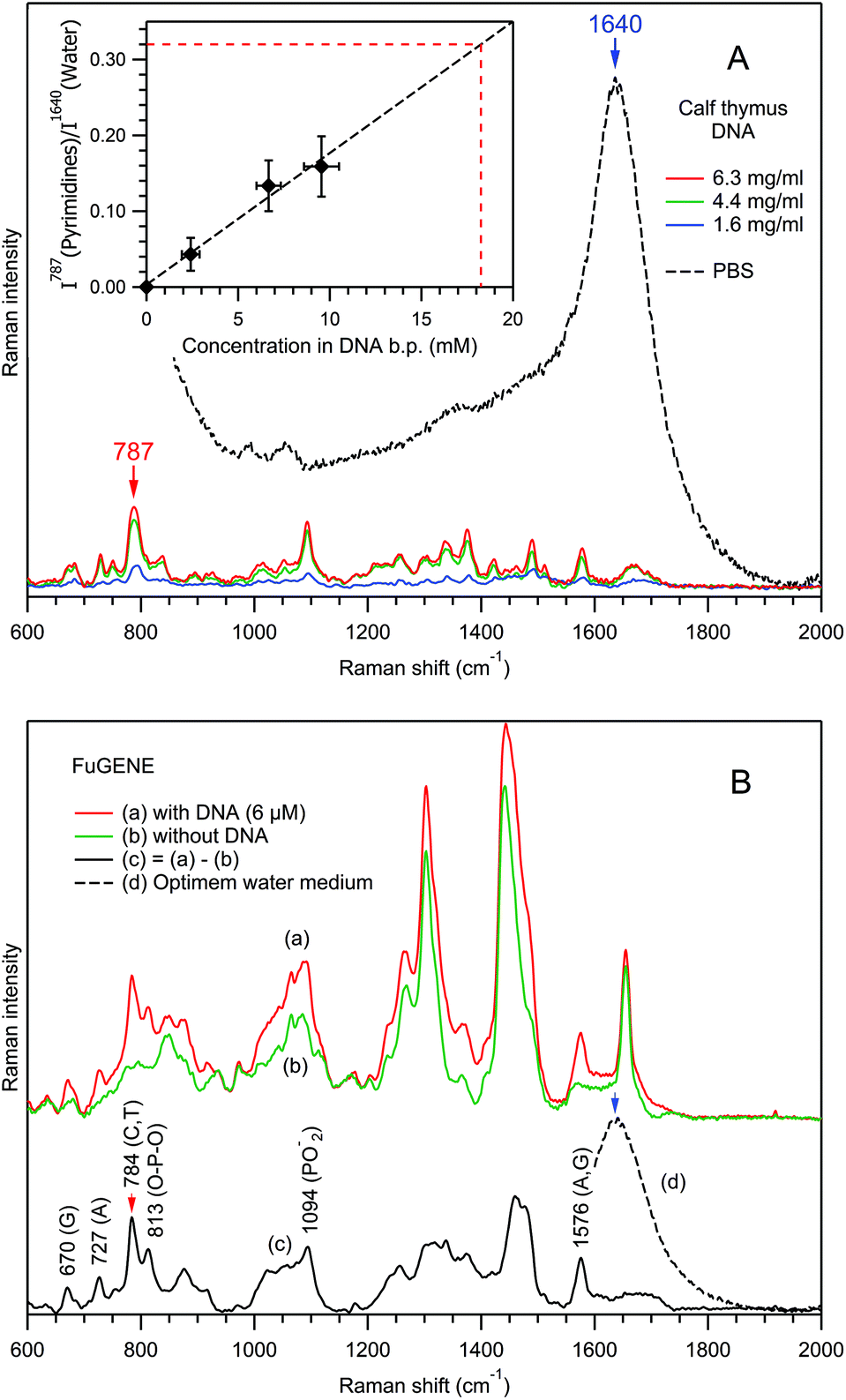 | ||
| Fig. 9 Explanation of biomolecular component analysis for nucleic acids. Panel A, Raman spectra of free calf thymus DNA in PBS (pH 7.4), after PBS contribution (dashed black curve) subtraction, at 3 different concentrations. Inset, the dependence I787(pyrimidines)/I1640(water) versus concentration in DNA base pairs: markers, experimental data; black dashed curve, best fit by a linear function. Panel B, Raman spectra of the transfection reagent FuGENE® in Optimem water medium: (a) with DNA; (b) without DNA; (c) (1:1) difference. Raman spectra of PBS and Optimem water medium were used for background subtraction and intensity normalization. | ||
First, using the example of the Raman spectra of free calf thymus DNA dissolved in PBS (Fig. 9A), the dependence of the normalized Raman intensity of the pyrimidine ring stretching mode at 787 cm−1 has been studied as a function of the concentration in DNA base pairs (CDNA, inset in Fig. 9A). Analysis of the blue spectrum recorded at the lowest CDNA shows that, for non-resonance Raman spectroscopy of nucleic acids, the detection limit is on the order of 1 mM, corresponding to I787/I1640 ≈ 0.02.
As a reference, for a spherical particle of 100 nm diameter, the RTM detection limit of 1 mM corresponds to 315 base pairs imbedded into the particle's volume. However, taking into account the extreme weakness of a single-exosome Raman signal, the detection of such small number of base pairs seems to us rather problematic. However, in the case of multi-vesicle trapping, the application of an appropriate scaling factor helps nucleic acid detection. Indeed, in the experiments of Fig. 7A and 8A, ∼10 and ∼5 exosomes have been trapped respectively.
Next, we studied the effect of a local concentration increase for a nucleic acid bound to a vesicle in the optical trap using the example of the transfection reagent FuGENE®, which is known to efficiently bind DNA molecules. A plasmid DNA of 4700 base pairs was dissolved in Optimem water medium and mixed with FuGENE® particles. It should be noted that the micromolar (6 μM) concentration of free DNA molecules precludes their detection in our RTM experiment, since the detection limit is ∼1 mM. Fig. 9B shows the Raman spectra of the trapped FuGENE® particles, with (curve (a)) and without (curve (b)) the presence of DNA. Spectrum 9B(a) is the average of 141 spectra recorded in 5 independent particle sets; spectrum 9B(b) is the average of 172 spectra from 6 particle sets. Differences between these two spectra are evident, reflecting the attachment of DNA molecules to the FuGENE® particles in the former case.
Indeed, the (1:1) Raman difference spectrum (c) = (a)–(b) contains many prominent DNA bands (Fig. 9B(c), Table 1), including a pyrimidine ring stretch at 784 cm−1. The value of its normalized intensity I784/I1640 ≈ 0.32 allows the estimation of the local concentration in base pairs to be CDNA ≈ 18 mM, from the calibration line in the inset of Fig. 9A. Of course, this value represents an estimation, since the calibration was performed for pyrimidines in calf thymus DNA, whereas in the FuGENE® experiment, we are studying a plasmid DNA. However, since the Raman scattering cross section of the pyrimidine ring stretch should not differ much between various DNAs, such an estimation error is rather small. We note that, if the provenance of the DNA in EVs is not known in advance, such type of discrepancy in the determination of CDNA is intrinsic to the RTM characterization of EVs.
The observed ∼3000-fold increase in local CDNA, from 6 μM in solution to ∼18 mM in the optical trap in the presence of the FuGENE® reagent, is remarkable. This suggests that the RTM technique is able to detect millimolar local concentrations of nucleic acids bound to/incorporated in the individual EVs, as was indeed observed for several preparations of exosomes from rat hepatocytes (Fig. 7A(c)) and human urine (Fig. 8A(c)). We estimate the local nucleic acid concentration to be CDNA ≈ 8 mM in the former case and CDNA ≈ 3 mM in the latter.
Conclusions
The RTM method for the global biomolecular content characterization of a single EV or a small number of EVs or other nanoscale (bio)particles in an aqueous environment in the challenging size range near 100 nm is described in detail. The particularities and potential of RTM are demonstrated using the examples of DOPC liposomes, exosomes from human urine and rat hepatocytes, and a mixed sample of the transfection reagent FuGENE® in diluted DNA solution.We demonstrate that for the exosomes from rat hepatocytes treated with the hepatotoxin Acetaminophen, RTM is sufficiently sensitive to detect the signatures of hepatotoxicity in EVs; therefore, RTM of EVs could be a valuable non-invasive diagnostic tool for liver damage.
We report repeatable observations of strong intra-sample biomolecular heterogeneity of individual exosomes from human urine and rat hepatocytes, due to variable contributions from nucleic acids and carotenoids in some preparations. The origin of the effect is not well understood yet and is still under study. At the same time, we have proven the usefulness of RTM for defining the different subpopulations that are present in an EV preparation. Importantly, RTM enables the characterization of these subpopulations by providing a global signature of biomolecular composition for each of them, at the level of a single EV or very few EVs.
We have proposed and successfully tested the approach of biomolecular component analysis for the estimation of the main EVs’ biomolecular contributions (proteins, lipids, nucleic acids, carotenoids, etc.). We report reliable detection of DNA with a millimolar local concentration for exosomes from human urine and rat hepatocytes, in some preparations.
For many decades, all the observed mammalian EVs were classified into three main categories: exosomes or exosome-like vesicles; microvesicles, also named ectosomes or microparticles; and apoptotic bodies.3 The possible heterogeneity of EVs in each of these main classes was not questioned. However, since 2012, after the creation of the International Society for Extracellular Vesicles (ISEV) devoted to deepening the knowledge on EVs, deciphering EV heterogeneity has become an increasing matter of concern. In fact, it is now one of the most significant challenges to solve to validate the appealing promises of liquid biopsy for early non-invasive diagnoses of many human diseases, including cancers.
Indeed, heterogeneity is everywhere in the human body. Many different types of healthy human cells secrete EVs into body fluids, with increased and varying contributions from sick cells. Furthermore, an important microbiota (there are as many bacteria as cells in the human body122) brings its own capacity to externalize EVs into body fluids, due to the universal process of EV secretion in all three kingdoms114 originating from LUCA, the last universal cell ancestor. Furthermore, viruses are much more numerous than human cells; their size analogy with exosomes suggests that viruses might also contribute to the multitude of the so-called “mammalian EVs”. Therefore, there is an urgent need to develop true robust procedures for classification and separation of the EV subpopulations to exploit all the potentialities arising in the field of EVs.
Many technologies have appeared for EV characterization, most of them addressing bulk concentrations. Therefore, they provide only a mean EV characterization, which is not very informative for the identification of EV subpopulations. However, very recent studies, presented during the ISEV-2018 conference, revealed a strong tendency towards microfluidic separation of only a few vesicles aimed at the clinical development of EVs towards diagnosis and prognosis of diseases. The need for a single-EV analysis is progressively increasing.
RTM is such a sensitive technique that can study a single EV or very few EVs. Although not pertinent for phenotyping the whole EV continuum panel, such as the developed EV arrays that use many specific antibodies,123 RTM is, however, very useful for verifying the overall EV heterogeneity of a given sample. However, the greatest asset of RTM might be the control, at the level of individual EVs, of the global biomolecular composition of the beforehand-separated EV fractions to precisely define their biological action(s) on recipient cells. Future possible association of RTM, NTA and Cryo-TEM methods with the AF4 separation technique77–79 seems to us to be very promising for the characterization of EV subpopulations, being one of the most interesting perspectives for RTM in the field of EV intercellular communications.
Possible RTM applications include, among others, searching for DNA biomarkers, cancer diagnosis, and discrimination between different subpopulations of EVs, lipid bodies, protein aggregates and viruses.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. G. K. would like to thank Raphael Voituriez for the helpful discussion of the dynamics of single-vesicle trapping. We acknowledge the donation of the MLP-29 cell line by Dr Enzo Medico. This work has been supported by the Health Basque Government (2015111149 to JMF), the Movember Foundation (GAP1 to JMF), the Ramón Areces Foundation (to JMF), the Instituto de Salud Carlos III (PI12/01604 to JMF), and the Spanish Ministry of Economy and Competitiveness MINECO (SAF2015-66312 to JMF), with all of them co-financed by the ERDF (FEDER) funds from the European Commission, “A way of making Europe”.References
- C. Thery, L. Zitvogel and S. Amigorena, Nat. Rev. Immunol., 2002, 2, 569–579 CrossRef CAS PubMed.
- S. Mathivanan, H. Ji and R. J. Simpson, J. Proteomics, 2010, 73, 1907–1920 CrossRef CAS PubMed.
- B. Gyorgy, T. G. Szabo, M. Pasztoi, Z. Pal, P. Misjak, B. Aradi, V. Laszlo, E. Pallinger, E. Pap, A. Kittel, G. Nagy, A. Falus and E. I. Buzas, Cell. Mol. Life Sci., 2011, 68, 2667–2688 CrossRef CAS PubMed.
- G. Raposo and W. Stoorvogel, J. Cell Biol., 2013, 200, 373–383 CrossRef CAS PubMed.
- C. Corrado, S. Raimondo, A. Chiesi, F. Ciccia, G. De Leo and R. Alessandro, Int. J. Mol. Sci., 2013, 14, 5338–5366 CrossRef CAS PubMed.
- I. Tatischeff, Cancer Res. Front., 2015, 1, 208–224 CrossRef.
- S. Stremersch, S. C. De Smedt and K. Raemdonck, J. Controlled Release, 2016, 244, 167–183 CrossRef CAS PubMed.
- K. W. Witwer, E. I. Buzas, L. T. Bemis, A. Bora, C. Lasser, J. Lotvall, E. N. Nolte-‘t Hoen, M. G. Piper, S. Sivaraman, J. Skog, C. Thery, M. H. Wauben and F. Hochberg, J. Extracell. Vesicles, 2013, 2, 20360 CrossRef PubMed.
- J. Kowal, G. Arras, M. Colombo, M. Jouve, J. P. Morath, B. Primdal-Bengtson, F. Dingli, D. Loew, M. Tkach and C. Thery, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E968–E977 CrossRef CAS PubMed.
- H. Valadi, K. Ekstrom, A. Bossios, M. Sjostrand, J. J. Lee and J. O. Lotvall, Nat. Cell Biol., 2007, 9, 654–659 CrossRef CAS PubMed.
- C. Thery, M. Ostrowski and E. Segura, Nat. Rev. Immunol., 2009, 9, 581–593 CrossRef CAS PubMed.
- P. D. Robbins and A. E. Morelli, Nat. Rev. Immunol., 2014, 14, 195–208 CrossRef CAS PubMed.
- F. Lavialle, S. Deshayes, F. Gonnet, E. Larquet, S. G. Kruglik, N. Boisset, R. Daniel, A. Alfsen and I. Tatischeff, Int. J. Pharm., 2009, 380, 206–215 CrossRef CAS PubMed.
- A. Tan, J. Rajadas and A. M. Seifalian, Adv. Drug Delivery Rev., 2013, 65, 357–367 CrossRef CAS PubMed.
- I. Tatischeff, C. R. Biol., 2012, 335, 103–106 CrossRef CAS PubMed.
- K. Al-Nedawi, B. Meehan and J. Rak, Cell Cycle, 2009, 8, 2014–2018 CrossRef CAS PubMed.
- E. Pap, E. Pallinger and A. Falus, Crit. Rev. Oncol. Hematol., 2011, 79, 213–223 CrossRef PubMed.
- J. Rak, Front. Pharmacol., 2013, 4, 21 Search PubMed.
- S. Jorfi and J. M. Inal, Biochem. Soc. Trans., 2013, 41, 293–298 CrossRef CAS PubMed.
- H. Peinado, M. Aleckovic, S. Lavotshkin, I. Matei, B. Costa-Silva, G. Moreno-Bueno, M. Hergueta-Redondo, C. Williams, G. Garcia-Santos, C. M. Ghajar, A. Nitadori-Hoshino, C. Hoffman, K. Badal, B. A. Garcia, M. K. Callahan, J. Yuan, V. R. Martins, J. Skog, R. N. Kaplan, M. S. Brady, J. D. Wolchok, P. B. Chapman, Y. Kang, J. Bromberg and D. Lyden, Nat. Med., 2012, 18, 883–891 CrossRef CAS PubMed.
- C. D'Souza-Schorey and J. W. Clancy, Genes Dev., 2012, 26, 1287–1299 CrossRef PubMed.
- A. Ashkin, Phys. Rev. Lett., 1970, 24, 156–159 CrossRef CAS.
- A. Ashkin, J. M. Dziedzic and T. Yamane, Nature, 1987, 330, 769–771 CrossRef CAS PubMed.
- A. Ashkin and J. M. Dziedzic, Science, 1987, 235, 1517–1520 CrossRef CAS PubMed.
- K. C. Neuman and S. M. Block, Rev. Sci. Instrum., 2004, 75, 2787–2809 CrossRef CAS PubMed.
- K. Dholakia, P. Reece and M. Gu, Chem. Soc. Rev., 2008, 37, 42–55 RSC.
- P. M. Bendix and L. B. Oddershede, Nano Lett., 2011, 11, 5431–5437 CrossRef CAS PubMed.
- I. R. Lewis and H. G. M. Edwards, Handbook of Raman Spectroscopy: From the Research Laboratory to the Process Line, Marcel Dekker, Inc., New York, Basel, 2001 Search PubMed.
- R. Thurn and W. Kiefer, Appl. Spectrosc., 1984, 38, 78–83 CrossRef CAS.
- K. Ajito and K. Torimitsu, Lab Chip, 2002, 2, 11–14 RSC.
- C. G. Xie, M. A. Dinno and Y. Q. Li, Opt. Lett., 2002, 27, 249–251 CrossRef PubMed.
- D. P. Cherney, J. C. Conboy and J. M. Harris, Anal. Chem., 2003, 75, 6621–6628 CrossRef CAS PubMed.
- J. M. Sanderson and A. D. Ward, Chem. Commun., 2004, 9, 1120–1121 RSC.
- J. W. Chan, A. P. Esposito, C. E. Talley, C. W. Hollars, S. M. Lane and T. Huser, Anal. Chem., 2004, 76, 599–603 CrossRef CAS PubMed.
- D. V. Petrov, J. Opt. A: Pure Appl. Opt., 2007, 9, S139–S156 CrossRef CAS.
- R. D. Snook, T. J. Harvey, E. C. Faria and P. Gardner, Integr. Biol., 2009, 1, 43–52 RSC.
- D. P. Cherney and J. M. Harris, Annu. Rev. Anal. Chem., 2010, 3, 277–297 CrossRef CAS PubMed.
- J. W. Chan, J. Biophotonics, 2013, 6, 36–48 CrossRef CAS PubMed.
- I. W. Schie and T. Huser, Appl. Spectrosc., 2013, 67, 813–828 CrossRef CAS PubMed.
- B. Redding, M. J. Schwab and Y. Pan, Sensors, 2015, 15, 19021–19046 CrossRef PubMed.
- C. Krafft and J. Popp, Anal. Bioanal. Chem., 2015, 407, 699–717 CrossRef CAS PubMed.
- S. Kumar, T. Verma, R. Mukherjee, F. Ariese, K. Somasundaram and S. Umapathy, Chem. Soc. Rev., 2016, 45, 1879–1900 RSC.
- K. Ajito and K. Torimitsu, Trends Anal. Chem., 2001, 20, 255–262 CrossRef CAS.
- K. Ajito and K. Torimitsu, Appl. Spectrosc., 2002, 56, 541–544 CrossRef CAS.
- H. Tang, H. Yao, G. Wang, Y. Wang, Y. Q. Li and M. Feng, Opt. Express, 2007, 15, 12708–12716 CrossRef PubMed.
- H. W. Wu, J. V. Volponi, A. E. Oliver, A. N. Parikh, B. A. Simmons and S. Singh, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3809–3814 CrossRef CAS PubMed.
- T. J. Moritz, J. A. Brunberg, D. M. Krol, S. Wachsmann-Hogiu, S. M. Lane and J. W. Chan, J. Raman Spectrosc., 2010, 41, 33–39 CrossRef CAS.
- D. P. Cherney, T. E. Bridges and J. M. Harris, Anal. Chem., 2004, 76, 4920–4928 CrossRef CAS PubMed.
- G. A. Myers and J. M. Harris, Anal. Chem., 2011, 83, 6098–6105 CrossRef CAS PubMed.
- J. W. Chan, D. Motton, J. C. Rutledge, N. L. Keim and T. Huser, Anal. Chem., 2005, 77, 5870–5876 CrossRef CAS PubMed.
- I. Tatischeff, E. Larquet, J. M. Falcon-Perez, P.-Y. Turpin and S. G. Kruglik, J. Extracell. Vesicles, 2012, 1, 19179 CrossRef PubMed.
- Z. I. Smith, C. Lee, T. Rojalin, R. P. Carney, S. Hazari, A. Knudson, K. Lam, H. Saari, E. Lasaro-Ibanez, T. Viitala, T. Laaksonen, M. Yliperttula and S. Wachsmann-Hogiu, J. Extracell. Vesicles, 2015, 4, 28533 CrossRef PubMed.
- E. I. Buzas, C. Gardiner, C. Lee and Z. J. Smith, Platelets, 2017, 28, 249–255 CrossRef CAS PubMed.
- R. P. Carney, S. Hazari, M. Colquhoun, T. Di, B. Hwang, M. S. Mulligan, J. D. Bryers, E. Girda, G. S. Leiserowitz, Z. J. Smith and K. S. Lam, Anal. Chem., 2017, 89, 5357–5363 CrossRef CAS PubMed.
- W. Lee, A. Nanou, L. Rikkert, F. A. W. Coumans, C. Otto, L. W. M. M. Terstappen and H. L. Offerhaus, Anal. Chem., 2018, 90, 11290–11296 CrossRef CAS PubMed.
- A. Gualerzi, S. Niada, C. Giannasi, S. Picciolini, C. Morasso, R. Vanna, V. Rossella, M. Masserini, M. Bedoni, F. Ciceri, M. E. Bernardo, A. T. Brini and F. Gramatica, Sci. Rep., 2017, 7, 9820 CrossRef PubMed.
- D. Cialla, S. Pollok, C. Steinbrucker, K. Weber and J. Popp, Nanophotonics, 2014, 3, 383–411 CAS.
- C. Lee, R. P. Carney, S. Hazari, Z. J. Smith, A. Knudson, C. S. Robertson, K. S. Lam and S. Wachsmann-Hogiu, Nanoscale, 2015, 7, 9290–9297 RSC.
- L. B. Kong, C. Lee, C. M. Earhart, B. Cordovez and J. W. Chan, Opt. Express, 2015, 23, 6793–6802 CrossRef CAS PubMed.
- F. R. Madiyar, S. Bhana, L. Z. Swisher, C. T. Culbertson, X. H. Huang and J. Li, Nanoscale, 2015, 7, 3726–3736 RSC.
- S. F. Zong, L. Wang, C. Chen, J. Lu, D. Zhu, Y. Z. Zhang, Z. Y. Wang and Y. P. Cui, Anal. Methods, 2016, 8, 5001–5008 RSC.
- B. Fazio, C. D'Andrea, A. Foti, E. Messina, A. Irrera, M. G. Donato, V. Villari, N. Micali, O. M. Marago and P. G. Gucciardi, Sci. Rep., 2016, 6, 26952 CrossRef CAS PubMed.
- S. Stremersch, M. Marro, B. E. Pinchasik, P. Baatsen, A. Hendrix, S. C. De Smedt, P. Loza-Alvarez, A. G. Skirtach, K. Raemdonck and K. Braeckmans, Small, 2016, 12, 3292–3301 CrossRef CAS PubMed.
- J. Park, M. Hwang, B. Choi, H. Jeong, J. H. Jung, H. K. Kim, S. Hong, J. H. Park and Y. Choi, Anal. Chem., 2017, 89, 6695–6701 CrossRef CAS PubMed.
- C. Lee, R. Carney, K. Lam and J. W. Chan, J. Raman Spectrosc., 2017, 48, 1771–1776 CrossRef CAS.
- F. Olson, C. A. Hunt, F. C. Szoka, W. J. Vail and D. Papahadjopoulos, Biochim. Biophys. Acta, 1979, 557, 9–23 CrossRef CAS.
- J. Conde-Vancells, E. Rodriguez-Suarez, E. Gonzalez, A. Berisa, D. Gil, N. Embade, M. Valle, Z. Luka, F. Elortza, C. Wagner, S. C. Lu, J. M. Mato and J. M. Falcon-Perez, Proteomics: Clin. Appl., 2010, 4, 416–425 CAS.
- F. Royo, L. Moreno, J. Mleczko, L. Palomo, E. Gonzalez, D. Cabrera, A. Cogolludo, F. P. Vizcaino, S. van-Liempd and J. M. Falcon-Perez, Sci. Rep., 2017, 7, 42798 CrossRef CAS PubMed.
- C. Thery, S. Amigorena, G. Raposo and A. Clayton, Curr. Protoc. Cell Biol., 2006, 30, 3.22.1–3.22.29 CrossRef PubMed.
- F. Royo, K. Schlangen, L. Palomo, E. Gonzalez, J. Conde-Vancells, A. Berisa, A. M. Aransay and J. M. Falcon-Perez, PLoS One, 2013, 8, e68693 CrossRef CAS PubMed.
- V. Wintgens, C. Le Coeur, C. Amiel, J. M. Guigner, J. G. Harangozo, Z. Miskolczy and L. Biczok, Langmuir, 2013, 29, 7682–7688 CrossRef CAS PubMed.
- C. J. De Grauw, M. Sijtsema, C. Otto and J. Greve, J. Microsc., 1997, 188, 273–279 CrossRef CAS.
- Y. Zimmels, Langmuir, 1992, 8, 2448–2454 CrossRef CAS.
- Y. Zimmels, J. Appl. Phys., 1994, 76, 5154–5160 CrossRef CAS.
- C. Song, P. Wang and H. A. Makse, Nature, 2008, 453, 629–632 CrossRef CAS PubMed.
- L. R. Huang, E. C. Cox, R. H. Austin and J. C. Sturm, Science, 2004, 304, 987–990 CrossRef CAS PubMed.
- A. Zattoni, D. C. Rambaldi, P. Reschiglian, M. Melucci, S. Krol, A. M. C. Garcia, A. Sanz-Medel, D. Roessner and C. Johann, J. Chromatogr. A, 2009, 1216, 9106–9112 CrossRef CAS PubMed.
- A. Zattoni, B. Roda, F. Borghi, V. Marassi and P. Reschiglian, J. Pharm. Biomed. Anal., 2014, 87, 53–61 CrossRef CAS PubMed.
- H. Zhang, D. Freitas, H. S. Kim, K. Fabijanic, Z. Li, H. Chen, M. T. Mark, H. Molina, A. B. Martin, L. Bojmar, J. Fang, S. Rampersaud, A. Hoshino, I. Matei, C. M. Kenific, M. Nakajima, A. P. Mutvei, P. Sansone, W. Buehring, H. Wang, J. P. Jimenez, L. Cohen-Gould, N. Paknejad, M. Brendel, K. Manova-Todorova, A. Magalhaes, J. A. Ferreira, H. Osorio, A. M. Silva, A. Massey, J. R. Cubillos-Ruiz, G. Galletti, P. Giannakakou, A. M. Cuervo, J. Blenis, R. Schwartz, M. S. Brady, H. Peinado, J. Bromberg, H. Matsui, C. A. Reis and D. Lyden, Nat. Cell Biol., 2018, 20, 332–343 CrossRef CAS.
- A. Sabelnikov and C. R. Kempf, Anal. Biochem., 2008, 383, 346–348 CrossRef CAS PubMed.
- M. Hedegaard, C. Matthaus, S. Hassing, C. Krafft, M. Diem and J. Popp, Theor. Chem. Acc., 2011, 130, 1249–1260 Search PubMed.
- T. W. Bocklitz, S. Guo, O. Ryabchykov, N. Vogler and J. Popp, Anal. Chem., 2016, 88, 133–151 CrossRef CAS PubMed.
- H. J. Byrne, P. Knief, M. E. Keating and F. Bonnier, Chem. Soc. Rev., 2016, 45, 1865–1878 RSC.
- T. Alempijevic, S. Zec and T. Milosavljevic, World J. Hepatol., 2017, 9, 491–502 CrossRef PubMed.
- N. C. Maiti, M. M. Apetri, M. G. Zagorski, P. R. Carey and V. E. Anderson, J. Am. Chem. Soc., 2004, 126, 2399–2408 CrossRef CAS PubMed.
- R. Tuma, J. Raman Spectrosc., 2005, 36, 307–319 CrossRef CAS.
- B. Hernandez, F. Pfluger, A. Adenier, S. G. Kruglik and M. Ghomi, J. Phys. Chem. B, 2010, 114, 15319–15330 CrossRef CAS PubMed.
- B. Hernandez, F. Pfluger, S. G. Kruglik and M. Ghomi, J. Raman Spectrosc., 2013, 44, 827–833 CrossRef CAS.
- A. Rygula, K. Majzner, K. M. Marzec, A. Kaczor, M. Pilarczyk and M. Baranska, J. Raman Spectrosc., 2013, 44, 1061–1076 CrossRef CAS.
- B. Hernandez, Y. M. Coic, F. Pfluger, S. G. Kruglik and M. Ghomi, J. Raman Spectrosc., 2016, 47, 210–220 CrossRef CAS.
- B. P. Gaber and W. L. Peticolas, Biochim. Biophys. Acta, 1977, 465, 260–274 CrossRef CAS.
- E. Kocisova, A. Antalik and M. Prochazka, Chem. Phys. Lipids, 2013, 172, 1–5 CrossRef PubMed.
- K. Czamara, K. Majzner, M. Z. Pacia, K. Kochan, A. Kaczor and M. Baranska, J. Raman Spectrosc., 2015, 46, 4–20 CrossRef CAS.
- Y. S. Huang, T. Karashima, M. Yamamoto, T. Ogura and H. Hamaguchi, J. Raman Spectrosc., 2004, 35, 525–526 CrossRef CAS.
- V. V. Pully and C. Otto, J. Raman Spectrosc., 2009, 40, 473–475 CrossRef CAS.
- L.-D. Chiu, F. Hullin-Matsuda, T. Kobayashi, H. Torii and H.-O. Hamaguchi, J. Biophotonics, 2012, 5, 724–728 CrossRef CAS PubMed.
- V. Kaliaperumal and H.-O. Hamaguchi, Chem. Rec., 2012, 12, 567–580 CrossRef CAS PubMed.
- Y. S. Huang, T. Karashima, M. Yamamoto and H. Hamaguchii, J. Raman Spectrosc., 2003, 34, 1–3 CrossRef CAS.
- R. C. Lord and G. J. Thomas, Spectrochim. Acta, Part A, 1967, A23, 2551–2591 CrossRef.
- M. Tsuboi, S. Takahashi, S. Muraishi, T. Kajiura and S. Nishimura, Science, 1971, 174, 1142–1144 CrossRef CAS.
- S. C. Erfurth, E. J. Kiser and W. L. Peticolas, Proc. Natl. Acad. Sci. U. S. A., 1972, 69, 938–941 CrossRef CAS.
- B. Prescott, W. Steinmetz and G. J. Thomas, Biopolymers, 1984, 23, 235–256 CrossRef CAS PubMed.
- G. Hui-Bon-Hoa, H. Kaddour, J. Vergne, S. G. Kruglik and M. C. Maurel, BMC Biophys., 2014, 7, 2 CrossRef PubMed.
- D. Gill, R. G. Kilponen and L. Rimai, Nature, 1970, 227, 743–744 CrossRef CAS.
- L. Rimai, M. E. Heyde and D. Gill, J. Am. Chem. Soc., 1973, 95, 4493–4501 CrossRef CAS.
- N. Tschirner, M. Schenderlein, K. Brose, E. Schlodder, M. A. Mroginski, C. Thomsen and P. Hildebrandt, Phys. Chem. Chem. Phys., 2009, 11, 11471–11478 RSC.
- L. Palomo, J. E. Mleczko, M. Azkargorta, J. Conde-Vancells, E. Gonzalez, F. Elortza, F. Royo and J. M. Falcon-Perez, Hepatol. Commun., 2018, 2, 1064–1079 CrossRef CAS PubMed.
- E. Willms, H. J. Johansson, I. Mager, Y. Lee, K. E. Blomberg, M. Sadik, A. Alaarg, C. I. Smith, J. Lehtio, S. El Andaloussi, M. J. Wood and P. Vader, Sci. Rep., 2016, 6, 22519 CrossRef CAS PubMed.
- B. Giebel, Ann. Transl. Med., 2017, 5, 150 CrossRef PubMed.
- D. W. Greening, R. Xu, S. K. Gopal, A. Rai and R. J. Simpson, Expert Rev. Proteomics, 2017, 14, 69–95 CrossRef CAS PubMed.
- D. Zabeo, A. Cvjetkovic, C. Lasser, M. Schorb, J. Lotvall and J. L. Hoog, J. Extracell. Vesicles, 2017, 6, 1329476 CrossRef PubMed.
- M. Tkach, J. Kowal and C. Thery, Philos. Trans. R. Soc., B, 2018, 373, 20160479 CrossRef PubMed.
- A. Di Meo, J. Bartlett, Y. Cheng, M. D. Pasic and G. M. Yousef, Mol. Cancer, 2017, 16, 80 CrossRef PubMed.
- D. Armstrong and D. E. Wildman, J. Pathol. Transl. Med., 2018, 52, 1–8 CrossRef PubMed.
- A. N. Kuzmin, A. Pliss and A. V. Kachynski, J. Raman Spectrosc., 2013, 44, 198–204 CrossRef CAS.
- H. P. Buschman, J. T. Motz, G. Deinum, T. J. Romer, M. Fitzmaurice, J. R. Kramer, A. van der Laarse, A. V. Bruschke and M. S. Feld, Cardiovasc. Pathol., 2001, 10, 59–68 CrossRef CAS PubMed.
- H. P. Buschman, G. Deinum, J. T. Motz, M. Fitzmaurice, J. R. Kramer, A. van der Laarse, A. V. Bruschke and M. S. Feld, Cardiovasc. Pathol., 2001, 10, 69–82 CrossRef CAS PubMed.
- K. E. Shafer-Peltier, A. S. Haka, M. Fitzmaurice, J. Crowe, J. Myles, R. R. Dasari and M. S. Feld, J. Raman Spectrosc., 2002, 33, 552–563 CrossRef CAS.
- A. S. Haka, K. E. Shafer-Peltier, M. Fitzmaurice, J. Crowe, R. R. Dasari and M. S. Feld, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12371–12376 CrossRef CAS PubMed.
- A. Pliss, A. N. Kuzmin, A. V. Kachynski and P. N. Prasad, Biophys. J., 2010, 99, 3483–3491 CrossRef CAS PubMed.
- N. Uzunbajakava, A. Lenferink, Y. Kraan, E. Volokhina, G. Vrensen, J. Greve and C. Otto, Biophys. J., 2003, 84, 3968–3981 CrossRef CAS PubMed.
- R. Sender, S. Fuchs and R. Milo, Cell, 2016, 164, 337–340 CrossRef CAS PubMed.
- M. Jorgensen, R. Baek, S. Pedersen, E. K. Sondergaard, S. R. Kristensen and K. Varming, J. Extracell. Vesicles, 2013, 2, 20920 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nr04677h |
| This journal is © The Royal Society of Chemistry 2019 |