
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Editing streptomycete genomes in the CRISPR/Cas9 age
Fabrizio
Alberti
 and
Christophe
Corre
*
and
Christophe
Corre
*
School of Life Sciences, Department of Chemistry, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK. E-mail: C.Corre@warwick.ac.uk
First published on 25th January 2019
Abstract
Covering: up to December 2018
This article aims to highlight advantages, drawbacks and issues that users should consider when implementing the use of CRISPR/Cas9-tools for genome editing in streptomycetes, the most prolific source of antimicrobial natural products to date. Here, we examine four toolkits that have so far been made available for streptomycete in vivo-engineering and one for in vitro-editing, and review how they have been applied over the last three years. Our critical evaluation of these toolkits intends to support potential users in determining what they could achieve, what they should consider and what system they should select/optimise for their application.
 Fabrizio Alberti | Fabrizio Alberti completed his Ph.D. in 2015 at the University of Bristol, where he studied the biosynthesis of fungal natural products under the supervision of Prof. Gary Foster and Dr Andy Bailey. He then joined Dr Christophe Corre's group at the University of Warwick as a postdoctoral research fellow to work on natural product discovery from streptomycetes and the development of whole-cell biocatalysts. In 2018 he became a Leverhulme Trust Early Career Fellow at the same institution, where he currently focuses on the elucidation of biosynthetic pathways of fungal meroterpenoid natural products. |
 Christophe Corre | Christophe Corre is an Associate Professor in Synthetic Biology; jointly appointed between the School of Life Sciences and the Department of Chemistry at the University of Warwick, UK. His research group is particularly interested in streptomycete transcriptional regulators, biocatalysts and natural products. Christophe received his education in Biochemistry and Molecular Biology at the University of Nice in France. He then carried out work on antibiotic biosynthesis for which he was awarded a PhD in Chemistry at the University of Exeter, UK. He then moved to Warwick in 2004 to work as a postdoctoral research fellow in Prof. Greg Challis group. In 2010, Christophe was awarded a Royal Society University Research Fellowship to unlock the production of novel microbial antibiotics and to start his independent research group at Warwick. |
1 Introduction
Streptomycetes are Gram-positive filamentous bacteria known for their ability to produce a wide variety of bioactive metabolites (Fig. 1) and recognised as the primary producers of the antibiotics currently used in human and veterinary medicine or undergoing clinical development.1 The cyclic lipopeptide daptomycin (1) is one the most prominent examples of antibiotics made by streptomycetes and recently exploited in the clinic. This natural product is used to treat complicated skin and skin structure infections, as well as bacteremia and endocarditis caused by Staphylococcus aureus.2 Streptomycetes are also acknowledged producers of useful bioactive molecules that find application in various other fields, such as the anthelmintic agents avermectins (2)3 and the herbicide bialaphos (3).4 Whole genome sequencing of streptomycetes has uncovered a plethora of cryptic biosynthetic gene clusters (BGCs), revealing that only a small fraction of the natural products encoded at the DNA level has been exploited.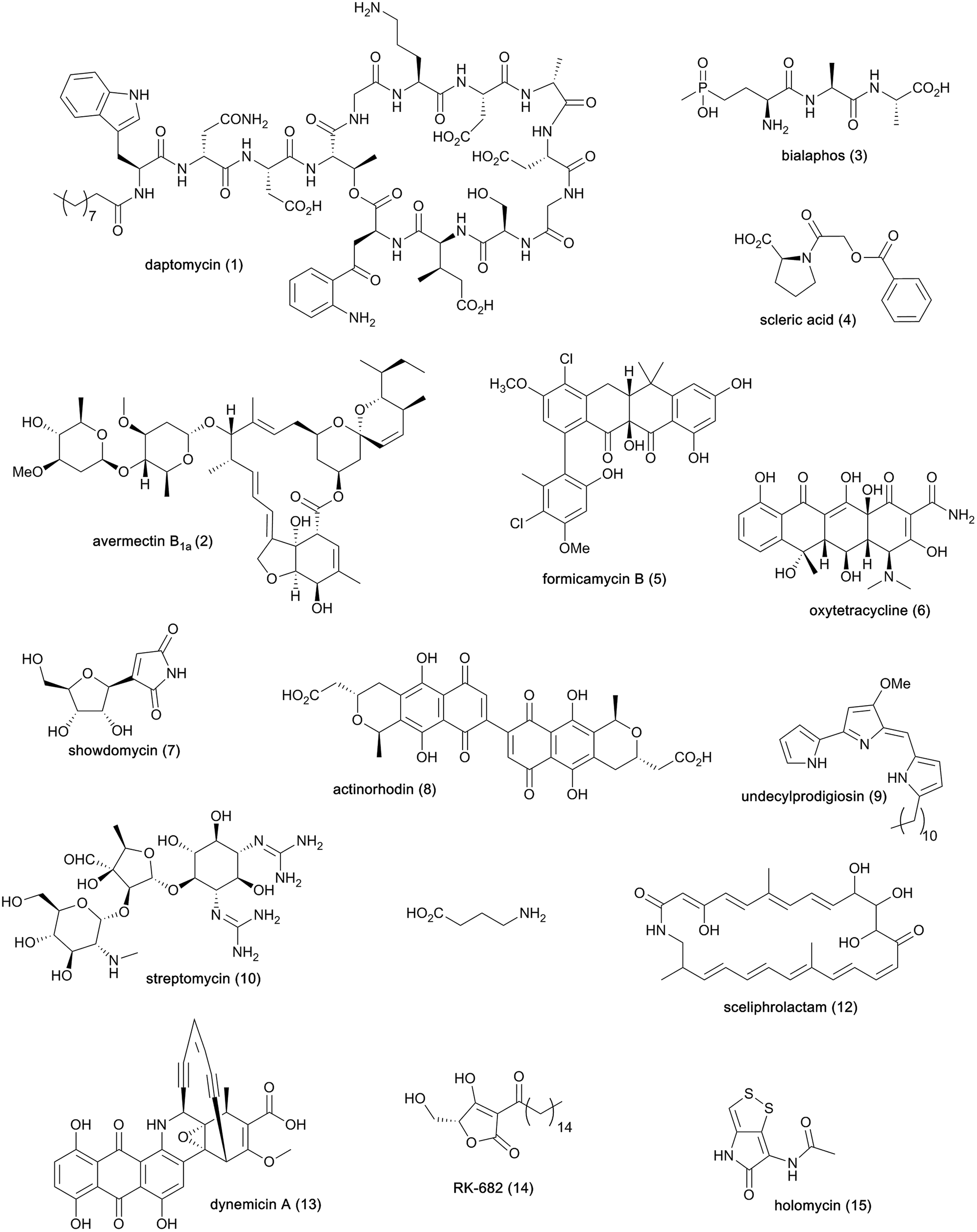 | ||
| Fig. 1 Chemical structures of natural products from streptomycetes and other bacteria mentioned in this article. | ||
In this context, genetic engineering of streptomycetes is recognised as a valuable approach to discover new bioactive compounds.5 Traditionally, editing of their genomes has been achieved using validated but often time-consuming protocols, such as RecA-mediated double-crossover homologous recombination using bespoke suicide plasmids or cosmids modified by λ-Red-mediated homologous recombination.6,7 More recently, targeted genome editing has been revolutionised by the advent of clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) systems, which form the basis for the CRISPR/Cas9-based genome editing technology.8 For a comprehensive overview of the mechanism behind CRISPR/Cas9-based genome editing we invite the reader to refer to the review by Tong et al.9
Following the advent of CRISPR/Cas systems, toolkits for editing streptomycete genomes have quickly been established,10–13 enabling researchers working on these microorganisms to overcome the issues associated with classical methods of gene disruption. This article will highlight the advantages, drawbacks and issues related to streptomycete genome editing through CRISPR/Cas systems, looking at all the instances in which the various CRISPR/Cas9-based toolkits have been used in the last three years for the purpose of natural product identification and characterisation.
2 Pre-CRISPR/Cas9 strategies to manipulate streptomycete genomes
Streptomycete genomes can be engineered by exploiting homologous recombination either via single or double crossover events.6 Insertion of a selectable marker using single crossover can be sufficient for the purpose of gene disruption. However, integration of the entire plasmid can result in polar effects, disrupting the expression of downstream genes. Other issues include reversion to the wild-type genotype in the absence of suitable selective pressure and the limited number of selectable markers (apramycin, hygromycin and kanamycin being the most reliable) effective in streptomycetes, which also prevents multiple events of genome editing using this approach. Double crossovers lead to more stable and “cleaner” mutations but require multiple steps, making the process more laborious (Fig. 2). First the DNA construct has to be integrated at the target locus (through single crossover), which is typically selected for using a selectable marker that is placed between the two homologous recombination arms; the second integration event (resulting in a double crossover) is then selected for via screening for loss of another selectable marker, which is instead located in the backbone vector; lastly, replica plating is used to discern between the mutants that derive from double crossover (and contain the desired genotype) and the strains that have instead reverted to the wild-type genotype.6 The intrinsic frequency of double crossover in streptomycetes is strain-dependent and can be extremely low.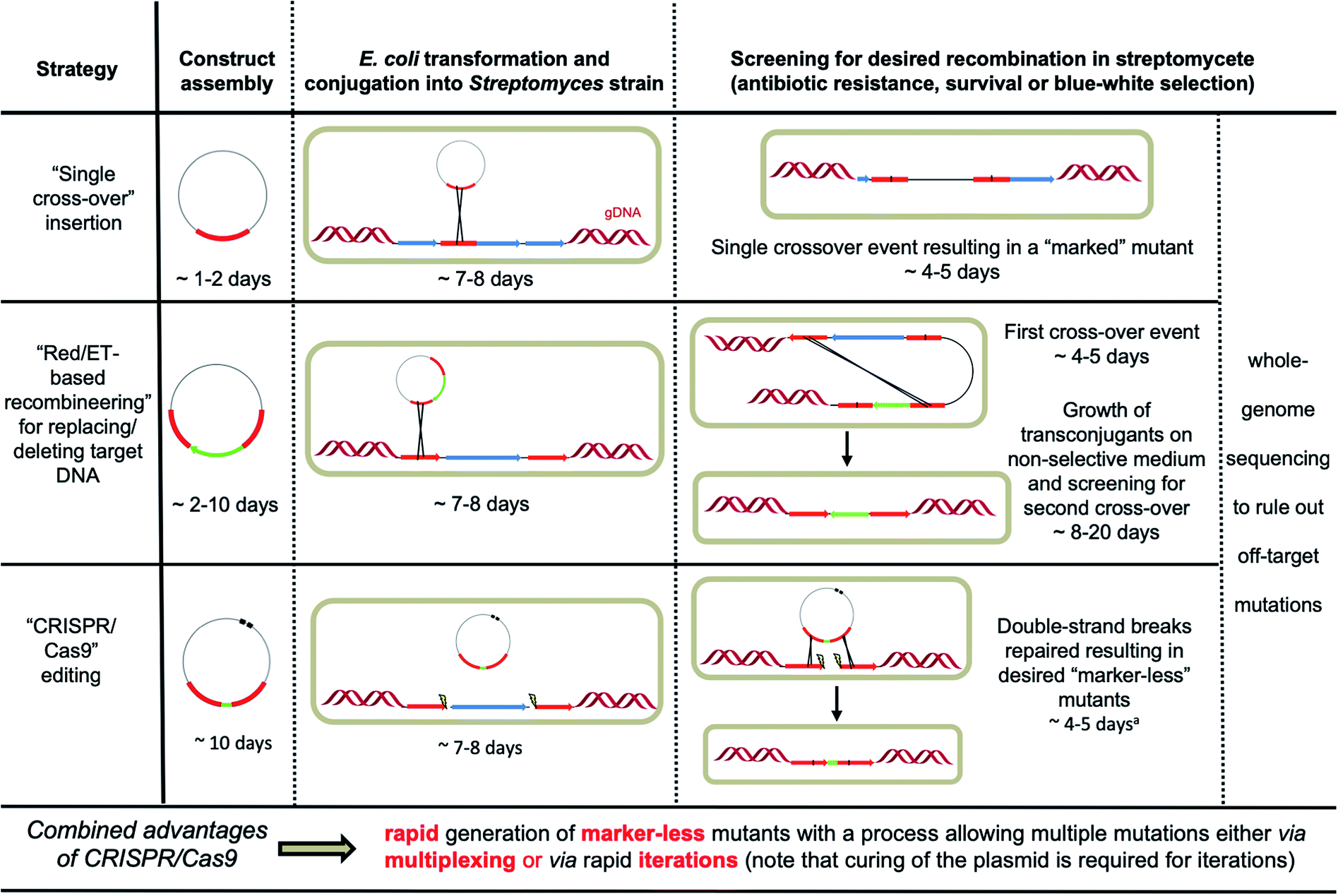 | ||
| Fig. 2 Overview of the different strategies available for streptomycete genome engineering. aClearance of the plasmid can take an additional 3–4 days through subculture at increased temperature (37–39 °C) when pSG5-based plasmids are used.10–12 | ||
A significant improvement for streptomycete genome engineering was achieved in 2003 with the introduction of a Red/ET-based recombineering approach. Cosmid clones could be rapidly edited using λ-Red-mediated homologous recombination in E. coli and introduced into streptomycetes for genome engineering.7 Generating and screening for the desired double crossover events in streptomycete DNA remained a relatively lengthy process (Fig. 2). However, a couple of strategies have been developed to improve the process of selecting double crossovers. For instance, introducing a unique 18 bp recognition site for the meganuclease I-SceI in the backbone of the DNA construct makes the integrated DNA (via single crossover) susceptible to double-stranded breaks (DSBs) when I-SceI is expressed in the strain of interest.14 Repair of the DNA break by homologous recombination provides a potent positive selection for double crossover mutants.14 It is important to note the potential toxicity of I-SceI and the possible presence of the 18 bp recognition site in the genome of the strain under study. Another improvement of the selection process came with the introduction of the gusA blue-white screening for single and double crossover.14 In this system, ex-conjugants are overlaid with X-Gluc (5-bromo-4-chloro-3-indolyl-β-D-glucuronide), which gives rise to a blue product when the β-glucuronidase gusA gene is still present in the plasmid backbone either in a free plasmid or in a single crossover configuration. When the double crossover has happened, and the plasmid backbone has been lost, the colonies with the desired mutation will instead appear white. This visual screening reduces the overall time of a Red/ET-based recombineering experiment but still compels to passage ex-conjugants 5 to 10 times in liquid medium to enrich for cells with double crossovers.14
3 Strategies for in vivo CRISPR/Cas9-based streptomycete genome engineering
When Charpentier and colleagues described the use of CRISPR/Cas9 to introduce DSBs in target DNA in Streptococcus pyogenes,8 they prompted scientists around the globe to adapt this system for the use in other hosts. Since then, protocols for CRISPR/Cas9-mediated genome editing have been developed for organisms across all domains of life, such as for the unicellular model organisms Escherichia coli and Saccharomyces cerevisiae, the archeon Methanosarcina acetivorans, the model plants Arabidopsis thaliana and Nicotiana benthamiana, zebrafish, mouse and human cell lines.15 In brief, CRISPR/Cas9-based genome engineering relies on the ability of the endonuclease Cas9 to cleave a target double-stranded DNA (dsDNA). Cas9 functionality depends upon the interaction with a dual-RNA structure that comprises of a CRISPR-RNA (crRNA) and a trans-activating crRNA (tracrRNA). These two can also be fused together, giving rise to a synthetic guide-RNA (sgRNA). The specific target site for the DSB is determined through a 20 bp protospacer seed sequence in the crRNA which must be followed in the target DNA by a protospacer adjacent motif (PAM, NGG for S. pyogenes, where N can be any nucleotide). The DSB in the target dsDNA occurs three bp upstream of the PAM sequence and can be repaired either through the error-prone non-homologous end joining (NHEJ) pathway or through the more efficient and accurate homology-directed repair (HDR). When HDR is chosen, precise genome editing can be achieved through introduction of a user-designed editing template or homologous recombination (HR) arms.Given the importance of streptomycetes in natural product discovery and the lack of a quick, efficient and programmable method for their genome engineering, different CRISPR/Cas9-based toolkits for the genome editing of these bacteria have been developed. Four different CRISPR/Cas9-based systems for in vivo editing of streptomycetes were made available between November 2014 and August 2015 and are quickly being implemented as a routine engineering strategy by many groups around the world to study natural product biosynthesis and regulation.
3.1 Key considerations
As with previously described genetic engineering approaches (Section 2), a pre-requisite of using the CRISPR-Cas9 system currently available is the genetic tractability of the streptomycete species under study. However, when investigating or exploiting natural product BGCs, one can clone and introduce the entire pathway into a genetically tractable heterologous host where specific toolkits have been validated (Table 1). Cloning of the BGC can be performed either via the classic generation of cosmid libraries, or through other more recently-developed techniques, such as transformation-associated recombination (TAR) cloning.16 For instance, using a combination of TAR cloning to capture a cryptic and silent BGC and pCRISPomyces-2 (Section 3.2) to inactivate one of the cluster-specific transcriptional repressors, our group have recently discovered and characterised the biosynthesis of the novel L-proline derivative scleric acid (4).17| pCRISPomyces-1 (ref. 10) | pCRISPomyces-2 (ref. 10) | pKCcas9dO11 | pCRISPR-Cas9 (ref. 12) | pCRISPR-dCas9 (ref. 12) | pWHU2653 (ref. 13) | |
|---|---|---|---|---|---|---|
| a Genome engineering in Paenibacillus polymyxa was achieved using a modified version of pCRISPomyces-2 where the promoter for expression of cas9 was replaced.31 b The efficiency of silencing with pCRISPR-dCas9 was expressed as the reduced absorbance at 530 nm (A530) detected on culture extracts of S. coelicolor M145 in which the actinorhodin BGC was targeted. Compared to the control strain (relative A530 = 100), the A530 was reduced to ∼3–25 when the promoter or the non-template strand of the coding region of the actIORF1 gene were targeted and ∼70–90 when the template strand of the coding region of the actIORF1 gene was targeted.12 | ||||||
| Streptomycete selectable marker | Apramycin | Apramycin | Apramycin | Apramycin | Apramycin | Apramycin |
| Promoter for cas9 expression | Constitutive: rpsLp | Constitutive: rpsLp | Inducible: tipAp | Inducible: tipAp | Inducible: tipAp | Constitutive: aac(3)IVp |
| Promoter for guide RNA expression | Constitutive: gapdhp for crRNA and rpsLp for tracrRNA | Constitutive: gapdhp for sgRNA | Constitutive: j23199p for sgRNA | Constitutive: ermE*p for sgRNA | Constitutive: ermE*p for sgRNA | Constitutive: ermE*p for sgRNA |
| Plasmid clearance | Temperature-sensitive rep origin from pSG5 | Temperature-sensitive rep origin from pSG5 | Temperature-sensitive rep origin from pSG5 | Temperature-sensitive rep origin from pSG5 | Temperature-sensitive rep origin from pSG5 | CodA counter-selectable marker |
| Species on which the plasmid was tested (% efficiency) | S. lividans (20–25%) | S. lividans (100%), S. viridochromogenes (67–100%), S. albus (67–100%), S. formicae,25S. rimosus,26S. showdoensis,27S. roseosporus,28S. venezuelae,28Actinoplanes sp. SE50/110,29Synechococcus elongatus,30Paenibacillus polymyxaa,31S. coelicolor M1152 (tested but not successful),17Streptomyces sp. KY 40-1 (tested but not successful)18 | S. coelicolor M145 (29–100%), S. pristinaespiralis (94%), S. cinnamonensis38 | S. coelicolor A3(2) (∼100%), Corynebacterium glutamicum,41Streptomyces SD-85,42Micromonospora chersina43 | S. coelicolor A3(2)b | S. coelicolor M145 (93–99%) |
| Addgene ref. # | 61![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 736 736 |
61737 |
62552 |
— | — | — |
Other potential and critical issues, more specific to CRISPR/Cas9 systems are: (i) toxicity of Cas9 in the specific strain used, as reported by Rohr and colleagues in Streptomyces sp. KY 40-1 (ref. 18) and (ii) poor expression of the cas9 gene or that of sgRNA(s).
Another concern to take into account when using CRISPR/Cas9 systems is that all four streptomycete toolkits currently available rely on the same selectable marker, apramycin. Since some streptomycete strains are known to be naturally resistant to apramycin, the lack of an alternative selectable marker is limiting the potential of these toolkits.
A similar consideration can be made on the ease of assembly of a construct for the specific genome engineering, which can affect the timeframe of the experiment. All four kits for in vivo streptomycete genome engineering require to insert both a 20 bp sgRNA and longer HR arms (normally between 1 and 2 kb). Plasmids developed by Zhao and colleagues10 combine a first efficient Golden Gate assembly19 step for the insertion of the sgRNA and a second Gibson assembly19 step for the insertion of the HR arms. Plasmid pKCcas9dO from the Hu, Lu and colleagues11 is assembled by the authors through a single Gibson assembly19 step between the backbone and a linear DNA string generated through overlapping extension PCR to include both sgRNA and HR arms. Lastly, toolkits developed by Weber, Lee and colleagues12 and Sun and colleagues13 require a first traditional cloning step for insertion of the sgRNA and a second Gibson assembly19 step for the insertion of the HR arms.
3.2 pCRISPomyces plasmids from Zhao and colleagues10
The first CRISPR/Cas9-based toolkit for streptomycetes that became widely available was developed in the Zhao lab, at the University of Illinois, Urbana (US). Two plasmids, named pCRISPomyces-1 and pCRISPomyces-2, were developed (Fig. 3). The first one contains separate crRNA and tracrRNA cassettes, whereas the second one includes an sgRNA cassette in which the tracrRNA and the crRNA have been fused to give a chimeric molecule. The sgRNA is known to have at least the same editing efficiency as if using separate tracrRNA and crRNA.8 These two plasmids include elements for maintenance and selection in E. coli and streptomycetes, such as the temperature sensitive replication (rep) region from pSG5,20 which allows for clearance of the plasmid once the genome editing has been accomplished (see Table 1 for a list of the main features of these and the other plasmids reviewed in this article). The streptomycetes codon-optimised cas9 gene included in these two plasmids is placed under control of the strong constitutive promoter rpsLp (CF, from Cellulomonas flavigena).21 Similarly, the sgRNA cassette in pCRISPomyces-2 (as well as the crRNA of pCRISPomyces-1 that has to be inserted by the user with the specific protospacer of choice) is placed under control of the gapdhp (EL, from Eggerthella lenta) strong promoter.21 Both promoters have been characterised as being able to drive strong expression in the heterologous host Streptomyces lividans, showing more than a 10-fold higher activity compared to ermE*p, which is a commonly used promoter for heterologous expression in streptomycetes.22 As well as in S. lividans, rpsLp (CF) and gapdhp (EL) have also shown the ability to drive heterologous gene expression at high levels in additional commonly used streptomycete hosts, such as Streptomyces albus J1074, Streptomyces venezuelae ISP5230, Streptomyces coelicolor M1146 and Streptomyces avermitilis SUKA16.22 Indeed, Zhao and colleagues tested the efficacy of their two CRISPR/Cas9 plasmids in different hosts: pCRISPomyces-1 was used to generate short deletions in S. lividans, whereas pCRISPomyces-2 was successfully employed to produce short and large deletions in S. lividans, S. albus and S. viridochromogenes.10 The efficiency of pCRISPomyces-1 was considerably lower than that of pCRISPomyces-2. The plasmid with separate tracrRNA and crRNA cassettes showed editing efficiency of around 20–25%, as opposed to an efficiency of 67 to 100% for the plasmid with sgRNA. The authors hypothesise that the pre-crRNA may not be processed very efficiently by the native RNase enzymes within streptomycetes, leading to a reduced editing efficiency for pCRISPomyces-1. As well as to generate individual short and large deletions from 20 bp to 31 kb with either one or two protospacer sequences (with two protospacers used for large deletions), worth noting is that pCRISPomyces-2 was also employed to successfully introduce simultaneous short deletions in two different loci, showing the potential to be used for multiplex genome editing.10 The authors reported that in one instance editing with pCRISPomyces-2 in S. albus aimed at deleting a 13 kb region resulted in production of a chimeric strain where a mixture of wild-type and mutated cells were present. Despite the relatively small size of streptomycete genomes and the consequent low probability of occurrence, the authors did not screen the mutants generated with their CRISPR/Cas9 plasmids for off-target effects of the activity of Cas9, which have been reported to be one of the main drawbacks of CRISPR/Cas9-based genome editing.23 Nevertheless, Zhao and collaborators reported to have chosen protospacers in which the last 12 bp plus protospacer adjacent motif (PAM) sequence (in total 15 bp) were unique in the genome of the target species, which is known to minimise off-target effects.10 Since its release, pCRISPomyces-2 has been successfully employed by other groups to edit the genome of various streptomycetes, demonstrating its potential for the engineering of both established hosts and other less commonly used strains.17,24–31 For instance, Hutchings and collaborators used it to generate both one-gene and whole-cluster deletions in Streptomyces formicae to confirm the identity of the antibiotic formicamycin (5) BGC and study the activity of the cluster-included halogenase ForV.25 The plasmid pCRISPomyces-2 was also employed by Zhang and colleagues to generate point-mutations and small deletions to increase oxytetracycline (6) production in Streptomyces rimosus,26 as well as by Metsä-Ketela and colleagues to investigate glycosylation of the nucleoside antibiotic showdomycin (7) in Streptomyces showdoensis.27 As well as for generating deletions,10 the same authors who developed pCRISPomyces-2 later used this plasmid for the precise insertion of single or bidirectional heterologous promoters to activate silent BGC in S. albus, S. lividans, Streptomyces roseosporus, S. venezuelae and S. viridochromogenes.28 Other than streptomycetes, pCRISPomyces-2 has also successfully been employed to edit the genome of other bacteria, such as Actinoplanes sp. SE50/110 (ref. 29) and the Gram-negative cyanobacterium Synechococcus elongatus,30 opening the way for the use of this CRISPR/Cas9 plasmid in a wider variety of organisms. Despite several examples of successful engineering of streptomycetes and other bacteria with pCRISPomyces-2, however, the promoters used to drive expression of the cas9 gene and that of the sgRNA may not currently be optimal in every streptomycete strains, let alone with other unrelated species, but could be optimised. Interestingly, Schmid and colleagues customised pCRISPomyces-2 for expression in Paenibacillus polymyxa by replacing the rpsLp (CF) promoter that guides expression of the cas9 gene, for a promoter that was previously characterised to drive strong expression in Paenibacillus, while keeping the gapdhp (EL) promoter for expression of the sgRNA.31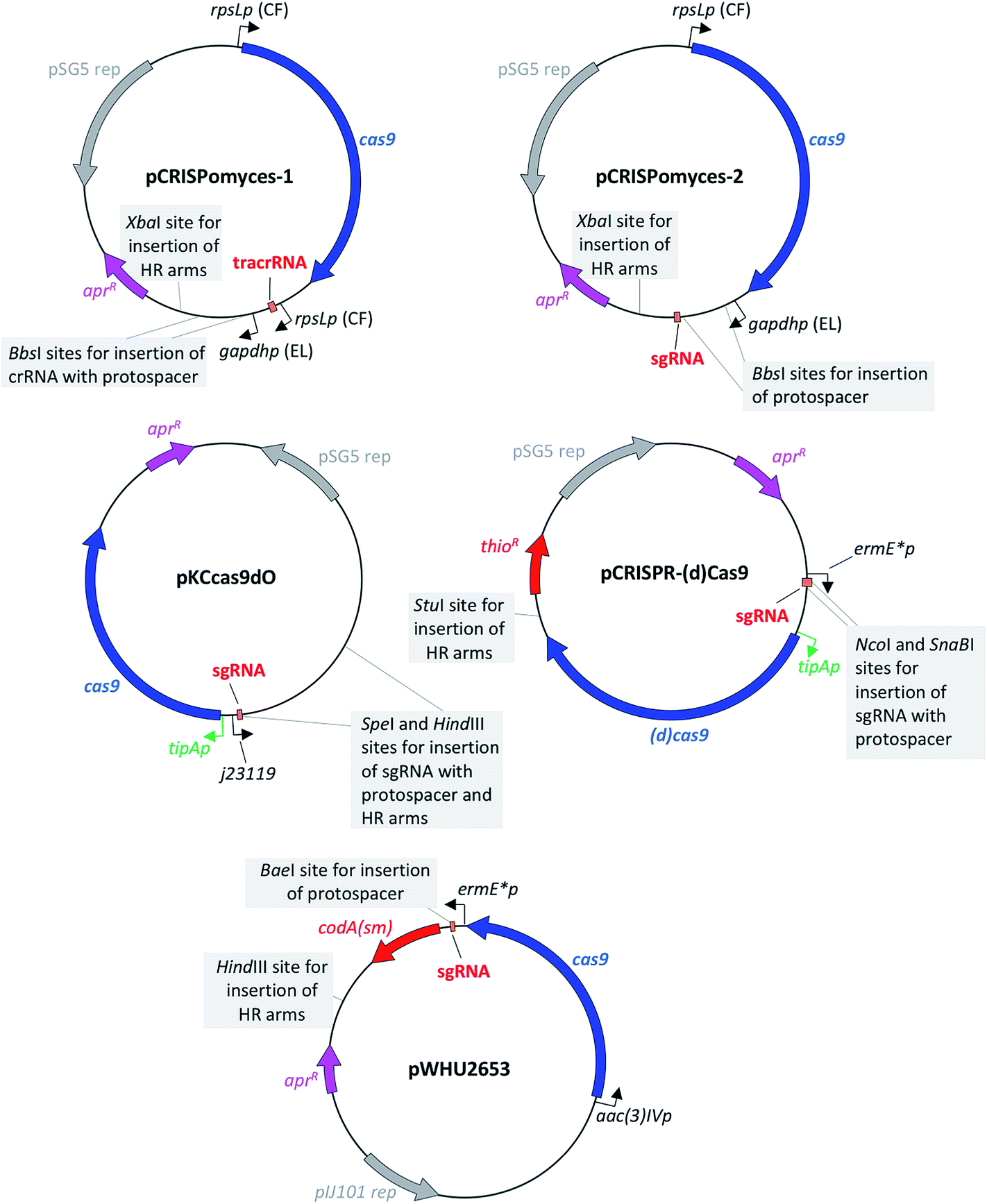 | ||
| Fig. 3 Maps of plasmids for in vivo CRISPR/Cas9-based engineering of streptomycetes.10–13 sgRNA: synthetic guide-RNA; aprR: aac(3)IV gene for apramycin resistance; HR arms: homologous recombination arms for homology-directed repair; thioR: gene for thiostrepton resistance; codA(sm): cytosine deaminase gene for the use of 5-fluorocytosine as a counter-selectable marker; tipAp is an inducible promoter represented in green, as opposed to all other constitutive promoters represented in black. | ||
3.3 pKCcas9dO plasmid from Hu, Lu and colleagues11
The second CRISPR/Cas9 toolkit that became available for streptomycete genome editing was developed by the Hu and Lu laboratories at the Shanghai Institutes for Biological Sciences and Pharmaceutical Industry, China. This plasmid features, as seen in pCRISPomyces-2, a chimeric sgRNA cassette, placed here under control of the synthetic constitutive promoter j23119, which has been designed and tested for expression in E. coli.32 The cas9 gene present in this plasmid was codon-optimised for the codon usage of Streptomyces coelicolor and placed under control of the inducible promoter tipAp.33 In the first place, pKCcas9dO (Fig. 3) was tested as a tool to engineer the genome of the model organism S. coelicolor M145, resulting in editing efficiency of 29–100% for single and multiplex gene deletions between 745 and 1053 bp, as well as of 38–100% for single and multiplex antibiotic BGC deletions of size ranging between 21.3 and 82.8 kb. Interestingly, the authors reported a higher deletion efficiency when editing the actinorhodin (8) BGC (100%) than when attempting to edit the undecylprodigiosin (9) BGC (29–71%). A similar problem was observed by the authors when attempting a simultaneous double deletion of BGCs for 8 and 9, which was resolved by using an alternative sgRNA to target cleavage within the BGC for 9. Moreover, a mixed phenotype was reported by the authors when attempting single-gene deletion in the BGC for 9, suggesting an inefficient cleavage for the sgRNA. As the cleavage efficiency did not seem to be related to the size of the desired deletion, it can be speculated that other factors may be the cause for this. For instance, potential secondary structures formed by the sgRNA could in turn affect its interaction with Cas9, decreasing the editing efficiency.13 As well as to generate deletions, the authors successfully used their CRISPR/Cas9 plasmid to introduce point-mutations into the rpsL gene of S. coelicolor, with the aim of confering resistance to streptomycin (10), similarly to the work from Zhang and colleagues using pCRISPomyces-2 to increase production of 6 in S. rimosus.26 The plasmid pKCcas9dO was also tested in Streptomyces pristinaespiralis, for which the authors reported a 25 kb BGC deletion with 94% efficiency, suggesting that their toolkit could be used also in non-model streptomycete species. Indeed, the same plasmid has been used by other research groups to edit streptomycete genomes.34–38 For instance, the same authors later used this toolkit on S. pristinaespiralis to study the regulation of the biosynthesis of the antibiotic pristinamycin I through generation of point-mutations,34 as well as to increase its production titres through deletion of the cluster-specific transcriptional repressors.35 In two other instances pKCcas9dO has been used to edit the genome of S. coelicolor: (i) to study the regulation mechanism of the two-component system AfsQ1/Q2 in coelimycin P2 biosynthesis,36 and (ii) to confirm the involvement of the global transcriptional regulator GlnR in antibiotic biosynthesis regulation.37 In addition to the two species in which pKCcas9dO was originally tested, Zhao and colleagues used this plasmid on Streptomyces cinnamonensis to generate a gene-deletion that proved the positive regulatory effect of DasR in biosynthesis of the polyether antibiotic monensin.383.4 pCRISPR-Cas9 and pCRISPR-dCas9 plasmids from Weber, Lee and colleagues12
The third CRISPR-Cas9 toolkit to become available for use in streptomycetes was developed by the groups of Weber and Lee at DTU, Denmark. Similarly to pKCcas9dO, pCRISPR-Cas9 (Fig. 3) also allows for an inducible expression of the cas9 gene (codon-optimised for the codon usage of S. coelicolor), through the promoter tipAp. However, as highlighted by the authors, this inducible system can only work in streptomycete strains that, like S. coelicolor, have a copy of the thiostrepton responsive activator TipA in their genome.39 The promoter that drives expression of the sgRNA in pCRISPR-Cas9 is instead ermE*p. This has been used for many years in heterologous expression of streptomycete genes, despite there being other promoters with increased strength.22 Weber, Lee and colleagues tested their pCRISPR-Cas9 plasmid on the model organism S. coelicolor A3(2), targeting six different positions for each of the two genes actIORF1 and actVB involved in the biosynthetic pathway for 8. Interestingly, the authors showed that the DSBs generated through their toolkit can be repaired, with different efficiencies, exploiting either the error-prone NHEJ pathway or through HDR. In the first case, no templates for homology-directed repair are provided with the plasmid but only the 20 bp protospacer within the sgRNA. This led to an editing efficiency that ranged between 3 and 54%, where the NHEJ pathway repaired the DSB giving insertions and deletions of various sizes but all at the desired target site. Introduction of a heterologous DNA Ligase D gene (LigD) resulted in a more efficient NHEJ pathway, which increased the positive outcome of CRISPR/Cas9-based deletion to 37–77%. In these strains, the extension of insertion or deletions by the target site decreased considerably in size compared to those strains generated through an incomplete NHEJ pathway. Lastly, the authors designed pCRISPR-Cas9 plasmids with 2.2 kb homologous recombination templates and used them to target the same two genes of the 8 pathway, resulting in correct deletion in almost 100% of the cases. The authors confirmed in all cases through whole-genome sequencing that no off-target effects had occurred when performing CRISPR/Cas9 engineering. As well as to generate genomic deletions, Weber, Lee and colleagues also used their toolkit to perform CRISPR interference (CRISPRi), similarly to what was shown previously in E. coli by Lim and colleagues.40 In order to do so, the authors inactivated both nuclease domains (RuvC1 and HNH) of the cas9 gene targeting a single amino acid position in each region, resulting in a catalytically inactive (or dead, dCas9) enzyme variant that lost its endonuclease activity. The dCas9 sterically blocked gene expression, leading to loss or considerably reduced production of 8, only when the sgRNA targeted the promoter region of the genes under study or the non-template strand of their coding sequence.The CRISPR-Cas9 systems developed by Weber, Lee and colleagues were successfully employed to knockout and knockdown genes in the industrially-relevant species Corynebacterium glutamicum, targeting genes involved in production of γ-aminobutyric acid (11).41 The plasmid pCRISPR-Cas9 was also employed by Liang and collaborators to confirm the identity of the BGC for the polyene macrolactam sceliphrolactam (12) from a streptomycete strain isolated from a mangrove sediment.42 Interestingly, the authors reported that only one of the five protospacers used to attempt deletion of the target polyketide synthase (PKS) module led to successful gene knockout, with the other four protospacers giving no genome editing in strain Streptomyces SD-85.42 Plasmid pCRISPR-Cas9 was also successfully employed in another non-streptomycete species by Cohen and Townsend.43 Here the authors used this plasmid to delete two cytochrome P450 genes and eight potential PKS genes from the genome of Micromonospora chersina, in the search for the PKS that would be responsible for production of the anthraquinone portion of the antitumor antibiotic dynemicin A (13). Deletion of a ninth putative PKS was not successful but this was explained by the potentially essential role of this gene rather than by the inefficacy of pCRISPR-Cas9.
3.5 pWHU2650 and derived plasmids from Sun and colleagues13
The most recent CRISPR-Cas9 toolkit for streptomycetes to become available was developed by Sun and collaborators, from the University of Wuhan, China. This includes the progenitor plasmid pWHU2650, which similarly to pCRISPR-Cas9, features a constitutive ermE*p promoter to control expression of the streptomycete codon-optimised cas9 gene. The sgRNA is instead under control of the aac(3)IVp promoter from the aminoglycoside 3-N-acetyltransferase type IV gene,44 which is commonly used as a selectable marker in E. coli/Streptomyces shuttle vectors to confer resistance to apramycin and is indeed the selectable marker found in all four CRISPR/Cas9-based toolkits for streptomycete genome editing reviewed here. As seen for pKCcas9dO and pCRISPR/Cas9, Sun and co-workers tested their pWHU2650 plasmid on the model species S. coelicolor M145, targeting genes involved in the biosynthesis of 8. The authors reported that introduction of their plasmid with a sgRNA, but without any template for homology-directed repair, into S. coelicolor resulted in generation of only seven ex-conjugants, which showed no changes at the target site. Adding HR arms of approximately 2 kb each led instead to the precise deletion of the actI-ORF2 gene (939 bp). The editing efficiency ranged between 93 and 99%, depending on which of the two different sgRNA templates (of 48 and 95 bp respectively) were used as a scaffold to clone the 20 bp protospacer. Interestingly, the authors also carried out a control experiment where they introduced the plasmid with cas9 gene and the homologous arms, without sgRNA, but this resulted in an editing efficiency of only 4%, supporting the essential role of the Cas9-generated DSB to enhance homology-directed repair. In order to accelerate the screening process and carry out more rounds of consecutive genome editing, Sun and co-workers introduced the cytosine deaminase gene codA(sm) in their CRISPR/Cas9 plasmid, generating the backbone plasmid pWHU2653 (Fig. 3) as well as plasmid pWHU2659 with sgRNA and HR arms to target the actI-ORF2 gene. When ex-conjugants are grown on plates that contain 5-fluorocytosine (5FC), CodA converts it into 5-fluorouracil (5FU), functioning as a counter-selectable marker.45 This allows to produce a marker-free progeny that can be further subjected to another round of CRISPR/Cas9-based engineering. The presence of such counter-selectable marker for plasmid clearance is a considerable advantage over those toolkits for CRISPR/Cas9-based in vivo engineering that rely on the temperature-sensitive pSG5 origin of replication. The authors showed that using this approach, 94% of the colonies screened contained the desired deletion and had lost the editing plasmid. Lastly, Sun and collaborators ruled out any off-target activity of Cas9 in few of their mutants, sequencing the eight most likely sites that could have been targeted by their sgRNA, as well as the PCR products generated when screening the correct deletions at the target site. Unlike the other CRISPR/Cas9-based streptomycete toolkits, the use of the method developed by Sun and co-workers has not yet been reported in the literature by other researchers.4 Strategies for in vitro CRISPR/Cas9-based streptomycete genome engineering
Soon after the release of their CRISPR/Cas9-based toolkit for in vivo genome engineering,13 Sun and collaborators also developed a strategy for in vitro CRISPR/Cas9-based DNA editing (ICE), which was applied to refactor streptomycete BGCs.46 In this toolkit, Cas9 is used as an in vitro programmable DNA endonuclease in combination with an in vitro-transcribed sgRNA with appropriate protospacer(s). The authors first confirmed the ability of Cas9 to generate DSBs in circular molecules when incubated with different sgRNAs, using as a model the circular cloning plasmid pUC18. This revealed that, due its 3′–5′ exonuclease activity, Cas9 produced in most cases small-size (5–14 bp) deletions upstream of the target site. However, the addition of T4 DNA polymerase to join the cohesive ends generated by Cas9 reduced the extent of these deletions. The design of the sgRNA and the choice of sgRNA combinations appeared however to influence considerably the efficiency of seamless deletions. Sun and collaborators used their in vitro CRISPR/Cas9 toolkit to generate gene deletions in a 20 kb plasmid that harboured the BGC for the protein phosphatase inhibitor RK-682 (14) and a 40 kb plasmid that contained the BGC for holomycin (15).47 In the first case a 975 bp deletion was achieved in 32% of the clones screened, whereas in the second case a 2817 bp deletion was accomplished only with a 16% efficiency. The authors then further tested the potential of their toolkit to carry out gene insertions. Combining a PCR-amplified gene for ampicillin resistance and the Cas9-linearised vector that contained the BGC for 14, they generated recombinant plasmids with ampicillin resistant. In this case, it was not possible to determine the efficiency of the insertion since the selection of clones was based on the acquired ampicillin resistance trait. To date, apart from the examples described by Sun and collaborators, no other application of the ICE system has been reported in the literature.5 Bioinformatic tools for designing CRISPR/Cas9 experiments
Different CRISPR/Cas9-mediated genome engineering tools and their effective use in streptomycete genomes have been described above. An important consideration to make is that when planning such experiments, the user has to design the 20 bp protospacer based on two primary requirements: (i) that the protospacer is directly upstream of a PAM sequence (NGG for S. pyogenes Cas9); (ii) that the protospacer + PAM sequence is unique in the genome. Streptomycete genomes are naturally rich in potential PAM (NGG) sequences due to their high GC content, so finding a protospacer within the target sequence is generally not difficult, depending obviously on the extension of the target region. Finding a unique protospacer + PAM sequence is, theoretically, also not challenging, given the relatively small size of streptomycete genomes (normally between 7 and 10 Mbp) compared to higher organisms. However, it is known that only the 13 bp sequence directly upstream of the PAM sequence is strictly required to direct binding of Cas9 to the target site, increasing considerably chances of off-target effects.8 Moreover, single and double mismatches in the core protospacer sequence are known to be tolerated to varying degrees by Cas9, increasing likelihood of off-target effects even further.23,48 Designing a protospacer that minimises chances of off-target effects requires therefore the use of bioinformatic tools. In order to help designing the best protospacer for the desired target sequence, several online and offline bioinformatic tools have been made available. One of these, which features a user-friendly interface, is CRISPy-web,49 a platform developed by Weber, Lee and collaborators. CRISPy-web first requires uploading the genome sequence of the specific strain of interest, which can be provided in the form of a GenBank file or an antiSMASH entry. The user then has to specify the target region within the genomic entry, which prompts to an output page with graphic interface that shows the location on the target sequence of all hits found, followed by a table-like analysis of the output. This includes, for each possible protospacer, the number of off-target hits on the whole genomic sequence that contain 0, 1, or 2 mismatches for the 13 bp core sequence upstream of the PAM site. Based on this information, the user can then choose whichever protospacer shows the lowest chances of off-target effects in the genome under study.6 Critical analysis of current toolkits and prospects
The advent of CRISPR/Cas9 has revolutionised the way researchers do genome engineering. Since the release of the four available toolkits for in vivo CRISPR/Cas9-based engineering for streptomycetes, several studies have been published in which these have successfully been used for the purpose of gene and whole BGC deletion, point mutations and insertions. Among these toolkits, pCRISPomyces-2 (ref. 10) is the plasmid that has seen the highest number of applications,17,24–31 showing also the potential to be used in non-streptomycete Gram-positive and even Gram-negative bacteria.29–31 It is interesting, however, that pCRISPomyces-2 has not yet been reported to work in S. coelicolor, which is considered the model species among streptomycetes. At the time of writing, there is only one published report of attempting its use in S. coelicolor, which was described as unsuccessful,17 similarly to what reported for the same plasmid in Streptomyces sp. KY 40-1.18 Out of the four toolkits for in vivo CRISPR/Cas9-based streptomycete engineering, two allow for inducible expression of cas9,11,12 which can increase chances of obtaining ex-conjugants before Cas9 is rendered active. Moreover, the one developed by Weber, Lee and collaborators allows reversible gene knockdowns, through CRISPRi,12 which can be useful for instance when aiming to carry out biosynthetic studies on natural product BGCs. Similar to pCRISPomyces-2, this toolkit has also been used in non-streptomycete hosts, showing a wide potential of applicability.41,43 It is worth mentioning that CRISPR/Cas9 is not the only CRISPR/Cas system to have been developed for the use in streptomycetes. Another class 2 CRISPR/Cas system called Cpf1 (CRISPR from Prevotella and Francisella 1) has recently been developed into a streptomycete CRISPR toolbox.50 This system can complement CRISPR/Cas9-based plasmids, giving the user the option to use a T-rich PAM (TTTN for Cpf1, as opposed to NGG for Cas9) and requiring only a crRNA (as opposed to crRNA and tracrRNA for Cas9). It can also be used for multiplex gene repression through CRISPRi, and it promises to work in streptomycete strains in which Cas9 may not work, such as Streptomyces hygroscopicus SIPI-KF.50One of the key advantages of using CRISPR/Cas9-based engineering is the reduced timeframe compared to traditional homologous recombination methods (Fig. 2).6,7 Several consecutive editing rounds can also be carried out in a reasonable time. This is particularly true when using the pWHU2653 plasmid that has a counter-selectable marker for plasmid loss after the editing event has taken place.13 Conversely, plasmid clearance for the other three in vivo CRISPR/Cas9-based toolkits can be achieved through few rounds of subculture at increased temperature (37–39 °C), somehow extending the experimental timeframe.
A general advantage of using CRISPR, in comparison to Red/ET-based recombineering system,7 is that one does not normally need to prepare a cosmid library but can instead directly work on the strain of interest. It should be noted that this is also not required when editing genomes through double crossover, which is however less efficient than CRISPR/Cas9-based engineering. Cosmid clones, or other BGC-captured plasmids generated for instance through TAR cloning, are still needed when using the in vitro CRISPR/Cas9-based ICE toolkit,46 or when dealing with genetically intractable strains.17 A minor issue that has been encountered in a few instances when using CRISPR/Cas9-based toolkits is the occurrence of strains with mixed phenotypes,10,11 which seems however to only account for a low percentage of the strains screened. Another drawback that can be faced when undertaking CRISPR/Cas9-based engineering, as opposed to classical homologous recombination methods, is that Cas9 can lead to off-target effects due to its ability to tolerate mismatches in the protospacer sequence.23,48 The risk of such undesired events can be reduced if using bioinformatic tools, such as CRISPy-web,49 to choose the best protospacer. In fact, a given protospacer may not produce the desired DSB, therefore often the use of more than one protospacer for each target may be advisable. In relation to off-target effects, given the affordable cost, whole-genome sequencing of the mutant strains generated may also be adopted to rule out any unpredicted off-target editing, not only for CRISPR/Cas9-mediated genome editing but also for other genome editing approaches, such as Red/ET-based recombineering system.
7 Conclusions
The CRISPR/Cas9-based toolkits developed for use in streptomycetes have revolutionised the way researchers in this field do genome engineering with the aim to characterise natural products and their biosynthesis. Since their release, successful use of these toolkits has been reported for 13 different Streptomyces species, as well as on other non-streptomycete Gram-positive and Gram-negative strains. Constraints related to toxicity of Cas9 in the species of interest and the risk of off-target effects are notable limitations of CRISPR/Cas9-based engineering. These can be addressed by cloning the BGC of interest into a genetically tractable Cas9-verified species and through an accurate protospacer design with the available bioinformatic tools. In conclusion, due to its flexibility and ability to produce seamless and marker-less mutations in a reduced timeframe, CRISPR/Cas9-based genome editing is expected to overtake conventional methods of genetic engineering in streptomycetes, not only for the purpose of natural product discovery but also for studies of developmental biology and synthetic biology applications such as engineering fusion proteins in streptomycetes.8 Conflicts of interest
There are no conflicts to declare.9 Acknowledgements
The authors would like to thank Audam Chhun and Dr Daniel Zabala Alvarez for proofreading this manuscript and providing valuable suggestions. Dr Yaojun Tong and Dr Tilmann Weber are also thanked for helpful discussion. Support for streptomycete research in the authors' lab, BBSRC grant BB/M022765/1 and the Warwick Integrative Synthetic Biology Centre (WISB), a UK Synthetic Biology Research Centre grant from EPSRC and BBSRC (BB/M017982/1), are gratefully acknowledged.10 References
- O. Genilloud, Nat. Prod. Rep., 2017, 34, 1203–1232 RSC.
- (a) R. D. Arbeit, D. Maki, F. P. Tally, E. Campanaro and B. I. Eisenstein, Clin. Infect. Dis., 2004, 38, 1673–1681 CrossRef CAS PubMed; (b) V. G. Fowler, H. W. Boucher, G. R. Corey, E. Abrutyn, A. W. Karchmer, M. E. Rupp, D. P. Levine, H. F. Chambers, F. P. Tally, G. A. Vigliani, C. H. Cabell, A. S. Link, I. DeMeyer, S. G. Filler, M. Zervos, P. Cook, J. Parsonnet, J. M. Bernstein, C. S. Price, G. N. Forrest, G. Fätkenheuer, M. Gareca, S. J. Rehm, H. R. Brodt, A. Tice and S. E. Cosgrove, N. Engl. J. Med., 2006, 355, 653–665 CrossRef CAS PubMed.
- R. W. Burg, B. M. Miller, E. E. Baker, J. Birnbaum, S. A. Currie, R. Hartman, Y.-L. Kong, R. L. Monaghan, G. Olson, I. Putter, J. B. Tunac, H. Wallick, E. O. Stapley, R. Oiwa and S. Ōmura, Antimicrob. Agents Chemother., 1979, 15, 361–367 CrossRef CAS PubMed.
- E. Bayer, K. H. Gugel, K. Hägele, H. Hagenmaier, S. Jessipow, W. A. König and H. Zähner, Helv. Chim. Acta, 1972, 55, 224–239 CrossRef CAS PubMed.
- P. J. Rutledge and G. L. Challis, Nat. Rev. Microbiol., 2015, 13, 509 CrossRef CAS PubMed.
- T. Kieser, M. J. Bibb, M. J. Buttner, K. F. Chater and D. A. Hopwood, Practical Streptomyces Genetics, John Innes Foundation, Norwich, U.K., 2000 Search PubMed.
- (a) Y. Zhang, J. P. P. Muyrers, G. Testa and F. Stewart, Nat. Biotechnol., 2000, 18, 1314–1317 CrossRef CAS PubMed; (b) B. Gust, G. L. Challis, K. Fowler, T. Kieser and K. F. Chater, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 1541–1546 CrossRef CAS PubMed.
- M. Jinek, K. Chylinski, I. Fonfara, M. Hauer, J. A. Doudna and E. Charpentier, Science, 2012, 337, 816–821 CrossRef CAS PubMed.
- Y. Tong, T. Weber and S. Y. Lee, Nat. Prod. Rep., 2019 10.1039/c8np00089a.
- R. E. Cobb, Y. Wang and H. Zhao, ACS Synth. Biol., 2015, 4, 723–728 CrossRef CAS PubMed.
- H. Huang, G. Zheng, W. Jiang, H. Hu and Y. Lu, Acta Biochim. Biophys. Sin., 2015, 47, 231–243 CrossRef CAS PubMed.
- Y. Tong, P. Charusanti, L. Zhang, T. Weber and S. Y. Lee, ACS Synth. Biol., 2015, 18, 1020–1029 CrossRef PubMed.
- H. Zeng, S. Wen, W. Xu, Z. He, G. Zhai, Y. Liu, Z. Deng and Y. Sun, Appl. Microbiol. Biotechnol., 2015, 99, 10575–10585 CrossRef CAS PubMed.
- (a) T. Siegl, L. Petzke, E. Welle and A. Luzhetskyy, Appl. Microbiol. Biotechnol., 2010, 87, 1525–1532 CrossRef CAS PubMed; (b) L. T. Fernández-Martínez and M. J. Bibb, Sci. Rep., 2014, 4, 7100 CrossRef PubMed; (c) M. Myronovskyi, E. Welle, V. Fedorenko and A. Luzhetskyy, Appl. Environ. Microbiol., 2011, 77, 5370–5383 CrossRef CAS PubMed; (d) M. Myronovskyi, B. Rosenkränzer, S. Nadmid, P. Pujic, P. Normand and A. Luzhetskyy, Metab. Eng., 2018, 49, 316–324 CrossRef CAS PubMed.
- (a) W. Jiang, D. Bikard, D. Cox, F. Zhang and L. A. Marraffini, Nat. Biotechnol., 2013, 31, 233–239 CrossRef CAS PubMed; (b) Z. Bao, H. Xiao, J. Liang, L. Zhang, X. Xiong, N. Sun, T. Si and H. Zhao, ACS Synth. Biol., 2014, 4, 585–594 CrossRef PubMed; (c) D. D. Nayak and W. W. Metcalf, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2976–2981 CrossRef CAS PubMed; (d) Q. Shan, Y. Wang, J. Li, Y. Zhang, K. Chen, Z. Liang, K. Zhang, J. Liu, J. J. Xi, J.-L. Qiu and C. Gao, Nat. Biotechnol., 2013, 31, 688–691 CrossRef PubMed; (e) W. Y. Hwang, Y. Fu, D. Reyon, M. L. Maeder, S. Q. Tsai, J. D. Sander, R. T. Peterson, J. R. Yeh and J. K. Joung, Nat. Biotechnol., 2013, 31, 227–229 CrossRef CAS PubMed; (f) L. Cong, F. A. Ran, D. Cox, S. Lin, R. Barretto, N. Habib, P. D. Hsu, X. Wu, W. Jiang, L. A. Marraffini and F. Zhang, Science, 2013, 339, 819–823 CrossRef CAS PubMed.
- (a) K. Yamanaka, K. A. Reynolds, R. D. Kerstena, K. S. Ryana, D. J. Gonzalez, V. Nizete, P. C. Dorrestein and B. S. Moore, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 1957–1961 CrossRef CAS PubMed; (b) X. Tang, J. Li, N. Millán-Aguiñaga, J. J. Zhang, E. C. O'Neill, J. A. Ugalde, P. R. Jensen, S. M. Mantovani and B. S. Moore, ACS Chem. Biol., 2015, 10, 2841–2849 CrossRef CAS PubMed.
- F. Alberti, D. J. Leng, I. Wilkening, L. Song, M. Tosin and C. Corre, Chem. Sci., 2019, 10, 453–463 RSC.
- S. M. Salem, S. Weidenbach and J. Rohr, ACS Chem. Biol., 2017, 12, 2529–2534 CrossRef CAS PubMed.
- (a) C. Engler, R. Kandzia and S. Marillonnet, PLoS One, 2008, 3, e3647 CrossRef PubMed; (b) D. G. Gibson, L. Young, R. Y. Chuang, J. C. Venter, C. A. Hutchison III and H. O. Smith, Nat. Methods, 2009, 6, 343–345 CrossRef CAS PubMed.
- G. Muth, B. Nußbaumer, W. Wohlleben and A. Pühler, Mol. Gen. Genet., 1989, 219, 341–348 CrossRef CAS.
- Z. Shao, G. Rao, C. Li, Z. Abil, Y. Luo and H. Zhao, ACS Synth. Biol., 2013, 2, 662–669 CrossRef CAS PubMed.
- (a) C. J. Wilkinson, Z. A. Hughes-Thomas, C. J. Martin, I. Bohm, T. Mironenko, M. Deacon, M. Wheatcroft, G. Wirtz, J. Staunton and P. F. Leadlay, J. Mol. Microbiol. Biotechnol., 2002, 4, 417–426 CAS; (b) G.-Y. Tan, K. Deng, X. Liu, H. Tao, Y. Chang, J. Chen, K. Chen, Z. Sheng, Z. Deng and T. Liu, ACS Synth. Biol., 2017, 6, 995–1005 CrossRef CAS PubMed; (c) C. Baia, Y. Zhanga, X. Zhaoa, Y. Hua, S. Xianga, J. Miaoa, C. Loua and L. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 12181–12186 CrossRef PubMed; (d) T. S. Freestone, K.-S. Ju, B. Wang and H. Zhao, ACS Synth. Biol., 2017, 6, 217–223 CrossRef CAS PubMed.
- X.-H. Zhang, L. Y. Tee, X.-G. Wang, Q.-S. Huang and S.-H. Yang, Mol. Ther.--Nucleic Acids, 2015, 4, e264 CrossRef CAS PubMed.
- (a) T. C. McLean, P. A. Hoskisson and R. F. Seipke, mSphere, 2016, 1, e00305–e00316 CrossRef CAS PubMed; (b) Y. Zhang, C.-Y. Lin, X.-M. Li, Z.-K. Tang, J. Qiao and G.-R. Zhao, J. Ind. Microbiol. Biotechnol., 2016, 43, 1681–1692 CrossRef CAS PubMed.
- Z. Qin, J. T. Munnoch, R. Devine, N. A. Holmes, R. F. Seipke, K. A. Wilkinson, B. Wilkinson and M. I. Hutchings, Chem. Sci., 2017, 8, 3218–3227 RSC.
- H. Jia, L. Zhang, T. Wang, J. Han, H. Tang and L. Zhang, Microbiology, 2017, 163, 1148–1155 CrossRef CAS PubMed.
- K. Palmu, P. Rosenqvist, K. Thapa, Y. Ilina, V. Siitonen, B. Baral, J. Mäkinen, G. Belogurov, P. Virta, J. Niemi and M. Metsä-Ketela, ACS Chem. Biol., 2017, 12, 1472–1477 CrossRef CAS PubMed.
- M. M. Zhang, F. T. Wong, Y. Wang, S. Luo, Y. H. Lim, E. Heng, W. L. Yeo, R. E. Cobb, B. Enghiad, E. L. Ang and H. Zhao, Nat. Chem. Biol., 2017, 13, 607–609 CrossRef CAS PubMed.
- T. Wolf, T. Gren, E. Thieme, D. Wibberg, T. Zemke, A. Pühler and J. Kalinowski, J. Biotechnol., 2016, 231, 122–128 CrossRef CAS PubMed.
- K. E. Wendt, J. Ungerer, R. E. Cobb, H. Zhao and H. B. Pakrasi, Microb. Cell Fact., 2016, 15, 115 CrossRef PubMed.
- M. Rütering, B. Cress, M. Schilling, B. Rühmann, M. A. G. Koffas, V. Sieber and J. Schmid, Synth. Biol., 2017, 2, ysx007 Search PubMed.
- M. H. Larson, L. A. Gilbert, X. Wang, W. A. Lim, J. S. Weissman and L. S. Qi, Nat. Protoc., 2013, 8, 2180–2196 CrossRef CAS PubMed.
- T. Murakami, T. G. Holt and C. J. Thompson, J. Bacteriol., 1989, 171, 1459–1466 CrossRef CAS PubMed.
- W. Wang, J. Tian, L. Li, M. Ge, H. Zhu, G. Zheng, H. Huang, L. Ruan, W. Jiang and Y. Lu, Appl. Microbiol. Biotechnol., 2015, 99, 7151–7164 CrossRef CAS PubMed.
- J. Meng, R. Feng, G. Zheng, M. Ge, Y. Mast, W. Wohlleben, J. Gao, W. Jiang and Y. Lu, Synth. Syst. Biol., 2017, 2, 130–136 Search PubMed.
- S. Chen, G. Zheng, H. Zhu, H. He, L. Chen, W. Zhang, W. Jiang and Y. Lu, FEMS Microbiol. Lett., 2016, 363, fnw160 CrossRef PubMed.
- J. M. He, H. Zhu, G. S. Zheng, P. P. Liu, J. Wang, G. P. Zhao, G. Q. Zhu, W. H. Jiang and Y. H. Lu, J. Biol. Chem., 2016, 291, 26443–26454 CrossRef CAS PubMed.
- Y. Zhang, C.-Y. Lin, X.-M. Li, Z.-K. Tang, J. Qiao and G.-R. Zhao, J. Ind. Microbiol. Biotechnol., 2016, 43, 1681–1692 CrossRef CAS PubMed.
- B. S. Yun, T. Hidaka, T. Kuzuyama and H. Seto, J. Antibiot., 2001, 54, 375–378 CrossRef CAS PubMed.
- L. S. Qi, M. H. Larson, L. A. Gilbert, J. A. Doudna, J. S. Weissman, A. P. Arkin and W. A. Lim, Cell, 2013, 152, 1173–1183 CrossRef CAS PubMed.
- J. S. Cho, K. R. Choi, C. P. S. Prabowo, J. H. Shin, D. Yang, J. Jang J and S. Y. Lee, Metab. Eng., 2017, 42, 157–167 CrossRef CAS PubMed.
- Z. J. Low, L. M. Pang, Y. Ding, Q. W. Cheang, K. L. M. Hoang, H. T. Tran, J. Li, X.-W. Liu, Y. Kanagasundaram, L. Yang and Z.-X. Liang, Sci. Rep., 2018, 8, 1584 CrossRef PubMed.
- D. R. Cohen and C. A. Townsend, Nat. Chem., 2018, 10, 231–236 CrossRef CAS PubMed.
- R. Allmansberger, B. Bräu and W. Piepersberg, Mol. Gen. Genet., 1985, 198, 514–520 CrossRef CAS PubMed.
- M. P. Dubeau, M. G. Ghinet, P. E. Jacques, N. Clermont, C. Beaulieu and R. Brzezinski, Appl. Environ. Microbiol., 2009, 75, 1211–1214 CrossRef CAS PubMed.
- Y. Liu, W. Tao, S. Wen, Z. Li, A. Yang, Z. Deng and Y. Sun, mBio, 2015, 6, e01714–e01715 CAS.
- (a) T. Hamaguchi, T. Sudo and H. Osada, FEBS Lett., 1995, 372, 54–58 CrossRef CAS PubMed; (b) S. Huang, Y. Zhao, Z. Qin, X. Wang, M. Onega, L. Chen, J. He, Y. Yu and H. Deng, Process Biochem., 2011, 46, 811–816 CrossRef CAS.
- Y. Fu, J. A. Foden, C. Khayter, M. L. Maeder, D. Reyon, J. K. Joung and J. D. Sander, Nat. Biotechnol., 2013, 31, 822–826 CrossRef CAS PubMed.
- K. Blin, L. E. Pederson, T. Weber and S. Y. Lee, Synth. Syst. Biol., 2016, 1, 118–121 Search PubMed.
- (a) B. Zetsche, J. S. Gootenberg, O. O. Abudayyeh, I. M. Slaymaker, K. S. Makarova, P. Essletzbichler, S. E. Volz, J. Joung, J. van der Oost, A. Regev, E. V. Koonin and F. Zhang, Cell, 2015, 163, 759–771 CrossRef CAS PubMed; (b) L. Li, K. Wei, G. Zheng, X. Liu, S. Chen, W. Jiang and Y. Lu, Appl. Environ. Microbiol., 2018, 84, e00827-18 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2019 |