DOI:
10.1039/C8NR03364A
(Paper)
Nanoscale, 2019,
11, 9341-9352
Utility of macrophages in an antitumor strategy based on the vectorization of iron oxide nanoparticles†
Received
24th April 2018
, Accepted 18th December 2018
First published on 5th April 2019
Abstract
Many solid tumors and their metastases are still resistant to current cancer treatments such as chemo- and radiotherapy. The presence of a small population of Cancer Stem Cells in tumors is held responsible for relapses. Moreover, the various physical barriers of the organism (e.g. blood–brain barrier) prevent many drugs from reaching the target cells. In order to alleviate this constraint, we suggest a Trojan horse strategy consisting of intravascular injection of macrophages loaded with therapeutic nanoparticles (an iron nanoparticle-based solution marketed under the name of FERINJECT®) to bring a high quantity of the latter to the tumor. The aim of this article is to assess the response of primary macrophages to FERINJECT® via functional assays in order to ensure that the macrophages loaded with these nanoparticles are still relevant for our strategy. Following this first step, we demonstrate that the loaded macrophages injected into the bloodstream are able to migrate to the tumor site using small-animal imaging. Finally, using synchrotron radiation, we validate an improvement of the radiotherapeutic effect when FERINJECT®-laden macrophages are deposited at the vicinity of cancer cells and irradiated.
1. Introduction
Cancer is the world's second leading cause of death, and was responsible for 8.8 million deaths in 2015. Nearly 1 in 6 deaths is due to cancer1 despite major progress in the development of anti-tumoral treatments such as surgery, chemotherapy, radiotherapy and immunotherapy. Solid tumors remain responsible for a high number of deaths due to the presence of cancer stem cells (CSCs) that are particularly resistant to current cancer treatments2,3 and are consequently responsible for relapses and metastases often turning out to be fatal. CSCs, like normal stem cells, are capable of both self-renewal and differentiation into multiple cells. The CSC population is generally small, but the number of CSCs depends on the type of tumor with a progression rate varying from <0.1% to 30%.4–6 However, they are responsible for high cellular heterogeneity in the tumor7—one of the main reasons explaining resistance to chemotherapy. CSC resistance is reinforced by the presence of protective mechanisms such as drug efflux transporters at their surface (e.g. ATP-binding cassette including P-glycoproteins), that prevent the accumulation of drugs within cells.6 Moreover, contrary to non-CSCs, CSCs have a high DNA repair capacity and anti-apoptotic mechanisms that jeopardize the efficiency of most current anticancer strategies.6,8 CSCs are also resistant to current radiotherapy9 because they are often in a quiescent state.10
To increase the efficiency of ionizing radiations in treating solid and resistant tumors, several pre-clinical studies have combined High-Z-element nanoparticles (HZE-NP) with radiation therapy in order to increase the intensity of radiation without damaging the surrounding healthy tissue. This strategy makes it possible to increase the contrast between healthy and cancerous tissues in absorbing ionizing radiation.11 The stimulation of HZE-NP liberates photoelectrons in situ. Their short-range allows these photoelectrons to kill, primarily, tumor cells with minimal damage to surrounding tissue.12,13 In current clinical experiments HZE-NP are generally injected directly into the tumor, with the most commonly used HZE-NP being gold NP.14 The three main constraints of this strategy are: (1) injecting HZE-NP into the tumor can cause injury and is therefore an invasive method. Moreover, some cancer cells have a low endocytosis capacity,15 and HZE-NP can spread outside the tumor, thereby limiting their specificity. (2) Since metastases are not always detectable with current imaging technologies, it is not possible to inject HZE-NP directly into non-detectable small tumors. (3) Gold is a rare and expensive material.16 (4) Persistent NP such as gold NP may cause adverse effects after the tumor has been treated. Another method consists of intravascular HZE-NP administration in the hope that they will spontaneously cross blood vessels to reach the tumor by means of the Enhanced Permeability and Retention (EPR) effect. Indeed, tumors are highly vascularized and have poor lymphatic drainage. This method has been considered in the case of many solid tumors, but the overall results have revealed that less than 2% of injected NP reached the tumor. In the particular case of glioblastoma (GBM), due to the blood brain barrier, the ratio of NP in the tumor can be even lower.17 However, the use of nanoparticles to deliver drugs, unlike the traditional use of drugs alone, improves the therapeutic effect. Moreover, functionalizing particles using antibodies or ligands makes it possible to increase the uptake and the specific targeting of the drug toward specific cells as in the case of cancer cells. This strategy is still controversial in comparison to non-functionalized nanoparticles.18,19
Often, active targeting by functionalized particles is effective in cell culture, but many studies have shown that, in vivo, functionalization is much less efficient20 due to the destruction and/or inactivation of ligands at the surface of particles (opsonization) and the difficulty of crossing the organism's various barriers. In the present article, we therefore suggest a new, Trojan horse strategy in order to protect particles, overcome the aforementioned constraints and treat resistant tumors using primary macrophages to specifically carry HZE-NP directly to the tumor. Inducing inflammation in the tumor makes it possible to recruit many circulating immune cells, such as neutrophils, lymphocytes21,22 and monocytes/macrophages.23 We chose to use monocytes/macrophages to transport NP-Fe2O3 for four reasons: (1) macrophages have a high phagocytic capacity and can store a large quantity of HZE NP-Fe2O3. (2) Macrophages are known to be present in tumors as Tumor-Associated Macrophages (TAM).24,25 (3) These cells are able to cross all body barriers and migrate toward the inflammatory tumor zone.26 (4) Monocytes are present in the bloodstream and are simple to harvest and differentiate in vitro into macrophages. Like titanium, gadolinium, quantum dots, etc., iron is an HZE, releasing photoelectrons when stimulated by X-rays. Beams at 18 KeV cause iron to release photoelectrons within an average range of 3 μm.27 Iron is a cheap and abundant metal, and iron oxide nanoparticles are easy to produce. Moreover, macrophages play an important role in iron homeostasis so that iron, unlike other metals, can easily be managed in large quantities by macrophages without inducing a strong risk of disrupting their viability and their functionalities.28 Consequently, we chose iron oxide nanoparticles to investigate tumor treatment by means of a Trojan horse strategy. In this study, we directly tested an NP-Fe2O3 source having marketing authorization,29,30i.e. FERINJECT®. The goal was to evaluate whether FERINJECT® impacted the macrophages in different ways.
Our strategy included six steps. We first focused on the characterization of FERINJECT®31,32 nanoparticles, using dynamic light scattering (DLS) and transmission electronic microscopy (TEM). We then evaluated the best working dose by performing viability assays, followed then by quantifying the amount of particles and iron loaded within the cells. Next, we assessed the effects of FERINJECT® on the functionality of macrophages, in vitro, to check whether our iron-loaded macrophages were still able to migrate toward the tumor (sensitivity and motility assessment). By using non-invasive small-animal imaging, the next step was to confirm that the iron-loaded primary macrophages intravenously injected into mice were able to migrate toward the tumor. Finally, we assessed the improved effects of radiotherapy on tumor cells when they were at the vicinity of FERINJECT®-laden macrophages.
2. Materials and methods
2.1. Nanoparticles
Iron carboxymaltose (FERINJECT®, 50 mg mL−1) was purchased from Vifor Pharma (Bern, Switzerland). The hydrodynamic diameter and particle size distribution were characterized by dynamic light scattering (DLS) after dilution in H2O or culture medium DMEM after 0 h or 24 h of incubation at 37 °C, 5% CO2. Nanoparticles were diluted at 10 μg mL−1 for measurement. Titanium oxide nanoparticles (21 nm) were purchased from Sigma-Aldrich (catalogue number 718467).
2.2. Transmission electron microscopy (TEM) for nanoparticle characterization
The negative stain Mica-carbon Flotation Technique (MFT) was used: samples were absorbed by the clean side of a carbon film on mica, stained and transferred to a 400-mesh copper grid. The images were captured under low-dose conditions (<10 e− Å−2) at a magnification of 11Kx, 13Kx, 23Kx and 30Kx with defocus values between 1.2 and 2.5 μm on a Tecnai 12 LaB6 electron microscope at 120 kV accelerating voltage using a CCD Camera Gatan Orius 1000.
2.3. Cell culture
Primary mouse macrophages were obtained from bone marrow as described by Schleicher and Bogdan,33 Tarakanova et al.34 and Triboulet et al.35 Bone marrow was aseptically collected from the femur and tibia of 6 weeks-old C57Bl6 mice sacrificed by cervical dislocation. The marrow plugs were dissociated by repeated flushing with the culture medium (DMEM supplemented with 20% v/v L929 cell culture supernatant, 10% v/v fetal bovine serum, 5% v/v horse serum, 2 mM glutamine and 1 mM sodium pyruvate), and cells were plated on non-adherent cell culture T175 flasks (Greiner bio-one CellStar). An equal volume of medium was added after 4 days in culture. After 7 days in culture, the cells were scraped off and replated at 1 million cells per mL in the same medium, lowering the L929 supernatant to 10% v/v. Mature macrophages were obtained after an additional 3 days, and were viable and stable up to 2 weeks, provided that the culture medium be renewed every 3 days. For treatment with iron oxide nanoparticles, the following protocol was used: cells were first seeded at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 cells per mL in 6-well plates (2 mL per well). They were exposed to nanoparticles on the following day and harvested after a further 24 h in culture. For all assays, cell viability was measured using either trypan blue exclusion or propidium iodide, the latter with a flow cytometer. Nanoparticles internalization was assessed using the light scattering of the cells in the flow cytometer (side scattering parameter).
000000 cells per mL in 6-well plates (2 mL per well). They were exposed to nanoparticles on the following day and harvested after a further 24 h in culture. For all assays, cell viability was measured using either trypan blue exclusion or propidium iodide, the latter with a flow cytometer. Nanoparticles internalization was assessed using the light scattering of the cells in the flow cytometer (side scattering parameter).
9L (from Sigma, catalog number: 94110705) and F98 glioma (from ATCC, catalog number: CRL-2397) were obtained, respectively, from Sigma (Darmstadt Germany) and ATCC (Middlesex UK). Both cell lines were grown in DMEM GlutaMAX high glucose with pyruvate, supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin at 37 °C, 5% CO2. Both cell lines were grown in monolayers and when they became confluent, trypsin was used for passaging.
2.4. Perls staining
Cells were incubated at 500000 cells per mL for 24 h at 37 °C, 5% CO2 in 6-well plates. They were then exposed to 1 mg mL−1 FERINJECT® for 24 h. Cells were rinsed twice with PBS and fixed with 4% PFA for 20 minutes at room temperature. Then, they were rinsed twice with PBS before adding 2 mL of 10 mg mL−1 potassium ferrocyanide(II) in 3% hydrochloric acid. Cells were then incubated for 15 min at 60 °C to obtain the blue color reaction. After two rinses with PBS, cells were counterstained for 5 minutes with safranine 2.5 mg mL−1 and rinsed twice with PBS before microscopic examination.
2.5. Quantitative bathophenanthroline assay
Cells were incubated at 500000 per mL for 24 h at 37 °C, 5% CO2 in 6 well plates. Then, they were treated with 1 mg mL−1 of FERINJECT® for 24 h. Cells were recovered using centrifugation at 290g and were washed twice with PBS. Cell pellets were dissolved in 500 μL of aqua regia (mix of HCL 3 M and HNO3 2.5 M) for 2 days. 180 μL of the cell lysate were taken, to which, in respective order, 380 μL of saturated ammonium acetate, 72 μL of 380 mg mL−1 sodium ascorbate and 144 μL of 17 mg mL−1 bathophenantroline dissulfonate were added. The mixture was incubated at room temperature for 30 minutes. Absorbance was measured at 535 nm. The quantity of iron was calculated with a calibration range from 0 to 500 μg mL−1 of iron (Mohr's salt).
2.6. Quantitative ICP-MS method
Cellular uptake of FERINJECT® was measured by inductively coupled plasma mass spectrometry (ICP-MS). 500000 cells were plated in 6-well plates. Twenty-four hours later, the media was changed with media containing different concentrations of FERINJECT®. After 24 h incubation, the wells were rinsed twice with PBS and were trypsinized. Cells were counted and samples centrifuged before removing the supernatants. Before ICP-MS measurement, samples were mineralized in 500 μL nitric acid 67% and for measure, diluted to 1/100 in ultrapure water containing Gallium (internal standard at 620 nmol L−1). Data depicted the average iron mass uptake per cell.
2.7. Phagocytosis assay
The phagocytic activity was measured using fluorescent latex beads (1 μm diameter, green labeled catalog number L4655 from Sigma-Aldrich) and flow cytometry, as described by Abel et al.36 and Triboulet et al.35
2.8. NO production
The cells were grown to confluence in a 6-well plate. Half of the wells were then treated with 100 ng mL−1 lipopolysaccharide (LPS, from salmonella), and arginine monohydrochloride was added to all wells (5 mM final concentration) to give a high concentration of substrate for nitric oxide synthases. After 24 h incubation, the cell culture medium was recovered, centrifuged at 10000g for 10 minutes to remove cells and debris, and the nitrite concentration was measured after adding an equal volume of Griess reagent (Griess’ reagent for nitrite, catalog number 03553 from Sigma-Aldrich). It was then incubated at room temperature for 30 minutes. Absorbance was measured at 540 nm.
2.9. Cytokines
Tumor necrosis factor (TNFα), interleukin 6 (IL-6), monocyte chemoattractant protein-1 (MCP-1) and interleukin 10 (IL10) concentrations were measured in the supernatant culture of cells exposed to NP and activated by LPS, using the Mouse Inflammation Cytometric Bead Array kit (BD Biosciences, Rungis, France) according to the manufacturer's instructions. Measurements were performed on a Facscalibur flow cytometer, and the data were analyzed using FCAP Array software.
2.10. F-Actin staining
Cells were cultured at 80000 cells per mL on coverslips placed in 6-well plates and exposed to NP for 24 h at 37 °C, 5% CO2. They were then washed twice in PBS before being fixed in 4% paraformaldehyde for 30 minutes at room temperature. Next, cells were permeabilized with Triton X 100 at 0.1% diluted in PBS for 5 minutes at room temperature, then washed twice in PBS and incubated with 500 nM Phalloidin-Atto 550 (Sigma-Aldrich) for 20 minutes at room temperature in the dark. Finally, the cells attached to the coverslips were washed twice in PBS and mounted with “Vectashield mounting medium for fluorescence with DAPI” (ThermoScientfic, Waltham, Massachusetts USA). Pictures were obtained using the LSM 800 confocal microscope (Zeiss, Oberkochen, Germany), lens 63×/1.4 oil and numerical zoom = 2. Colors were added using FIJI software.
2.11. Transwell migration assay
Cells were seeded at 200000 per Transwell filter (5 μm, Sigma-Aldrich) in the upper compartment and incubated for 24 h at 37 °C, 5% CO2. The next day, nanoparticles were added for 24 h in both compartments of the transwells. At the end of incubation period, culture medium was removed from the upper compartment. Transwell filters were washed in two successive baths of PBS, fixed with cold ethanol 50% for 20 min and rinsed once with PBS. Cells having adhered to Transwell filters were colored with 0.1% crystal violet for 15 to 30 min. Transwell filters were washed again in two successive baths of PBS. Cells that had not migrated across the Transwell filters were removed by wiping the upper side compartment with a cotton swab. Transwell filters were washed again in two successive baths of PBS. Cells having migrated were observed and counted by light microscopy.
2.12. Macrophage polarization
After inducing differentiation in macrophages over 10 days, cells were activated (M1 condition) with 100 ng mL−1 LPS for 24 h. In the case of M2 polarization, cells were activated on Day 7 for 72 h with 20 ng mL−1 IL-4. After M0, M1 or M2 polarization or incubation with FERINJECT® for 24 h, 1 × 106 cells per condition were harvested and rinsed with PBS. Cells were resuspended and incubated with 10 μg mL−1 FC block (catalogue number 553141) for 5 minutes at room temperature. Next, cells were stained using antibodies diluted in “PBS-Fluo” (containing 3% fetal calf serum and 0.16% sodium azide) for 45 minutes at room temperature. The antibodies used were anti-CD38 coupled with FITC (catalogue number 558813) and anti-CD206 combined with Alexa Fluor® 647 (catalogue number 565250) at, respectively, 5 μg mL−1 and 2 μg mL−1. Data were acquired and analyzed using a FacsCalibur flow cytometer equipped with CellQuest cytometry software.
2.13. Animal experiments
All animal experiments were conducted in agreement with the Principles of Laboratory Animal Care (National Institutes of Health publication no. 86-23, revised 1985) and approved by the regional ethics committee.
2.13.1. Mammary tumor model.
Female 10-week-old BALB/c mice (Janvier, Le Genest-Isle, France) were anesthetized (isoflurane/air 4% for induction and 2% thereafter), and a suspension of 4T1 cells (ATCC®-CRL-2539™, 1 × 106 in 200 μL phosphate-buffered saline) was subcutaneously injected into the right flank. Ten days later tumor volumes had reached 156 ± 54 mm3, and mice were distributed into three groups so that the mean tumor volumes and standard deviations would be similar in all groups. The mice were then intravenously injected with a suspension of macrophages (2.5 × 106 macrophages in 200 μL phosphate-buffered saline) that had been pre-loaded overnight with Fluospheres 400 μg mL−1 (Dark Red 0.2 μm; Thermofisher) or Fluospheres 400 μg/mL + FERINJECT® 1 mg mL−1.
2.13.2.
In vivo whole body fluorescence imaging.
Fluorescence imaging was performed with an NIR 2D-fluorescence reflectance-imaging device (Fluobeam®700, Fluoptics, France). The excitation was provided by a class 1 expanded laser source at 680 nm, and the fluorescence signal collected via a CCD through a high pass filter >700 nm. Mice were anesthetized (isoflurane/air 4% for induction and 2% thereafter), and whole body fluorescence imaging was performed at 1, 2, 3, 5, 24 and 48 h in the tumor model. For image analysis, regions of interest were defined at the tumor site, contralaterally and on the liver, lung, spleen and femur. Results were expressed as tumor/contralateral skin site ratios. At the final time point, the mice were euthanized and isolated organs were harvested for ex vivo fluorescence imaging of the isolated organs.
2.14. X-ray irradiation
Before irradiation, 10 × 106 FERINJECT®-laden primary macrophages and 10 × 106 F98 cells were mixed in a 0.2 μL PCR tube to obtain cell pellets. The pellets were scanned with an 18 KeV X-ray beam, obtained by synchrotron radiation (ESRF, FAME beamline). Radiation doses, measured using the Fricke method were equivalent to 4 or 8 Gy.37,38 The cells were then seeded in a 6-well adherent plate with 200000 cells per well. They were incubated for 48 h at 37 °C, 5% CO2 (the first 24 h allowing for cell adherence and the next 24 h allowing for cell division). The cell proliferation assay was performed using crystal violet. After removing the culture medium, cells were rinsed twice with PBS and fixed with an acid–alcohol solution (mix of 1% acetic acid, 49% ethanol and 50% water) for 20 minutes. Cells were then stained with crystal violet (4 μg mL−1) for 30 minutes and eluted with 1 mL of the same acid–alcohol solution. Absorbance of the colored eluate is proportionate to the number of cells and was measured at 590 nm.39
3. Results and discussion
3.1. Nanoparticle behavior
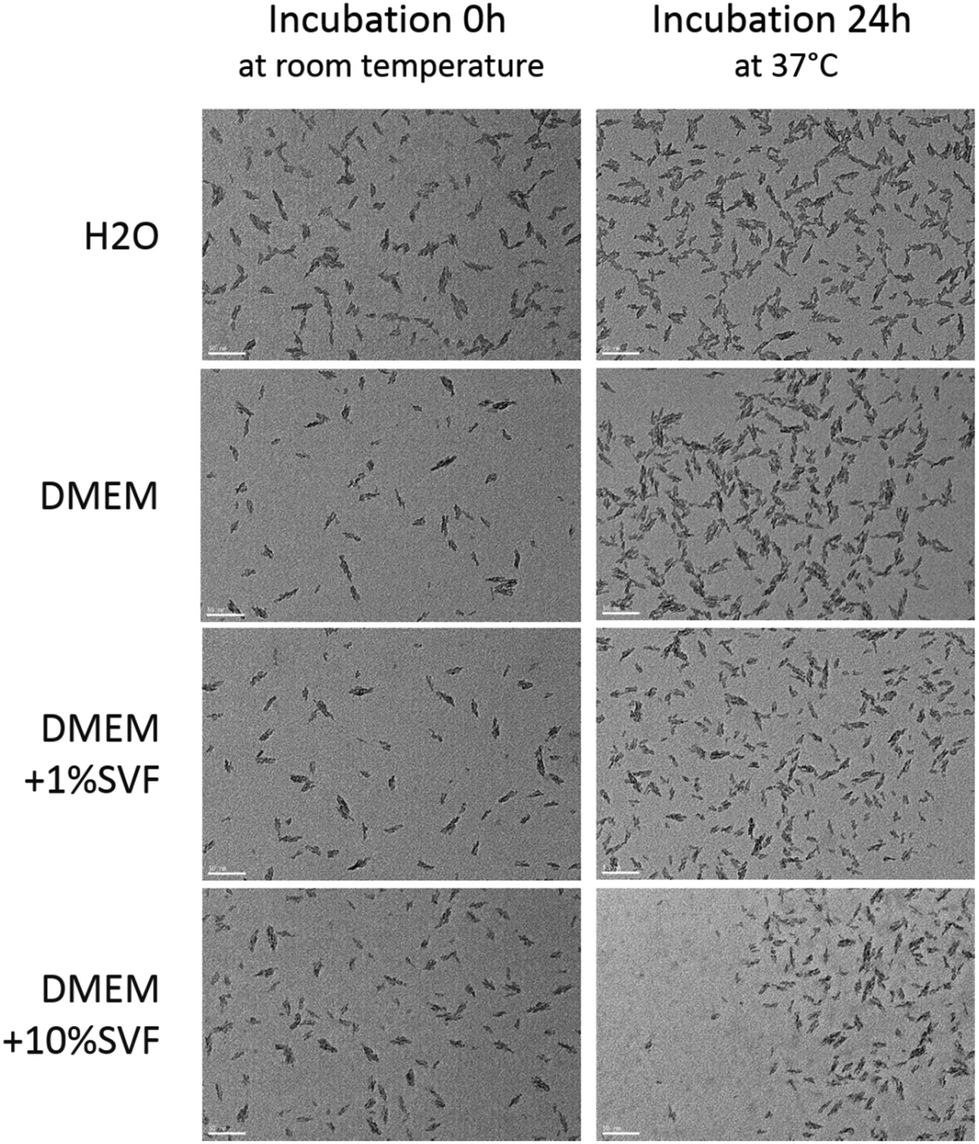
NP dissolution was characterized using DLS. In H2O and DMEM, FERINJECT® was considered monodispersed (percentage of dispersity <15%) and in DMEM supplemented with 1% or 10% SVF, FERINJECT® appeared as very polydispersed due to the presence of serum, i.e., the dispersity was not measurable (Table 1). FERINJECT® had a hydrodynamic diameter comprised between 21.6 nm and 25 nm in H2O or DMEM medium. After 24 h of incubation at 37 °C, 5% CO2, in H2O and DMEM, FERINJECT® was still considered monodispersed but with a higher percentage of dispersity. Overall, after incubation 24 h at 37 °C, the hydrodynamic diameter decreased very slightly (NP diameter decreasing by 1 nm to 3.4), suggesting minimal dissolution (Table 1). Examination by Transmission Electron Microscopy (TEM) revealed that FERINJECT® NP have an irregular stick shape with a length between 18.9 nm and 23.9 nm. No aggregation is visualized under any of the conditions (Fig. 1, Table 2). Thus, for DMEM with SVF, the high dispersity observed with DLS measurement is due to the proteins present in serum. Size measurement and the slight dissolution of FERINJECT® observed by TEM were similar to DLS characterization.
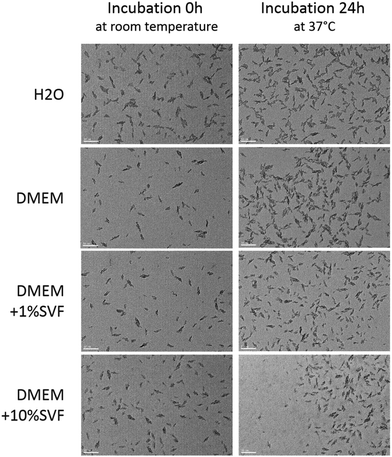 |
| | Fig. 1 Characterization of FERINJECT® by TEM. Nanoparticles were incubated in H2O or DMEM for two incubation times (0 h of incubation or 24 h of incubation at 37 °C, 5% CO2). Scale bar = 50 nm. | |
Table 1 Characterization of FERINJECT® by DLS at t0 and after 24 h of incubation in H2O or DMEM with or without SVF
| Ferinject® |
| Medium |
H2O |
DMEM |
DMEM + 1%SVF |
DMEM + 10%SVF |
| Time |
0 h |
24 h |
0 h |
24 h |
0 h |
24 h |
0 h |
24 h |
| Size (nm) |
23.4± 0.4 |
22.4 ± 0.4 |
24.4 ± 0.3 |
21.9 ± 0.4 |
23.8 ± 1.3 |
24.9 ± 0.5 |
25.0 ± 0.5 |
21.6 ± 0.6 |
| Dispersity (%) |
8.8 ± 6.9 |
11.7 ± 6.9 |
8.5 ± 2.4 |
12.2 ± 3.3 |
Multi-modal |
Multi-modal |
Multi-modal |
Multi-modal |
Table 2 Characterization of FERINJECT® by TEM at t0 and after 24 h of incubation in H2O or DMEM with or without SVF
| Ferinject® |
| Medium |
H2O |
DMEM |
DMEM + 1%SVF |
DMEM + 10%SVF |
| Time |
0 h |
24 h |
0 h |
24 h |
0 h |
24 h |
0 h |
24 h |
| Length (nm) |
22.7 ± 5.6 |
18.9 ± 4.3 |
23.9 ± 5.9 |
22.5 ± 6.5 |
23.1 ± 6.7 |
21.6 ± 6.1 |
22.0 ± 5.6 |
21.4 ± 5.6 |
3.2. Specificity of cell type regarding iron uptake
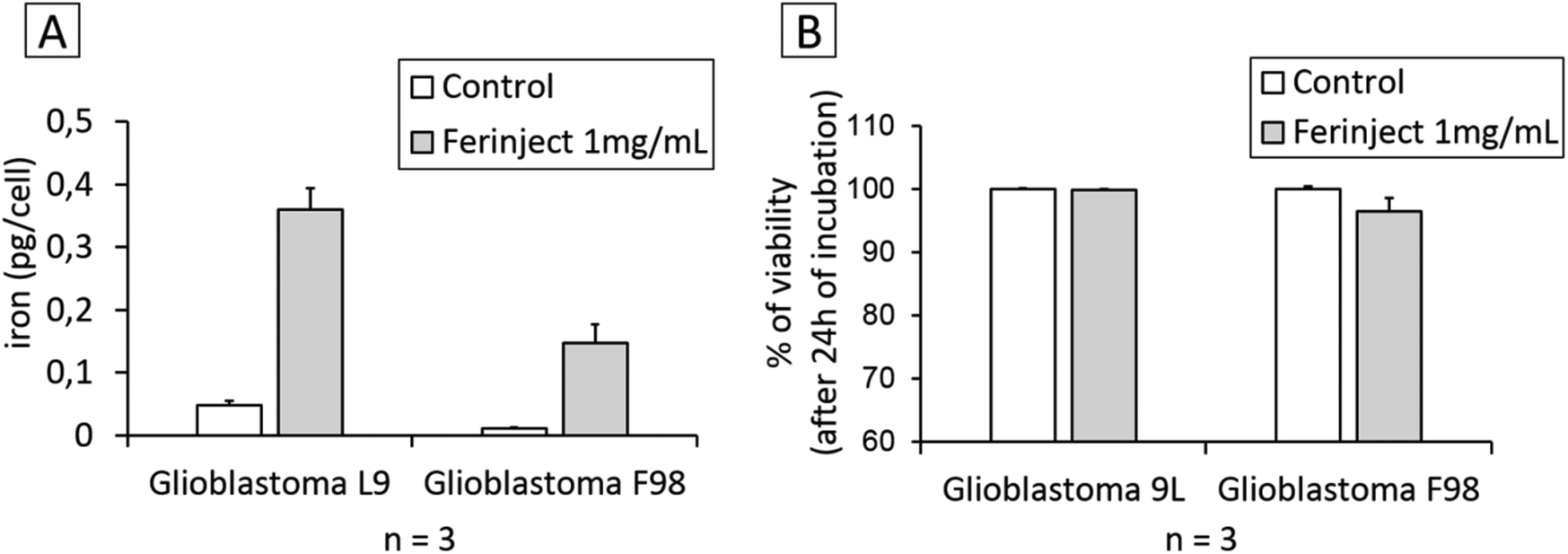
We wanted to assess the capacity of cancer cells such as GBM to engulf iron in large quantities when in contact with 1 mg mL−1 of FERINJECT®. This assessment was performed using ICP-MS, proving that L9 and F98 GBM cells cannot store FERINJECT® (only 0.36 and 0.15 pg per cell of iron was stored) (Fig. 2A). This low percentage of FERINJECT® internalization is not due to a high cell mortality (Fig. 2B). In our experiment, the lack of iron accumulation in typical tumor cells explains the importance of using macrophages to enhance maintaining iron at the tumor site, aside from the ability of targeting tumors and crossing body barriers. Without macrophages, these particles cannot accumulate in a tumor and, therefore, may diffuse outside the tumor zone.
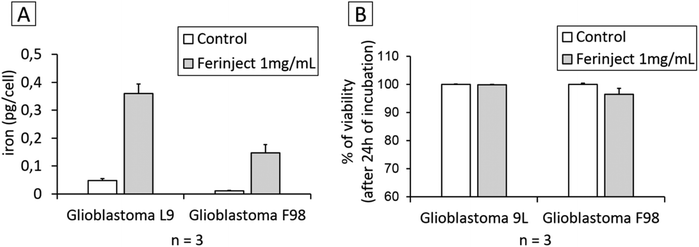 |
| | Fig. 2 FERINJECT® uptake by tumor cells. Panel A: Quantity of iron engulfed by tumor cell lines using ICP-MS method (condition with or without FERINJECT® exposure). Panel B: Viability of glioblastoma L9 and F98 cells with or without FERINJECT®. Viability was measured using propidium iodide (1 μg mL−1). | |
3.3. Working dose determination of FERINJECT® on primary macrophages
In a therapeutic protocol, the concentration of FERINJECT® must not induce high lethality on primary macrophages. We considered 20% lethality to be the upper limit for the compromise between the cellular loading in NP and their viability. Results displayed in Fig. 3 show that FERINJECT® is only slightly toxic to primary macrophages. LD20 was only reached at a concentration of over 1 mg mL−1. Consequently, at this stage of our research, FERINJECT® could be used at this high concentration (1 mg mL−1) in order to store a maximum amount of iron within macrophages without killing them. We considered that 1 mg mL−1 is a high concentration compared to other articles studying the impact of zinc oxide,40,41 silver42 and copper oxide43 nanoparticles on macrophages.
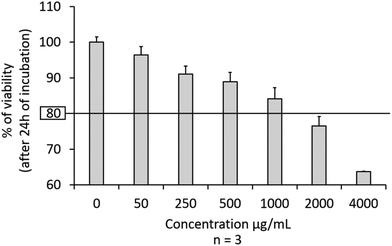 |
| | Fig. 3 Viability of primary mouse macrophages. Cells were exposed for 24 h to increasing doses of FERINJECT®. Viability was measured using propidium iodide (1 μg mL−1). | |
3.4. Iron uptake by primary macrophages
We first performed qualitative experiments. Perls staining, commonly used in optical microscopy to reveal the presence of ferric elements inside cells, revealed blue iron deposits in primary macrophages (Fig. 4A). Iron loading in the cells was accurately quantified by bathophenanthroline assay, which showed a significant quantity of iron in the macrophages exposed to FERINJECT®, contrary to GBM cells (Fig. 2A). Indeed, after 24 h of incubation, 8.3 pg per cell of iron was internalized by primary macrophages (Fig. 4B)—a result 23 times greater than L9 cells and 56 times greater than F98 cells. However, when cells were left in culture for an additional 24 h incubation period in the absence of FERINJECT®, we observed a slight release of iron (decrease to 6.5 pg per cell) that became even more marked after 72 h recovery (decrease to 2.7 pg per cell). This decrease is due to the progressive dissolution of FERINJECT®. Indeed, the evolution of granulometry (corresponding to the amount of NP stored within the cells) of FERINJECT®-laden cells was compared with same-size indissoluble NP-TiO2-laden cells using a flow cytometer. For FERINJECT®, we observed a significant decrease of granulometry (−11%) after 72 hours recovery. On the contrary, the granulometry values of NP-TiO2 remained unchanged over time (ESI 1†). Thus, the loss of iron from macrophages proves that treating a tumor with low-doses of X-rays should take place no later than 24 h after injection of loaded macrophages.
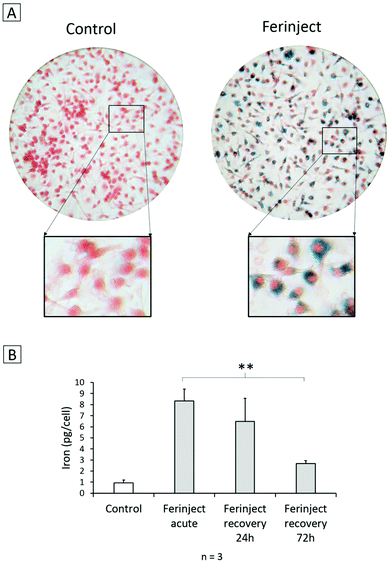 |
| | Fig. 4 Presence of iron in primary macrophages. Panel A: Perls staining. Red staining: Safranin cytosolic staining; blue staining: complex KFe[Fe(CN)6] named Prussian blue. Panel B: Quantitative assessment of the iron engulfed by primary macrophages using bathophenanthroline method. Statistical confidence (student T-test) is indicated as follows **p ≤ 0.01. | |
3.5. Functional studies of primary macrophages
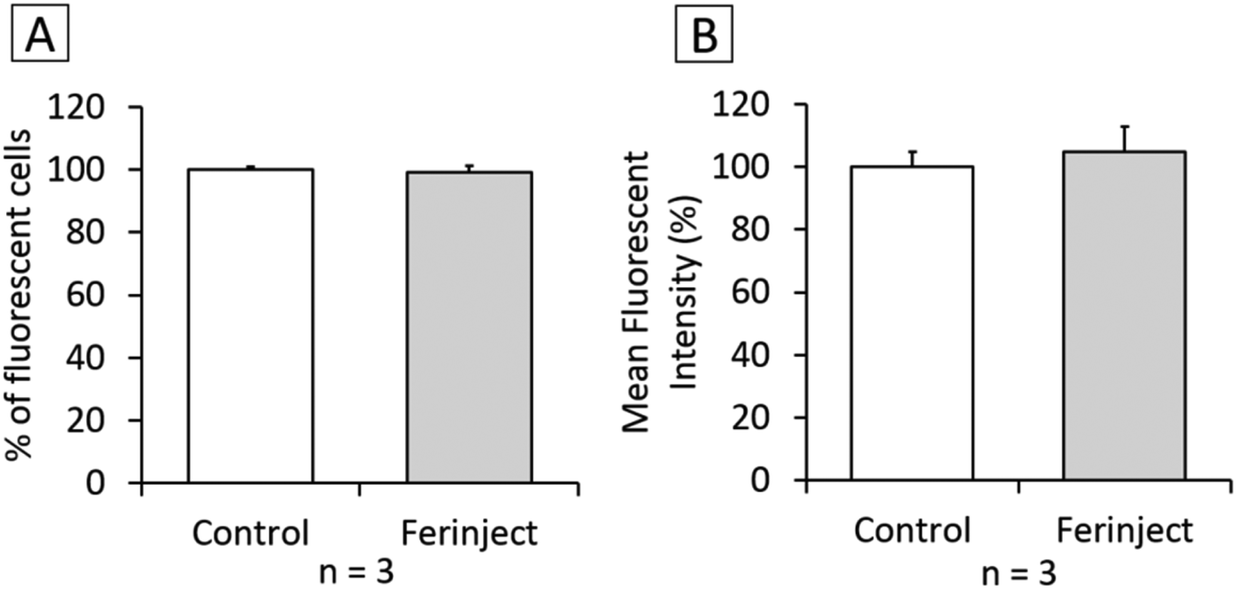
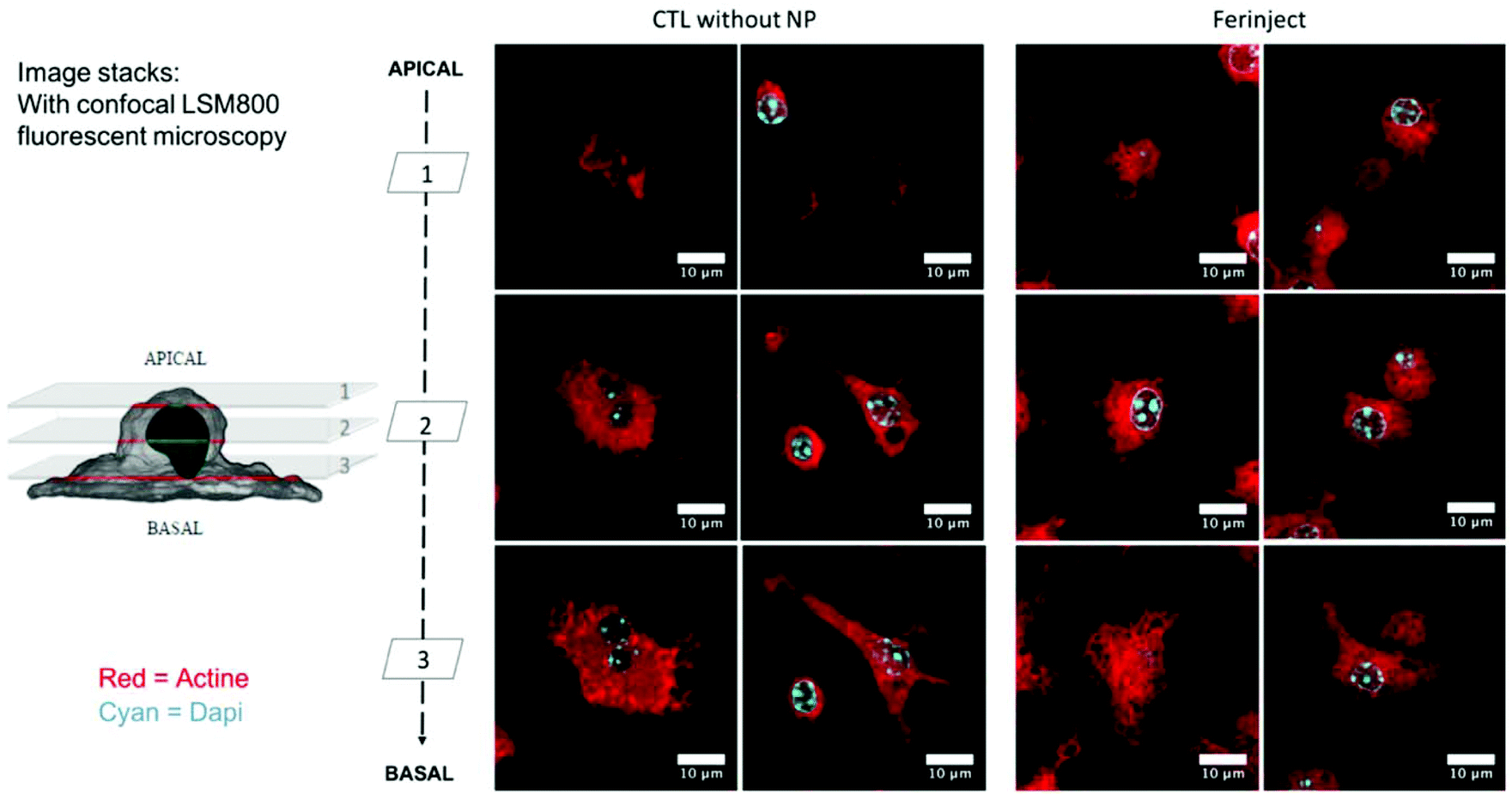
Functional studies were set up to determine if macrophages loaded with iron particles were still able to respond to stimuli and move towards inflammatory zones such as wounds, infections and, more importantly, tumors. We tested the impact of FERINJECT® on the classical functions of macrophages such as phagocytic ability, modulation of the LPS-induced production of cytokines, and NO. Regarding phagocytosis, flow cytometry enables investigation of two parameters, i.e., the proportion of cells that remain phagocytic after being exposed to NPs and the intensity of the phagocytic activity for phagocytosis-positive cells. When primary macrophages were exposed to FERINJECT®, the cell proportion and the intensity of the phagocytic intensity were unchanged compared to controls without FERINJECT® (Fig. 5A and B). Results did not show a FERINJECT® effect on the phagocytosis capacity of primary macrophages. These results were confirmed by visualizing the integrity of the F-actin cytoskeleton. No difference was observed between cells incubated with FERINJECT® compared to control cells, without NP (Fig. 6). For NO production, after stimulation with LPS, secretion decreased very slightly in the presence of FERINJECT®, i.e. by a non-significant 11% (Fig. 7A). Flow cytometric analysis revealed that FERINJECT® did not affect the production of IL-6 and TNFα pro-inflammatory cytokines, nor was secretion of IL-10 anti-inflammatory significantly modified. However, secretion of pro-inflammatory MCP-1 significantly decreased by 26.6% (Fig. 7B). All these results showed that FERINJECT® induces only a marginal effect on the functionality of macrophages. FERINJECT® thus appears to be a good candidate for therapeutic strategy.
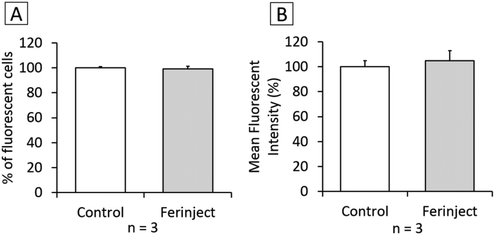 |
| | Fig. 5 Phagocytic ability. Panel A: Percentage of cells able to phagocyte fluorescent FITC-labeled latex beads (positive cells). Panel B: Phagocytic ability of positive cells. | |
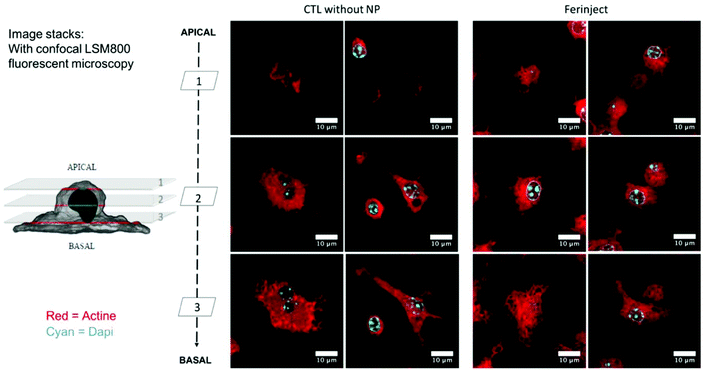 |
| | Fig. 6 Confocal microscopy: Observation of actin filaments with phalloidin labeled in red (Atto 560). Cell nucleus is colored blue by Dapi. Upper section = apical microscopy view; middle section = center of cell; lower section = basal microscopy view. | |
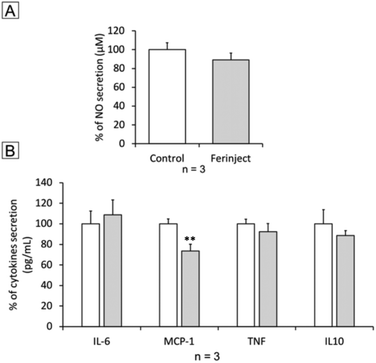 |
| | Fig. 7 Inflammation ability. Panel A: NO secretion with LPS stimulation. Panel B: Secretion of inflammatory cytokines with LPS stimulation. Statistical confidence (student T-test) is indicated as follows **p ≤ 0.01. | |
3.6. Transwell migration assay
We set up the Transwell migration assay to evaluate the ability of primary macrophages to migrate toward inflammatory stimuli. This assessment confirmed the phagocytosis assays. FERINJECT® neither inhibited the motility of primary macrophages, nor prevented macrophages from being able to respond to inflammatory stimuli such as LPS. We showed that, under both conditions (with and without exposure to FERINJECT®), macrophages were able to cross the membrane and adhere to the basal side of the Transwell. Moreover, cells were still responsive to a chemotactic stimulus such as LPS (Fig. 8).
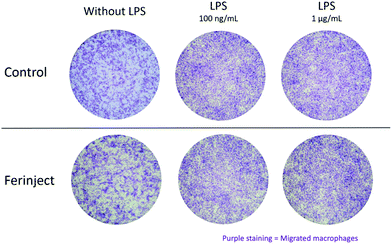 |
| | Fig. 8 Migratory ability using 5 nm pores Transwell membranes. Macrophages which cross the membrane (basal side) were stained with crystal violet 0.1%. | |
3.7. Small animal imaging
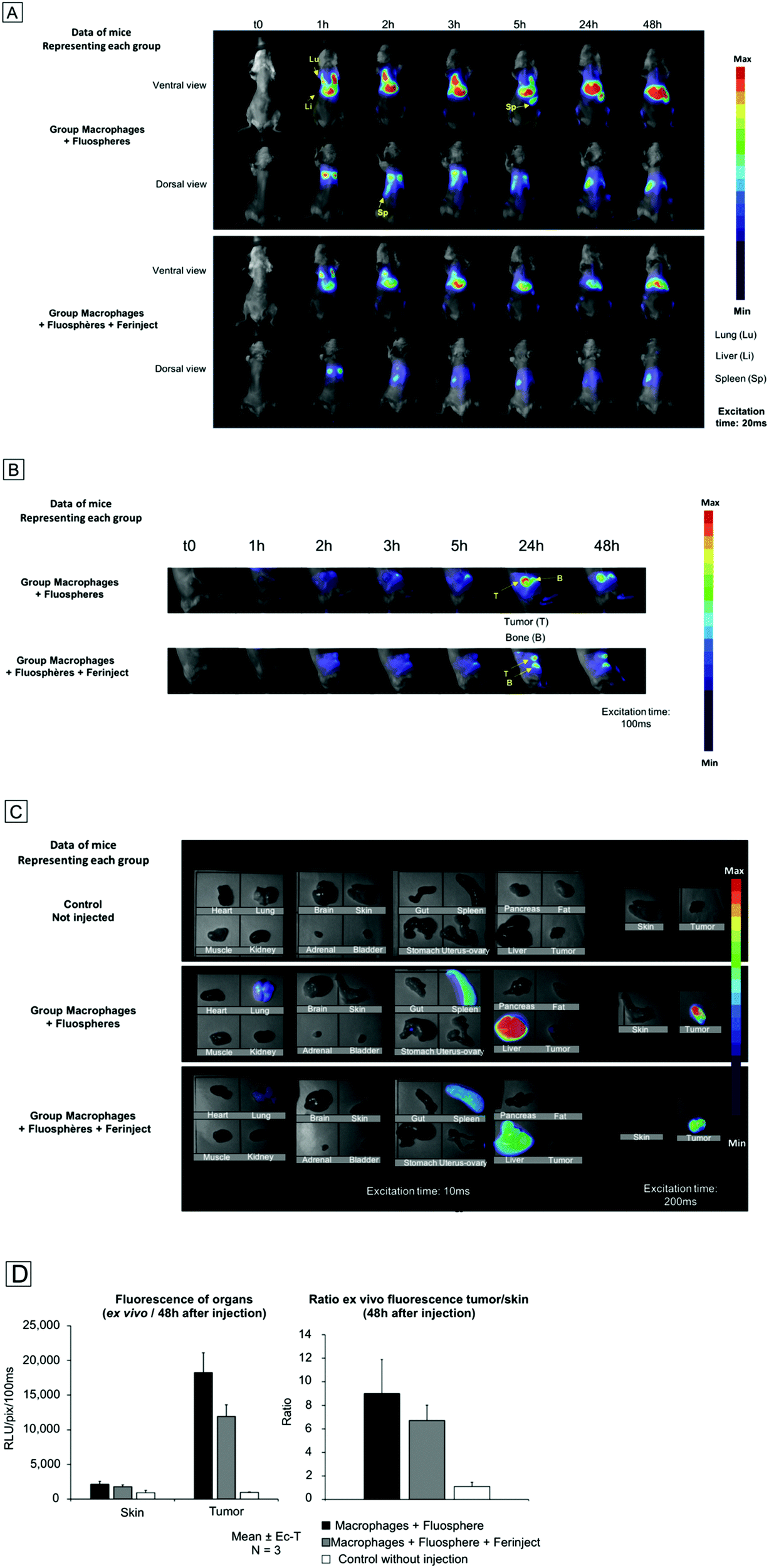
We wanted to approach real-use conditions to confirm that primary labeled macrophages injected into the bloodstream were able to migrate specifically to the tumor zone. Small-animal imaging was used to determine the in vivo biodistribution of loaded macrophages and to assess their tumor uptake kinetics. Non-invasive fluorescence imaging was performed over a span of 48 h and showed that injected macrophages quickly accumulated (1 h after injection) in macrophage-rich organs, including the liver and lungs. Interestingly, at 24 h post injection, the presence of labeled macrophages in the lungs had drastically decreased. On the contrary, we observed an uptake of labeled macrophages in bones and a massive accumulation in the spleen after 5 h (Fig. 9A). Small-animal imaging revealed a moderate, but significant, recruitment of labeled macrophages to the tumor site 24 h after injection (Fig. 9B). These results were similar under both conditions, i.e. macrophages loaded with fluospheres only and those loaded with Fluospheres and FERINJECT®, thus proving that FERINJECT® did not alter macrophage sensitivity to stimuli and ability to migrate toward the tumor. However, the fluorescence signal in the tumor slightly decreased between 24 h and 48 h after injection, a factor that offers insight into a potential clinical strategy. Indeed, we observed that the optimal rate of macrophage accumulation in the tumor should occur no later than 24 h after injection. After 48 h, mouse organs were retrieved and individually imaged (Fig. 9C). Results confirmed the massive and lasting accumulation of macrophages in detoxifying organs—lungs, and to a lesser extent, lymph nodes. They also confirmed the very significant accumulation of loaded macrophages at the tumor site. Measurement of the ex vivo fluorescence signal in the tumor compared to healthy contralateral skin showed a tumor/skin ratio of 9 for the fluosphere-only loaded macrophages and 6.7 for the simultaneously-loaded fluosphere and FERINJECT® macrophages (Fig. 9D). Recruitment of macrophages in the tumor was slightly lower when charged with FERINJECT®, but this difference may be explained by the fact that macrophages were more heavily loaded in the fluorescent probe+FERINJECT® condition than in the fluosphere-only condition. However, these results clearly illustrate the capacity of injected primary macrophages to be recruited by tumors, thereby promoting the concept of using iron particles-loaded macrophages to treat solid and resistant tumors. Moreover, this strategy may also be applied to other types of particles and/or drugs.
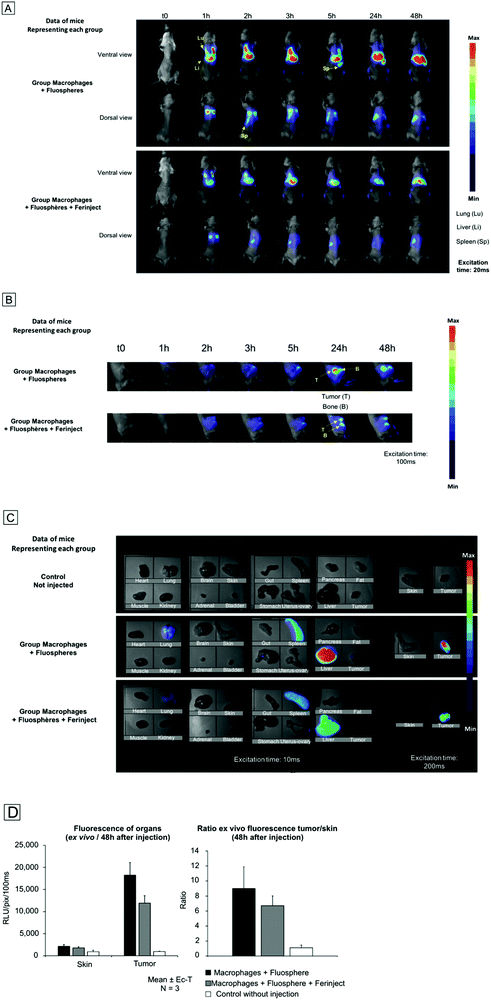 |
| | Fig. 9 Small animal imaging, BalB/c mice with subcutaneous mammary tumor. Panel A: Whole body in vivo fluorescence imaging. Panel B: Whole body in vivo fluorescence imaging, focus on the tumor site. Panel C: Ex vivo fluorescence imaging on isolated tissue samples 48 h post injection. Panel D: Ex vivo fluorescence imaging on isolated tissue samples 48 h post injection. Left graph: Quantitative measurement of the fluorescence of tumor in comparison to contralateral skin, 48 h after intravascular injection of labeled macrophages. Right graph: Ratio of fluorescence intensity between contralateral skin and tumor. | |
3.8. X-Ray irradiation
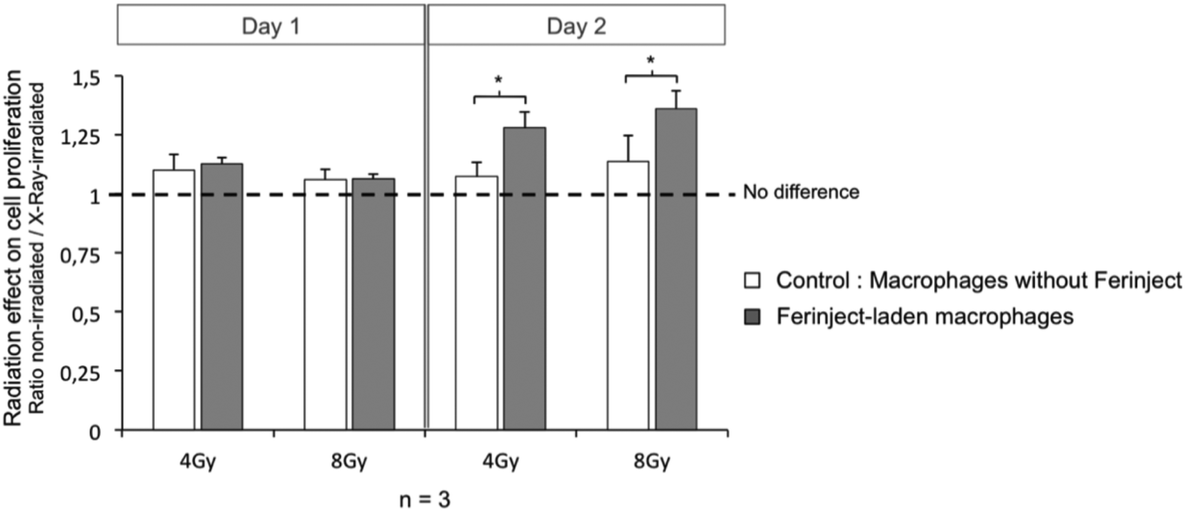
Two days following radiation exposure (4 or 8 Gy at 18 KeV), the F98 cell proliferation assay showed a significant decrease of cell multiplication for the cells that had been irradiated in the presence of FERINJECT®-laden macrophages. For the cells that had been exposed to the same radiation dose but in the presence of unloaded macrophages, the decrease was less marked. Indeed, radiation efficiency was enhanced by 19% for 4 Gy and 20% for 8 Gy (Fig. 10). This effect is only observed 2 days after radiation exposure, certainly because of cell adhesion before first divisions. This in vitro assay proves that the use of FERINJECT®-laden macrophages to enhance radiation therapy is effective. Furthermore, it also demonstrates that direct internalization of nanoparticles by cancer cells, as classically described12,13 is not an absolute requisite for HZE-enhanced radiotherapy. In the future, we aim to reinforce the effect of photoelectron emissions and, consequently, of radiotherapy. A possible approach would be to increase the size of the NP contained in FERINJECT®. This would facilitate their phagocytosis and thus their accumulation in macrophages. It should also decrease the rate of iron loss over time after macrophage loading with iron. Moreover, we could irradiate cells with higher-energy X-rays (e.g. 40 Kev or 80 KeV instead of 18 Kev). This would imply moving away from the appropriate wavelength to excite iron (7 KeV), meaning that fewer electrons would be emitted. However, they would travel a longer distance (e.g. 40 KeV ≈ 30 μm, 80 Kev ≈ 100 μm and 120 KeV ≈ 200 μm)44,45 and thus be more likely to kill neighboring cells, the average size of a macrophage being 30 μm. The next goal will be to find a level of energy offering a suitable compromise between the amount of photoelectrons produced by FERINJECT® and those released out of the macrophages.
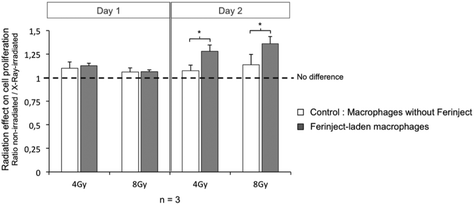 |
| | Fig. 10 Cell proliferation of F98 Glioblastoma after exposure to radiation at 18 KeV (4 Gy or 8 Gy). Statistical confidence (student T-test) is indicated as follows *p ≤ 0.05. | |
4. Conclusions
In summary, despite ever more effective anticancer treatments, the mortality rate due to solid tumor relapses remains too high. The concept of CSCs can explain most of these therapeutic failures. The development of NP offers new possibilities for targeted therapies, although efficient targeting and NP internalization by cancer cells are still controversial. In order to increase the specific accumulation of NP such as HZE-NP (e.g. NP-Fe2O3) at the tumor site, a Trojan horse strategy using macrophages shows promise. The present results illustrated that FERINJECT® loaded at 1 mg mL−1 did not alter (or very marginally) primary macrophage function, and that recruitment of injected primary macrophages at tumor sites could be successfully visualized by in vivo small-animal imaging. Finally, results demonstrated that macrophages are promising tools to accumulate and maintain a large amount of anticancerous particles (HZE nanoparticles). In the future, additional assays must be carried out to enhance the attraction of injected macrophages into the tumor and to increase the quantity of particles engulfed, while limiting damage to macrophages. One possibility would be to inject macrophages that have been pre-activated in vitro by inflammatory stimuli. The second possibility would be to induce a better inflammatory signal22 in the tumor zone, e.g. using a first wave of low-intensity X-rays.
We consider this strategy to be a very promising approach to resistant tumors such as GBM. After assessing the capacity of tumors to attract injected macrophages loaded with particles, our next objective is to assess the capacity of loaded macrophages to migrate to GBM, and thus cross the additional constraint of the blood brain barrier. Zanganeh et al. have recently demonstrated that intravenous injection of NP-Fe2O3 used to treat anemia proved to be a useful tool in reducing tumor growth.46 The anti-tumor effect is due to a change in tumor microenvironment without necessarily being aware of all the phenomena involved. Indeed, tumors are largely composed of anti-inflammatory M2 macrophages (TAM), favoring cancerous development. Some research explains that NP-Fe2O3 induce macrophage (M1 macrophages) inflammatory polarization,46 which could explain why NP-Fe2O3-laden macrophages would be more efficient to detect and interrupt cancer development. However, this interpretation is far too restrictive. Many as yet unidentified phenomena might explain the ability of NP-Fe2O3-laden macrophages to more efficiently attack tumors. Moreover, the M1-polarization is not visualized in our experiment with FERINJECT® (ESI 2†). Therefore, this effect is not general to all NP-Fe2O3 usable.
Our Trojan horse strategy offers two main advantages: (1) the use of FERINJECT® will enhance the efficiency of radiotherapy thanks to the release of photoelectrons in the tumor. (2) Contrary to strategies relying on other nanoparticles such as gold, the properties of FERINJECT® appear to be major assets in developing NP-based anti-tumor strategies, given their dissolution ability and therefore their low biopersistence in organs treated (i.e., the brain in the case of GBM). Moreover, in addition to being a metal easily managed by macrophages, iron is an essential element already present in the body, between 3 and 4 g. The daily intake from food is estimated to be between 10 and 20 mg, 10% being absorbed by the intestine.47 Thus, the iron supplied via our therapeutic strategy is minimal compared to the high quantity already present in the body, thus limiting side effects. Finally, as regards other Trojan horse strategies with macrophages, our work is not limited to in vitro therapeutic effects.48 We also assessed the safety of FERINJECT® and validated the strategy up to preclinical experimentation. Thus, treating cancers combining intravascular injection of laden macrophages and radiotherapy is a practicable solution. Even if the therapeutic (X-ray) effects were slight but still significant, this is the first time that killing neighboring cells without those having directly accumulated NP has been shown to be possible. Some publications assess the therapeutic efficacy only on NP-laden cells,48 other strategies target CSCs with functionalized NP49 or attempt to limit angiogenesis with different types of NP.50 In the future, our results could be optimized in in vivo assays using one or several laden-macrophages injections and radiotherapy.
Author contributions
B.D., C.A-G performed the DLS, phagocytosis, NO and cytokines experiments. In addition, B.D. performed the F-Actin and Perls staining and migratory assay. M.G., J.V. and V.J. performed small animal imaging. S.R. and J.A. performed ICP-MS. In addition, S.R. performed viability assays. D.F. and G.S. performed TEM microscopy visualization. DT, TR and BD performed radiation experiments. TR, DT, OP, IT and MC design Synchrotron X-Ray radiation experiments. V.J. designed small animal imaging. C.A-G and BD (to a lesser extent) supervised the overall research. The manuscript was written by B.D, T.R and V.J. All authors have given approval to the final version of the manuscript.
Abbreviations
| CSC | Cancer stem cell |
| F-actin | Actin's filamentous form |
| GBM | Glioblastoma |
| HZE | High-Z-element |
| ICP-MS | Inductively coupled plasma mass spectrometry |
| IL | Interleukin |
| LPS | Lipopolysaccharide |
| MCP-1 | Monocyte chemoattractant protein-1 |
| NP | Nanoparticle |
| TNF | Tumor necrosis factor |
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This paper is dedicated to the memory of Catherine Aude-Garcia, who played a crucial role in this research project and died Nov. 21st, 2018. Her scientific creativity, enthusiasm for science and friendly presence in the lab will be dearly missed.
We would like to thank Dr. Gail Taillefer, Emeritus professor of English (native speaker), Université Toulouse-1 Capitole and Maxime Petit (PRAG), Université Toulouse 1-Capitole for proofreading the manuscript, Bernard Verrier (Institute of Biology and Protein Chemistry, IBCP, LBTI) for suggestions concerning in vivo experiments, Patrick Colomp for ESRF radioprotection service, and Serge Candéias for his advice and help in the preliminary radiation tests.
Finally, BD gratefully acknowledges the funding received towards his Ph.D. from the French National Center for Scientific Research (CNRS) through their National Fellowship for Students with Disabilities.
This work was supported by CEA transverse toxicology program.
Small animal imaging experiments were performed on the OPTIMAL platform (Institute for Advanced Biosciences, Grenoble, France) and optical imaging systems were acquired thanks to France Life Imaging (French program “Investissement d'Avenir” grant; “Infrastructure d'avenir en Biologie Sante”, ANR-11-INBS-44 0006).
This work used the platforms of the Grenoble Instruct centre (ISBG; UMS 3518 CNRS-CEA-UJF-EMBL) with support from FRISBI (ANR-10-INSB-05-02) and GRAL (ANR-10-LABX-49-01) within the Grenoble Partnership for Structural Biology (PSB). The electron microscope facility is supported by the Rhône-Alpes Region, the Fondation Recherche Medicale (FRM), the fonds FEDER, the Centre National de la Recherche Scientifique (CNRS), the CEA, the University of Grenoble, EMBL, and the GIS-Infrastrutures en Biologie Sante et Agronomie (IBISA).
References
- World Health Organization Programmes Cancer Page. http://www.who.int/cancer/en/access Nov 10 2017.
- J. E. Visvader and G. J. Lindeman, Nat. Rev. Cancer, 2008, 8, 755–768 CrossRef CAS PubMed
 .
.
- D. Beier, J. B. Schulz and C. P. Beier, Mol. Cancer, 2011, 10, 128 CrossRef CAS PubMed .
- M. Dean, T. Fojo and S. Bates, Nat. Rev. Cancer, 2005, 5, 275–284 CrossRef CAS PubMed .
- S. Vinogradov and X. Wei, Nanomedicine, 2012, 7, 597–615 CrossRef CAS PubMed .
- J. M. Adams and A. Strasser, Cancer Res., 2008, 68, 4018–4021 CrossRef CAS PubMed .
- D. Nassar and C. Blanpain, Annu. Rev. Pathol.: Mech. Dis., 2016, 11, 47–76 CrossRef CAS PubMed .
- S. Skvortsov, P. Debbage, P. Lukas and I. Skvortsova, Semin. Cancer Biol., 2015, 31, 36–42 CrossRef CAS PubMed .
- K. Rycaj and D. G. Tang, Int. J. Radiat. Biol., 2014, 90, 615–621 CrossRef CAS PubMed .
- F. Pajonk, E. Vlashi and W. H. McBride, Stem Cells, 2010, 28, 639–648 CrossRef CAS PubMed .
- S. J. McMahon, M. H. Mendenhall, S. Jain and F. Currell, Phys. Med. Biol., 2008, 53, 5635–5651 CrossRef PubMed .
- M. Yamada, M. Foote and T. W. Prow, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 428–445 CAS .
- M.-Y. Chang, A.-L. Shiau, Y.-H. Chen, C.-J. Chang, H. H.-W. Chen and C.-L. Wu, Cancer Sci., 2008, 99, 1479–1484 CrossRef CAS PubMed .
- S. Jain, D. G. Hirst and J. M. O'Sullivan, Br. J. Radiol., 2012, 85, 101–113 CrossRef CAS PubMed .
- S. S. Chauhan, X. J. Liang, A. W. Su, A. Pai-Panandiker, D. W. Shen, J. A. Hanover and M. M. Gottesman, Br. J. Cancer, 2003, 88, 1327–1334 CrossRef CAS PubMed .
- D. R. Cooper, D. Bekah and J. L. Nadeau, Front. Chem., 2014, 2, 86 Search PubMed .
- Y. H. Bae and K. Park, J. Controlled Release, 2011, 153, 198–205 CrossRef CAS PubMed .
- T. Lammers, F. Kiessling, W. E. Hennink and G. Storm, J. Controlled Release, 2012, 161, 175–187 CrossRef CAS PubMed .
- S. Nie, Nanomedicine, 2010, 5, 523–528 CrossRef PubMed .
- B. Dalzon, C. Lebas, G. Jimenez, A. Gutjahr, C. Terrat, J.-Y. Exposito, B. Verrier and C. Lethias, PLoS One, 2016, 11, e0167663 CrossRef PubMed .
- M. J. Moravan, J. A. Olschowka, J. P. Williams and M. K. O'Banion, Radiat. Res., 2011, 176, 459–473 CrossRef CAS PubMed .
- M. Mason, C. Maurice, M. G. McNamara, M. T. Tieu, Z. Lwin, B.-A. Millar, C. Menard, N. Laperriere, M. Milosevic, E. G. Atenafu, W. Mason and C. Chung, J. Neuro-Oncol., 2017, 132, 463–471 CrossRef PubMed .
- S. B. Lee, H. W. Lee, T. D. Singh, Y. Li, S. K. Kim, S. J. Cho, S.-W. Lee, S. Y. Jeong, B.-C. Ahn, S. Choi, I.-K. Lee, D.-K. Lim, J. Lee and Y. H. Jeon, Theranostics, 2017, 7, 926–934 CrossRef CAS PubMed .
- J. Condeelis and J. W. Pollard, Cell, 2006, 124, 263–266 CrossRef CAS PubMed .
- T. Chanmee, P. Ontong, K. Konno and N. Itano, Cancers, 2014, 6, 1670–1690 CrossRef CAS PubMed .
- R. K. Gherardi, H. Eidi, G. Crépeaux, F. J. Authier and J. Cadusseau, Front. Neurol., 2015, 6, 4 Search PubMed .
-
M. J. Berger, J. H. Hubbell, S. M. Seltzer, J. S. Coursey and D. S. Zucker, ESTAR, PSTAR, and ASTAR: Computer Programs for Calculating Stopping-Power and Range Tables for Electrons, Protons, and Helium Ions (Version 1.2.2), Gaithersburg, MD, National Institute of Standards and Technology, 2000. https://physics.nist.gov/cgi-bin/Star/e_table.pl Search PubMed .
- T. Ganz, J. Innate Immun., 2012, 4, 446–453 CrossRef CAS PubMed .
- Vifor Pharma Media Press releases Page. http://www.viforpharma.com/en/media/press-releases/201307/1719021 (accessed Nov 11 2017).
- D. Cada, T. Levien and D. Baker, Hosp. Pharm., 2014, 49, 52–69 CrossRef CAS PubMed .
- J. Toblli and M. Angerosa, Drug Des., Dev. Ther., 2014, 2475 CrossRef PubMed .
- M. R. Jahn, H. B. Andreasen, S. Fütterer, T. Nawroth, V. Schünemann, U. Kolb, W. Hofmeister, M. Muñoz, K. Bock, M. Meldal and P. Langguth, Eur. J. Pharm. Biopharm., 2011, 78, 480–491 CrossRef CAS PubMed .
-
U. Schleicher and C. Bogdan, in Macrophages and Dendritic Cells, ed. N. E. Reiner, Humana Press, Totowa, NJ, 2009, vol. 531, pp. 203–224 Search PubMed .
- V. L. Tarakanova, V. Leung-Pineda, S. Hwang, C.-W. Yang, K. Matatall, M. Basson, R. Sun, H. Piwnica-Worms, B. P. Sleckman and H. W. Virgin, Cell Host Microbe, 2007, 1, 275–286 CrossRef CAS PubMed .
- S. Triboulet, C. Aude-Garcia, M. Carrière, H. Diemer, F. Proamer, A. Habert, M. Chevallet, V. Collin-Faure, J.-M. Strub, D. Hanau, A. Van Dorsselaer, N. Herlin-Boime and T. Rabilloud, Mol. Cell. Proteomics, 2013, 12, 3108–3122 CrossRef CAS PubMed .
- G. Abel, J. Szöllösi and J. Fachet, Eur. J. Immunogenet., 1991, 18, 239–245 CrossRef CAS PubMed .
- D. Saffré, E. Atinault, S. Pin, J. P. Renault, J. L. Hazemann and G. Baldacchino, J. Phys.: Conf. Ser., 2011, 261, 012013 CrossRef .
-
H. Fricke and E. J. Hart, Radiation chemistry, Vil.II, Academic Press, 1966 Search PubMed .
- O. Proux, X. Biquard, E. Lahera, J. J. Menthonnex, A. Prat, O. Ulrich, Y. Soldo, P. Trvisson, G. Kapoujyan, G. Perroux, P. Taunier, D. Grand, P. Jeantet, M. Deleglise, J. Roux and J. Hazemann, Phys. Scr., 2005, 970 CrossRef CAS .
- S. Triboulet, C. Aude-Garcia, L. Armand, A. Gerdil, H. Diemer, F. Proamer, V. Collin-Faure, A. Habert, J.-M. Strub, D. Hanau, N. Herlin, M. Carrière, A. Van Dorsselaer and T. Rabilloud, Nanoscale, 2014, 6, 6102–6114 RSC .
- C. Aude-Garcia, B. Dalzon, J.-L. Ravanat, V. Collin-Faure, H. Diemer, J. M. Strub, S. Cianferani, A. Van Dorsselaer, M. Carrière and T. Rabilloud, J. Proteomics, 2016, 134, 174–185 CrossRef CAS PubMed .
- C. Aude-Garcia, F. Villiers, V. Collin-Faure, K. Pernet-Gallay, P.-H. Jouneau, S. Sorieul, G. Mure, A. Gerdil, N. Herlin-Boime, M. Carrière and T. Rabilloud, Nanotoxicology, 2016, 10, 586–596 CrossRef CAS PubMed .
- S. Triboulet, C. Aude-Garcia, L. Armand, V. Collin-Faure, M. Chevallet, H. Diemer, A. Gerdil, F. Proamer, J.-M. Strub, A. Habert, N. Herlin, A. Van Dorsselaer, M. Carrière and T. Rabilloud, PLoS One, 2015, 10, e0124496 CrossRef PubMed .
-
C. Boudou, PHD thesis, Joseph Fourier University, Grenoble1, 2006, page 20, https://tel.archives-ouvertes.fr/tel-00119516/document Search PubMed .
- C. Boudou, J. Balosso, F. Estève and H. Elleaume, Phys. Med. Biol., 2005, 50, 4841–4851 CrossRef PubMed .
- S. Zanganeh, G. Hutter, R. Spitler, O. Lenkov, M. Mahmoudi, A. Shaw, J. S. Pajarinen, H. Nejadnik, S. Goodman, M. Moseley, L. M. Coussens and H. E. Daldrup-Link, Nat. Nanotechnol., 2016, 11, 986–994 CrossRef CAS PubMed .
- P. Mladenka, R. Hrdina, M. Hübl and T. Simůnek, Acta Med., 2005, 48, 127–135 CAS .
- M.-R. Choi, K. J. Stanton-Maxey, J. K. Stanley, C. S. Levin, R. Bardhan, D. Akin, S. Badve, J. Sturgis, J. P. Robinson, R. Bashir, N. J. Halas and S. E. Clare, Nano Lett., 2007, 7, 3759–3765 CrossRef CAS PubMed .
- S. S. An, G.-B. Jang, H.-Y. Lee, I.-S. Hong and J.-S. Nam, Int. J. Nanomed., 2015, 251 CrossRef PubMed .
- N. Hashemi Goradel, F. Ghiyami-Hour, S. Jahangiri, B. Negahdari, A. Sahebkar, A. Masoudifar and H. Mirzaei, J. Cell. Physiol., 2018, 233, 2902–2910 CrossRef CAS PubMed .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nr03364a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence g,
Marie
Carrière
g,
Marie
Carrière