
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCell factory engineering for improved production of natural products
Jens
Nielsen
 abc
abc
aBioInnovation Institure, Ole Maaløes Vej 3, DK2200 Copenhagen, Denmark
bDepartment of Biology and Biological Engineering, Chalmers University of Technology, SE41296 Gothenburg, Sweden
cNovo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, DK2800 Kgs. Lyngby, Denmark. E-mail: jni@novo.dk
First published on 18th April 2019
Abstract
Natural product biosynthesis is inherently linked with the primary metabolism in terms of providing precursors and co-factors. Thus, even though it is important to improve the performance of natural product biosynthetic enzymes in building efficient cell factories, it is equally important to engineer the central carbon metabolism, such that this is adjusted to meet the biosynthetic demand of the desired product.
 Jens Nielsen | Jens Nielsen is CEO of the BioInnovation Institute, Denmark and Professor at Chalmers University of Technology, Sweden. His research is to study and engineer metabolism. He has supervised more than 120 PhD students and more than 75 post doctoral researchers. He has published so far more than 700 papers that have been cited more than 53 |
1 Introduction
Over the last 70 years natural products have served as a rich source of pharmaceuticals, and as these chemicals often can provide novel chemical scaffolds enabling new bioactive properties, there is much interest in developing platforms for efficient production of them. Traditionally, bioactive molecules have been produced by the cell factory that naturally produces the molecule, e.g. microorganism or plant, and in many cases breeding has been used to improve the production level. This is best illustrated in the production of penicillin, where strain improvement programs have resulted in more than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times improvement of the production level by the fungus Penicillium chrysogenum. However, with the introduction of genetic engineering, and more recently of synthetic biology tools that has enabled efficient genome editing, it has become possible to use a more directed approach to strain improvement. Moreover, it has been possible to express biosynthetic pathways in heterologous hosts, which has enabled decoupling the choice of host (or chassis) from the biosynthetic pathway of interest. Furthermore, this has enabled generation of so-called platform cell factories1 that are optimized for producing a class of compounds. An excellent early example of this was demonstrated by the Dutch company DSM, that recruited one of their very efficient penicillin producing strains for production of adipoyl-7-aminodeacetoxycephalosporanic acid (adipoyl-7-ADCA) by expressing a heterologous expandase that could convert adipoyl-6-aminopenicillanic acid (adipoyl-6-APA) into adipoyl-7-ADCA. Adipoyl-7-ADCA is an important precursor for chemical synthesis of cephalexin, a widely used broad-spectrum antibiotics.
000 times improvement of the production level by the fungus Penicillium chrysogenum. However, with the introduction of genetic engineering, and more recently of synthetic biology tools that has enabled efficient genome editing, it has become possible to use a more directed approach to strain improvement. Moreover, it has been possible to express biosynthetic pathways in heterologous hosts, which has enabled decoupling the choice of host (or chassis) from the biosynthetic pathway of interest. Furthermore, this has enabled generation of so-called platform cell factories1 that are optimized for producing a class of compounds. An excellent early example of this was demonstrated by the Dutch company DSM, that recruited one of their very efficient penicillin producing strains for production of adipoyl-7-aminodeacetoxycephalosporanic acid (adipoyl-7-ADCA) by expressing a heterologous expandase that could convert adipoyl-6-aminopenicillanic acid (adipoyl-6-APA) into adipoyl-7-ADCA. Adipoyl-7-ADCA is an important precursor for chemical synthesis of cephalexin, a widely used broad-spectrum antibiotics.
In more recent years this has been taken much further, resulting in refactoring of complex biosynthetic pathways in heterologous hosts, as illustrated by the recent reconstruction of a 23 step enzymatic pathway for production of hydrocodone in Saccharomyces cerevisiae.2 Here there has been focus on a few different cell factory platforms for production of natural products: (1) the yeast S. cerevisiae, which is beneficial as it is easy to genetically engineer and enables expression of plant P450 enzymes; (2) the Gram-negative bacterium Escherichia coli, which allows for high-level expression of heterologous enzymes and has a flexible metabolism that can adjust easily to heterologous pathways; (3) the Gram-positive actinomycetes Streptomyces coelicolor, which produces several natural products and hence has the capacity to support production of many other natural products, in particular gene clusters from other actinomycetes. However, the list of cell factory platforms is expanding, and many would probably add a filamentous fungus to this list, but here there is less consensus, as both P. chrysogenum and various Aspergilli compete for this spot. The advantage of using cell factory platforms is that the central carbon metabolism can be tailored to production of a class of molecules that use the same precursor, e.g. acetyl-CoA often used for polyketide synthases.
Here I will discuss some of the key points to consider when natural product biosynthesis has to be improved in a given microbial cell factory: (1) the central carbon metabolism as it provides the precursors and the co-factors for natural product biosynthesis; (2) choice of microbial cell factory; and (3) optimization of the enzymes in the biosynthetic pathway in order to avoid a proteome constraint issue.
2 Link between primary and secondary metabolism
Penicillin production by the filamentous fungus P. chrysogenum is a good example of how primary and secondary metabolism is closely linked. Penicillin is produced in a three-step biosynthetic pathway. In the first step, the non-ribosomal peptide synthetase (NRPS) L-α-aminoadipoyl-L-cysteinyl-D-valine synthetase (ACVS) condenses L-α-aminoadipic acid, L-cystein, and L-valine into the tripeptide L-α-aminoadipoyl-L-cysteinyl-D-valine (ACV). In the next step, isopenicillin N synthase (IPNS) converts ACV into isopenicillin (IPN) with the use of molecular oxygen as electron acceptor. In the final step, the L-α-aminoadipoyl side chain of IPN is cleaved off resulting in formation of 6-APA that can be further converted to penicillin V or penicillin G depending on whether the cells are fed with phenoxyacetic acid or phenylacetic acid. Naturally, P. chrysogenum produces a minute amount of penicillin, and thus only a small amount of the three amino acids (with L-α-aminoadipic acid being an intermediate of L-lysine biosynthesis) are used for penicillin production. However, in connection with strain improvement programs, where the flux towards penicillin was significantly improved, increasing the supply of the three amino acids was essential. Furthermore, as cysteine biosynthesis requires reduction of sulfate provided in the medium to hydrogen sulfate, penicillin biosynthesis indirectly requires large amounts of NADPH. Thus, whereas the first strain improvement initiatives (all done by classical mutagenesis and screening) most likely resulted in improvement of the activity of the penicillin biosynthetic enzymes as well as increased expression through increasing the copy number of the biosynthetic gene cluster, the later strain improvement efforts clearly required mutations supporting efficient provision of the three amino acids and sufficient NADPH. Another example is that actinorhodin (and to some extend undecylprodigiosin) production by S. coelicolor could be significantly improved by deletion of one of the isoforms of phosphofructokinase (pfkA2).3 From metabolic modelling it was identified that the production of the polyketide actinorhodin requires large amounts of NADPH, and through deletion of one of the phosphofructokinase isoenzymes flux through the glycolysis was attenuated resulting in an increased flux through the pentose phosphate pathway. This led to increased NADPH supply for secondary metabolite production, and hence increased production of actinorhodin.At the more general level, the key links between primary and secondary metabolism are (Fig. 1A): (1) precursor metabolites, with acetyl-CoA being the most notable as it is used as precursor for biosynthesis of polyketides as well as isoprenoids; (2) amino acids, that are used as precursors for production of non-ribosomal peptides; (3) aromatic amino acids, which are used as precursors for flavonoids and alkaloids. Many other intermediates of the central metabolism can serve as precursors for natural product biosynthesis, but those listed above represent important ones. Besides the precursor, there is also a strong link between central metabolism and natural product biosynthesis through the requirement for redox power, often in the form of NADPH, and Gibbs free energy, often in the form of ATP. These links are very well illustrated in studies where natural product pathways have been reconstructed in a heterologous host, and in order to achieve high level production it has been necessary to engineer central metabolism. Examples are the production by yeast of artemisinic4 acid, farnesene5 and resveratrol.6 Artemisinic acid is an anti-malarial drug and farnesene is a hydrocarbon that can be used as jet-fuel or for synthesis of squalene. Both chemicals are derived from farnesyl-pyrophosphate (FPP) by expressing a specific sesquiterpene synthase, and whereas farnesene is produced directly by one enzyme, artemisinic acid biosynthesis requires a few additional biosynthetic steps. However, for production of both chemicals it is necessary to have efficient supply of FPP, and this required engineering of not only the mevalonate pathway leading to FPP, but also central carbon metabolism to ensure efficient provision of acetyl-CoA and NADPH.5 To ensure high-yields, a heterologous pathway involving phosphoketolase was expressed in yeast.5 This pathway takes xylulose-5-phosphate, an intermediate of the pentose phosphate pathway, and converts it into acetyl-phosphate and glyceraldehyde-3-phosphate. Acetyl-phosphate can be converted directly to acetyl-CoA by another enzyme, and hereby a new pathway for providing acetyl-CoA was installed. For improving resveratrol biosynthesis, which is synthesized from phenylalanine or tyrosine as well as malonyl-CoA, it was necessary to engineer the aromatic amino acid biosynthetic pathway to allow for high flux, but also to improve the provision of malonyl-CoA.6 Also this pathway requires extensive NADPH, so steps to ensure efficient provision of this co-factor also had to be considered.
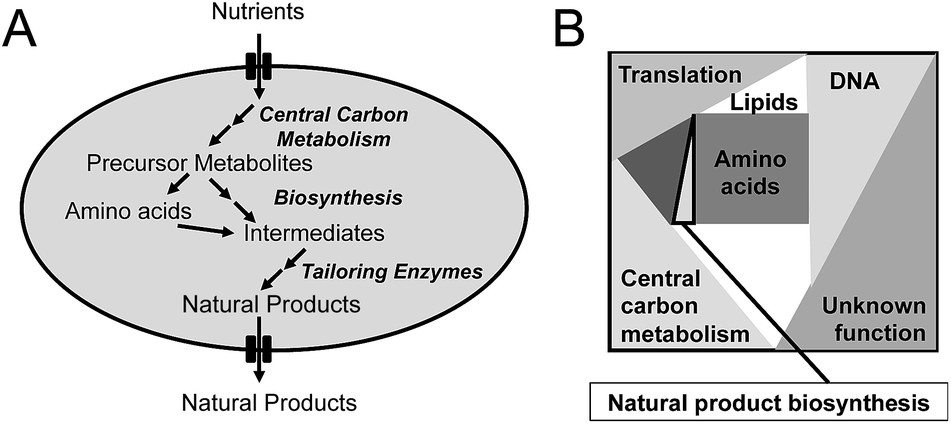 | ||
| Fig. 1 Simplified illustration of how primary and secondary metabolism is connected and the concept of proteome constraints. (A) Overview of metabolism. Nutrients, e.g. glucose, are metabolized via central carbon metabolism and hereby generates precursor metabolites. These are converted into amino acids and other cellular building blocks, but can also be used directly by secondary metabolism, e.g. by polyketide synthetases. In secondary metabolism amino acids, precursor metabolites (or other intracellular metabolites) are converted into natural products via a series of intermediates, and this part of metabolism is often distributed into initial biosynthetic steps and tailoring steps. A typical example is biosynthesis of an non-ribosomal peptide by an NRPS followed by modifications as mentioned for penicillin biosynthesis in the text. Finally the natural products are secreted out of the cell. (B) Illustration of the concept of proteome constraints. The total cellular proteome has an upper bound, which for many cell factories is even relatively constant across different environmental conditions. The cellular proteome can therefore be defined as allocation for different fractions within a defined box as illustrated in the figure. The total proteome is allocated for translation, DNA metabolism and transcription, central carbon metabolism, amino acid metabolism, lipid biosynthesis and natural product biosynthesis (normally a small fraction). A certain part of the proteome is also allocated to proteins with unknown function. The proteome constraints mean that if one part of the proteome requires additional proteome mass it has to be at the expense of proteome mass for other functions. | ||
3 Choice of cell factory
The second critical factor for optimizing the production of natural products is the chose the right cell factory. This can be very difficult, but the following points should be considered: (1) is the pathway compatible with the host, i.e. can the host deliver the precursors required for the biosynthetic pathway and if the pathway spans different compartments in its endogenous host, can this be reproduced in the heterologous host. An excellent example of pathway compatibility with the host is the production of adipoyl-7-ADCA mentioned earlier, as here the host already had a very efficient pathway for biosynthesis of adipoyl-6-APA. Often it is, however, more difficult to judge, but there some of the following points can probably assist; (2) is it easy to engineer the cell factory that is going to host the heterologous pathway? The answer is for sure yes for E. coli and S. cerevisiae, but these two cell factories are not always optimal for production of natural products. E.g. it may be better to use a Streptomyces species to express pathways discovered in Gram-positive actinomycetes and a fungus to express pathways discovered in other fungi. This is due to differences in G/C-content of the gene clusters but also compatibility of the promoter regions. Here the final choice may depend on which tools are available for genetic engineering of the host; (3) even though it can be an advantage of expressing pathways for fungal natural products in a fungus, there are drawbacks of this approach. Thus, most filamentous fungi that produce natural products, contain a large number of biosynthetic gene clusters, i.e. up to 50 clusters have been discovered through genome-sequencing of several Penicillia.7 This means that there is a risk that enzymes from other biosynthetic pathways are expressed in the chosen host, and these enzymes may interfere with the heterologous biosynthetic pathway. This holds true in particular for glycosyltransferases, but also for many other types of so-called tailoring enzymes that decorate the aglycone. This can of course be an advantage if one is aiming for product diversity, but it does complicate pathway discovery and will also complicate optimizing production of a specific molecule. Here use of hosts that have no or limited secondary biosynthesis capabilities, such as E. coli and yeast, have benefits, but the trade-off is poor pathway compatibility; (4) the ability to express functional pathway enzymes in the host is a very important factor. In some cases this is hard to judge at the outset, but if the pathway e.g. contains P450 enzymes it does not makes a lot of sense to use E. coli as a host, as this class of enzymes express poorly in bacteria, whereas they generally express well in yeast. This is mainly due to their membrane association, i.e. they can be expressed to function in the endoplasmic reticulum membrane of yeast. This, however, also means that it is important to consider the proteome constraints discussed below (see also Fig. 1B), as this may now apply not only to the total proteome, but the proteome of the specific membrane compartment where the enzyme(s) need(s) to be expressed; and (5) is the natural product toxic to the cell factory? This is of course a very important factor. Often natural producers also express a gene encoding for an enzyme or transporter that confer resistance towards the chemical, and this may then also be expressed in the cell factory of choice. However, it is not always the case, and this then becomes part of the cell factory and natural product biosynthesis compatibility evaluation.4 Enzyme activity and flux control
The third critical factor for improving natural product biosynthesis is the catalytic efficiency of the biosynthetic enzymes. Enzymes of secondary metabolism generally have lower kcat values and are generally larger than enzymes of the primary metabolism.8 This generally means that it may be hard to increase the flux through the biosynthetic pathway leading towards the natural product for predominantly two reasons.First, with low kcat values of the enzymes it is generally necessary to express these enzymes to very high levels in order to ensure sufficient enzymatic capacity required for a given flux. Even though this can easily be achieved through using strong promoters and multi-copy expression (as has occurred naturally in high penicillin producing strains of P. chrysogenum that carries multiple copies of the biosynthetic gene cluster), there is a trade-off with this approach. High-level expression of the biosynthetic enzymes, which generally have a high molecular weight as mentioned above, results in an increased requirement for allocation of proteome mass to natural product biosynthesis. As the cellular proteome is finite (see Fig. 1B), this means that there have to be reduced allocation of proteome for other cellular processes. As the majority of the proteome in a cell is normally allocated for central metabolism and protein biosynthesis, the consequence is that there is a reduction in proteome allocated for these processes. The result is a reduction in growth rate. It may be possible to accept this trade-off, but it is important to keep in mind, and as it is difficult to control which part of the proteome should be “sacrificed”, the trade-off may well result in unwanted phenotypes. In order to overcome this problem it is therefore important to quantify the kcat of the biosynthetic enzymes and evaluate how much of the proteome is required for supporting a certain flux through the pathway. If this fraction is large it is important to focus on improving the catalytic efficiency of the enzymes in the pathway.
Second, not only does the natural product pathway enzymes generally have low kcat-values, but there may also be large differences along the pathway (most natural product pathways have multiple steps). This can cause large differences in flux control along the pathway (Fig. 2), and in order to identify whether this is a problem it is important to measure pathway intermediates when the pathway enzymes are expressed at different levels. It is difficult to quantify the level of flux control along a pathway, but with the development of biosensors that enable measurements of intracellular metabolite levels, it has become possible to screen for large libraries of cells where the pathway enzymes are expressed at different levels. The issue of flux control is particularly challenging for very long biosynthetic pathways, but in some cases one can benefit from breaking the pathway into different segment and then optimize flux through each segment separately before the whole pathway is assembled.
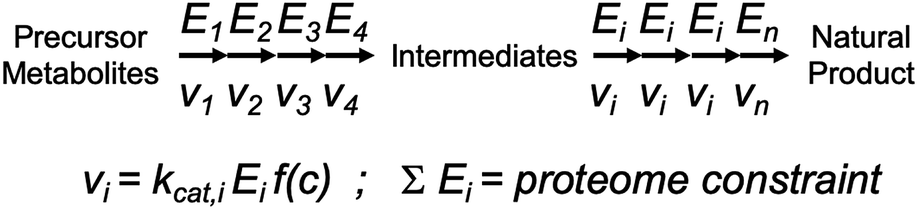 | ||
| Fig. 2 Illustration of how kcat impacts flux control through a biosynthetic pathway. Flux through each enzymatic step is determined by the product of three terms: (1) kcat; (2) enzyme concentration Ei; and (3) the concentration of substrates, products and allosteric regulators of the enzyme, here captured in the non-specified function f(c). The sum of enzyme concentrations have an upper value, i.e. determined by how much proteome mass that can be allocated for this specific pathway, and therefore it is important to distribute the enzyme mass, which can be controlled through varying gene expression, appropriately to the different steps in the pathway such that flux through each step is balanced. The distribution of mass to each enzyme should clearly be determined based on knowledge of the kcat values of each enzyme. | ||
5 Conclusions
I here argue that in connection with optimization of cell factories for production of natural products there are in particular three aspects that needs to be considered: (1) engineering of the central metabolism in order to ensure sufficient supply of precursors and co-factors; (2) choice of cell factory; and (3) improving the properties of the enzymes in the biosynthetic pathway, particularly to overcome the problem of a proteome constraint within the cell (or specific compartment where the pathway is expressed). Having all these three aspects in mind, it should be possible to design a strategy for developing an efficient cell factory for production of natural products.6 Conflicts of interest
There are no conflicts to declare.7 Acknowledgements
I acknowledge funding from the Novo Nordisk Foundation (grant no. NNF10CC1016517) and the Knut and Alice Wallenberg Foundation.8 References
- J. Nielsen, Science, 2015, 349, 1050 CrossRef CAS PubMed.
- S. Galanie, K. Thodey, I. J. Tenchard, M. F. Interrante and C. D. Smolke, Science, 2015, 349, 1095 CrossRef CAS PubMed.
- I. Borodina, J. Siebring, J. Zhang, C. P. Smith, G. van Keulen, L. Dijkhuizen and J. Nielsen, J. Biol. Chem., 2008, 283, 25186 CrossRef CAS PubMed.
- D.-K. Ro, E. M. Paradise, M. Ouellet, K. J. Fisher and K. L. Newman, et al. , Nature, 2006, 440, 940 CrossRef CAS PubMed.
- A. L. Meadows, K. M. Hawkins, Y. Tsegaye, E. Antipov and Y. Kim, et al. , Nature, 2016, 537, 694 CrossRef CAS PubMed.
- M. Li, K. Schneider, M. Kristensen, I. Borodina and J. Nielsen, Sci. Rep., 2016, 6, 36827 CrossRef CAS PubMed.
- J. C. Nielsen, S. Grijseels, S. Prigent, B. Ji, J. Dainat, K. F. Nielsen, J. C. Frisvad, M. Workman and J. Nielsen, Nat. Microbiol., 2017, 2, 17044 CrossRef CAS PubMed.
- A. Bar-Even, E. Noor, Y. Savir, W. Liebermeister, D. Davidi, D. S. Tawfik and R. Milo, Biochemistry, 2011, 50, 4402 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |