
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEn route to terpene natural products utilizing supramolecular cyclase mimetics
Qi
Zhang
 a,
Lorenzo
Catti
b,
Leonidas-Dimitrios
Syntrivanis
c and
Konrad
Tiefenbacher
*cd
a,
Lorenzo
Catti
b,
Leonidas-Dimitrios
Syntrivanis
c and
Konrad
Tiefenbacher
*cd
aKey Laboratory of Green Chemistry and Technology of Ministry of Education, College of Chemistry, Sichuan University, China
bLaboratory for Chemistry and Life Science, Institute of Innovative Research, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama 226-8503, Japan
cDepartment of Chemistry, University of Basel, St. Johanns-Ring 19, CH-4056 Basel, Switzerland. E-mail: konrad.tiefenbacher@unibas.ch
dDepartment of Biosystems Science and Engineering, ETH Zürich, Mattenstrasse 26, CH-4058 Basel, Switzerland. E-mail: tkonrad@ethz.ch
First published on 25th April 2019
Abstract
Covering: literature up to 2018
Terpenes are a class of natural products characterized by remarkable structural diversity. Much of this diversity arises biosynthetically from a handful of linear precursors through the so-called tail-to-head terpene cyclization reaction. This reaction is one of the most complex observed in nature, and historically attempts to replicate it with non-enzymatic means have met with little success. In recent years, however, the development of manmade binding pockets that allow such reactions to take place has been reported. This Highlight provides an overview of this nascent field, and outlines the challenges that need to be overcome moving forward.
 Qi Zhang | Qi Zhang obtained his master degree in 2012 at the Technical University of Munich and began his Ph.D. studies under the supervision of Prof. Konrad Tiefenbacher in the same year. After receiving his Ph.D degree at the Technical University of Munich in 2016, he re-joined the research group of Prof. Konrad Tiefenbacher at the University of Basel as a postdoctoral fellow. In late 2018, he started his independent career at Sichuan University in Chengdu, China. |
 Lorenzo Catti | Lorenzo Catti received his master degree in organic chemistry from the Technical University of Munich in 2013 under the supervision of Prof. Konrad Tiefenbacher. In 2016, he moved with the Tiefenbacher group to the University of Basel and, in 2017, completed his Ph.D. research on the application and development of resorcinarene-based supramolecular catalysts. He is currently a JSPS/AvH postdoctoral fellow in the Yoshizawa research group at the Tokyo Institute of Technology. |
 Leonidas-Dimitrios Syntrivanis | Leonidas-Dimitrios Syntrivanis received his master's degree from the University of Bologna in 2013. In the same year he moved to Oxford to join the Oxford Innovative Organic Synthesis for Cancer Research doctoral programme. There he worked with Prof. Jeremy Robertson and Prof. Luet Wong on the synthesis of eleuthoside structures and their selective hydroxylation through biocatalytic means, obtaining his DPhil in 2017. He is currently a postdoctoral researcher in the group of Prof. Konrad Tiefenbacher at the University of Basel. |
 Konrad Tiefenbacher | Konrad Tiefenbacher is a dual tenure track Assistant Professor of Chemistry at the University of Basel and the ETH Zurich. He obtained his master degree at the Technical University of Vienna in 2004 and the Ph.D. degree at the University of Vienna in the group of Prof. Johann Mulzer in 2009. He conducted postdoctoral research with Prof. Julius Rebek at The Scripps Research Institute in La Jolla in 2010/2011. In December 2011 he joined the faculty of TU Munich as assistant professor (no tenure track) before switching to his current position in 2016. His main research interest is supramolecular catalysis. |
1 Introduction
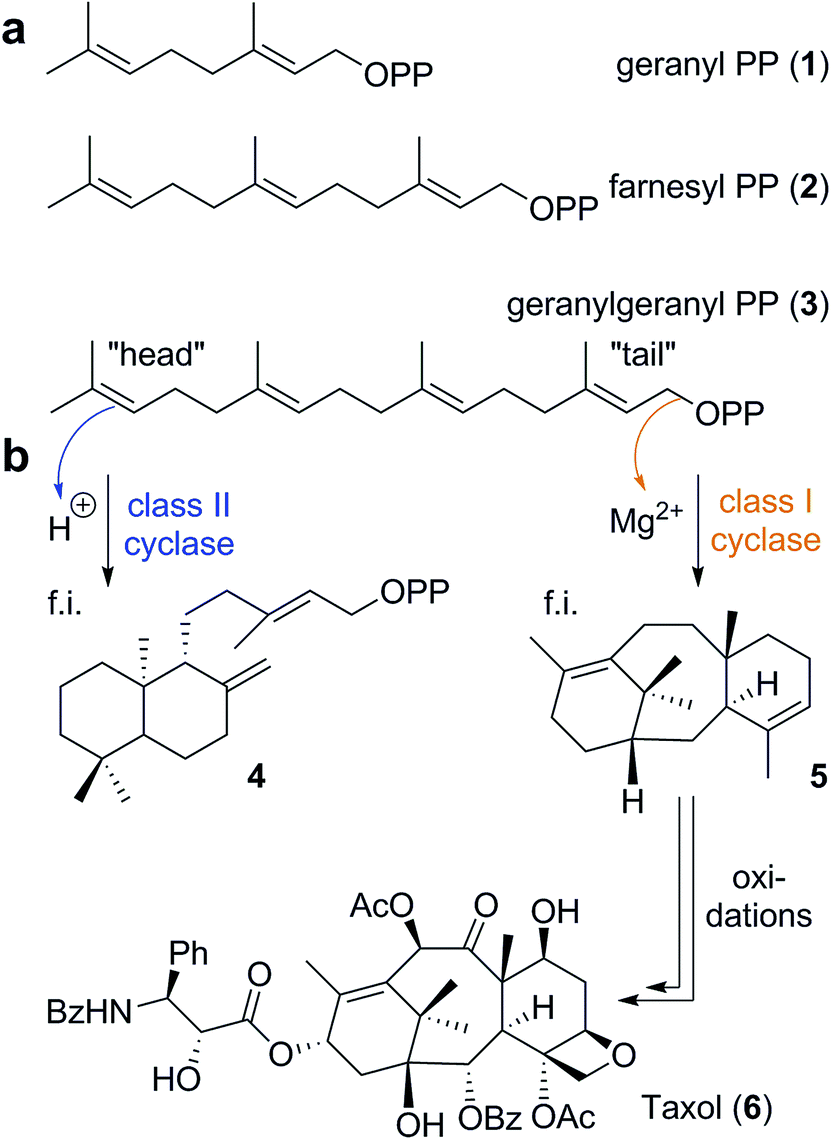
Nature's way of synthesizing natural products has fascinated and inspired chemists for decades. One of the most complex organic transformations performed in nature is the terpene cyclization. This kind of reaction provides the chemically and structurally most diverse class of natural products, the terpenoids.1 More than 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 members of the terpenoid family have been described so far. Nevertheless, the currently known structures constitute just a fraction of potential structures, as was recently highlighted for monoterpenes in a computational study.2 Interestingly, this whole variety is produced from just a handful of relatively simple acyclic terpenes via cyclization and oxidation processes. Geranyl pyrophosphate (GPP 1, Scheme 1a), farnesyl pyrophosphate (FPP 2), and geranylgeranyl pyrophosphate (GGPP 3) are the three most common substrates utilized for cyclizations. These linear isoprenoid diphosphates are formed in nature by prenyltransferases utilizing the simple C5 building blocks dimethylallyl diphosphate and isopentenyl diphosphate.1
000 members of the terpenoid family have been described so far. Nevertheless, the currently known structures constitute just a fraction of potential structures, as was recently highlighted for monoterpenes in a computational study.2 Interestingly, this whole variety is produced from just a handful of relatively simple acyclic terpenes via cyclization and oxidation processes. Geranyl pyrophosphate (GPP 1, Scheme 1a), farnesyl pyrophosphate (FPP 2), and geranylgeranyl pyrophosphate (GGPP 3) are the three most common substrates utilized for cyclizations. These linear isoprenoid diphosphates are formed in nature by prenyltransferases utilizing the simple C5 building blocks dimethylallyl diphosphate and isopentenyl diphosphate.1
 | ||
| Scheme 1 Structures of the linear substrates (a) utilized by class I and class II cyclases and examples of cyclization products (b) generated by such enzymes. | ||
In nature, two classes of enzymes are responsible for the subsequent cyclization processes.3 Class I cyclase enzymes initiate the reaction by cleaving the pyrophosphate residue of the molecule via a trinuclear metal cluster. Class II cyclases, on the other hand, trigger reactions via protonation of the terminal olefin or a previously installed epoxide moiety. Ryan Shenvi proposed the term “tail-to-head” cyclization for chemical reactions similar to reactions observed in class I cyclase enzymes since the reaction is initiated at the tail end of the molecule and propagates towards the head group (Scheme 1b).4 Accordingly, the term “head-to-tail” cyclization refers to a process similar to those catalyzed by class II enzymes.4 This terminology is very well suited to differentiate between these two reaction pathways in a non-enzymatic setting, the main focus of this article. Interestingly, the structural variety of products formed by class II cyclase enzymes is rather limited. Mainly six-membered rings are formed from the energetically most preferred chair conformation of the flexible substrate. As an example, the reaction product ent-copalyl diphosphate5 (4) is displayed in Scheme 1b. Chemists have had some success in mimicking the head-to-tail cyclization mode in solution,6 and even some enantioselective cyclizations using chiral Lewis acids have been reported. The reproducibility of this cyclization mode in solution using abiotic catalysts is likely due to the reliance on the energetically preferred substrate conformation, which translates into the product structure. Nevertheless, the head-to-tail cyclization mode is still far away from being generally applicable in a solution setting.
In contrast to class II cyclases, class I enzymes produce a much larger variety of products. This is due to their ability to bind a specific conformation of the flexible precursor, enabling cyclization modes not observed with the unrestrained conformations in solution. Additionally, the tail-to-head cyclization is stepwise and the cationic intermediates formed can undergo a variety of reactions like rearrangements, hydride shifts, proton transfers and eliminations. Again, the shape of the active site determines the accessible conformations of these carbocationic intermediates, thereby determining the reaction outcome. The active site of the enzyme, additionally, stabilizes cationic intermediates and transition states via a range of interactions: coulombic, cation–dipole and cation–π interactions.7 As an example of a tail-to-head cyclization product, taxadiene (5), the carbon framework of the anti-cancer drug Taxol (6),8 is depicted in Scheme 1b. This cyclization involves several cationic intermediates, an intramolecular proton transfer and an elimination reaction as the terminating step.1 Computational chemistry is increasingly utilized to learn more about the mechanisms involved in terpene cyclizations.9–14 Such studies have provided insight into how cyclase enzymes exert control over the cyclization outcome, via specific interactions between active site moieties and carbocations.9,11,14,15 The enzyme, however, not necessarily has to intervene throughout the whole cascade reaction; some steps can be driven by the intrinsic reactivity of the cationic intermediates.16 Nevertheless, in solution the tail-to-head cyclization mode of class I enzymes proved to be very challenging to reproduce.4 This may not be too surprising, since even a very large Lewis or Brønsted acid is hardly able to influence the conformation of the flexible acyclic terpene substrate. Moreover, nucleophiles in solution may intercept cationic intermediates, thereby preventing the formation of polycyclic products.
2 Tail-to-head terpene cyclizations in solution
There have been some reports about the tail-to-head cyclizations of monoterpene alcohols (Scheme 2a) in solution using for instance phosphoric acid17 or ferric chloride.18 These studies did not employ internal standards for the quantification of the products formed and reported the relative percentage yields only. Reproduction of the reaction conditions19 indicated that only traces of tail-to-head products were formed in solution under such conditions and that substitution products dominated. In fact, in the case of phosphoric acid catalysis, the head-to-tail product cyclogeranyl acetate 8 was formed as the major species (Scheme 2b). Interestingly, there are additional reports that describe the formation of head-to-tail products in solution. Geraniol or geranyl acetate (7) produced cyclogeranyl derivatives 8 as main products employing either zeolites or chlorosulfuric acid.20–22 Related head-to-tail cyclizations have also been observed with sesquiterpene substrates using the conditions reported for monoterpenes20,22 or employing superacids.23 Strong acids seem to favor protonation of the head olefin in solution.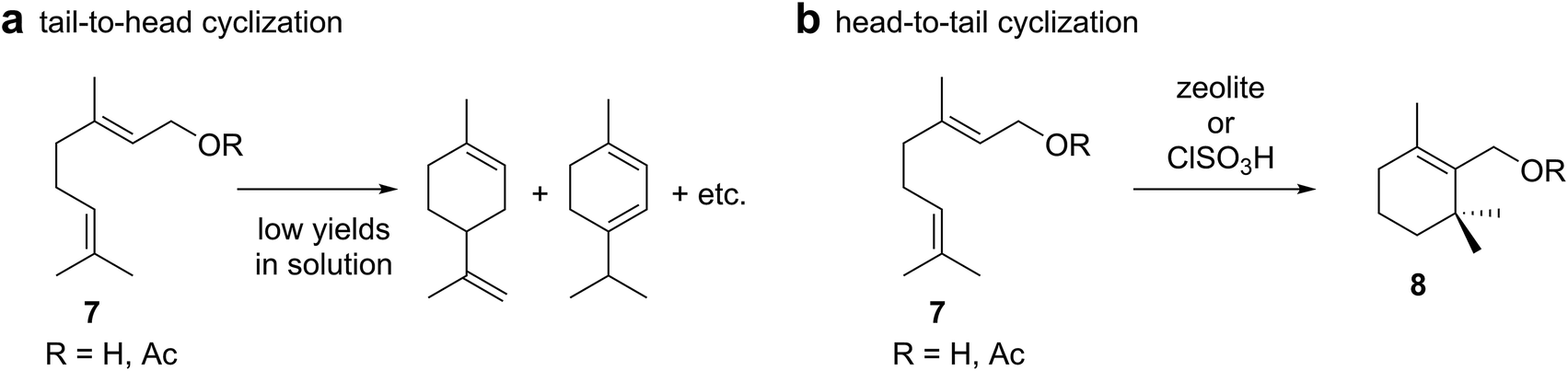 | ||
| Scheme 2 Examples of (a) tail-to-head and (b) head-to-tail cyclizations of monoterpenes using traditional Lewis or Brønsted acids in solution. | ||
Although there have been reports about tail-to-head cyclizations of sesquiterpenes in solution, these reactions again suffer from the competition of elimination and/or substitution reactions and produce cyclic terpenes in low yields only.4 The Shenvi group attributed these problems to the presence of counteranions that can function as a base and/or nucleophile. Due to coulombic attraction, this negatively charged counteranion is held in close proximity to the formed cation, resulting in fast elimination and substitution reactions. Shenvi and coworkers devised a strategy that prevented the mobility of the cleaved leaving group. They synthesized a modified sesquiterpene substrate 9 (Scheme 3a), containing a vinyl epoxide, in four steps. The addition of aluminum Lewis acids activates the epoxide and allows 1,6-ring closure to form the bisabolyl cation 10. Due to the covalent bond between the substrate and the former Lewis acid, the anionic leaving group is immobilized. Therefore, the cationic charge is able to freely propagate and undergo a reaction cascade consisting of hydride shifts, cyclizations and elimination, to produce the cedrene (11, 12) and funebrene (13) skeletons rather efficiently in a combined yield of 44%.
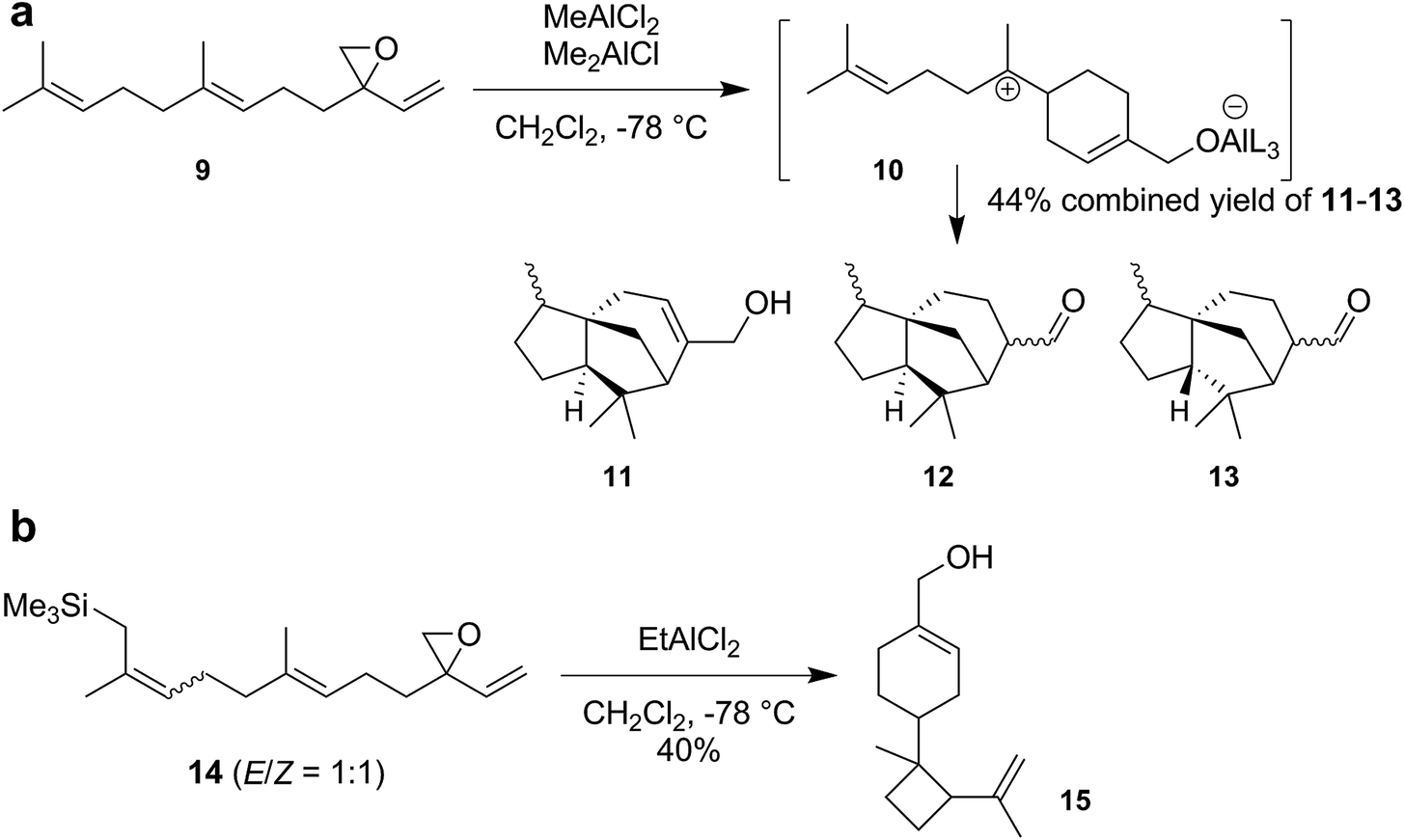 | ||
| Scheme 3 Selective tail-to-head sesquiterpene cyclizations using Lewis acids reported by the Shenvi group. This strategy gives access to (a) cedrene/funebrene and (b) cumacrene/dunniene skeletons and relies on the immobilization of anionic species, which allows the free propagation of the positive charge in the reactive intermediates. | ||
Moreover, the Shenvi group synthesized a similar sesquiterpene substrate 14 (Scheme 3b) that features a trimethylsilyl (TMS) group at the head end, in five steps. The TMS group should increase the nucleophilicity of the terminal alkene and allow the direct reaction between the terminal double bond and the initially formed bisabolyl cation 10. Indeed, exposure of 14 to aluminum Lewis acids produced the cumacrene/dunniene skeleton 15 in 40% yield as a mixture of isomers.
With these two examples, the Shenvi group clearly demonstrated that strained and thermodynamically unstable sesquiterpenes were accessible via this strategy. Nevertheless, two main limitations remain: (1) the unnatural substrates have to be synthesized in four to five linear steps. The ideal starting material for a chemical cyclization would be the commercially available and inexpensive farnesol itself. (2) The use of a Lewis acid in solution does not allow control over the conformation of the flexible sesquiterpene substrate. To achieve such control, uptake of the substrate into a binding pocket would be required.
3 Tail-to-head terpene cyclizations inside man-made binding pockets
Mimicking the active site of cyclase enzymes is obviously very challenging. In an early report, the groups of Janda and Lerner described the concept of raising antibodies for cationic terpene-like cyclizations using transition state analogs.24 In the antibody-catalyzed cyclization of monoterpene-like substrate 16 (Scheme 4a), the cyclic alkene products 17 and 18 were formed in a ratio of 80:20. Regular uncatalyzed hydrolysis produced mainly the tertiary alcohol product 19 as a diastereomeric mixture. Interestingly, but maybe not too surprisingly, the cavity of such active antibodies is lined with several aromatic residues to stabilize cationic species, much like in natural cyclase enzymes.25,26 The reaction catalyzed by the antibody, however, does not correspond to a tail-to-head cyclization that is characterized by the initial formation of an allylic cation. It shares distant similarities to a head-to-tail cyclization and this strategy was later indeed used for the formation of class II cyclase-like decalin structures.27
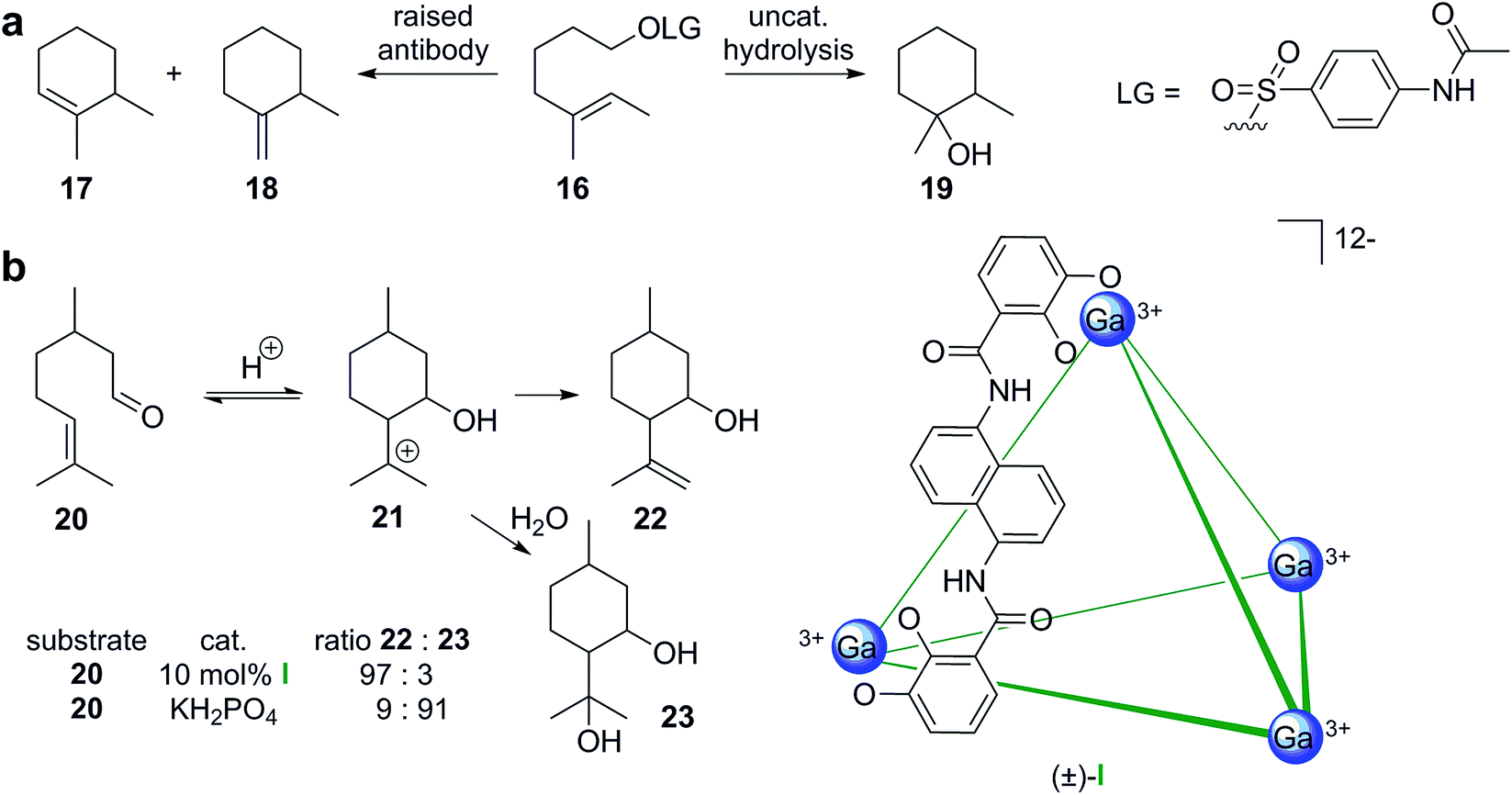 | ||
| Scheme 4 Terpene-like cyclization reactions catalyzed inside binding pockets. (a) Cyclization reaction occurring in a raised antibody. (b) Prins-cyclization of a monoterpene-like substrate catalyzed inside the metal–ligand assembly I. In both examples, the cyclization is likely facilitated by the stabilization of the reactive intermediate inside the binding pocket. | ||
More recently, investigations of supramolecular structures as simple enzyme mimetics have intensified.28–32 Supramolecular capsules share some basic similarities to cyclase enzymes. They feature a binding pocket that can entrap guest molecules temporarily. Most capsules reported in the literature feature aromatic walls that can potentially stabilize cationic species inside the binding pocket via cation–π interactions. A main difference to enzymes is the general absence of a pyrophosphate-binding motif, which is essential for strong substrate binding in cyclase enzymes. However, the Lledó group reported a supramolecular cavitand equipped with an anion binding motif,33 but was not able to initiate cyclizations so far. The groups of Toste, Bergman and Raymond reported monoterpene-like Prins-cyclizations inside the anionic metal–ligand cage (±)-I (Scheme 4b).34 Inside the supramolecular cavity (10 mol% of I, 60 °C, aqueous phosphate buffer, pH = 7.5), citronellal (20) was cyclized mainly to alkenol 22. In solution much more acidic conditions (pH = 3.2) were required to observe conversion. Interestingly, under these conditions diol 23 was formed preferentially. The observed differences in reactivity are likely due to the stabilization of cationic species and the hydrophobic environment inside the cavity of I. Utilizing an optically active host related to I, the authors were able to produce enantiomerically enriched products (up to 69% ee) with substrates closely related to the monoterpene 20.35
The Tiefenbacher group reported the first tail-to-head terpene cyclization inside a supramolecular container,19 utilizing the hexameric resorcinarene capsule II36–38 as the catalyst. This capsule self-assembles from six subunits 24 and eight water molecules in apolar solvents via the formation of a complex hydrogen bond network (Scheme 5a). Despite being completely closed, guest uptake into II is usually facile and occurs most likely via the dissociation of one subunit.39 Optimized reaction conditions turned out to be 10 mol% capsule II and 3 mol% HCl in chloroform at 30 °C.40 Under these conditions, commercially available nerol (25) was cyclized to eucalyptol in 40% yield (Scheme 5b). The reaction was initiated by the protonation of the alcohol, followed by cyclization to produce the α-terpinyl cation 26. By utilizing an 18O-labeled substrate, it was shown that the cleaved water molecule, not the water in the hydrogen bond network of II, was the preferred nucleophile for the introduction of the alcohol functionality of 27. Subsequently, the alkene is activated by protonation, to facilitate the cyclization leading to the formation of the bicyclic monoterpene eucalyptol (28). Interestingly, this report marked the first direct access to eucalyptol from an acyclic terpene precursor utilizing a man-made catalyst. Further studies revealed that the interception of cation 26 can be avoided by using less nucleophilic leaving groups. For instance, the conversion of geranyl acetate (29) under otherwise identical conditions produced α-terpinene (30) as the main product (35%, Scheme 5c). A series of control experiments indicated that the reaction indeed took place inside the cavity of II. For instance, without capsule, 3 mol% HCl did not initiate any conversion. The same was true when the cavity of II was blocked by a high affinity guest molecule (nBu4NBr). In addition, a very high size selectivity was observed, indicating that substrates that are encapsulated well react much faster than more bulky substrates.
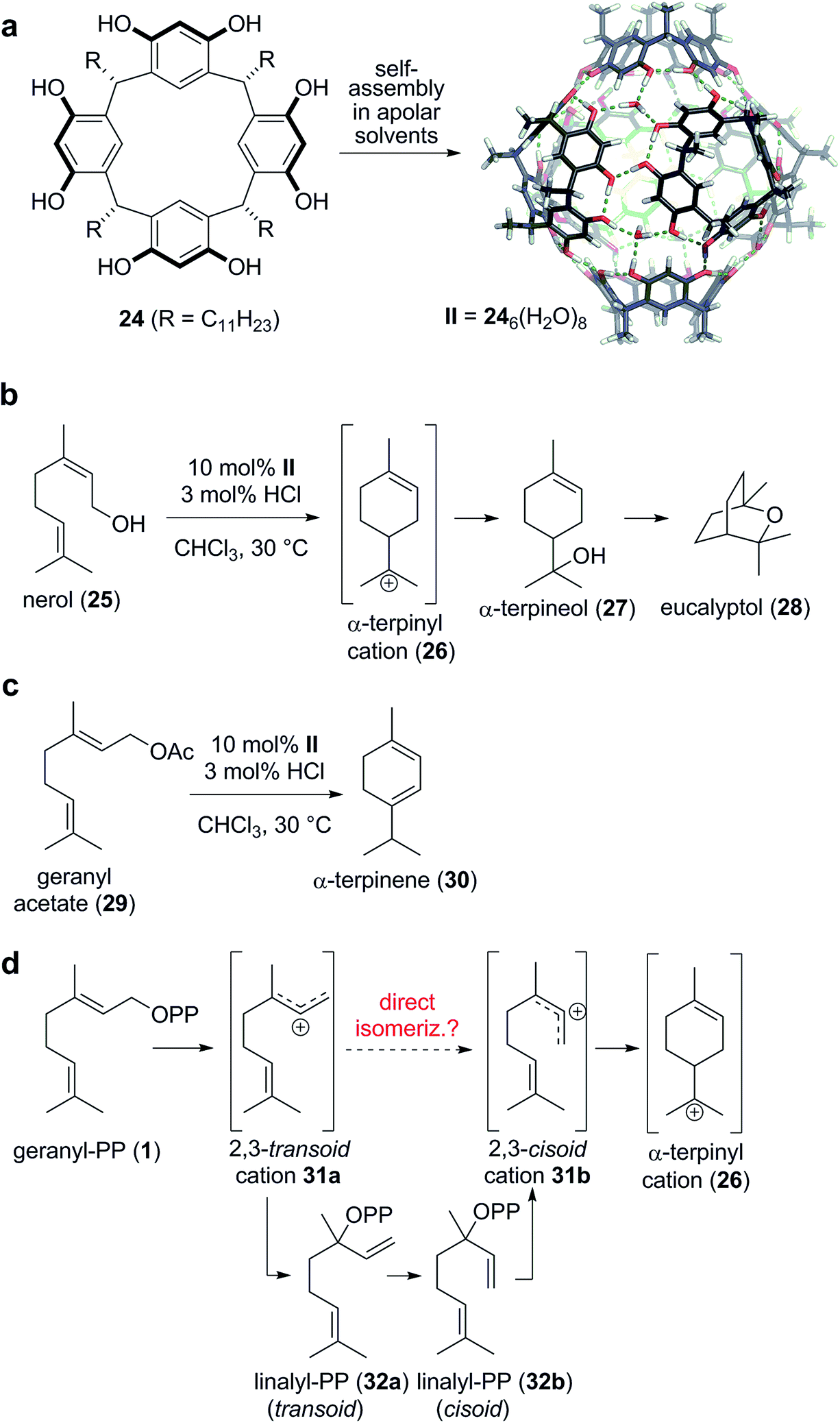 | ||
| Scheme 5 Tail-to-head monoterpene cyclization catalyzed inside a supramolecular catalyst. (a) In the presence of trace amount of water, the resorcinarene monomers self-assemble into the hexameric capsule in apolar solvent. (b and c) Capsule II-catalyzed cyclization reactions using nerol and geranyl acetate as the substrate. (d) According to the observations obtained from the supramolecular system, a direct isomerization of the transoid cation to its cisoid counter-part should not be excluded for the terpene biosynthesis. | ||
The reaction mechanism for the formation of α-terpinene (30) from geranyl acetate (29) must involve the α-terpinyl cation 26, the same intermediate as in the formation of eucalyptol. How is this cation generated from 29? A direct cyclization like in the reaction of nerol (25) is prevented by the trans geometry of the C2–C3 alkene in geranyl acetate (29). In the proposed biosynthesis (Scheme 5d), the leaving group of geranyl pyrophosphate (1) is first cleaved to generate the transoid allylic cation 31a. The recombination of the leaving group at the more hindered allylic position of the cation then produces linalyl-PP 32a. The free rotation about the C2–C3 sigma bond allows for the facile conversion to the cisoid isomer 32b, required for the cyclization to 26. After another cleavage of the leaving group, the cisoid cation is generated, which is believed to produce the key intermediate 26via cyclization. This isomerization mechanism is generally accepted in the literature1 for terpene cyclizations and is in very good agreement with experimental evidence. Nevertheless, the release of the formed tertiary pyrophosphate (32a/b in case of monoterpenes) has not been observed so far.41 The direct isomerization of the initially formed transoid allylic cation 31a was excluded due to gas phase calculations which indicated that the energy barrier for such a process is relatively high (approx. 55 kJ mol−1).42,43 However, in the capsule-catalyzed cyclization of geranyl acetate (29), a stepwise isomerization mechanism involving linalyl derivatives can be excluded for two reasons: (1) the reaction profiles for geranyl acetate and linalyl acetate do not match, indicating that geranyl acetate predominantly reacts via an alternative pathway that does not involve linalyl acetate as intermediate. (2) More importantly, in the cyclization of geranyl acetate, no intermediates – like for instance linalyl acetate – can be detected. In contrast to cyclase enzymes that bind formed intermediates tightly, capsule II does not exhibit a strong affinity to intermediates. Therefore, intermediates – for instance α-terpineol (Scheme 5b) – can be readily detected via GC and NMR. In light of these observations, a direct isomerization of the transoid cation 31a to the cisoid cation 31b was considered highly likely. Although this result does not necessarily imply that a direct isomerization is also operational in the terpene biosynthesis, the direct isomerization should not be categorically excluded anymore.
The reaction mechanism for the conversion of geranyl acetate (29) was investigated in detail.40 Kinetic investigations indicated that the cleavage of the protonated leaving group was the rate-determining step of the cyclization cascade, indicating that substrate uptake into capsule II is not the slowest step.
Recently, the Tiefenbacher group also reported the first examples of sesquiterpene cyclizations inside the capsule II.44,45 Utilizing the optimized conditions for monoterpene cyclizations, all alkene isomers of farnesol and farnesol acetate were investigated (Scheme 6a). Compared to monoterpenes, formation of more complex product mixtures was observed. This was not too surprising, since the additional prenyl unit and the higher flexibility of the farnesol precursor enable a much greater diversity in cyclization outcomes. Five main products were identified in these experiments (Scheme 6a and b): δ-selinene (35), 10-epi-zonarene (37), α-cedrene (38), 2-epi-α-cedrene (39), and ε-patchoulene (40). Interestingly, δ-selinene (35) was only observed with substrates containing a (2E)-alkene. Its formation is initiated by a 1,10-ring connection to form cation 34 (Scheme 6a).46 This ring closure seems to be only operational when the 1,6-ring formation (Scheme 6b) is attenuated by the (2E)-alkene geometry. The most selective conversion to δ-selinene (35) was observed in the case of (2E,6Z)-farnesyl acetate (18% yield). It is likely that the (6Z)-alkene favors substrate conformations suitable for the required 1,10-ring closure. This difficile control over the substrate conformation is not possible with capsule II yet, since its cavity is too large and too symmetric. Therefore, it was decided to further restrict the conformational freedom of the sesquiterpene substrate by incorporating a six-membered ring. The cyclofarnesyl acetate 41 (Scheme 6c) is readily available in three steps from an inexpensive starting material. Cyclization of this conformationally restricted substrate led to a relatively selective formation of isolongifolene (42) in 24% yield (GC analysis). Purification at this step was difficult and required repeated chromatography on silica, which reduced the yield to 11% (85% purity according to GC analysis). Analytically pure material was obtained by direct allylic oxidation of the crude cyclization mixture of 42 (after the removal of resorcinarene), to deliver the natural product isolongifolenone (43). It was isolated in 20% yield from starting material 41 over two steps. This route (total of four steps for isolongifolene; five steps for isolongifolenone) is much shorter than previously reported total synthesis approaches which required ten or more linear steps.44 This highlights the great potential of terpene cyclizations inside supramolecular catalysts. Nevertheless, it is only a starting point. Several advances are required to transform this proof of principle study to a generally useful terpene cyclase mimic.
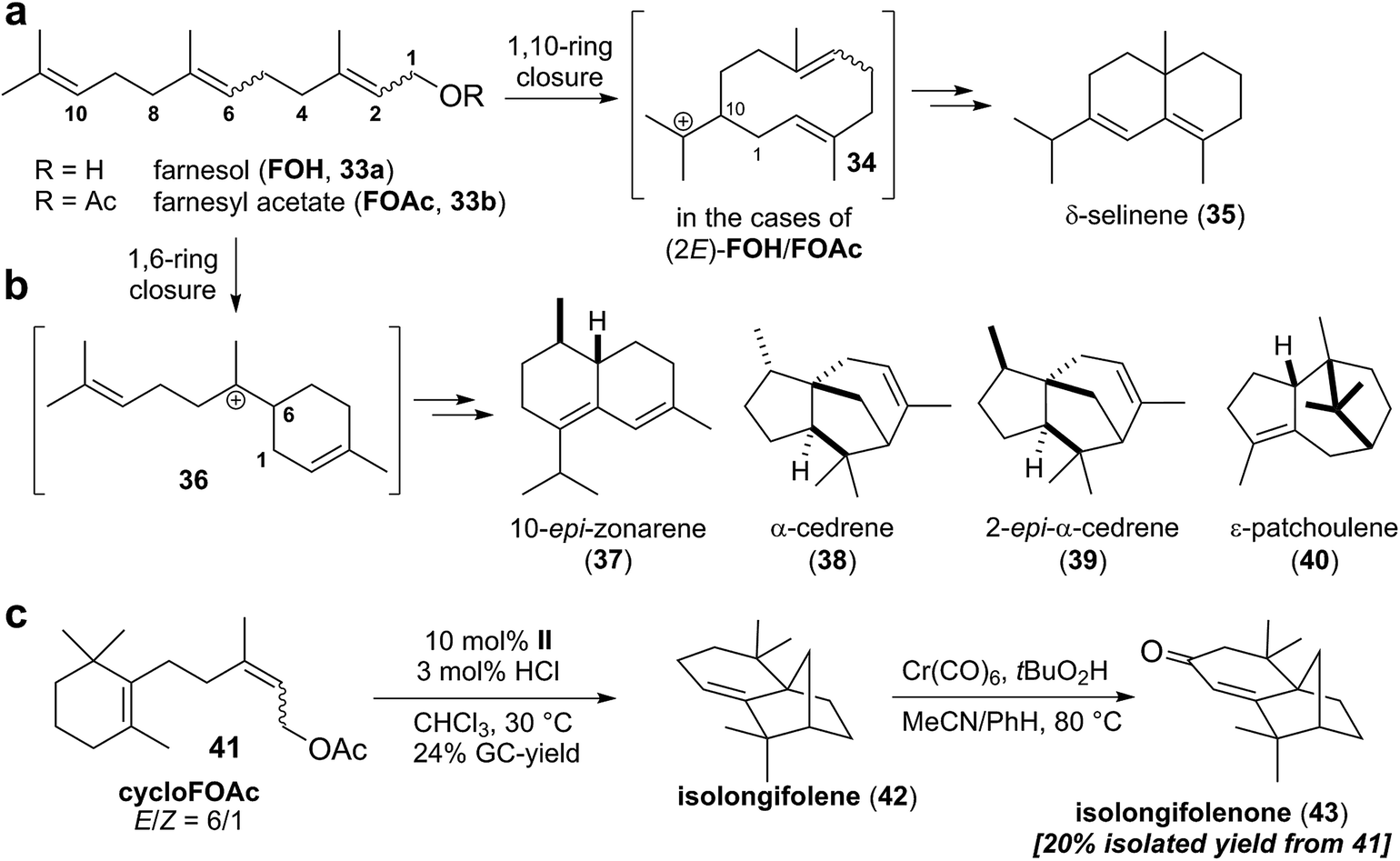 | ||
| Scheme 6 Tail-to-head sesquiterpene cyclization catalyzed inside a supramolecular catalyst. Depending on the double bond geometry of the substrate, the reactions of farnesyl substrates proceed via a 1,10- (a) or 1,6-cyclization mechanism (b). (c) A six-membered ring is incorporated into the cyclization precursor to restrict the conformational freedom of the substrate. This modification enabled a relatively selective conversion of the substrate to the tricyclic sesquiterpene natural product isolongifolene. | ||
4 Remaining challenges
Although some first examples for tail-to-head terpene cyclizations inside man-made catalysts have appeared in literature, several huge challenges remain. The main obstacle, that prevents useful synthetic applications, certainly concerns selectivity issues. Natural cyclase enzymes are able to control the substrate conformation via binding into a complementarily shaped cavity. This has not been possible utilizing man-made catalysts so far. However, in contrast to regular Lewis or Brønsted acids, supramolecular containers offer this possibility. While the large size and the high symmetry of the supramolecular capsule utilized for the cyclizations so far did not restrict the conformational freedom of acyclic terpenes well, alternative systems might do so. Therefore, the exploration of other supramolecular containers is highly desirable. Initial studies, however, demonstrated that not all systems are catalytically active,47 while the prerequisites for catalytic activity are not fully understood yet. Elucidating these requirements will enable the construction of more selective systems. In the even more distant future, emphasis might shift to the predictability of cyclization products of such cyclase mimetics. Molecular modelling that has helped understanding terpene cyclizations will certainly be helpful in these future endeavors.5 Conclusion
This highlight article summarizes the progress made in the field of biomimetic tail-to-head terpene cyclizations. First successful examples have been reported over the last years. In solution, modified terpene precursors were cyclized to complex terpene products in a biomimetic fashion. The strategy employed enabled the formation of strained sesquiterpene products. However, the substrate conformation cannot be controlled in cyclization reactions occurring in solution. This can be addressed through the temporary encapsulation into a supramolecular capsule, which potentially allows restriction of the substrate conformation. First examples of tail-to-head terpene cyclizations inside a supramolecular container have been reported. Nevertheless, due to the large size and the high symmetry of the cavity, sufficient control over the substrate conformation is still lacking. Further studies are required to elucidate the prerequisites for catalytic activity in such systems, to enable construction of more selective supramolecular catalysts.One could ask why mimicking enzymes that nature has already optimized close to perfection would be a worthwhile endeavor. The power and utility of the biomachinery for the production of tail-to-head terpene products is undisputed, as for instance demonstrated by the efficient production of artemisinic acid, the precursor to the anti-malaria artemisinin.48 Nevertheless, there are several (potential) advantages for man-made catalysts: (1) there are cases, especially for natural products from marine sources, where the producer organism is unknown and, therefore, the biomachinery is not available to us. (2) Learning how to construct artificial cyclase enzymes may allow chemists to access completely novel terpene structures, yet unknown to nature. Modified cyclization substrates might be especially of interest, since natural enzymes do not tolerate large variations of the substrate structure.49 (3) Such studies also may help chemists to better understand the working principles of natural enzymes.
In addition, the intrinsic challenge of performing one of the most complex reaction of nature in a man-made catalyst is very motivating in itself. Therefore, we believe that this field will continue to receive an increasing amount of attention over the next decade.
6 Conflicts of interest
There are no conflicts to declare.7 Acknowledgements
This work was supported by funding from the European Research Council Horizon 2020 Programme [ERC Starting Grant 714620-TERPENECAT], the Swiss National Science Foundation as part of the NCCR Molecular Systems Engineering and the Bayerische Akademie der Wissenschaften (Junges Kolleg). L. C. thanks JSPS and the Alexander von Humboldt Foundation for a postdoctoral fellowship.8 References
- D. W. Christianson, Structural and Chemical Biology of Terpenoid Cyclases, Chem. Rev., 2017, 117(17), 11570–11648 CrossRef CAS PubMed.
- B. Tian, C. D. Poulter and M. P. Jacobson, Defining the Product Chemical Space of Monoterpenoid Synthases, PLoS Comput. Biol., 2016, 12(8), e1005053 CrossRef PubMed.
- K. U. Wendt and G. E. Schulz, Isoprenoid biosynthesis: manifold chemistry catalyzed by similar enzymes, Structure, 1998, 6, 127–133 CrossRef CAS PubMed.
- S. V. Pronin and R. A. Shenvi, Synthesis of highly strained terpenes by non-stop tail-to-head polycyclization, Nat. Chem., 2012, 4, 915–920 CrossRef CAS PubMed.
- D. Morrone, J. Chambers, L. Lowry, G. Kim, A. Anterola, K. Bender and R. J. Peters, Gibberellin biosynthesis in bacteria: Separate ent-copalyl diphosphate and ent-kaurene synthases in Bradyrhizobium japonicum, FEBS Lett., 2009, 583(2), 475–480 CrossRef CAS PubMed.
- R. A. Yoder and J. N. Johnston, A Case Study in Biomimetic Total Synthesis: Polyolefin Carbocyclizations to Terpenes and Steroids, Chem. Rev., 2005, 105, 4730–4756 CrossRef CAS PubMed.
- J. C. Ma and D. A. Dougherty, The Cation–π Interaction, Chem. Rev., 1997, 97, 1303–1324 CrossRef CAS PubMed.
- D. G. I. Kingston, Recent Advances in the Chemistry of Taxol, J. Nat. Prod., 2000, 63, 726–734 CrossRef CAS.
- T. Ansbacher, Y. Freud and D. T. Major, Slow-Starter Enzymes: Role of Active-Site Architecture in the Catalytic Control of the Biosynthesis of Taxadiene by Taxadiene Synthase, Biochemistry, 2018, 57(26), 3773–3779 CrossRef CAS PubMed.
- H. Sato, K. Narita, A. Minami, M. Yamazaki, C. Wang, H. Suemune, S. Nagano, T. Tomita, H. Oikawa and M. Uchiyama, Theoretical Study of Sesterfisherol Biosynthesis: Computational Prediction of Key Amino Acid Residue in Terpene Synthase, Sci. Rep., 2018, 8(1), 2473 CrossRef PubMed.
- R. Driller, S. Janke, M. Fuchs, E. Warner, A. R. Mhashal, D. T. Major, M. Christmann, T. Brück and B. Loll, Towards a comprehensive understanding of the structural dynamics of a bacterial diterpene synthase during catalysis, Nat. Commun., 2018, 9(1), 3971 CrossRef PubMed.
- C. H. McCulley and D. J. Tantillo, Secondary Carbocations in the Biosynthesis of Pupukeanane Sesquiterpenes, J. Phys. Chem. A, 2018, 122(40), 8058–8061 CrossRef CAS PubMed.
- T. E. O'Brien, S. J. Bertolani, Y. Zhang, J. B. Siegel and D. J. Tantillo, Predicting Productive Binding Modes for Substrates and Carbocation Intermediates in Terpene Synthases—Bornyl Diphosphate Synthase As a Representative Case, ACS Catal., 2018, 8(4), 3322–3330 CrossRef PubMed.
- N. G. H. Leferink, K. E. Ranaghan, V. Karuppiah, A. Currin, M. W. van der Kamp, A. J. Mulholland and N. S. Scrutton, Experiment and Simulation Reveal How Mutations in Functional Plasticity Regions Guide Plant Monoterpene Synthase Product Outcome, ACS Catal., 2018, 8(5), 3780–3791 CrossRef CAS.
- D. T. Major, Y. Freud and M. Weitman, Catalytic control in terpenoid cyclases: multiscale modeling of thermodynamic, kinetic, and dynamic effects, Curr. Opin. Chem. Biol., 2014, 21, 25–33 CrossRef CAS PubMed.
- D. J. Tantillo, Importance of Inherent Substrate Reactivity in Enzyme-Promoted Carbocation Cyclization/Rearrangements, Angew. Chem., Int. Ed., 2017, 56(34), 10040–10045 CrossRef CAS PubMed.
- J. P. McCormick and D. L. Barton, Studies in 85% H3PO4—II: On the role of the α-terpinyl cation in cyclic monoterpene genesis, Tetrahedron, 1978, 34, 325–330 CrossRef CAS.
- W. Yu, M. Wen, L. Yang and Z. L. Liu, Ferric chloride catalyzed isomerization and cyclization of geraniol, linalool and nerol, Chin. Chem. Lett., 2002, 13, 495–496 CAS.
- Q. Zhang and K. Tiefenbacher, Terpene cyclization catalysed inside a self-assembled cavity, Nat. Chem., 2015, 7, 197–202 CrossRef CAS PubMed.
- C. Tsangarakis and M. Stratakis, Biomimetic cyclization of small terpenoids promoted by zeolite NaY: Tandem formation of alpha-ambrinol from geranyl acetone, Adv. Synth. Catal., 2005, 347, 1280–1284 CrossRef CAS.
- W. Yu, F. Bian, Y. Gao, L. Yang and Z.-L. Liu, Y-Zeolite-Catalyzed Cyclizations of Terpenols, Adv. Synth. Catal., 2006, 348, 59–62 CrossRef CAS.
- C. Raptis, I. N. Lykakis, C. Tsangarakis and M. Stratakis, Acid-Catalyzed Cyclization of Terpenes Under Homogeneous and Heterogeneous Conditions as Probed Through Stereoisotopic Studies: A Concerted Process with Competing Preorganized Chair and Boat Transition States, Chem.–Eur. J., 2009, 15(44), 11918–11927 CrossRef CAS PubMed.
- M. P. Polovinka, D. V. Korchagina, Y. V. Gatilov, I. Y. Bagrianskaya, V. A. Barkhash, V. B. Perutskii, N. D. Ungur, P. F. Vlad, V. V. Shcherbukhin and N. S. Zefirov, Cyclization and Rearrangements of Farnesol and Nerolidol Stereoisomers in Superacids, J. Org. Chem., 1994, 59(6), 1509–1517 CrossRef CAS.
- J. Hasserodt, K. D. Janda and R. A. Lerner, Antibody Catalyzed Terpenoid Cyclization, J. Am. Chem. Soc., 1996, 118, 11654–11655 CrossRef CAS.
- X. Zhu, A. Heine, F. Monnat, K. N. Houk, K. D. Janda and I. A. Wilson, Structural Basis for Antibody Catalysis of a Cationic Cyclization Reaction, J. Mol. Biol., 2003, 329(1), 69–83 CrossRef CAS PubMed.
- C. M. Paschall, J. Hasserodt, T. Jones, R. A. Lerner, K. D. Janda and D. W. Christianson, Convergence of Catalytic Antibody and Terpene Cyclase Mechanisms: Polyene Cyclization Directed by Carbocation–π Interactions, Angew. Chem., Int. Ed., 1999, 38(12), 1743–1747 CrossRef CAS PubMed.
- J. Hasserodt, K. D. Janda and R. A. Lerner, Formation of Bridge-Methylated Decalins by Antibody-Catalyzed Tandem Cationic Cyclization, J. Am. Chem. Soc., 1997, 119(26), 5993–5998 CrossRef CAS.
- M. D. Pluth, R. G. Bergman and K. N. Raymond, Proton-Mediated Chemistry and Catalysis in a Self-Assembled Supramolecular Host, Acc. Chem. Res., 2009, 42, 1650–1659 CrossRef CAS.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Functional Molecular Flasks: New Properties and Reactions within Discrete, Self-Assembled Hosts, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS PubMed.
- S. H. A. M. Leenders, R. Gramage-Doria, B. de Bruin and J. N. H. Reek, Transition metal catalysis in confined spaces, Chem. Soc. Rev., 2015, 44, 433–448 RSC.
- D. Ajami and J. Rebek, More Chemistry in Small Spaces, Acc. Chem. Res., 2012, 46, 990–999 CrossRef PubMed.
- L. Catti, Q. Zhang and K. Tiefenbacher, Advantages of Catalysis in Self-Assembled Molecular Capsules, Chem.–Eur. J., 2016, 22, 9060–9066 CrossRef CAS PubMed.
- A. Lledó and A. Soler, Binding of ion pairs in a thiourea-functionalized self-folding cavitand, Org. Chem. Front., 2017, 4(7), 1244–1249 RSC.
- W. M. Hart-Cooper, K. N. Clary, F. D. Toste, R. G. Bergman and K. N. Raymond, Selective Monoterpene-like Cyclization Reactions Achieved by Water Exclusion from Reactive Intermediates in a Supramolecular Catalyst, J. Am. Chem. Soc., 2012, 134, 17873–17876 CrossRef CAS.
- C. Zhao, Q.-F. Sun, W. M. Hart-Cooper, A. G. DiPasquale, F. D. Toste, R. G. Bergman and K. N. Raymond, Chiral Amide Directed Assembly of a Diastereo- and Enantiopure Supramolecular Host and its Application to Enantioselective Catalysis of Neutral Substrates, J. Am. Chem. Soc., 2013, 135, 18802–18805 CrossRef CAS PubMed.
- L. R. MacGillivray and J. L. Atwood, A chiral spherical molecular assembly held together by 60 hydrogen bonds, Nature, 1997, 389, 469–472 CrossRef CAS.
- A. Shivanyuk and J. Rebek, Reversible encapsulation by self-assembling resorcinarene subunits, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 7662–7665 CrossRef CAS PubMed.
- L. Avram and Y. Cohen, Spontaneous Formation of Hexameric Resorcinarene Capsule in Chloroform Solution as Detected by Diffusion NMR, J. Am. Chem. Soc., 2002, 124, 15148–15149 CrossRef CAS.
- M. Yamanaka, A. Shivanyuk and J. Rebek, Kinetics and Thermodynamics of Hexameric Capsule Formation, J. Am. Chem. Soc., 2004, 126, 2939–2943 CrossRef CAS PubMed.
- Q. Zhang, L. Catti, J. Pleiss and K. Tiefenbacher, Terpene Cyclizations inside a Supramolecular Catalyst: Leaving-Group-Controlled Product Selectivity and Mechanistic Studies, J. Am. Chem. Soc., 2017, 139, 11482–11492 CrossRef CAS PubMed.
- D. E. Cane, G. Yang, Q. Xue and J. H. Shim, Trichodiene Synthase. Substrate Specificity and Inhibition, Biochemistry, 1995, 34(8), 2471–2479 CrossRef CAS PubMed.
- R. Croteau, Biosynthesis and catabolism of monoterpenoids, Chem. Rev., 1987, 87, 929–954 CrossRef CAS.
- N. L. Allinger and J. H. Siefert, Organic quantum chemistry. XXXIII. Electronic spectra and rotational barriers of vinylborane, allyl cation, and related compounds, J. Am. Chem. Soc., 1975, 97, 752–760 CrossRef CAS.
- Q. Zhang, J. Rinkel, B. Goldfuss, J. S. Dickschat and K. Tiefenbacher, Sesquiterpene cyclizations catalysed inside the resorcinarene capsule and application in the short synthesis of isolongifolene and isolongifolenone, Nat. Catal., 2018, 1(8), 609–615 CrossRef PubMed.
- D. T. Major, Complex terpenes in one pot, Nat. Catal., 2018, 1(8), 567–568 CrossRef.
- C. L. Steele, J. Crock, J. Bohlmann and R. Croteau, Sesquiterpene Synthases from Grand Fir (Abies grandis): comparison of constitutive and wound-induced activities, and cDNA isolation, characterization, and bacterial expression of δ-selinene synthase and γ-humulene synthase, J. Biol. Chem., 1998, 273(4), 2078–2089 CrossRef CAS.
- Q. Zhang, L. Catti, V. R. I. Kaila and K. Tiefenbacher, To catalyze or not to catalyze: elucidation of the subtle differences between the hexameric capsules of pyrogallolarene and resorcinarene, Chem. Sci., 2017, 8, 1653–1657 RSC.
- C. J. Paddon, P. J. Westfall, D. J. Pitera, K. Benjamin, K. Fisher, D. McPhee, M. D. Leavell, A. Tai, A. Main, D. Eng, D. R. Polichuk, K. H. Teoh, D. W. Reed, T. Treynor, J. Lenihan, H. Jiang, M. Fleck, S. Bajad, G. Dang, D. Dengrove, D. Diola, G. Dorin, K. W. Ellens, S. Fickes, J. Galazzo, S. P. Gaucher, T. Geistlinger, R. Henry, M. Hepp, T. Horning, T. Iqbal, L. Kizer, B. Lieu, D. Melis, N. Moss, R. Regentin, S. Secrest, H. Tsuruta, R. Vazquez, L. F. Westblade, L. Xu, M. Yu, Y. Zhang, L. Zhao, J. Lievense, P. S. Covello, J. D. Keasling, K. K. Reiling, N. S. Renninger and J. D. Newman, High-level semi-synthetic production of the potent antimalarial artemisinin, Nature, 2013, 496, 528–532 CrossRef CAS PubMed.
- D. J. Miller, F. Yu, N. J. Young and R. K. Allemann, Competitive inhibition of aristolochene synthase by phenyl-substituted farnesyl diphosphates: evidence of active site plasticity, Org. Biomol. Chem., 2007, 5, 3287–3298 RSC.
| This journal is © The Royal Society of Chemistry 2019 |