
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemistry, bioactivity and biosynthesis of cyanobacterial alkylresorcinols
Teresa P.
Martins
 a,
Caroline
Rouger†
b,
Nathaniel R.
Glasser
c,
Sara
Freitas
a,
Nelly B.
de Fraissinette
a,
Emily P.
Balskus
c,
Deniz
Tasdemir
b and
Pedro N.
Leão
*a
a,
Caroline
Rouger†
b,
Nathaniel R.
Glasser
c,
Sara
Freitas
a,
Nelly B.
de Fraissinette
a,
Emily P.
Balskus
c,
Deniz
Tasdemir
b and
Pedro N.
Leão
*a
aInterdisciplinary Centre of Marine and Environmental Research (CIIMAR/CIMAR), University of Porto, Matosinhos, Portugal. E-mail: pleao@ciimar.up.pt
bResearch Unit Marine Natural Products Chemistry, GEOMAR Centre for Marine Biotechnology (GEOMAR-Biotech), GEOMAR Helmholtz Centre for Ocean Research Kiel, Germany
cDepartment of Chemistry & Chemical Biology, Harvard University, Cambridge, MA, USA
First published on 31st January 2019
Abstract
Covering: up to 2019
Alkylresorcinols are amphiphilic metabolites, well-known for their diverse biological activities, produced by both prokaryotes and eukaryotes. A few classes of alkylresorcinol scaffolds have been reported from the photoautotrophic cyanobacteria, ranging from the relatively simple hierridins to the more intricate cylindrocyclophanes. Recently, it has emerged that cyanobacteria employ two different biosynthetic pathways to produce unique alkylresorcinol scaffolds. However, these convergent pathways intersect by sharing biosynthetic elements which lead to common structural motifs. To obtain a broader view of the biochemical diversity of these compounds in cyanobacteria, we comprehensively cover the isolation, structure, biological activity and biosynthesis of their mono- and dialkylresorcinols. Moreover, we provide an overview of the diversity and distribution of alkylresorcinol-generating biosynthetic gene clusters in this phylum and highlight opportunities for discovery of novel alkylresorcinol scaffolds. Because some of these molecules have inspired notable syntheses, different approaches used to build these molecules in the laboratory are showcased.
 Teresa P. Martins | Teresa Martins obtained her BSc and MSc in Biochemistry from the University of Porto in 2017. During her MSc studies, under the supervision of Dr Pedro Leão, she focused on the natural products chemistry of cyanobacteria. Teresa has recently been granted with a PhD scholarship to study the biosynthesis of unusual cyanobacterial secondary metabolites, in the Cyanobacterial Natural Products group at CIIMAR (University of Porto). |
 Caroline Rouger | Caroline Rouger graduated in pharmacy from the University of Angers where she received her PhD in 2015, studying prenylated polyphenols and their anti-inflammatory and immunomodulatory activities under the guidance of Prof. Pascal Richomme and Dr Séverine Derbré. After one year as research and teaching assistant at the University of Limoges, she joined the group of Prof. Deniz Tasdemir (2016–2018) as natural product chemist/project manager in the field of marine biotechnology. She recently got a position of assistant professor at the University of Bordeaux and her current research focuses on the characterization of bioactive polyphenols from vine and wine. |
 Nathaniel R. Glasser | Nathaniel Glasser received his PhD in 2017 from the California Institute of Technology under the supervision of Dr Dianne Newman, where he studied the biochemistry of extracellular electron shuttling. In 2018 he joined the group of Dr Emily Balskus as a post-doctoral fellow at Harvard University where he is researching new halogenases from cyanobacteria. His research interests include the biosynthesis of natural products as well as their biological and ecological functions. |
 Sara Freitas | Sara Freitas obtained her BSc in Biology in 2008 followed by a MSc in Toxicology and Environmental Contamination in 2012, both from the University of Porto. During her MSc thesis Sara studied the molecular mechanisms associated with natural product cytotoxicity in human cancer cell lines. She is currently undertaking PhD studies focused on the discovery of new natural products at CIIMAR (University of Porto), under the supervision of Dr Pedro Leão. |
 Nelly B. de Fraissinette | Nelly Brugerolle de Fraissinette is a post-graduate member of the Cyanobacterial Natural Products group at CIIMAR (University of Porto). Nelly recently obtained a MSc in Applied Blue Biotechnology in 2018 (University of La Rochelle), and is currently a project technician. Her research interests include natural product discovery for biotechnological applications to the environment and cyanobacterial cultivation. |
 Emily P. Balskus | Emily Balskus received her BA from Williams College and her PhD in synthetic organic chemistry from Harvard University, where she worked with Eric Jacobsen. Her postdoctoral studies at Harvard Medical School with Christopher T. Walsh focused on deciphering the biosynthesis of cyanobacterial sunscreens. She began her independent career at Harvard University in 2011 and is now a Professor of Chemistry and Chemical Biology. Her research program explores the intersection of organic chemistry, biochemistry, and microbiology, with a major focus on understanding unusual natural product biosynthetic pathways and metabolic activities of the human microbiota. |
 Deniz Tasdemir | Deniz Tasdemir obtained her PhD degree in phytochemistry in 1997 at ETH-Zurich. She then carried out post-doctoral research with Prof. Chris M. Ireland at University of Utah on anticancer marine sponge metabolites (1998–2001). She was senior lecturer at University of London (2006–2011), before taking up a full professorship in marine biodiscovery at National University of Ireland Galway (2011–2014). Currently she is full professor of marine natural product chemistry at GEOMAR Helmholtz Centre for Ocean Research Kiel. Her research interests include marine biotechnology, drug discovery, metabolomics and chemical ecology of marine invertebrates, seaweeds, seagrasses and microorganisms. |
 Pedro N. Leão | Pedro Leão obtained his PhD from the University of Porto in 2010, where he studied allelopathy in cyanobacteria under the supervision of Prof. Vitor Vasconcelos. For his postdoctoral studies at CIIMAR (U. Porto), Pedro focused on marine natural products chemistry and biosynthesis with research stays at the Gerwick lab (Scripps Institution of Oceanography) and Balskus group (Harvard University). In 2015 he became Auxiliary Researcher at CIIMAR and in 2018 he started the Cyanobacterial Natural Products group in the same institution. His research interests lie in the chemistry, biosynthesis and chemical ecology of cyanobacterial secondary metabolites. |
1. Introduction
Alkylresorcinols represent a large structural group of amphiphilic phenolic lipids. These scaffolds are widespread in Nature—alkylresorcinols have been isolated from higher plants, insects, fungi, algae, slime molds, mosses and bacteria.1–3 They play structural roles in the membranes of the producing organisms, and have been associated with many biological activities including antioxidant, antibacterial, cytotoxic, dermatotoxic, genotoxic, and signaling properties.1–3 The chemistry, structural diversity and associated biological activity of alkylresorcinols, mostly from the plant world, has been thoroughly reviewed by Kozubek and co-workers.1,2It has recently emerged that alkylresorcinols are generated by different pathways that sometimes intersect to create further chemical diversity. The type III polyketide synthase (PKS), which elongate and cyclize different fatty-acyl precursors give rise to monoalkylresorcinols (MARs) in plants and bacteria and have been studied for the past few decades.4,5 However, the biosynthesis of dialkylresorcinols (DARs, so far only encountered in bacteria) was not fully understood until the recent report of two different DAR-generating pathways. In 2013, the Balskus lab showed that the unusual paracyclophane DARs derive from dimerization of type III PKS-derived MAR units.6 In 2014, the Bode group revealed that a new type of ketosynthase generated 2,5-DARs from two different fatty acid primary metabolism-derived precursors.7
Cyanobacterial alkylresorcinols, in particular DARs, are structurally distinct from those of heterotrophic bacteria, in that they commonly feature halogenation, glycosylation, long alkyl chains and intra- or inter-molecular cyclization. Our current understanding of alkylresorcinol diversity in cyanobacteria is limited, as many orphan gene clusters that lead to MARs and DARs are found in their genomes.8 Despite this small number of cyanobacterial alkylresorcinols, the structural connections among them are evident and reflect their intertwined biosynthetic pathways. This realization has been the main motivation for the present review. We begin by covering the structural diversity of the cyanobacterial alkylresorcinols and their biological activities, before detailing the biosynthetic pathways that generate such structures. We then peer into the genomes of cyanobacteria and look into the distribution of alkylresorcinol-related biosynthetic pathways, including those without any associated natural product. Since these structures have inspired a number of total syntheses that contrast with their natural biosynthesis, some of the strategies that have been used to obtain cyanobacterial alkylresorcinol scaffolds in the laboratory are briefly discussed.
2. Structural variety and biological activity of alkylresorcinols in cyanobacteria
2.1. Monoalkylresorcinols (MARs)
MARs have been reported from a variety of organisms, mainly from plants, bacteria and fungi, but also from protists and bryozoans.1–3 Biological activities associated with these metabolites include anticancer, antifungal, anti-inflammatory, antimicrobial, antiparasitical, antioxidant, genotoxic, among others.1,2 In higher plants, MARs have been obtained, for example, from rye and wheat bran oils, mango peals as well as from leaves and seeds used in ancient traditional medicines.1,2 Structural variations in plant MARs are usually related to the alkyl substituent; for example, DNA-polymerase inhibiting bis-5-alkylresorcinols isolated from the angiosperm Panopsis rubescens9 bear two terminal resorcinol rings on each side of the alkyl chain. Some additional structural features are noteworthy in non-plant MARs: for example, monochasiols isolated from the slime mold Dictyostelium monochasioides contain a chlorinated phenolic ring, while adeninealkylresorcinol, isolated from the fungus Lasiodiplodia sp.,10 features a nucleobase moiety. O-Methylation is also common in MARs.1In heterotrophic bacteria, MARs have been reported from Actinobacteria and alpha-, delta- and gamma-Proteobacteria11–13 which also include sulfated, methylated, and glycosylated resorcinol variants (Fig. 1). Interestingly, Azotobacter MARs were found to be the main components of its cyst membranes14,15 and are essential for the encystment process.16 In Streptomyces sp., MARs were found to be associated with the cytoplasmic membrane and to confer β-lactam antibiotic resistance.17
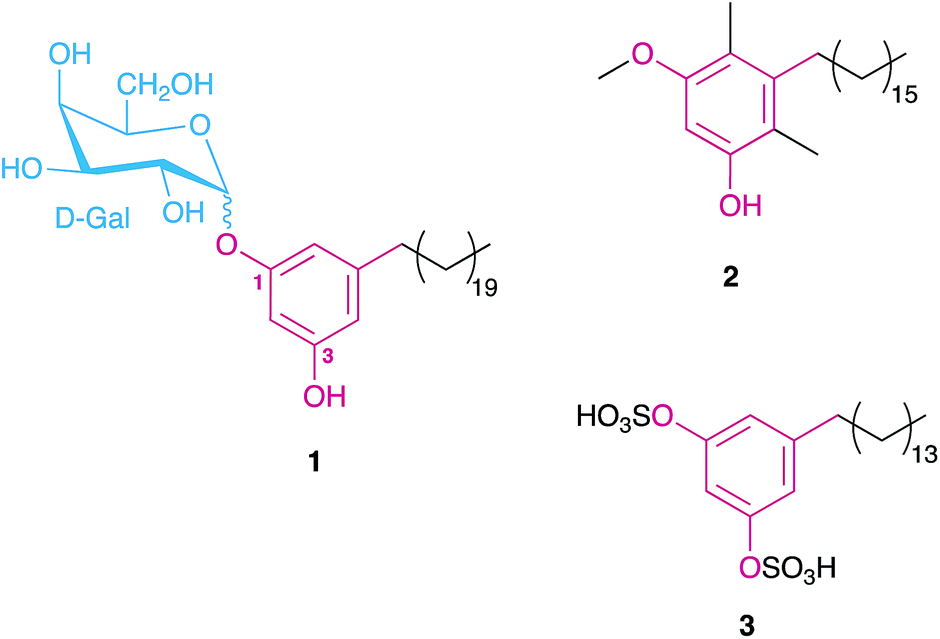 | ||
| Fig. 1 Examples of MARs isolated from different heterotrophic bacteria, namely a glycolipid from the cysts of Azotobacter vinelandii (1),15 the main component of the beta-leprosol mixture from Mycobacterium leprae (2)21 and one of the components of panosialin from Streptomyces spp. (3).22 Compound 1 shows the numbering scheme used throughout the text to refer to resorcinol ring substituents. | ||
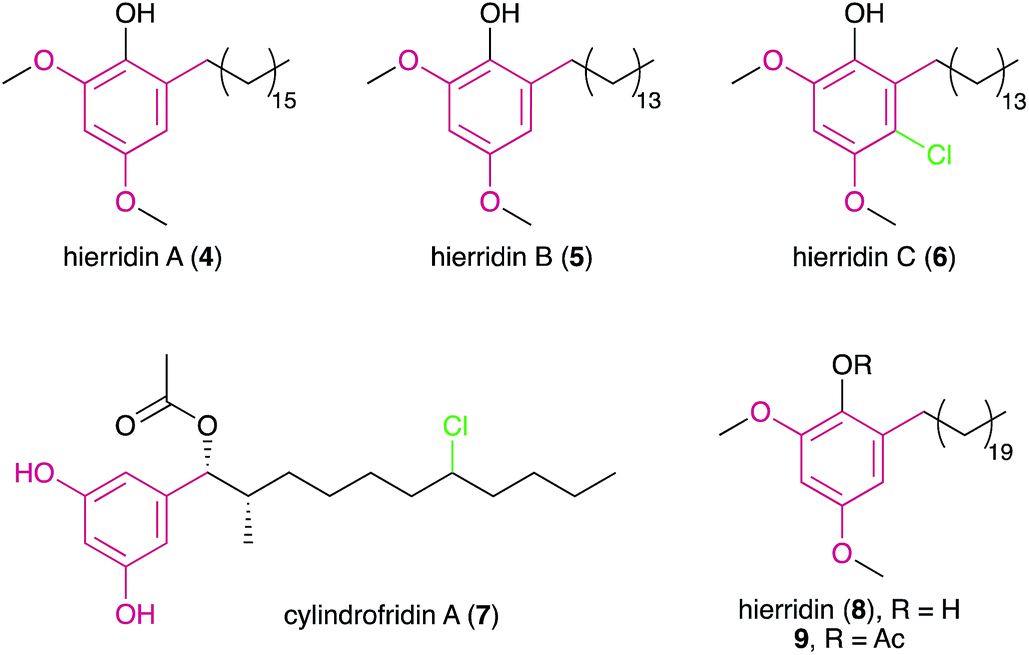
Despite the large number of type III PKS-containing biosynthetic gene clusters (BGCs) found in cyanobacterial genomes (see Section 3), only four cyanobacterial MARs (hierridins A–C18,19 and cylindrofridin A,204–7) are known to date.
| Alkylresorcinol class | Congener(s) | Reported biological activity | Potencya (in μM, unless otherwise stated) |
|---|---|---|---|
| a As reported in the cited references. b Compounds 35 and 36 only. c From MABA (microplate alamar blue assay). d From LORA (low-oxygen recovery assay). e MIC value corresponds to compound 40, compound 41 was reported to have a MIC > 10 μM. | |||
| MARs | |||
| Hierridins | A (4) and B (5), mixture | Antiplasmodial (Plasmodium falciparum, D6 and W2 strains)18 | 3.7–5.2 μg mL−1 (IC50) |
| B (5) | Cytotoxic (HT-29 cells)26 | ∼100 (IC50) | |
| B (5) and C (6) | Antiplasmodial (P. falciparum, 3D7 and Dd2 strains)19 | 1.5–2.3 (IC50) | |
| Cylindrofridins | A (7) | Antibacterial (Gram-positive: MRSA, Streptococcus pneumoniae)20 | 8.6–17 (MIC) |
| Cytotoxic (HaCaT cells)20 | ∼100 (IC50) | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| DARs | |||
| Bartolosides | A (58) | Cytotoxic (HT-29 cells)51 | 21 (IC50) |
| A (58), E (62) and I (66) | Cytotoxic (T-47D, MG-63, RKO cells)52 | 22–60 (IC50) | |
| B (59) | Cytotoxic (T-47D, MG-63, HepG2, SH-SY5Y, PC-3, HT-29, RKO cells)51 | 9.5–26 (IC50) | |
| Carbamidocyclophanes | A–C (35–37) | Antibacterial (Staphylococcus aureus)44 | 40–100 (MIC) |
| Cytotoxic (FI and MCF7 cells)44 | 0.86–5.1 (IC50) | ||
| Antibacterialb (S. pneumoniae)45 | 5.6–8.2 (MIC) | ||
| A–C (35–37), F (40) and G (41) | Antibacterial (Enterococcus faecalis, S. aureus)45 | 0.1–1.1 (MIC) | |
| A (35), B (36) and F (40) | Antifungal (Candida albicans)45 | 1.3–5.5 (MIC) | |
| F (40) and G (41) | Antimycobacterial (Mycobacterium tuberculosis)45 | 0.8–1.8 (MIC)c, 5.4 (MIC)d,e | |
| A–F (35–40) and H–L (42–46) | Antibacterial (Gram positive: MRSA and S. pneumoniae)46 | 0.10–0.32(MIC) | |
| Cytotoxic (HaCaT cells)46 | 2.8–7.6 (IC50) | ||
| A (35) and F (40) | Cytotoxic (MDA-MB-435, MDA-MB-231 and OVCAR3 cells)48 | 1.0–4.3 (IC50) | |
| Cylindrocyclophanes | A (16) and A4–A1 (26–29) | Antibacterial (Gram positive: MRSA and S. pneumoniae)46 | 0.43–2.10 (MIC) |
| Cytotoxicity (HaCaT cells)46 | 5.0–11.5 (IC50) | ||
| A–F (16, 21–25) | Cytotoxic (KB cells)39 | 0.5–5 μg mL−1 (IC50) | |
| A (16), C (22), A4–A1 (26–29), C4 (30), C2 (32) and F4 (34) | 20S proteasome inhibition42 | 2.55–59.3 (IC50) | |
| A4–A2 (26–28), C4 (30) and C2 (32) | Cytotoxic (HT-29 cells)42 | 0.5–2.8 (EC50) | |
| D (23) | Cytotoxic (MDA-MB-435 and MDA-MB-231 cells)49 | 2.3–5.2 (IC50) | |
| F (25) | Cytotoxic (MDA-MB-435, MDA-MB-231 and OVCAR3 cells)48 | 10–13 (IC50) | |
| Merocyclophanes | A (47) and B (48) | Cytotoxic (HT-29 cells)47 | 1.7–3.3 (IC50) |
| A (47), C (49) and D (50) | Cytotoxic (MDA-MB-435, MDA-MB-231 and OVCAR3 cells)48 | 0.9–9.8 (IC50) | |
| Nostocyclophanes | A–D (18–20, 17) | Cytotoxic (KB and LoVo cells)43 | 0.5–2 μg mL−1 (IC50) |
| Nostocyclyne | A (70) | Antibacterial (S. aureus and Bacillus subtilis)54 | 10–12.5 μg/disk (MIC) |
| Ribocyclophanes | A–D (51–54) | Cytotoxic (MDA-MB-435 and MDA-MB-231 cells)49 | 0.6–6.4 (IC50) |
Hierridin B was later reported from a marine picocyanobacterium (Cyanobium sp. LEGE 06113), collected from the Portuguese coast.26 To further interrogate its biological properties, metabolite 5 was tested against eight human cell lines showing selective albeit very mild cytotoxicity to a colon adenocarcinoma cell line HT-29, with an IC50 value of ∼100 μM. This selective bioactivity was further investigated by Freitas et al.27 Compound 5 seems to interfere with VDAC-1 (voltage-dependent anion-selective channel 1) in mitochondria as well as some cell cycle and structural genes responsible for the cell cycle checkpoints, leading to apoptosis in the HT-29 cells.
During the re-isolation of 5, a halogenated analogue, hierridin C (6) was discovered.19 The aromatic ring of this metabolite has a chlorine atom adjacent to the alkyl-substituted carbon, confirmed through NMR and MS experiments. Compound 6 showed antimalarial activity against Plasmodium falciparum strains (3D7 and Dd2) with IC50 values of 1.5 μM and 2.3 μM, respectively. This is the first bacterial example of a MAR featuring ring halogenation. Curiously, the structurally-related monochasiols possessing a chlorination in position 2 of the resorcinol ring were recently reported from a Dictyostelium slime mold28 and are related to Dictyostelium differentiation-inducing (DIF) polyketides.29 Similar to hierridin B, the DIF-family compound DIF-3 targets mitochondria in cancer cells.30
2.2. Dialkylresorcinols
DARs have been known for a few decades, with flexirubin (10)31 and 2-n-hexyl-5-n-propylresorcinol (also referred as DB-2073, 11)32 being the first described compounds of this family. While flexirubin-type pigments have so far only been reported from Flexibacter, Flavobacteria and Cytophaga strains,33 the description of the potent antibiotic DB-2073 marked the discovery of a series of DARs from Pseudomonas spp. Examples include resorstatin (2-hexyl-5-pentylresorcinol, 12), showing a strong inhibitory activity against lipid peroxidation induced by free radicals in rat brain homogenate (IC50 = 2.06 μM)34 and other short-chain DARs such as 2-butyl-5-propylresorcinol (13) and 2-hexyl-5-methylresorcinol (14) that show antibacterial or antifungal activity.35 More recently, arcuflavin (15), a xanthomonadin-DAR hybrid that bears resemblance to the flexirubin pigments was reported from an Azoarcus sp. β-proteobacterium.36 The structures, biosynthesis and biological role of bacterial DARs (mostly from heterotrophic bacteria) have been reviewed by Schöner et al.37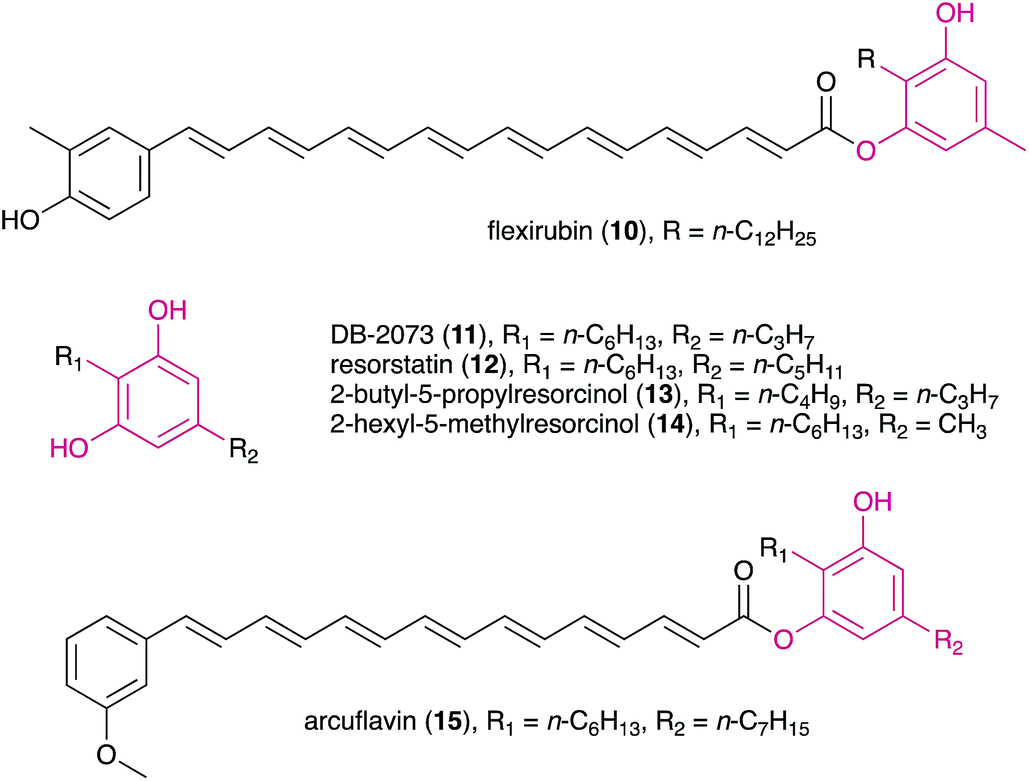
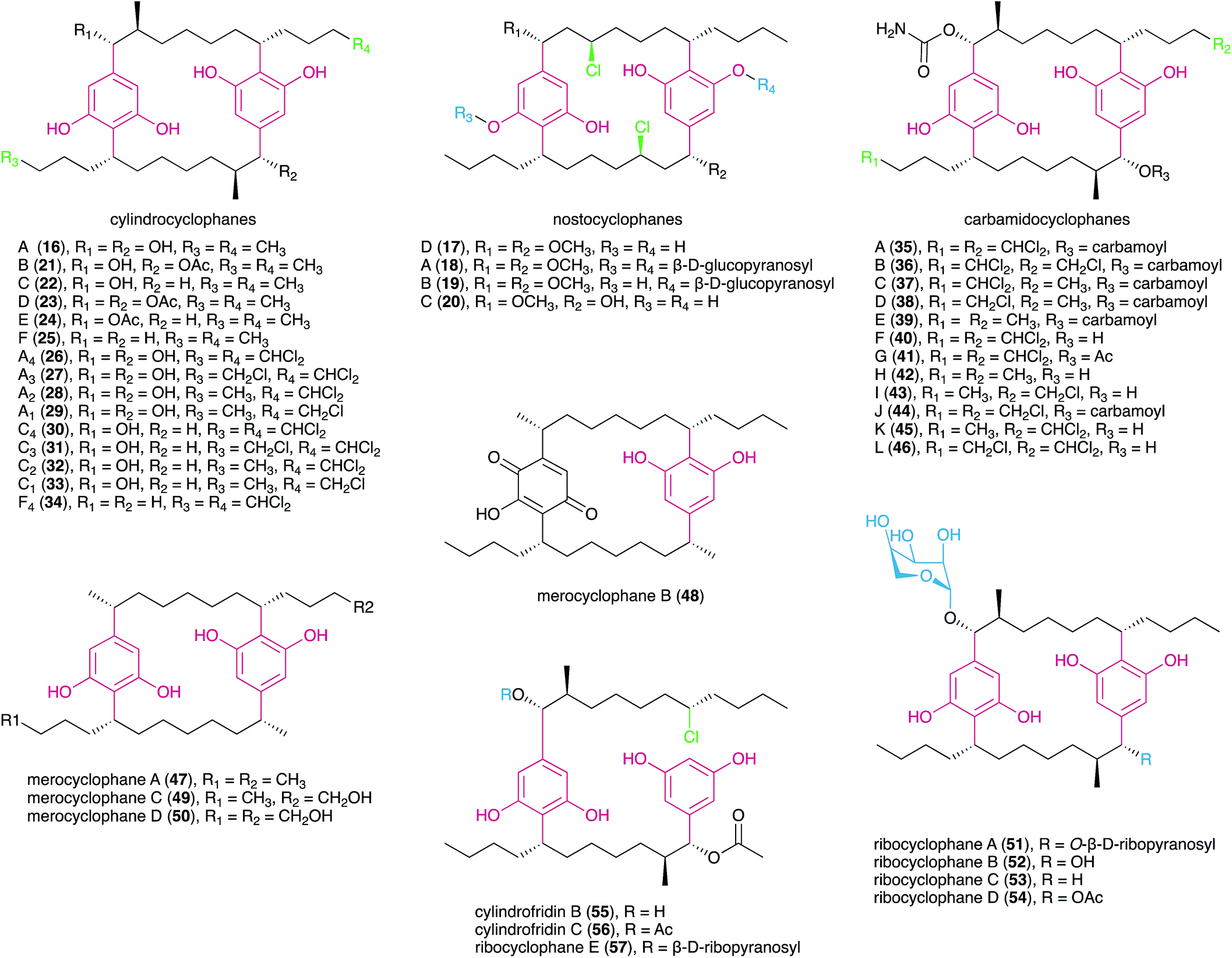
The first cyanobacterial DARs were discovered in the early 1990s when Richard Moore's lab described the highly unusual structures of a new class of naturally-occurring cyclic DARs: a series of cyclophanes isolated from Nostocales strains.38,39 Since then, a number of DARs have been reported from cyanobacteria, most of which differ structurally from their non-cyanobacterial counterparts and have a variety of biological activities. Many structural features are shared by different classes of cyanobacterial DARs, including chlorination, alkyne moieties and glycosylations, which reflects common biosynthetic elements (see Section 3.2) but also suggests that these compounds share biological roles.
 | ||
| Fig. 2 Cyanobacterial [7.7]paracyclophanes (16–47, 49–54) and related metabolites (48, 55–57). | ||
The carbamidocyclophanes are characterized by carbamate substituents at both benzylic positions. Carbamidocyclophanes A–E (35–39) were isolated from a Vietnamese Nostoc sp. strain, guided by antibacterial activity towards Gram-positive bacteria as well as antifungal (Candida maltosa) activity.44 These metabolites showed mild potency towards Staphylococcus aureus ATCC 6538 but had stronger, low micromolar cytotoxicity towards MCF7 breast adenocarcinoma cells indicating that cyanobacterial paracyclophanes exhibit generalized cytotoxicity.44 The congeners F (40) and G (41) are similar to 35 but feature hydroxyl and acetyl substituents, respectively, in one of the benzylic positions. Both metabolites showed sub-micromolar cytotoxicity towards human cancer cell lines.45 Notably, compound 40 exhibited potent anti-Mycobacterium activity and was also reported to strongly inhibit the growth of S. aureus and Enterococcus faecalis.45 Five new carbamidocyclophanes (42–46) with minor structural differences to known congeners were later reported by the same research group,46 and exhibited submicromolar antibacterial (Gram-positive) activity and low-micromolar cytotoxicity in a human keratinocyte cellular assay.
Merocyclophanes have a benzylic methyl substituent and lack the beta methyl group found in the cylindrocyclophanes. Merocyclophane A (47) was isolated from a terrestrial Nostoc sp. strain (UIC 10062) together with the hydroxyquinone-containing analogue merocyclophane B (48).47 Merocyclophanes C (49) and D (50) were later isolated from the freshwater Nostoc sp. UIC 10110 and are analogues of 47 with hydroxylation at one or both terminal alkyl positions.48 The merocyclophanes, as with most cyanobacterial paracyclophanes, are strongly cytotoxic (low-to-sub micromolar IC50) towards human cancer cell lines.47,48
Recently, the Orjala group reported members of a new class of paracyclophanes, namely ribocyclophanes A–D (51–54), that contain β-D-ribopyranose as substituent at one or both benzylic positions.49 These sugar moieties were found to be in the rare 1C4 conformation; the authors suggest that this is due to steric hindrance from the cylindrocyclophane core not allowing the more common (and typically more stable) 4C1 conformation. The ribocyclophanes were co-isolated with cylindrocyclophane D (23) suggesting a common biosynthetic origin.49
The isolation of pharacine, a highly symmetrical paracyclophane (a cyclic terephthalate), from the gliding marine Gram-negative bacterial strain Cytophaga sp. (AM13.1), suggested that the occurrence of [n.n]cyclophanes is not restricted to the phylum Cyanobacteria.50 Still, the presence of traces of this molecule in ubiquitous phthalate plasticizers has raised doubts as to the natural origin of the compound.50
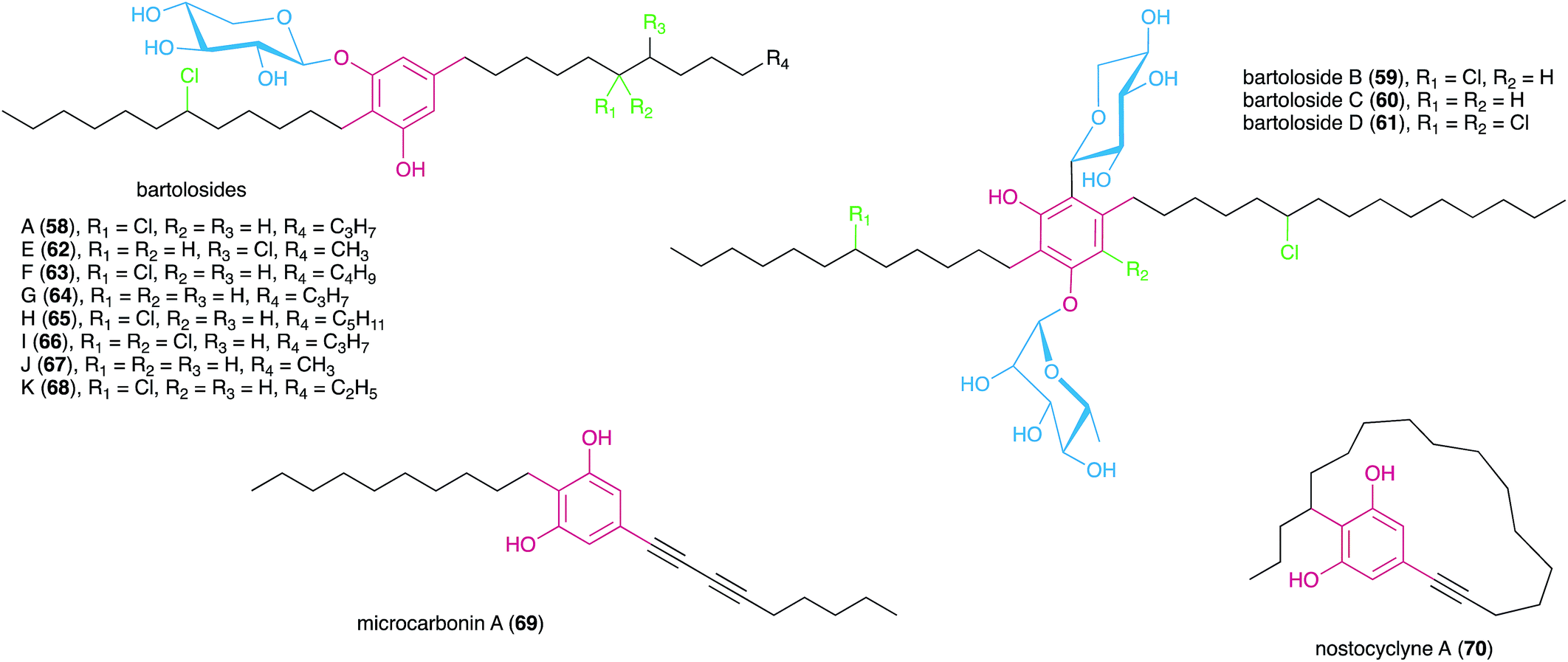
In 2015, Preisitsch and collaborators20 isolated two new cylindrocyclophane-related DARs, cylindrofridins B (55) and C (56). These compounds are dimers of the co-isolated MAR 7 or its putative deacetylated monomer (that has not been isolated to date) and are reminiscent of the Friedel–Crafts alkylation events that are involved in cyanobacterial paracyclophane biosynthesis8 (see Section 3.2.1.). This is evident from the chlorination at the ω-5 carbon atom in cylindrofridins, the site of [7.7]paracyclophane macrocycle formation, and therefore compounds 55 and 56 represent linearized biosynthetic intermediates of the cylindrocyclophanes.20 Likewise, the recently described ribocyclophane E (57), isolated from the ribocyclophane-producing strain Nostoc sp. UIC 10366,49 is a linearized version of compound 54. No significant bioactivity has been reported for compounds 55–57, indicating that the open versions of the cyclophane macrocycles do not conserve their cytotoxic and antibacterial properties.20,49
In a later study using the cyanobacterium S. salina LEGE 06099, closely related to the bartolosides B–D (59–61) producer (S. salina LEGE 06155), Afonso et al.52 reported new analogues of metabolite 58, bartolosides E–K (62–68) by following cytotoxic activity towards cancer cell lines. Most of the structural differences of these new analogues related to the length of the alkyl chain and the presence or absence of a chlorine substituent in the 5-alkyl chain. A notable exception is compound 66 that features a rare, mid-chain gem-dichloro moiety.52 Interestingly, this same strain produced bartoloside A in elevated amounts (reaching up to 0.6% of its dry weight), suggesting a potential structural role.52 The bartolosides have been tested for cytotoxicity towards several cancer cell lines but showed only mild activity with compound 59 being the most potent of the series (IC50 value 9 μM against PC-3 human prostate adenocarcinoma).51,52
3. Biosynthesis of alkylresorcinols in cyanobacteria
3.1. Monoalkylresorcinols from type III PKS pathways
MARs are, to date, known only to be biosynthesized via type III PKS enzymes. Homodimeric and iterative, type III PKSs are typically smaller and simpler than type I and II PKS, but they retain nonetheless elongation and cyclization functionalities.4 Generally, type III PKSs use a thioester as the starter unit and perform elongation through the condensation of extender units, usually malonyl-CoA, to form a polyketide chain that is then intramolecularly cyclized via aldol condensation, Claisen condensation or lactonization.56 Despite the simplicity of their reactivity, by diversifying the starter and extender units, number of condensation steps and cyclization mechanisms, these enzymes are responsible for the biosynthesis of a breadth of compounds, not only alkylresorcinols but also alkylpyrones, chalcones, stilbenes, quinolones and acylphloroglucinols, among other.57,58 MARs are generated by a specialized group of type III PKS, the alkylresorcinol synthases (ARS), which catalyze the condensation between fatty acyl-CoA (or- ACP) thioesters (starter units) and malonyl-CoA thioesters (or other extender units), followed by an aldol-type cyclization. Most of the currently characterized ARSs are bacterial, and only few plant and fungi-derived examples are known.5 The first ARS enzyme, ArsB, which was characterized in 2006 from Azotobacter vinelandii, is involved in the biosynthesis of cyst envelope alkylresorcinols (e.g. compound 1).16 Since then, a few other homologs have been characterized, namely SrsA from Streptomyces griseus17 (that produces 6-methyl-5-alkylresorcinols), MMAR_2470 and MMAR_2474 from Mycobacterium marinum which provide methylated alkylresorcinols,59 the alkylresorcylic acid-generating FtpA from Myxococcus xanthus,13 BpsA from Bacillus subtilis which produces long-chain MARs,60 as well as DpgA61 and DhpaS57 which produce the MAR (S)-3,5-dihydroxyphenylglycine (a common moiety in actinobacterial glycopeptide antibiotics).Apart from the MAR-mediated biosynthesis of cyanobacterial cyclophanes6 (see Section 3.2.1.), only very recently the first cyanobacterial biosynthetic pathway leading to MAR end products (hierridins 5 and 6) has been reported.19 The hid BGC was found in the genome of the hierridin-producer Cyanobium sp. LEGE 06113 by mining it for type III PKSs. This small BGC codes for three proteins with functions consistent with hierridin biosynthesis: a type-III PKS (HidC), a SAM-dependent methyltransferase (HidA) and a FAD-dependent oxidoreductase (HidB) (Fig. 3). Based on in vivo assays and the biosynthetic logic associated with Hid enzymes, hierridin biosynthesis putatively involves four main steps. First, HidC accepts thioester-activated palmitic acid and performs four elongation steps with malonyl-CoA and finally a decarboxylative condensation (C2-to-C7 aldol condensation) forms the alkylresorcinol ring (Fig. 3). Methylation of resorcinol phenol groups by HidA likely ensues, since a recombinant version of HidA was able to methylate 5-pentadecylresorcinol.19 This observation not only established experimentally the involvement of the hid BGC in hierridin biosynthesis, but also suggested that methylation precedes HidB-mediated hydroxylation of the resorcinol ring to yield hierridin B (5). The last biosynthetic step involves regioselective chlorination of 5 generating hierridin C (6). However, as observed for the resorcinol-halogenated bartolosides, the hid BGC does not encode a halogenase and no halogenase was found in the genome of Cyanobium sp. LEGE 06113, and so the nature of this halogenation is still unknown. Leão et al.19 report that hid-like BGCs are present in many cyanobacterial genomes, most prevalently in those of picocyanobacteria (see also Section 3.3).
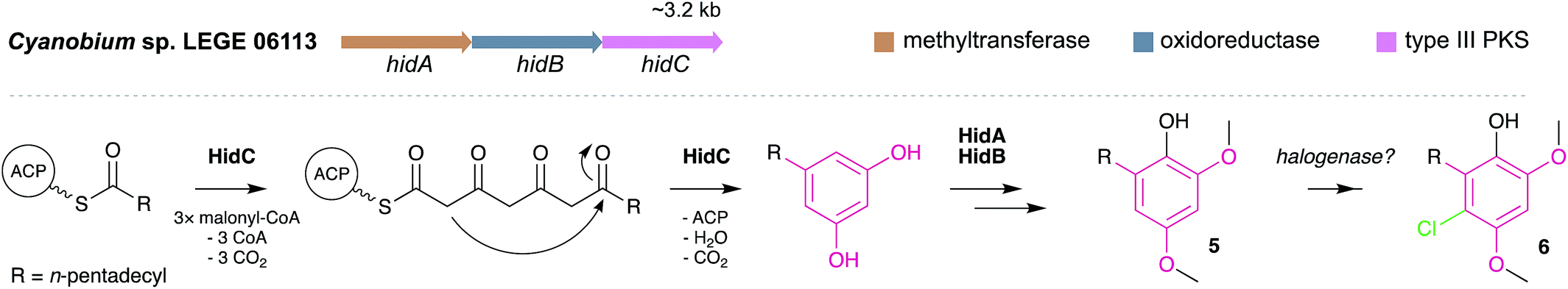 | ||
| Fig. 3 Biosynthesis of hierridins. Top – Hierridin biosynthetic gene cluster (hid). Bottom – Proposed19 biosynthesis of hierridins B (5) and C (6). | ||
3.2. Dialkylresorcinols
The first insights into paracyclophane biosynthesis came from feeding studies with isotopically labeled acetate. Bobzin and Moore fed a mixture of unlabeled and [1,2-13C2]-labeled acetate to the cylindrocyclophane producer Cylindrospermum licheniforme ATCC 29204.62 The resulting cylindrocyclophane D contained adjacent 13C atoms in specific positions (Fig. 4), indicating that acetate is incorporated into the final product. Feeding of [1-13C,2H3]acetate and [2-13C,2H3]acetate further indicated that the C-1 and C-2 positions of acetate are incorporated as expected for an aromatic polyketide (Fig. 4). Similar results were also obtained from the nostocyclophane producer Nostoc linckia UTEX B1932.62 From these data, and the 2-fold symmetry of the paracyclophane scaffold, Bobzin and Moore correctly proposed that natural paracyclophanes are biosynthesized from a polyketide-derived alkylresorcinol precursor that undergoes dimerization and that the β-methyl groups in compound 23 were derived from acetate and not from methionine.62 This realization was key to the Balskus' group strategy for the discovery of the biosynthetic gene cluster for cylindrocyclophanes (cyl).6,63 Because β-methyl groups are introduced into polyketides by a highly-conserved pathway that includes a homolog of hydroxymethylglutaryl-CoA (HMG-CoA) synthase, using degenerate PCR and whole-genome sequencing, Nakamura et al.6 identified an HMG-CoA synthase homolog in the cylindrocyclophane producer C. licheniforme ATCC 29412. The HMG-CoA synthase homolog gene (cylF) was found to be part of a gene cluster (cylA-P) that encodes other enzymes whose predicted activities appeared related to cylindrocyclophane biosynthesis.6 The cluster's biosynthetic role was confirmed by heterologous expression and purification of the type III PKS CylI, which was shown to convert a diketide into an alkylresorcinol that resembles the putative precursor of cylindrocyclophane biosynthesis.6
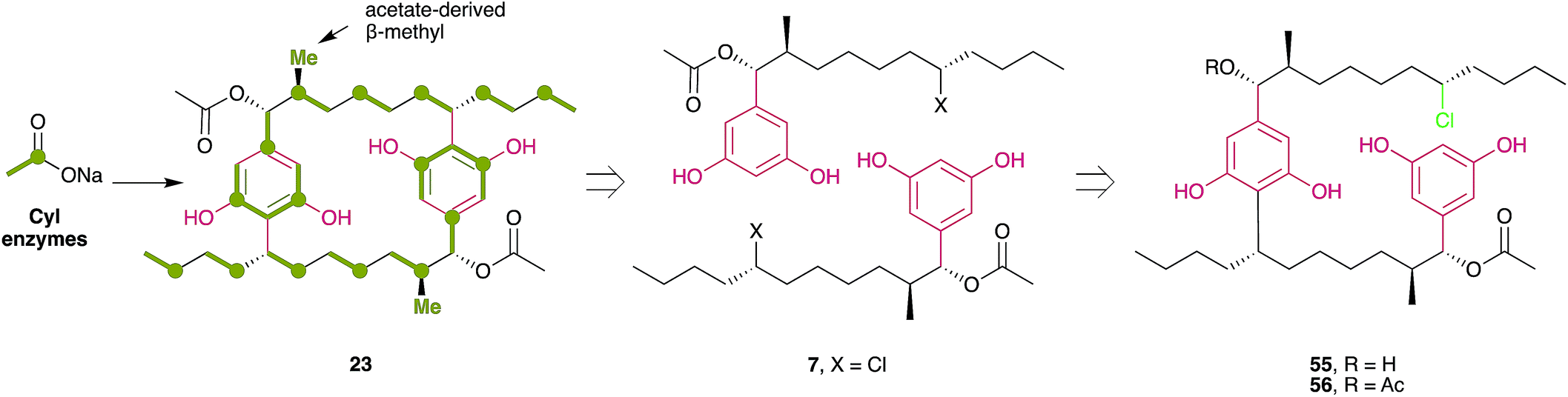 | ||
| Fig. 4 Key findings that provided insight into the overall biosynthesis of cylindrocyclophanes. (A) Feeding experiments with 13C-labeled acetate produce cylindrocyclophanes with a distinctive labeling pattern. [1,2-13C]acetate produces adjacent 13C atoms in the positions indicated in green, with the 1- and 2-positions of acetate incorporated into specific positions. (B) Discovery of the cylindrofridins, which contain a chlorine atom at the site of carbon–carbon bond formation, matched a retrosynthetic hypothesis for the origin of cylindrocyclophanes. Cylindrofridin A (7) is the monomeric precursor to cylindrocyclophane D (23), while cylindrofridins B (55) and C (56) likely represent the product of partial macrocyclization. | ||
The cyl cluster contains an abbreviated set of polyketide synthases that perform only four elongation reactions. This activity is insufficient to explain the biosynthesis of the paracyclophane alkyl chain. Feeding studies with d19-decanoic acid led to the production of d18- and d36-cylindrocyclophane F,6 indicating that the alkyl tail originates from fatty acids instead of from a polyketide, which is nonetheless consistent with the original feeding studies of Bobzin and Moore.62 Purification and characterization of CylA and CylB, a fatty acid adenylating enzyme and acyl carrier protein (ACP), respectively, further confirmed that cylindrocyclophane biosynthesis begins with the recruitment of free decanoic acid.6 Because the alkyl chain of decanoic acid is incorporated into cylindrocyclophanes in toto, the dimerizing macrocyclization event necessarily requires functionalization of an unactivated carbon center.6 However, the true identity of the activated precursor remained elusive because it was not initially detected in culture extracts.6,63
The dimerization mechanism was clarified with the discovery of the cylindrofridins from other cylindrocyclophane producers.20 Cylindrofridins 55 and 56 are nearly chemically identical to the cylindrocyclophanes, except that one of the dialkylresorcinol motifs is disconnected. Instead, a chlorine atom occupies the position of C–C bond formation in one alkyl tail (Fig. 4). Chlorination had been proposed as an activating step that might allow dimerization using a Friedel–Crafts-style alkylation,63 suggesting the cylindrofridins represent an incomplete macrocyclization or intermediate of cylindrocyclophane biosynthesis. The corresponding halogenase, CylC, was identified based on a bioinformatic analysis which revealed that CylC homologs are present in the biosynthetic gene clusters for certain halogenated products from cyanobacteria.8 Using heterologous protein expression and purification, Nakamura et al.8 demonstrated that CylC chlorinates ACP-bound decanoic acid early in cylindrocyclophane biosynthesis.
The macrocyclizing enzyme was initially thought to be CylP,63,64 a predicted Rieske oxygenase that shares homology to a C–C bond-forming enzyme in streptorubin B biosynthesis,65 but in vitro assays demonstrated that CylP instead installs the α-hydroxy group of cylindrocyclophanes.8 Further enzyme purification and in vitro assays showed that CylK, initially annotated as a hemolysin-type calcium-binding protein, is in fact the macrocyclizing C–C bond-forming enzyme that completes cylindrocyclophane biosynthesis.8 CylK uses the chlorinated alkyl tail to perform a Friedel–Crafts-style alkylation reaction with the monoalkylresorcinol to form the final DAR, a reaction that removes the chlorine group from the final product.
Together, a combination of bioinformatics and in vitro enzymology6,8 provides a comprehensive picture of cylindrocyclophane biosynthesis (outlined in Fig. 5). Biosynthesis starts with free decanoic acid, presumably recruited from fatty acid biosynthesis or degradation. The fatty-acyl AMP ligase CylA loads free decanoic acid onto the ACP CylB. The halogenase CylC then chlorinates the ACP-loaded decanoic acid, and the chlorination is maintained until the final macrocyclization event. Following an initial extension by the PKS CylD (module 1), the HMG-CoA synthase homolog CylF installs the functionality that eventually becomes the β-methyl group, likely working with CylE to incorporate malonyl-CoA, leading to an acetate-derived β-methyl group. A substituted olefin is briefly introduced by the enoyl-CoA hydratase CylG. The PKS CylH (module 2) performs decarboxylation and olefin reduction to complete the β-methyl group and then further extends the precursor by one ketide subunit. The assembly line is terminated by the type III PKS CylI, which performs two more elongations and releases the product through ketide cyclization, forming a completed monoalkylresorcinol precursor. The precursor can be further decorated by the hydroxylase CylP to install an α-hydroxy group and then acetylated, likely by the predicted acetylase CylO, to create the diversity of cylindrocyclophanes observed from live cultures. Decoration at this stage, rather than following macrocyclization, is supported by the isolation of cylindrofridin A (7) which is predecorated with an α-acetyl group.20 Finally, the calcium-dependent enzyme CylK forms the final cylindrocyclophane product by dimerizing two precursors in a Friedel–Crafts-style alkylation reaction.
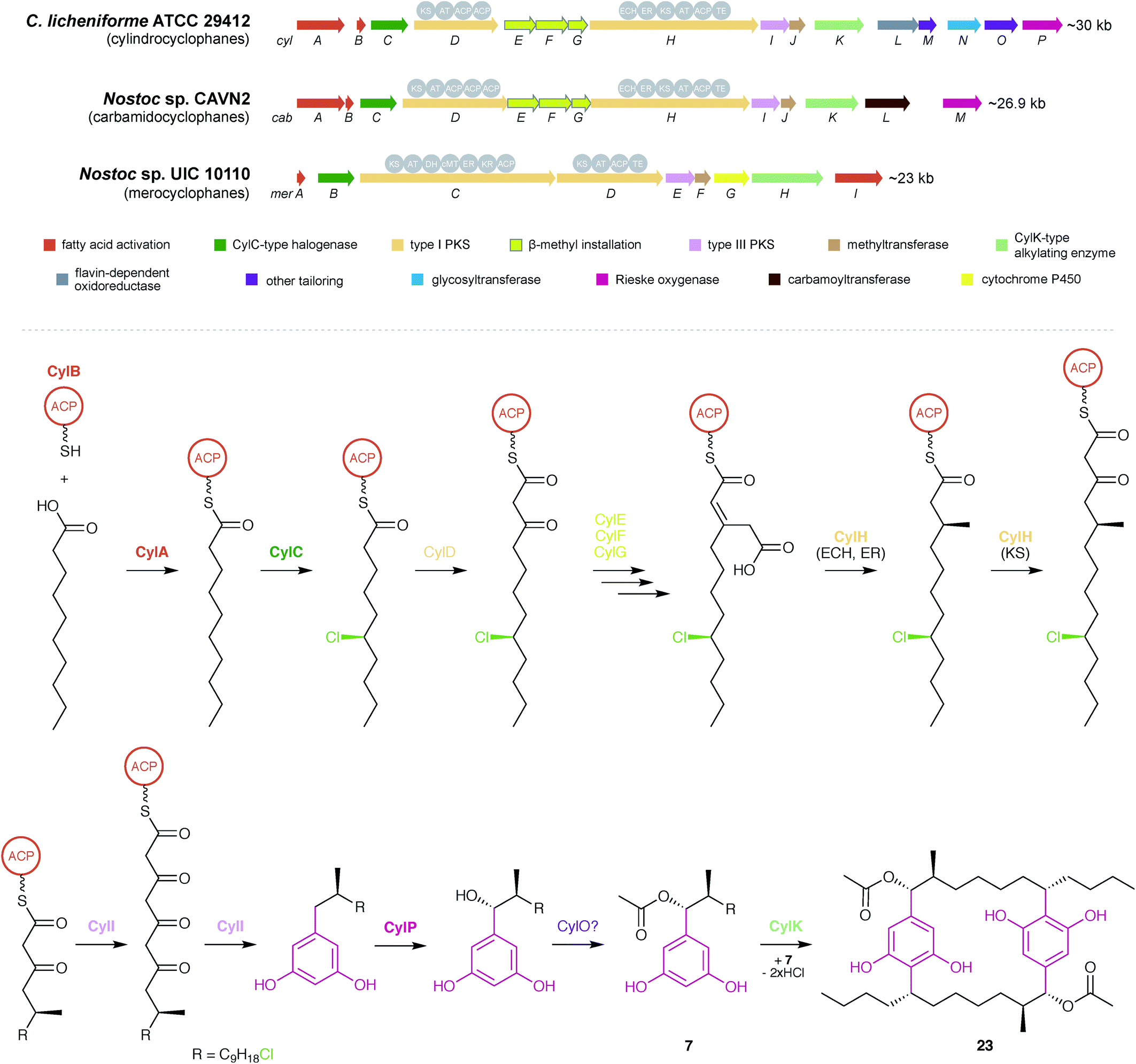 | ||
| Fig. 5 Biosynthesis of cyanobacterial cyclophanes. Overview of the BGCs for different cyclophane classes (top) and biosynthetic pathway for the cylindrocyclophanes (bottom), as illustrated by cylindrocylophane D (23). Enzymes that have been purified and characterized in vitro6,8 are shown in bold. | ||
Biosynthesis of the other natural paracyclophanes, including nostocyclophanes, carbamidocyclophanes, and merocyclophanes, likely follows a similar scheme, and producers of the latter two are known to contain homologs of the key biosynthetic enzymes including CylC, CylK, and CylI48,64 (Fig. 5). The primary difference in biosynthesis is the enzymes that decorate the monoalkylresorcinol precursor. The biosynthetic gene cluster for carbamidocyclophanes contains a predicted carbamoyltransferase that likely installs the amide group.64 The biosynthetic gene cluster for merocyclophanes lacks homologs of CylE–G, the enzymes responsible for β-methyl incorporation, and the CylH homolog lacks the ECH and ER domains involved in completing the β-methyl group. Instead, the CylD homolog contains a predicted methyltransferase domain that likely incorporates the α-methyl group of the merocyclophanes.48 The only known producer of nostocyclophanes,38Nostoc linckia UTEX B1932, is no longer available from the UTEX culture collection, and consequently a genetic analysis has yet to be performed. However, it is anticipated that nostocyclophane biosynthesis is essentially equivalent to cylindrocyclophane biosynthesis, with the internal chlorine atom being introduced by a decorating halogenase prior to macrocyclization and a glycosyltransferase adding the glucose moiety to the resorcinol ring (for compounds 18 and 19). The other uncharacterized enzymes in the cylindrocyclophane biosynthetic gene cluster (CylJ, CylL, CylM, and CylN) may provide other as-yet undiscovered decorations to the paracyclophane scaffold. For example, the ribocyclophanes (51–54, 57)49 might originate from the action of CylN, annotated as a glycosyltransferase.8
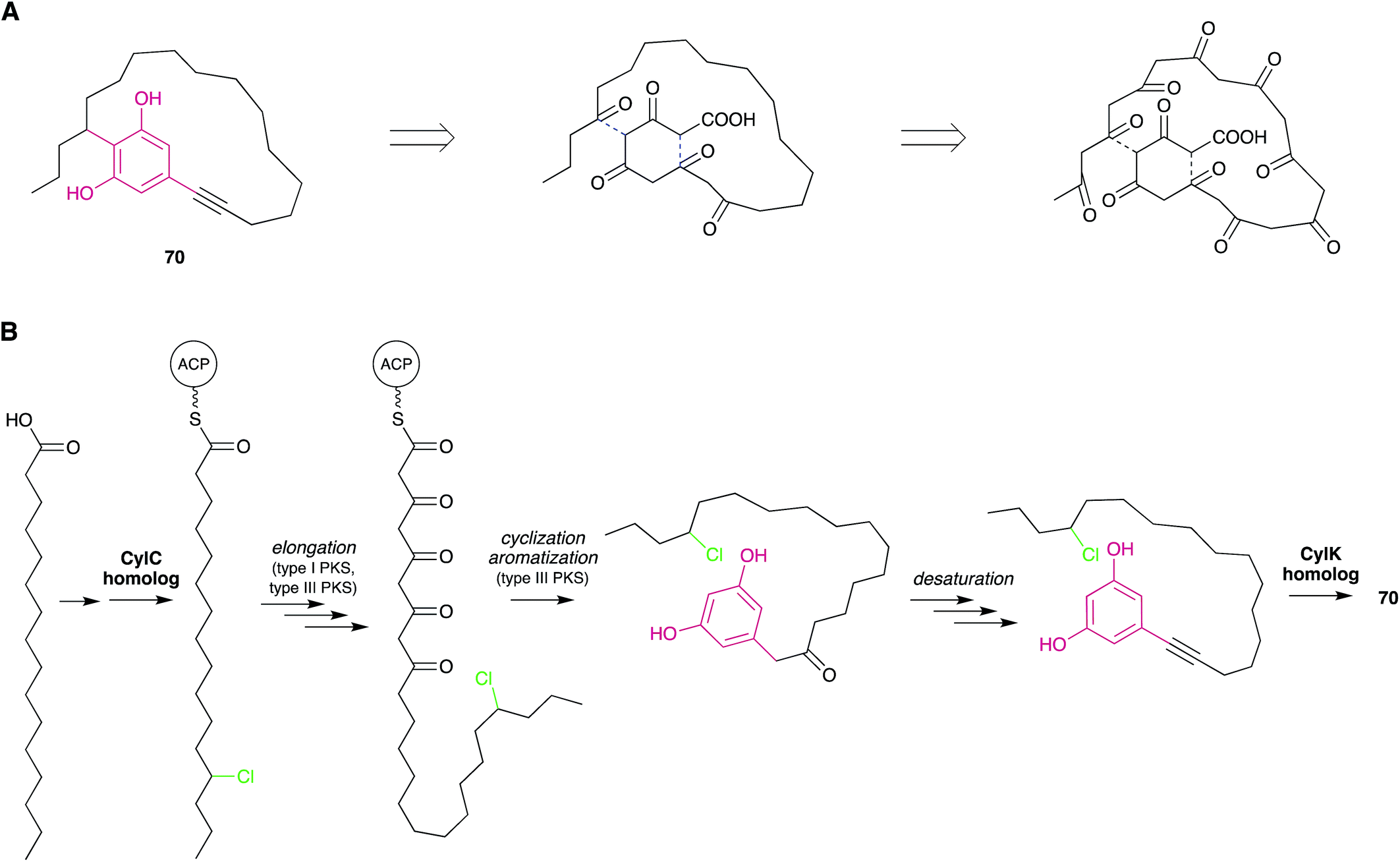 | ||
| Fig. 6 Biosynthetic hypotheses for the origin of nostocyclyne A. (A) The original hypothesis by Ploutno and Carmeli.54 Dashed lines indicate eventual ring-closing events. (B) An alternative hypothesis based on the elucidation of cylindrocyclophane biosynthesis. | ||
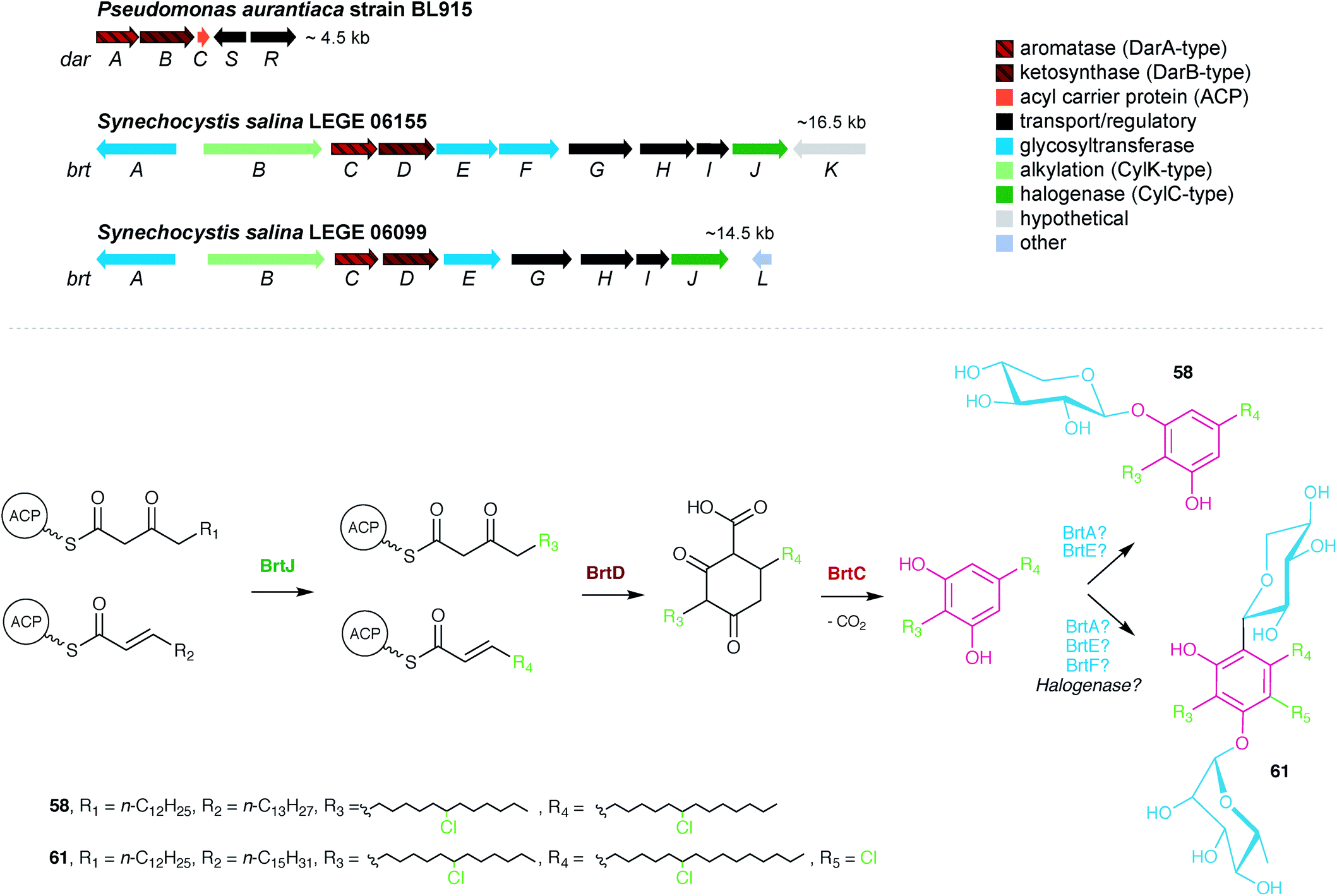 | ||
| Fig. 7 Biosynthesis of the bartolosides. Top – prototypical DAR-generating dar gene cluster from Pseudomonas aurantiaca and the two brt BGCs found in Synechocystis salina strains. Bottom – the brt pathway encodes the production of mono- and di-glycosylated bartolosides. | ||
The first characterized cyanobacterial DarAB-like pathway was the bartoloside BGC (brt) from the cyanobacterium Synechocystis salina LEGE 06155.51 This gene cluster (Fig. 7) is involved in the biosynthesis of diglycosylated 59–61 and encodes, apart from DarA and DarB, three glycosyltransferases, a glycolipid export operon, a CylC homolog and a CylK homolog, as well as a hypothetical protein. An additional brt BGC was later reported by Afonso et al.52 from the closely-related cyanobacterium S. salina LEGE 06099 and is involved in the biosynthesis of the monoglycosylated 58 and 62–68. Just as in the previously characterized DarAB system,7 the biosynthesis of bartolosides is initiated by BrtD (DarB homolog) that connects the two fatty acid derivative thioesters via Claisen condensation and simultaneous Michael addition, yielding a carboxy-CHD (Fig. 7).51 All diglycosylated bartolosides (B–D) are synthetized by condensation of C16 and C18 fatty acyl-derivatives, namely 2-octadecenoyl-ACP and 3-oxo-hexadecanoyl-ACP thioesters. Regarding monoglycosylated bartolosides, the initial substrates likely have different chain lengths (C14–C18), including odd-chain C15 and C17 α,β-unsaturated fatty acyl-ACP thioesters. The origin of these odd-chain precursors is still unclear.52 Although brt lacks a dedicated ACP, unlike previous characterized DAR clusters, it is foreseen that the ACP-bound fatty acyl derivatives are recruited from primary metabolism, especially considering the unlikely participation of acyl-CoA substrates due to the apparent absence of a canonical β-oxidation pathway in cyanobacteria.68 By using heterologously-expressed BrtC (DarA homolog) and BrtD in a coupled assay, Leão et al.51 provided the first biochemical evidence of DarA enzymes acting as aromatases, as conversion of the fatty acyl derivative surrogates to the DAR backbone of 59–61 was observed only in the presence of BrtC. Backed by crystallographic data, Mori et al.69 showed that in bacterial stilbene biosynthesis by a DarAB system (StlC/StlD from Photorhabdus luminescens), decarboxylation occurs during aromatization, clarifying that carboxy-CHDs are the substrates for DarA aromatases. Following DAR backbone generation, the biosynthesis of the bartolosides should proceed with the decoration of the resorcinol with sugar moieties, namely xylose and rhamnose by glycosyltransferases BrtA, E and F (diglycosylated bartolosides) or Brt A and E (monoglycosylated bartolosides), although it is unclear whether all these enzymes are involved in the glycosylations. Halogenation (including gem-dichlorination) in the alkyl chains is predicted to be carried out by the CylC homolog BrtJ. The timing of halogenation in bartoloside biosynthesis in unclear, but it is foreseeable that BrtJ—like CylC—acts on ACP tethered fatty acyl derivatives, i.e., before DAR formation. As for hierridin C (6), bartoloside D (61) is halogenated in the resorcinol ring but no additional halogenase has been found in the genome of S. salina LEGE 06155 that could explain this functionality.51 Intriguingly, BrtB enzymes, found in both brt clusters, are CylK homologs that could alkylate nucleophilic substrates using the mid-chain halogenated positions in the bartolosides as electrophiles. Still, such reactivity is not required to explain the biosynthesis of currently known bartolosides and bartolosides do not have a free C2 position as do known BrtK nucleophiles.8 Apart from the biosynthetic genes, the brt cluster also encodes a DevBCA-like glycolipid export system,70 BrtGHI, which could be responsible for the presence of bartolosides in spent culture medium or for potential incorporation into the outer membrane.51
Fuchs et al. reported7 that in Microcystis sp. PCC 9808, a pair of DarA and DarB homologs were part of a small BGC that also encoded two desaturases, a DevBCA exporter system and an oxygenase (Fig. 8). Taking into consideration such predicted reactivity, the cluster was proposed to encode microcarbonin A (69), produced by a Microcystis sp. strain, although there is no direct experimental evidence supporting this proposal. Hypothetically, the carboxy-CHD resulting from the DarB-mediated condensation of C14 and C12 fatty acyl derivatives is aromatized by the DarA homolog (Fig. 8). Like bartolosides, microcarbonin A BGC also lacks an ACP, suggesting that the fatty-acyl precursors are obtained from primary metabolism. Potentially after DAR formation, the two desaturases likely catalyze the dehydrogenation of the fatty-acid chains, creating the two internal alkyne moieties. A similar alkyne system is found in 70 and these are of considerable biosynthetic interest. The DevBCA transporter, as in the case of bartolosides, suggests export to the outer membrane or to the extracellular medium. The oxygenase has no corresponding homolog in characterized DAR clusters and it is unclear what its role might be in the biosynthesis of 69.
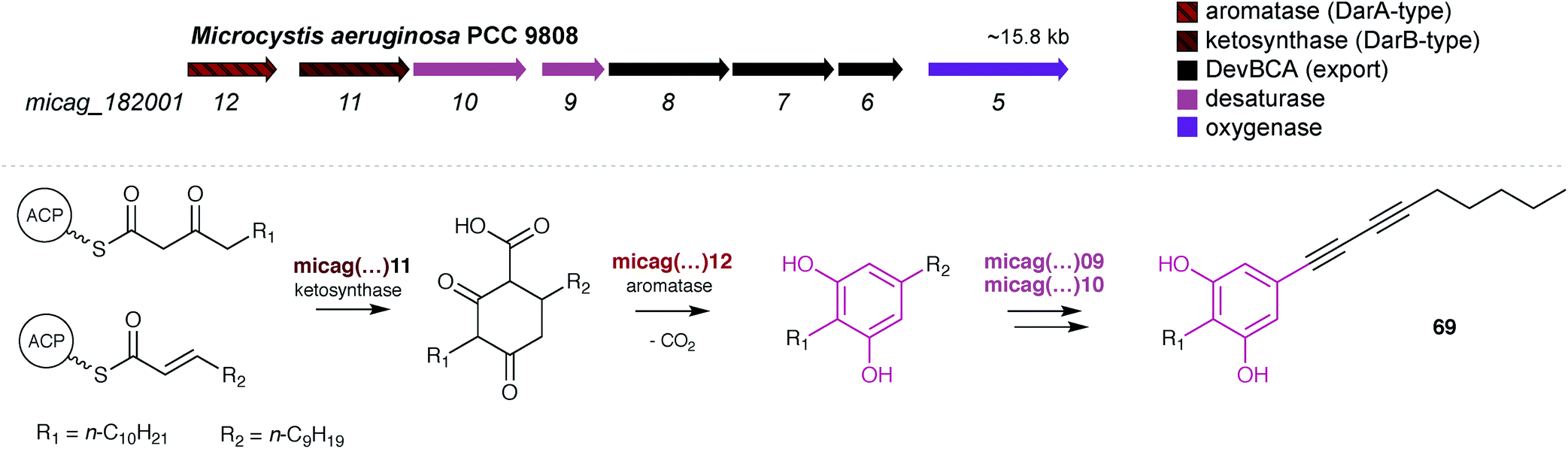 | ||
| Fig. 8 The DAR microcarbonin (69) is hypothesized7 to be produced by a BGC found in Microcystis aeruginosa PCC 9808 (top). The biosynthetic events eventually leading to 69 are shown (bottom). | ||
3.3. Diversity of alkylresorcinol pathways in cyanobacterial genomes
Genomic data for the phylum Cyanobacteria have been increasing over the past years, mostly as a result of a few major sequencing efforts,71–73 but most genera have either none or a single assembly available publicly. As of August 2018, over a thousand cyanobacterial genomes were publicly available in the GenBank database, more than half of which corresponding to the smaller-genome genera Prochlorococcus and Synechococcus picocyanobacteria. This wealth of genomic data can be used to obtain a predictive overview of MAR and DAR diversity (according to currently understood biosynthesis) in the phylum Cyanobacteria. In fact, Nakamura et al.8 have done this exercise for CylC/CylK containing cyanobacterial genomes and found a series of different orphan BGC archetypes that likely correspond to unprecedented natural product classes. Likewise, Costa et al.19 who looked into the distribution of hid-like BGCs in cyanobacteria observed that these are widely distributed in picocyanobacteria. Within the scope of this review, we carried out a distribution analysis of MAR- and DAR-encoding pathways among currently available cyanobacterial amino acid sequence data (mostly from genome-sequencing studies) available in the GenBank. We performed phylogenetic analysis of type III PKSs (Fig. 9) and DarB homologues (Fig. 10) which overall suggest that most scaffolds of MAR and DAR types are still to be uncovered in this phylum.‡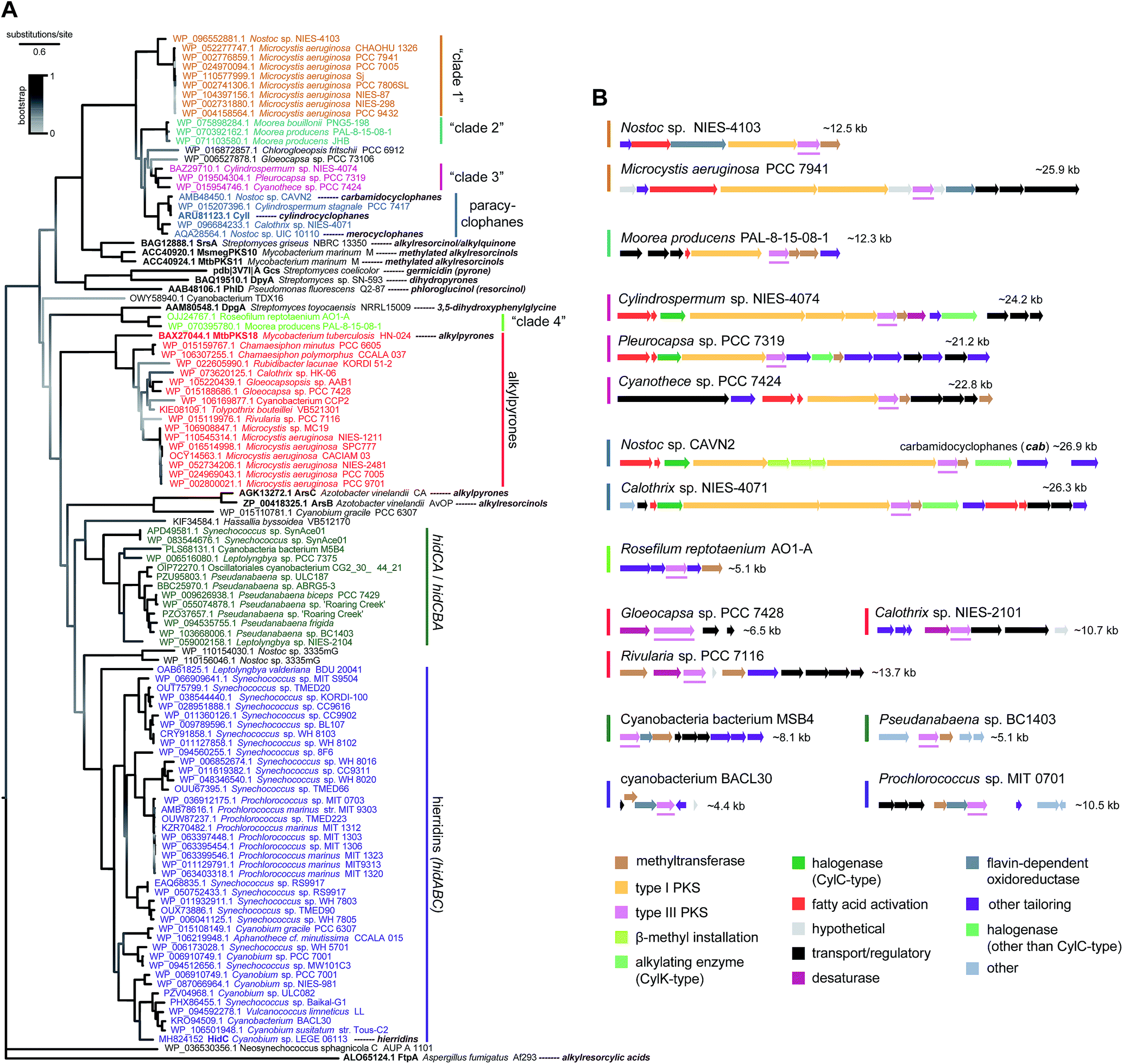 | ||
| Fig. 9 Phylogenetic diversity of type III PKS enzymes in cyanobacteria. (A) Maximum-likelihood tree (JTT + G model) computed‡ from an amino acid alignment containing sequences of type III PKS homologs from cyanobacteria (retrieved from the GenBank) as well as characterized bacterial type III PKSs (shown in bold). (B) – Selected BGCs from each clade (as represented in the phylogenetic tree) of cyanobacterial type III PKSs, the position of the type III PKS-encoding gene within each BGC is underlined. | ||
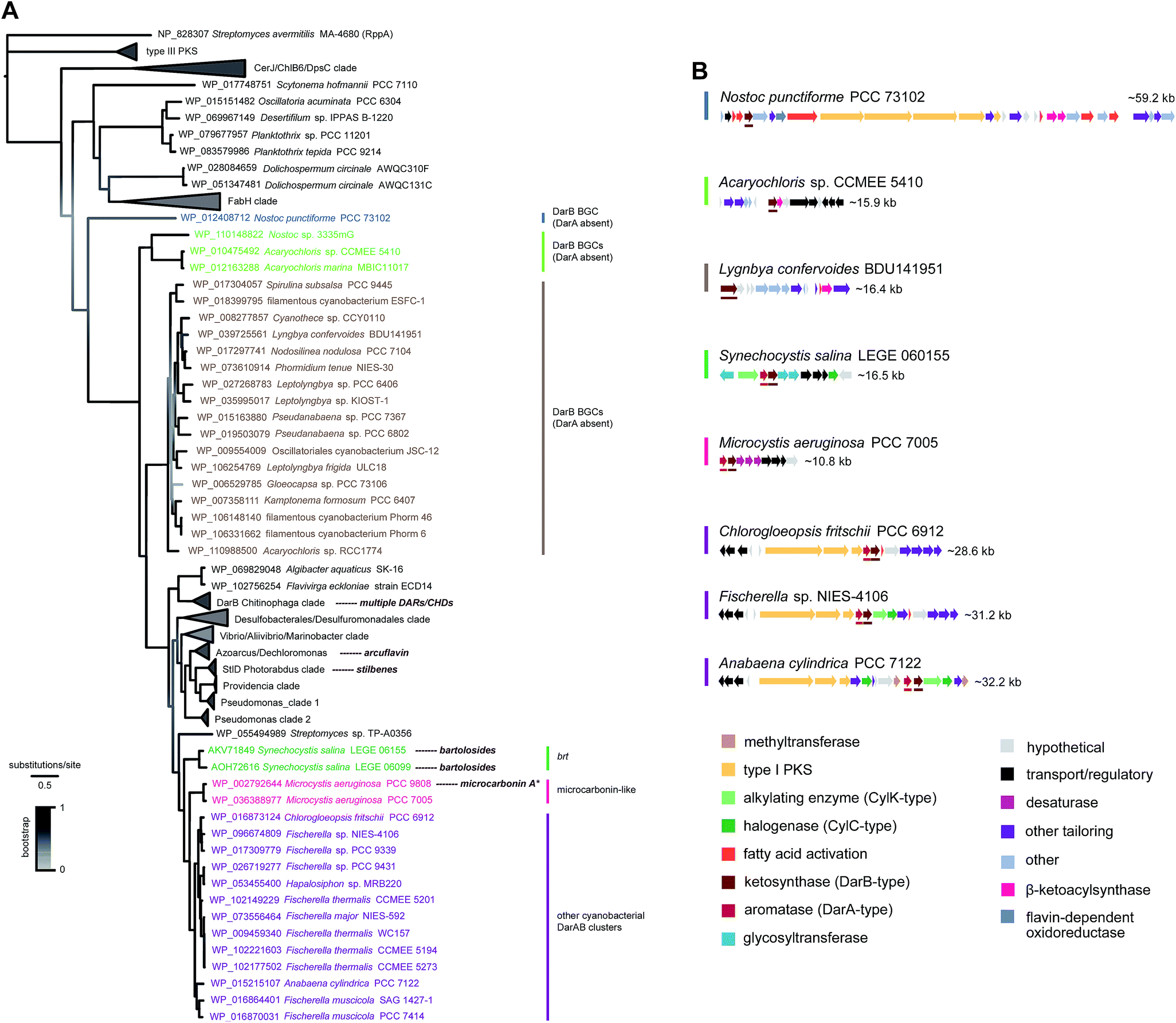 | ||
| Fig. 10 Phylogenetic diversity of BrtD enzymes in cyanobacteria. (A) Maximum-likelihood tree (JTT + G model) computed‡ from an amino acid alignment containing sequences of the top BrtD (DarB) homologs from bacteria (retrieved from the GenBank), characterized DarB homologs (corresponding natural products shown next to taxa), and sequences of other ketosynthases from bacteria (selected based on Fuchs et al.).7 (B) Selected BGCs from each clade depicted in the phylogenetic tree, darA and darB homologs are underlined * the putative microcarbonin A BGC assignment is not supported by direct biochemical evidence. | ||
Type III PKSs in cyanobacteria are distributed among eight well-supported clades (Fig. 9). The majority of these enzymes are HidC homologs and are found among picocyanobacteria (hidABC clade) and other Synechococcales (hidCA/hidCBA clade). These are part of short clusters that include always a methyltransferase homolog of HidA and are likely to correspond exclusively to MAR end-products. The paracyclophane-associated type III PKSs clade together, as expected, and are, in turn, closely-related to three additional clades (clades “1”, “2” and “3”, Fig. 9). One such clade is composed of Microcystis and Nostoc enzymes, and they are part of clusters that include fatty acid activation enzymes and type I PKSs. Three Moorea spp. type III PKSs compose “clade 2” and are part of BGCs encoding type I PKSs and methyltransferases. “Clade 3” is composed of type III PKS homologs from Cylindrospermum, Cyanothece and Pleurocapsa strains. These are encoded within heterogeneous BGCs that share fatty acid activation and type I PKSs. Interestingly, from all the BGCs extracted for the three clades closest to the paracyclophanes clade, only in Cylindrospermum sp. NIES-4074 (“clade 3”) a pair of CylC/CylK homologs was detected, suggesting that all other pathways encode MARs. An additional cyanobacterial type III PKS clade observed in the phylogenetic tree (Fig. 9) is composed only of a Moorea and a Roseofilum strain. These homologs are closely related to the actinobacterial DpgA, that is involved in the biosynthesis of the non-proteinogenic amino acid 3,5-hydroxyphenylglycine,61 and are found in small (∼5 kb) clusters that also encode DpgB (dehydratase that is associated with DpgA). The remaining well-supported clade of cyanobacterial type III PKSs includes homologs of MtbPKS18, that takes part in alkylpyrone biosynthesis in Mycobacterium tuberculosis.74 The potential alkylpyrone-generating cyanobacterial homologs are part of relatively small gene clusters (<15 kb), most of which contain a putative cytochrome b5 (desaturase) adjacent to the type III PKS.
The DarB phylogeny shows a less diverse topology for cyanobacteria, with three related clades with strong bootstrap support that are closely-related and likely involved in DAR biosynthesis since they have adjacent DarA homologs (Fig. 10). One of these clades includes only the two BrtD homologs from S. salina. Another small clade is composed of two DarB homologs from Microcystis aeruginosa that correspond to potential microcarbonin-generating BGCs7 (see also Fig. 8). Considerably more diversity of BGC archetypes is found within the third cluster, which comprises homologs from heterocystous Nostocales and Stigonematales strains. The associated BGCs are relatively large (>25 kb) and encode diverse functionalities including type I PKSs, DevBCA transporters, acetyltransferases and oxidoreductases. Most also include CylC/CylK pairs.8 Interestingly, other cyanobacterial DarB homologs have no DarA homolog in an adjacent position (nor in their immediate genomic context). These DarB homologs are nevertheless evolutionarily more distant from their DarA-associated cyanobacterial counterparts than are those from other bacteria (including the biochemically characterized DarB and StlD), which suggests that their reactivity might be different from DarB and, consequently, that their reaction products may not be carboxy-CHDs. These “DarA-less” DarB enzymes are grouped in two clades (one with homologs from two Acaryochloris sp. strains and a larger that encompasses homologs from a wider variety of genera), as well as a loner Nostoc punctiforme homolog. The latter (Nostoc) homolog is encoded in a large BGC (∼60 kb) that encodes multiple type I PKSs and different fatty acid activating- or modifying functionalities. The BGCs associated with the two clades are more simple and do not contain type I PKSs (Fig. 10).
Overall, the phylogenetic analysis and annotation of the genomic context of cyanobacterial type III PKS and DarB homologs shows that MARs and DARs are produced by a wide range of cyanobacteria, from the simpler picocyanobacteria to the cell-differentiating Nostocales and Stigonematales. For the overwhelming majority of BGCs that encode these MAR and DAR-generating enzymes, we cannot currently assign a natural product. Moreover, these orphan BGCs are diverse in size and encoded functionalities. Still, and as expected, several fatty acid activating and modifying enzymes are present in the BGCs. Interestingly, the presence of CylC/CylK homolog pairs seems punctuated among the phylogenetic clades and among BGCs of similar architecture. Taking into account that these enzyme pairs are involved in dimerization or eventually polymerization, their incorporation in BGCs might result in gain of new functions for the encoded MARs or DARs. Also of considerable interest is the relatively widespread occurrence of DarB homologs in BGCs without an associated aromatase, whose end-products remain to be discovered.
4. Synthetic approaches to cyanobacterial alkylresorcinols
The time-consuming growth culture experiments and low yields obtained for a number of bioactive alkylresorcinols have raised the need for alternative and more effective means to access and study these secondary metabolites. Due to their simple and planar structure, relatively short and simple organic syntheses of the monoalkyl derivatives hierridins have been achieved already in the 1990s. The uniqueness of the [7.7]paracyclophane scaffold has further drawn the attention of the synthetic community. This has culminated in the development of admirable series of reactions towards the preparation of the dimeric macrocycles cylindrocyclophanes A (16) and F (25).4.1. Monoalkylresorcinols (MARs)
Many studies have described the synthesis of natural MARs, employing several approaches to accomplish the critical step of carbon–carbon bond formation between the resorcinol ring and the alkyl side chain. In some cases, additions using a Gilman reagent,75 a Suzuki76 or a Sonogashira3 coupling were adopted; however, Wittig and Grignard reactions are the most typically used.77,78 The few procedures dedicated to the synthesis of hierridins relied on one of the two latter alkylation strategies, using a readily available brominated or methoxylated salicylaldehyde derivative as starting material.In 1998, Jinno and Okita79 reported the total synthesis of the yeast-derived alkylphenol phaffiaol (also known as hierridin A,184, see Section 2.1.1.). To this end, 3,5-dibromosalicylaldehyde (71) was subjected to a Wittig olefination to produce the key intermediate 72 as a 1:1 mixture of E/Z isomers (57% yield). Reduction of the alkenyl phenol 72 was then carried out by hydrogenation, providing the alkyl phenol 73. Finally, the nucleophilic aromatic substitution of the dibromide 73 with sodium methoxide afforded the expected MAR 4 (67% yield from 73) (Scheme 1).
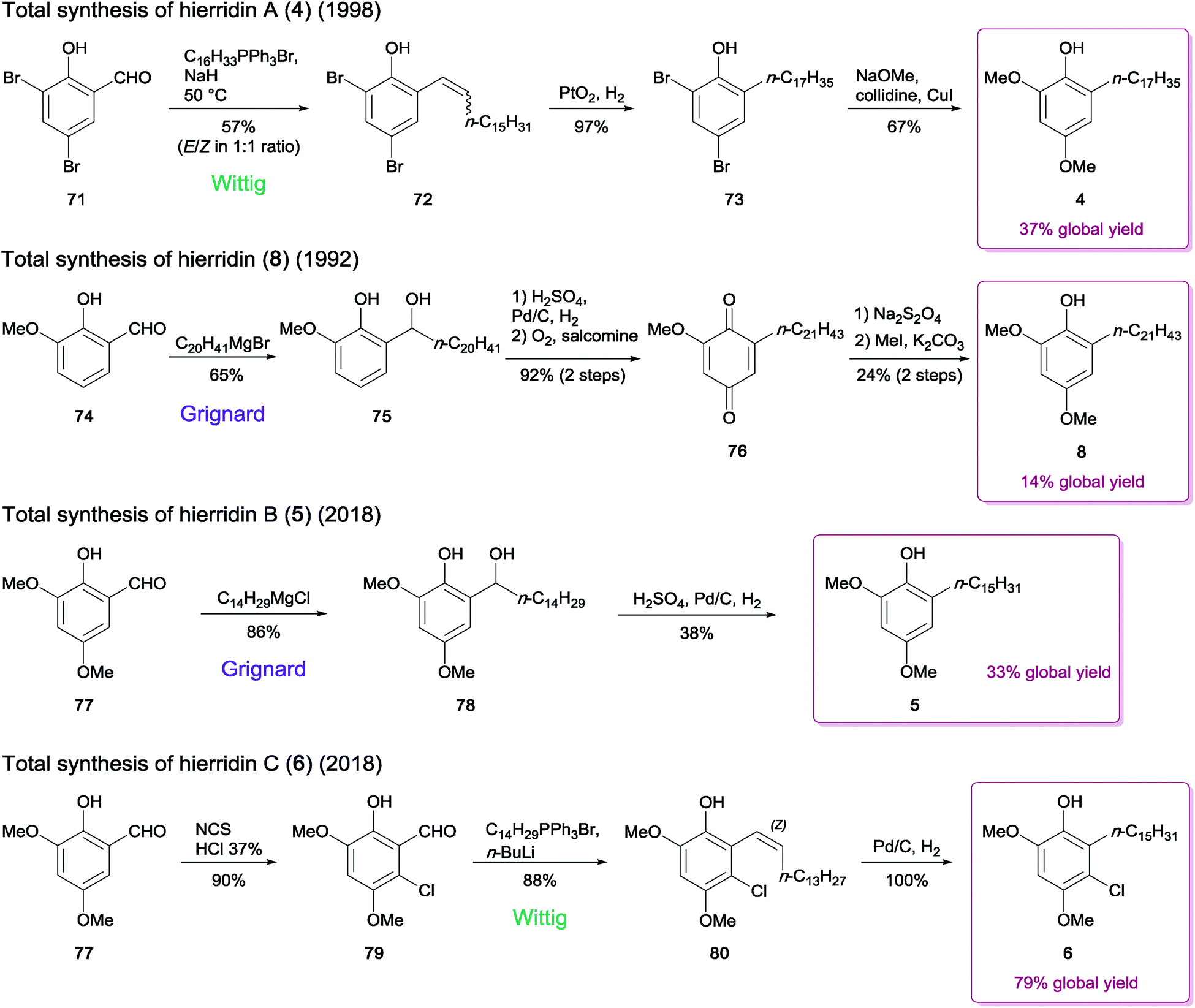 | ||
| Scheme 1 Synthesis of hierridins 4–6 and 8.19,79,80 | ||
The extended chain analogue hierridin (8), isolated from the lichen Ramalina hierrensis, was synthesized in five steps by González and co-workers.80 The phenolic aldehyde precursor o-vanillin (74) was first alkylated in the presence of an n-eicosanyl Grignard reagent to generate compound 75 (yield 65%). Subsequent hydrogenation followed by oxidation in the presence of salcomine under a stream of oxygen provided the quinone intermediate 76, which was further subjected to sodium dithionite reduction to afford the corresponding hydroquinone. The methylation of the sterically most accessible hydroxy group by methyliodide and K2CO3 under reflux for 24 h provided hierridin (8) (Scheme 1).
Costa and co-workers19 recently achieved the total synthesis of hierridin B (5), which contains a 15-carbon length alkyl side chain moiety. In contrast to the synthetic routes described above for hierridins 4 and 8, their method used 2-hydroxy-3,5-dimethoxybenzaldehyde (77) as the initial building block, avoiding a subsequent methoxylation step in position 5. The synthesis was thus achieved in only two steps, via Grignard reaction of 77 to produce the intermediate 78, which was then hydrogenated to afford 5 in moderate yield (38% from 78) (Scheme 1). The authors suggested that the methoxy group in position 5 negatively influences the efficiency of the reduction step, since the product yield obtained was significantly lower than that reported in the synthesis of compound 8 (38% versus > 90%).80
The chlorinated derivative hierridin C (6) has been synthesized concurrently to hierridin B (5).19 Since direct chlorination of 5 in the appropriate position failed, a procedure consisting in monohalogenation of 77 prior to the Grignard reaction was undertaken, but the outcome of the subsequent hydrogenation step was unsuccessful. An alternate synthetic route for the carbon–carbon bond formation was applied, using a similar approach employed for compound 4. This included the regioselective chlorination of 77 to afford the compound 79. Wittig olefination of 79 followed by palladium-catalyzed hydrogenation led to monoalkylresorcinol 6 (Scheme 1).
These strategies provided 14% to 79% overall recovery of hierridins. Despite the efficiency of the Grignard addition, the subsequent reactions leading to hierridins 5 or 8 led to substantial decrease of the global yield. Furthermore, using the precursor 2-hydroxy-3,5-dimethoxybenzaldehyde (77) as starting material enabled shorter synthesis of 5 and 6. Thus it may be worthwhile to revisit the synthesis of hierridins 4, 5 and 8 by involving a Wittig reaction directly on compound 77 to improve the overall yields.
4.2. Dialkylresorcinols (DARs)
Effective synthetic routes to simple natural DARs have been designed.81,82 In these studies, alkylated α,β-unsaturated ketones and diethylmalonate were subjected to Michael addition/Dieckmann condensation followed by saponification/decarboxylation to generate alkylcyclohexane-1,3-diones. Different strategies were then employed to form 2,5-dialkylresorcinols, involving first a 2-alkylation, if necessary, prior to aromatization. However, to the best of our knowledge, the total synthesis of the functionalized cyanobacterial DARs bartolosides and microcarbonin A (69) has not been reported yet.Cyclophanes possess a more complex architecture, with a macrocyclic system representing a rigid cavity of great interest in host–guest chemistry.83 For this reason, their synthesis has been extensively investigated since the 1950s.40,84,85 From a synthetic perspective, closure of the ring core accounts for the most challenging step, which has led to the consideration of multiple strategies for the formation of such key carbon–carbon bonds in cyclophanes.83,84 A few of these strategies have been applied to the total synthesis of compounds from the [7.7]paracyclophane series (Scheme 2). Mascal and co-workers86 proceeded to the synthesis of a diester precursor prior to cyclization in [7.7]paracyclophane via intramolecular acyloin condensation. In an attempt to prepare benzocyclononanone, Schubert et al.87 used high dilution Friedel–Crafts reaction between monoalkylated monomers, which resulted in the fortuitous synthesis of a [7.7]paracyclophanedione (5% yield). Another study by Staab and co-workers88 used bis(dithiane) alkylation for preparation of a tetramethoxy[7.7]paracyclophane-4,17-dione. As for the synthesis of the C2-symmetric cylindrocyclophanes A (16) and F (25), head-to-tail cyclodimerizations were performed via more advanced procedures, furnishing diene macrocycles as intermediates. Smith and co-workers89 as well as Yamakoshi et al.90 employed a tandem cross metathesis/ring-closing metathesis (RCM). Macrocyclic dimerization by a double Horner–Wadsworth–Emmons olefination was reported by Hoye et al.91 while Nicolaou and co-workers92 envisioned the Ramberg-Bäcklund olefination reaction to successfully obtain the [7.7]paracyclophane scaffold leading to the formation of 16 and 25.
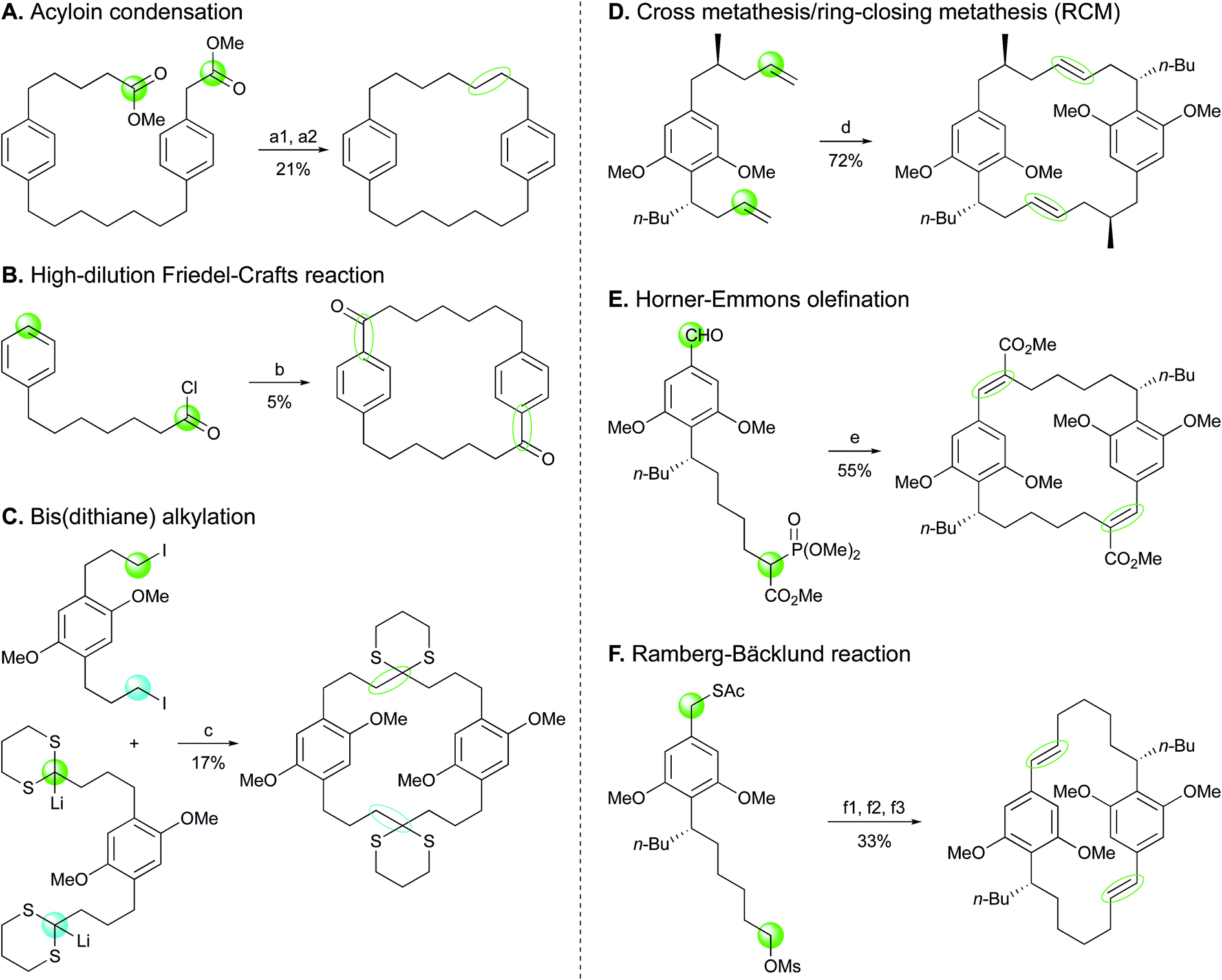 | ||
| Scheme 2 Ring closing approaches implemented in the syntheses of [7.7]paracyclophanes. Reagents and conditions:86–89,91,92 (a1) Na, Me3SiCl, heat; (a2) Zn–Hg, HCl, AcOH, heat; (b) AlCl3; (c) −25 °C to 20 °C, then HCl; (d) schrock cat.; (e) NaH, cat. 15-crown-5; (f1) NaOMe; (f2) H2O2, cat. (NH4)6Mo7O24; (f3) KOH/Al2O3, CF2Br2, 0 °C to 23 °C. | ||
The natural compounds (−)-cylindrocyclophane A (16) and (−)-cylindrocyclophane F (25) possess six and four stereocenters, respectively. Two strategies were used to obtain the desired configurations of (−)-16 and (−)-25. While two groups, Smith et al.89,93 and Yamakoshi and co-workers,90 introduced all stereocenters before the dimerization step, two other groups, Hoye et al.91 and Nicolaou and co-workers92 preferred establishing only the stereogenic center bearing the n-butyl unit prior to macrocyclization. Asymmetric syntheses were therein required at the end of the process to settle the desired stereochemistry for hydroxy- and methyl-bearing carbons.
Smith and co-workers reported the first total synthesis of cylindrocyclophane F (25) in 1999,93 through a stepwise procedure to prepare the dimer structure before closing the macro-ring (Scheme 3). The imide 81 was used as starter unit to construct intermediate 82, ensuring stereocontrol in the initial step introducing the allyl chain. A Danheiser benzannulation between 82 and cyclobutenone 83 and subsequent methylation furnished the resorcinol intermediate 84, which was then converted to iodide 85 and tosyl hydrazone 86. The assembly of 85 and 86 using lithium–halogen exchange followed by Myers reductive coupling led to compound 87 in 73% yield. Conversion of the latter to diene 88 followed by olefin metathesis using a Grubbs ruthenium catalyst provided the paracyclophane 89, which after hydrogenation and removal of the methyl ethers afforded cylindrocyclophane F (25).
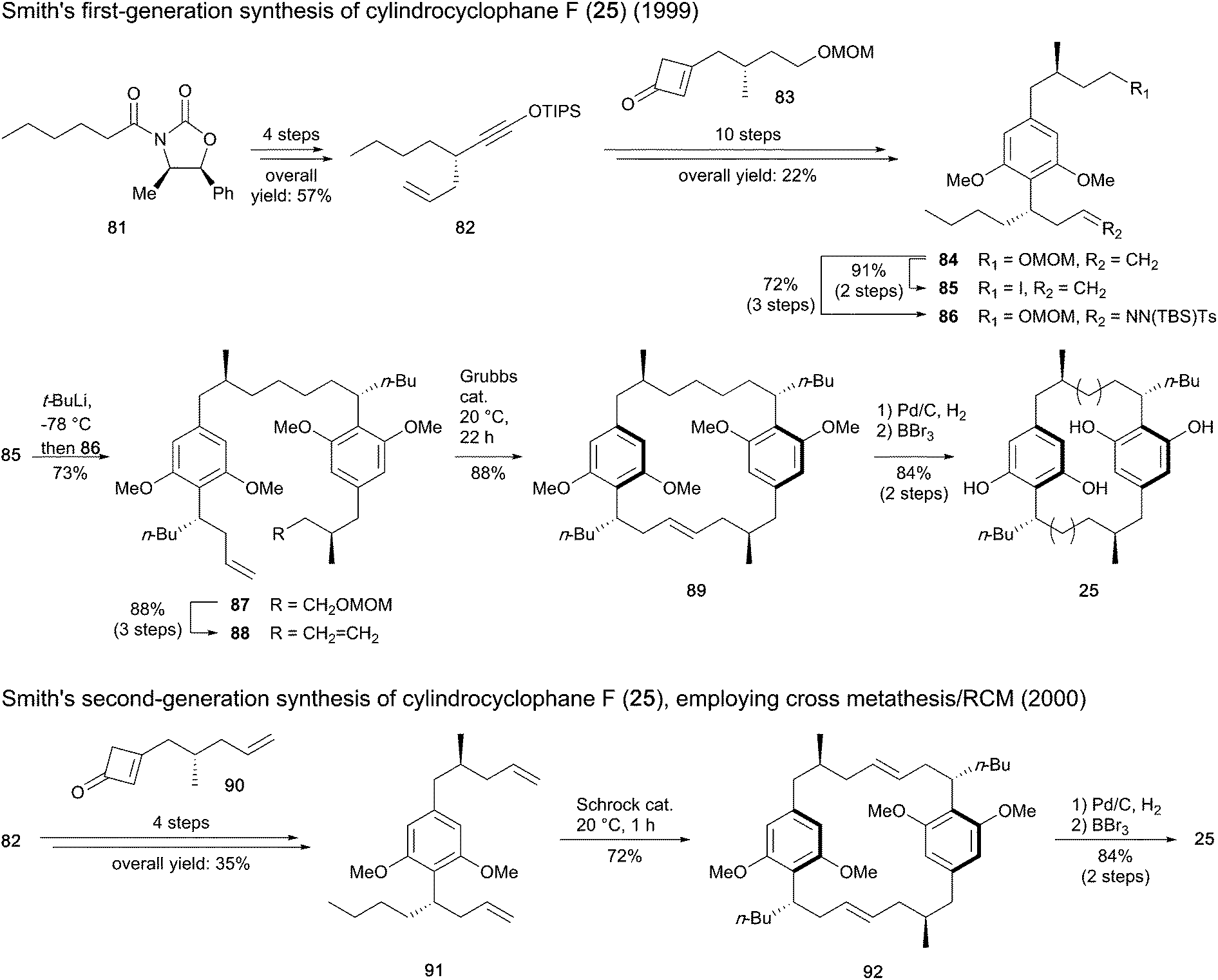 | ||
| Scheme 3 Synthesis of cylindrocyclophane F (25) by Smith and co-workers.89,93,95 | ||
The same authors published a second-generation synthesis of compound 25,89 revisiting the macrocycle assembling strategy through the one-pot cross metathesis/RCM (Scheme 3). To this end, instead of fragment 83, cyclobutenone 90 was synthesized and united to siloxyalkyne 82 to form the paracyclophane monomer 91. For dimerization of 91, substantial improvements in reactivity were found in the presence of Schrock molybdenum catalyst in comparison to Grubbs ruthenium catalysts. Hydrogenation of the diene macrocycle 92 and cleavage of the methyl ethers afforded cyclindrocyclophane F (25). While the first-generation synthesis with 20 steps gave 8.3% overall yield of targeted compound 25, only 11 steps were required with the second method to result in 22% global yield.94
Since the cross olefin metathesis provides the thermodynamically most favored isomer possessing double bonds with trans geometry and seven-carbon atom tethers,95 this reaction was also exploited for the synthesis of the natural hydroxylated analogue 16. A disconnection study revealed a monomer hydroxylated on the side alkyl chain, which was synthesized by Smith and co-workers89 as follows. Stannyl cyclobutenone 93 underwent a Danheiser annulation with compound 82 to provide aryl iodide 94. Addition of the amide 95via lithium–halogen exchange and conversion of the ketone intermediate obtained to the corresponding alcohol in a stereoselective manner provided compound 96 (Scheme 4A). Finally, dimerization of monomer 96 using Schrock catalyst gave the bis(olefin) 97, which afforded cylindrocyclophane A (16) after alcohol deprotection, hydrogenation and ether demethylation (Scheme 4B). An overall yield of 8.1% was recovered from a total of 16 steps.
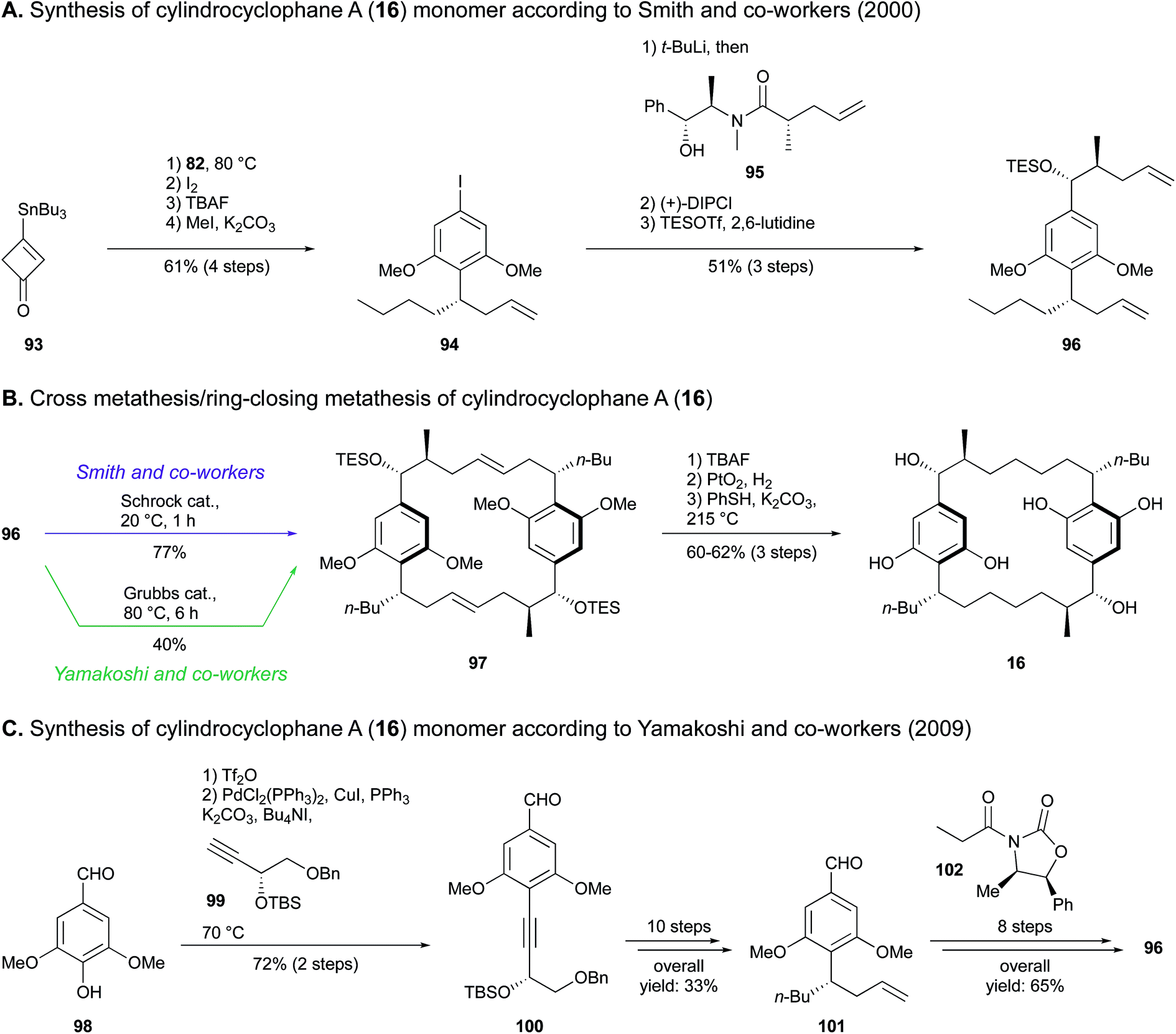 | ||
| Scheme 4 Total synthesis of cylindrocyclophane A (16) by Smith and co-workers (A and B) and Yamakoshi and co-workers (C and B).89,90 | ||
The tandem dimerization-macrocyclization scheme developed by Smith and co-workers in 2000 (ref. 89) was also implemented by Yamakoshi and co-workers in 2009,90 who synthesized both cylindrocyclophane A (16) and its non-natural dextrorotatory enantiomer (Scheme 4C). A Sonogashira coupling of syringaldehyde (98) and alkyne 99 prepared from L-diethyl tartrate resulted in the tetrasubstituted resorcinol 100. In the synthetic route leading to 16, a diastereocontrolled reduction of the alkyne led to an intermediary E-alkene, which was subjected to a Johnson–Claisen rearrangement prior to a Wittig reaction to establish the n-butyl group. This afforded benzaldehyde 101 in ten steps with 33% yield. Within eight subsequent steps, compound 101 underwent a diastereoselective aldolisation in the presence of oxazolidinone 102, followed by extension of the side alkyl chain via Horner–Wadsworth–Emmons olefination to furnish the paracyclophane monomer 96. The final steps were common to the procedure adopted by Smith and co-workers, apart from the choice of ring-closing metathesis catalyst, being a Grubbs second-generation complex (Scheme 4B). Overall, 3.5% yield of compound 16 was obtained in a 20-step synthesis.
Hoye and co-workers91 also used a resorcinol derivative, 4-bromo-3,5-dimethoxybenzyl alcohol (103), as starting material for the total synthesis of cylindrocyclophane A (16) (Scheme 5). The synthetic route targeted the paracyclophane monomer 106, which possesses a seven-carbon length side chain bearing the butyl moiety. To this end, alkene 104 was synthesized in four steps, prior to enzyme-assisted conversion of the allylic alcohol to (R)-acetate 105. A [3,3]-sigmatropic rearrangement followed by reduction of the alkene provided the n-butyl group, while the ester side chain formed was elongated via Horner–Wadsworth–Emmons reaction. Transformation to a phosphono ester and oxidation of the benzylic alcohol furnished the bifunctional monomeric unit 106, which yielded E,E-diene 107 through Horner–Wadsworth–Emmons macrocyclic dimerization. In order to introduce the four stereocenters holding the methyl and the hydroxy groups, the ester groups were converted to methyls and stereoselective hydroboration followed by H2O2 oxidation led to the requisite enantiomer 108 (53% yield over four steps). The final step entailed an efficient demethylation to release cylindrocyclophane A (16). This alternate synthetic route revealed an overall yield of 1.6% obtained in 22 steps. Furthermore, the olefination reaction that used the saturated monomer 106 was found to be stereoselective, in contrast to its (E)-4,5-alkene homologue that led to the formation of a mixture of stereoisomers.
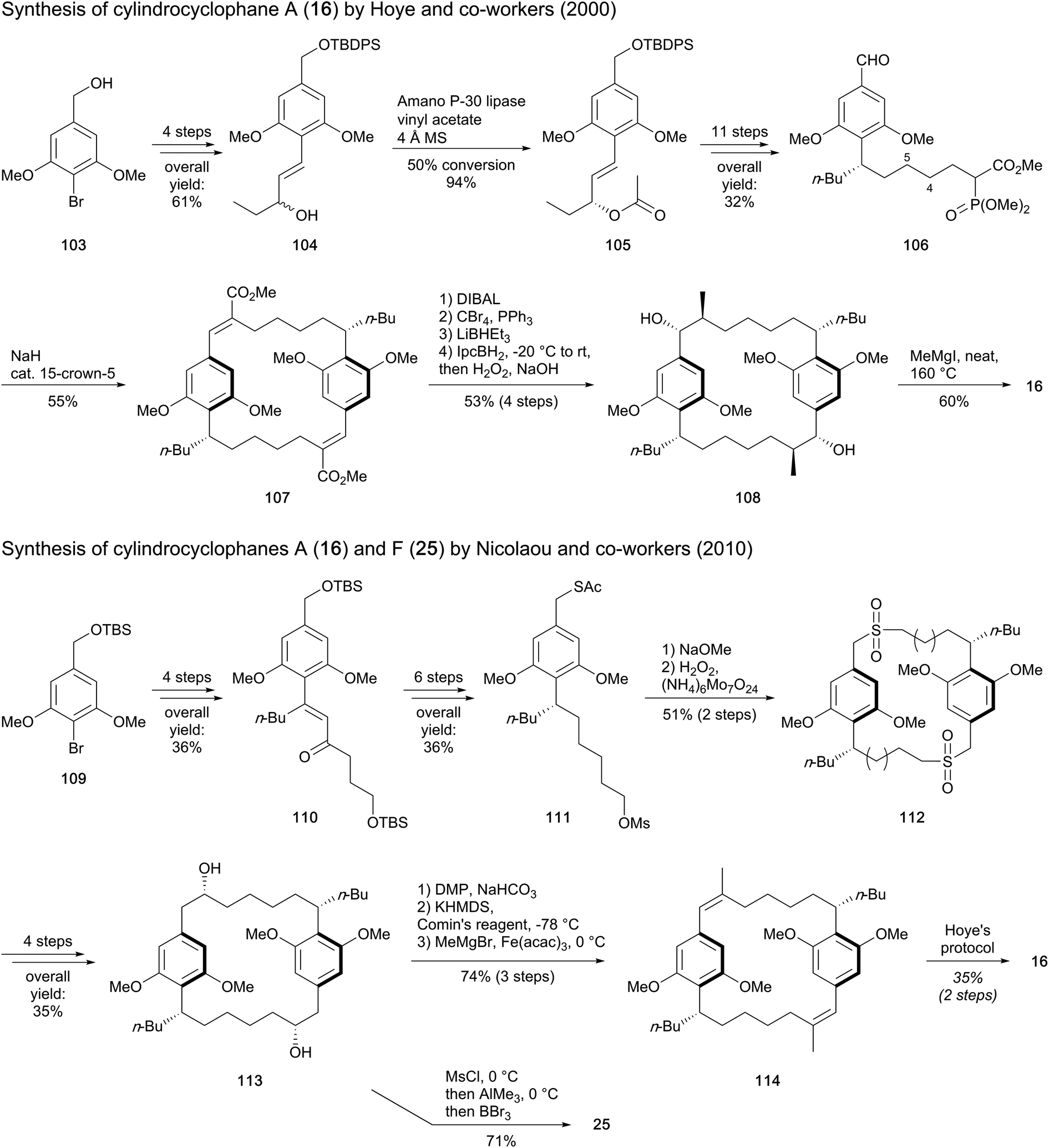 | ||
| Scheme 5 Synthesis of cylindrocyclophane A (16) by Hoye and co-workers and Nicolaou's approach to synthesise cylindrocyclophanes A (16) and F (25).91,92 | ||
Nicolaou and co-workers employed a similar building block to construct the monomer 111 from bromide 109 (Scheme 5).92 Reaction with pentanal and nucleophilic addition of a vinyl lithium followed by allylic transposition-oxidation of the resulting tertiary alcohol provided enone 110. This product (110) bears an n-butyl residue and the five-atom side chain intended to connect the two resorcinol units. Subsequent enantioselective ketone reduction and catalytic olefin hydrogenation established the stereochemistry of the bifunctional monomer precursor 111. The macrocyclization occurred via displacement of the mesylate substituent to generate a (bis)thioether, which was further oxidized to bis(sulfone) 112. With the macrocyclization completed, a Ramberg–Bäcklund reaction of 112 with Chan's modification provided a bis(olefin), which after asymmetric dihydroxylation afforded the macrocyclic diol 113, the common precursor for the synthesis of both cylindrocyclophanes A (16) and F (25). Cylindrocyclophane F (25) was obtained with good yield (71%) via a one-pot sequential reaction including methylation of the side chains and demethylation of the methoxy groups. Concomitantly, the route to cylindrocyclophane A (16) was envisaged through oxidation of alcohol to ketone, enol triflate formation and cross coupling with a Grignard reagent to furnish bis(olefin) 114. Compound 114 was then subjected to hydroboration, H2O2 oxidation and ether demethylation according to Hoye's procedure. Nicolaou's approach gave 1.7% (19 steps) and 0.6% (21 steps) overall yields of paracyclophanes 25 and 16, respectively.
In summary, the syntheses of MARs 4–6 and 8 were based on alkylation of a salicylaldehyde precursor via Wittig or Grignard reaction. The synthetic approaches leading to cylindrocyclophanes 16 and 25 first consisted in the preparation of a tetrasubstituted resorcinol monomer through Danheiser annulation, Sonogashira coupling followed by Johnson–Claisen rearrangement, or asymmetric functionalization. Head-to-tail cyclodimerization then occurred via olefin metathesis, double Horner–Emmons olefination or Ramberg–Bäcklund reaction. Final minor synthetic steps enabled to release or install functional groups. Thus, conventional organic synthesis methods have been employed to prepare the aforementioned compounds (16, 25). In contrast to the assumed biosynthetic pathways, the resorcinol ring was either constructed by reaction of a cyclobutenone and an acetylene, or it was introduced through a commercially available functionalized aromatic derivative. Moreover, the macrocycle assembling step required p-substituted monomers, in contrast to the suggested natural dimerization of the resorcinol ring and the non-functionalized alkyl chain. As an alternative to conventional syntheses of natural products, biomimetic approaches have become popular in the last twenty years, revealing new powerful synthetic routes and enabling to confirm or invalidate biosynthetic hypotheses.96 Such a strategy has, for instance, been applied to the naturally occurring paracyclophane dimer longithorone A.97 Despite the challenging linkage formation at C(7) and C(20) and the configuration of the stereocenters, similar approaches mimicking natural biosynthetic pathways may open up new prospects towards efficient organic synthesis and stereochemical elucidation of cyanobacterial DAR series.
5. Conclusions
Only a small fraction of cyanobacterial alkylresorcinol diversity has been documented, but it is now evident that these structural motifs are present throughout the phylum. In particular, the DAR motif seems to be so relevant for cyanobacterial ecophysiology that two fundamentally different biosynthetic pathways have converged to create it in the same bacterial phylum. This is remarkable since there are limited known examples of mechanistically-distinct natural product biosynthetic pathways that have converged to create the same scaffold, and even then this usually occurs in separate kingdoms.69,98,99 Despite the dissimilar biochemistry involved in creating the DAR moiety through MAR biosynthesis followed by Friedel–Crafts alkylation or by head-to-head condensation of fatty acyl precursors, a number of decorations to the DAR scaffold are installed for both pathways, including internal alkynes, glycosylation and halogenation. Illustrative of this are the pairs of CylC and CylK homologs that are found in a considerable diverse set of cyanobacterial gene clusters of DarAB but also type III PKS biosynthetic origin. These shared features likely reflect evolutionary pressure associated with the DAR skeleton in cyanobacteria and are probably reminiscent of related ecological roles. Still, and contrary to what is known for MARs and DARs produced by other bacteria15,100 current understanding of the functions that cyanobacterial alkylresorcinols may play in the natural setting is minimal, although many have potent biological activity in pharmacologically-relevant assays. The uncovered diversity of alkylresorcinols in cyanobacteria represents an opportunity to perform genome-guided isolation efforts that have the potential to reveal new compound classes and important bioactivities; the study of their ecological roles will nevertheless remain a challenge.6. Conflicts of interest
There are no conflicts to declare.7. Acknowledgements
This work was funded by the BLUEandGREEN project (H2020 TWINNING, Grant Agreement no. 692419) and was further supported by an European Research Council (ERC) Starting Grant (759840) to PNL and National Science Foundation (NSF) through grant CHE-1454007 to EPB. We acknowledge funding by Fundação para a Ciência e a Tecnologia (FCT) through projects PTDC/BIA-BQM/29710/2017 (co-financed by COMPETE 2020, Portugal 2020, ERDF) UID/Multi/04423/2013, as well as through scholarship SFRH/BD116009/2016 to SF and grant IF/01358/2014 to PNL.8. Notes and references
- A. Kozubek and J. H. P. Tyman, Chem. Rev., 1999, 99, 1–26 CrossRef CAS PubMed.
- M. Stasiuk and A. Kozubek, Cell. Mol. Life Sci., 2010, 67, 841–860 CrossRef CAS PubMed.
- H. Kikuchi, I. Ito, K. Takahashi, H. Ishigaki, K. Iizumi, Y. Kubohara and Y. Oshima, J. Nat. Prod., 2017, 80, 2716–2722 CrossRef CAS PubMed.
- M. B. Austin and J. P. Noel, Nat. Prod. Rep., 2003, 20, 79–110 RSC.
- S. R. Baerson, J. Schröder, D. Cook, A. M. Rimando, Z. Pan, F. E. Dayan, B. P. Noonan and S. O. Duke, Plant Signaling Behav., 2010, 5, 1286–1289 CrossRef CAS PubMed.
- H. Nakamura, H. A. Hamer, G. Sirasani and E. P. Balskus, J. Am. Chem. Soc., 2012, 134, 18518–18521 CrossRef CAS PubMed.
- S. W. Fuchs, K. A. J. Bozhüyük, D. Kresovic, F. Grundmann, V. Dill, A. O. Brachmann, N. R. Waterfield and H. B. Bode, Angew. Chem., Int. Ed., 2013, 52, 4108–4112 CrossRef CAS PubMed.
- H. Nakamura, E. E. Schultz and E. P. Balskus, Nat. Chem. Biol., 2017, 13, 916–921 CrossRef CAS PubMed.
- J.-Z. Deng, S. R. Starck and S. M. Hecht, J. Nat. Prod., 1999, 62, 477–480 CrossRef CAS PubMed.
- Y.-M. Gao, T.-Y. Sun, M. Ma, G.-D. Chen, Z.-Q. Zhou, C.-X. Wang, D. Hu, L.-G. Chen, X.-S. Yao and H. Gao, Fitoterapia, 2016, 112, 254–259 CrossRef CAS PubMed.
- N. Tsuge, M. Mizokami, S. Imai, A. Shimazu and H. Seto, J. Antibiot., 1992, 45, 886–891 CrossRef CAS PubMed.
- A. Kozubek, S. Pietr and A. Czerwonka, J. Bacteriol., 1996, 178, 4027–4030 CrossRef CAS PubMed.
- T. Hayashi, Y. Kitamura, N. Funa, Y. Ohnishi and S. Horinouchi, ChemBioChem, 2011, 12, 2166–2176 CrossRef CAS PubMed.
- R. N. Reusch and H. L. Sadoff, J. Bacteriol., 1979, 139, 6 Search PubMed.
- R. N. Reusch and H. L. Sadoff, Nature, 1983, 302, 268–270 CrossRef CAS PubMed.
- N. Funa, H. Ozawa, A. Hirata and S. Horinouchi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 6356–6361 CrossRef CAS PubMed.
- M. Funabashi, N. Funa and S. Horinouchi, J. Biol. Chem., 2008, 283, 13983–13991 CrossRef CAS PubMed.
- O. Papendorf, G. M. König and A. D. Wright, Phytochemistry, 1998, 49, 2383–2386 CrossRef CAS PubMed.
- M. Costa, I. E. Sampaio-Dias, R. Castelo-Branco, H. Scharfenstein, R. Rezende de Castro, A. Silva, M. P. C. Schneider, M. J. Araújo, R. Martins, V. F. Domingues, F. Nogueira, V. M. Vasconcelos and P. N. Leão, ChemRxiv, 2018 DOI:10.26434/chemrxiv.7090526.v1.
- M. Preisitsch, T. H. J. Niedermeyer, S. E. Heiden, I. Neidhardt, J. Kumpfmüller, M. Wurster, K. Harmrolfs, C. Wiesner, H. Enke, R. Müller and S. Mundt, J. Nat. Prod., 2016, 79, 106–115 CrossRef CAS PubMed.
- J. D. Bu'Lock and A. T. Hudson, J. Chem. Soc. C, 1969, 61–63 RSC.
- M. Kumagai, Y. Suhara, T. Aoyagi and H. Umezawa, J. Antibiot., 1971, 24, 870–875 CrossRef CAS PubMed.
- A. G. González, J. B. Barrera, E. M. R. Perez and C. E. H. Padron, Planta Med., 1992, 58, 214–218 CrossRef PubMed.
- W. Horper and F. J. Marner, Nat. Prod. Lett., 1995, 6, 163–170 CrossRef CAS.
- S. Jinno, K. Hata, N. Shimidzu and T. Okita, J. Antibiot., 1998, 51, 508–511 CrossRef CAS PubMed.
- P. N. Leão, M. Costa, V. Ramos, A. R. Pereira, V. C. Fernandes, V. F. Domingues, W. H. Gerwick, V. M. Vasconcelos and R. Martins, PLoS One, 2013, 8, e69562 CrossRef PubMed.
- S. Freitas, R. Martins, M. Costa, P. N. Leão, R. Vitorino, V. Vasconcelos and R. Urbatzka, Mar. Drugs, 2016, 14, 158 CrossRef PubMed.
- H. Kikuchi, I. Ito, K. Takahashi, H. Ishigaki, K. Iizumi, Y. Kubohara and Y. Oshima, J. Nat. Prod., 2017, 80, 2716–2722 CrossRef CAS PubMed.
- M. Berks, D. Traynor, I. Carrin, R. H. Insall and R. R. Kay, Dev. Suppl., 1991, 1, 131–139 CAS.
- A. Dubois, C. Ginet, N. Furstoss, A. Belaid, M. A. Hamouda, W. El Manaa, T. Cluzeau, S. Marchetti, J. E. Ricci, A. Jacquel, F. Luciano, M. Driowya, R. Benhida, P. Auberger and G. Robert, OncoTargets Ther., 2016, 7, 26120–26136 Search PubMed.
- H. Reichenbach, H. Kleinig and H. Achenbach, Arch. Microbiol., 1974, 101, 131–144 CrossRef CAS.
- T. Kitahara and N. Kanda, J. Antibiot., 1975, 28, 943–946 CrossRef CAS PubMed.
- H. Reichenbach, W. Kohl, A. Böttger-Vetter and H. Achenbach, Arch. Microbiol., 1980, 126, 291–293 CrossRef CAS.
- S. Kato, K. Shindo, H. Kawai, M. Matsuoka and J. Mochizuki, J. Antibiot., 1993, 46, 1024–1026 CrossRef CAS PubMed.
- A. Pohanka, J. Levenfors and A. Broberg, J. Nat. Prod., 2006, 69, 654–657 CrossRef CAS PubMed.
- T. A. Schöner, S. W. Fuchs, B. Reinhold-Hurek and H. B. Bode, PLoS One, 2014, 9, e90922 CrossRef PubMed.
- T. A. Schöner, D. Kresovic and H. B. Bode, Appl. Microbiol. Biotechnol., 2015, 99, 8323–8328 CrossRef PubMed.
- B. S. Moore, J. L. Chen, G. M. L. Patterson, R. E. Moore, L. S. Brinen, Y. Kato and J. Clardy, J. Am. Chem. Soc., 1990, 112, 4061–4063 CrossRef CAS.
- B. S. Moore, J.-L. Chen, G. M. L. Patterson and R. E. Moore, Tetrahedron, 1992, 48, 3001–3006 CrossRef CAS.
- D. J. Cram and H. Steinberg, J. Am. Chem. Soc., 1951, 73, 5691–5704 CrossRef CAS.
- Modern Cyclophane Chemistry, ed. R. Gleiter and H. Hopf, Wiley-Blackwell, 2005, pp. i–xx Search PubMed.
- G. E. Chlipala, M. Sturdy, A. Krunic, D. D. Lantvit, Q. Shen, K. Porter, S. M. Swanson and J. Orjala, J. Nat. Prod., 2010, 73, 1529–1537 CrossRef CAS PubMed.
- J. L. Chen, R. E. Moore and G. M. L. Patterson, J. Org. Chem., 1991, 56, 4360–4364 CrossRef CAS.
- H. T. N. Bui, R. Jansen, H. T. L. Pham and S. Mundt, J. Nat. Prod., 2007, 70, 499–503 CrossRef CAS PubMed.
- S. Luo, H.-S. Kang, A. Krunic, G. E. Chlipala, G. Cai, W.-L. Chen, S. G. Franzblau, S. M. Swanson and J. Orjala, Tetrahedron Lett., 2014, 55, 686–689 CrossRef CAS PubMed.
- M. Preisitsch, K. Harmrolfs, H. T. Pham, S. E. Heiden, A. Füssel, C. Wiesner, A. Pretsch, M. Swiatecka-Hagenbruch, T. H. Niedermeyer, R. Müller and S. Mundt, J. Antibiot., 2015, 68, 165–177 CrossRef CAS PubMed.
- H.-S. Kang, B. D. Santarsiero, H. Kim, A. Krunic, Q. Shen, S. M. Swanson, H. Chai, A. D. Kinghorn and J. Orjala, Phytochemistry, 2012, 79, 109–115 CrossRef CAS PubMed.
- D. S. May, W.-L. Chen, D. D. Lantvit, X. Zhang, A. Krunic, J. E. Burdette, A. Eustaquio and J. Orjala, J. Nat. Prod., 2017, 80, 1073–1080 CrossRef CAS PubMed.
- D. S. May, H.-S. Kang, B. D. Santarsiero, A. Krunic, Q. Shen, J. E. Burdette, S. M. Swanson and J. Orjala, J. Nat. Prod., 2018, 81, 572–578 CrossRef CAS PubMed.
- M. Shaaban, R. P. Maskey, I. Wagner-Döbler and H. Laatsch, J. Nat. Prod., 2002, 65, 1660–1663 CrossRef CAS.
- P. N. Leão, H. Nakamura, M. Costa, A. R. Pereira, R. Martins, V. Vasconcelos, W. H. Gerwick and E. P. Balskus, Angew. Chem., Int. Ed., 2015, 54, 11063–11067 CrossRef PubMed.
- T. B. Afonso, M. S. Costa, R. Rezende de Castro, S. Freitas, A. Silva, M. P. C. Schneider, R. Martins and P. N. Leão, J. Nat. Prod., 2016, 79, 2504–2513 CrossRef CAS PubMed.
- D. Beresovsky, O. Hadas, A. Livne, A. Sukenik, A. Kaplan and S. Carmeli, Isr. J. Chem., 2006, 46, 79–87 CrossRef CAS.
- A. Ploutno and S. Carmeli, J. Nat. Prod., 2000, 63, 1524–1526 CrossRef CAS.
- B. Shen, Hindra, X. Yan, T. Huang, H. Ge, D. Yang, Q. Teng, J. D. Rudolf and J. R. Lohman, Bioorg. Med. Chem. Lett., 2015, 25, 9–15 CrossRef CAS PubMed.
- Y. Shimizu, H. Ogata and S. Goto, ChemBioChem, 2017, 18, 50–65 CrossRef CAS PubMed.
- T.-L. Li, O. W. Choroba, H. Hong, D. H. Williams and J. B. Spencer, Chem. Commun., 2001, 20, 2156–2157 RSC.
- D. Yu, F. Xu, J. Zeng and J. Zhan, IUBMB Life, 2012, 64, 285–295 CrossRef CAS PubMed.
- A. Parvez, S. Giri, G. R. Giri, M. Kumari, R. Bisht and P. Saxena, Sci. Rep., 2018, 8, 6529 CrossRef PubMed.
- C. Nakano, H. Ozawa, G. Akanuma, N. Funa and S. Horinouchi, J. Bacteriol., 2009, 191, 4916–4923 CrossRef CAS PubMed.
- V. Pfeifer, G. J. Nicholson, J. Ries, J. Recktenwald, A. B. Schefer, R. M. Shawky, J. Schröder, W. Wohlleben and S. Pelzer, J. Biol. Chem., 2001, 276, 38370–38377 CrossRef CAS PubMed.
- S. C. Bobzin and R. E. Moore, Tetrahedron, 1993, 49, 7615–7626 CrossRef CAS.
- H. Nakamura and E. P. Balskus, Synlett, 2013, 24, 1464–1470 CrossRef CAS.
- M. Preisitsch, S. Heiden, M. Beerbaum, T. Niedermeyer, M. Schneefeld, J. Herrmann, J. Kumpfmüller, A. Thürmer, I. Neidhardt, C. Wiesner, R. Daniel, R. Müller, F.-C. Bange, P. Schmieder, T. Schweder and S. Mundt, Mar. Drugs, 2016, 14, 21 CrossRef PubMed.
- P. K. Sydor, S. M. Barry, O. M. Odulate, F. Barona-Gomez, S. W. Haynes, C. Corre, L. Song and G. L. Challis, Nat. Chem., 2011, 3, 388–392 CrossRef CAS PubMed.
- U. Sankawa, H. Shimada and K. Yamasaki, Chem. Pharm. Bull., 1981, 29, 3601–3605 CrossRef CAS.
- B. Nowak-Thompson, P. E. Hammer, D. S. Hill, J. Stafford, N. Torkewitz, T. D. Gaffney, S. T. Lam, I. Molnár and J. M. Ligon, J. Bacteriol., 2003, 185, 860–869 CrossRef CAS PubMed.
- S. von Berlepsch, H.-H. Kunz, S. Brodesser, P. Fink, K. Marin, U.-I. Flügge and M. Gierth, Plant Physiol., 2012, 159, 606–617 CrossRef CAS PubMed.
- T. Mori, T. Awakawa, K. Shimomura, Y. Saito, D. Yang, H. Morita and I. Abe, Cell Chem. Biol., 2016, 23, 1468–1479 CrossRef CAS PubMed.
- G. Fiedler, M. Arnold, S. Hannus and I. Maldener, Mol. Microbiol., 1998, 27, 1193–1202 CrossRef CAS PubMed.
- P. M. Shih, D. Wu, A. Latifi, S. D. Axen, D. P. Fewer, E. Talla, A. Calteau, F. Cai, N. Tandeau de Marsac, R. Rippka, M. Herdman, K. Sivonen, T. Coursin, T. Laurent, L. Goodwin, M. Nolan, K. W. Davenport, C. S. Han, E. M. Rubin, J. A. Eisen, T. Woyke, M. Gugger and C. A. Kerfeld, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 1053–1058 CrossRef CAS PubMed.
- T. Dagan, M. Roettger, K. Stucken, G. Landan, R. Koch, P. Major, S. B. Gould, V. V. Goremykin, R. Rippka, N. Tandeau de Marsac, M. Gugger, P. J. Lockhart, J. F. Allen, I. Brune, I. Maus, A. Pühler and W. F. Martin, Genome Biol. Evol., 2013, 5, 31–44 CrossRef PubMed.
- S. J. Biller, P. M. Berube, J. W. Berta-Thompson, L. Kelly, S. E. Roggensack, L. Awad, K. H. Roache-Johnson, H. Ding, S. J. Giovannoni, G. Rocap, L. R. Moore and S. W. Chisholm, Sci. Data, 2014, 1, 140034 CrossRef CAS PubMed.
- P. Saxena, G. Yadav, D. Mohanty and R. S. Gokhale, J. Biol. Chem., 2003, 278, 44780–44790 CrossRef CAS PubMed.
- S. Combes and J.-P. Finet, Synth. Commun., 1997, 27, 3769–3778 CrossRef CAS.
- A. Fürstner and G. Seidel, J. Org. Chem., 1997, 62, 2332–2336 CrossRef.
- Y. Zhu, D. N. Soroka and S. Sang, J. Agric. Food Chem., 2012, 60, 8624–8631 CrossRef CAS PubMed.
- M. Ortega, J. Pantoja, C. de los Reyes, E. Zubía, M. J. Ortega, J. J. Pantoja, C. de los Reyes and E. Zubía, Mar. Drugs, 2017, 15, 344 CrossRef PubMed.
- S. Jinno and T. Okita, Chem. Pharm. Bull., 1998, 46, 1688–1694 CrossRef CAS.
- A. G. González, J. Bermejo Barrera and E. M. Rodriguez Perez, Phytochemistry, 1992, 31, 1436–1439 CrossRef.
- M. Kronenwerth, C. Dauth, M. Kaiser, I. Pemberton and H. B. Bode, Eur. J. Org. Chem., 2014, 2014, 8026–8028 CrossRef CAS.
- R. Madhavachary and D. B. Ramachary, Eur. J. Org. Chem., 2014, 2014, 7317–7323 CrossRef CAS.
- T. Gulder and P. S. Baran, Nat. Prod. Rep., 2012, 29, 899–934 RSC.
- S. Kotha, M. E. Shirbhate and G. T. Waghule, Beilstein J. Org. Chem., 2015, 11, 1274–1331 CrossRef CAS PubMed.
- P. G. Ghasemabadi, T. Yao and G. J. Bodwell, Chem. Soc. Rev., 2015, 44, 6494–6518 RSC.
- M. Mascal, J.-L. Kerdelhué, A. S. Batsanov and M. J. Begley, J. Chem. Soc., Perkin Trans. 1, 1996,(1), 1141–1151 RSC.
- W. M. Schubert, W. A. Sweeney and H. K. Latourette, J. Am. Chem. Soc., 1954, 76, 5462–5466 CrossRef CAS.
- H. A. Staab, G. Matzke and C. Krieger, Chem. Ber., 1987, 120, 89–91 CrossRef CAS.
- A. B. Smith, S. A. Kozmin, C. M. Adams and D. V. Paone, J. Am. Chem. Soc., 2000, 122, 4984–4985 CrossRef CAS.
- H. Yamakoshi, F. Ikarashi, M. Minami, M. Shibuya, T. Sugahara, N. Kanoh, H. Ohori, H. Shibata and Y. Iwabuchi, Org. Biomol. Chem., 2009, 7, 3772–3781 RSC.
- T. R. Hoye, P. E. Humpal and B. Moon, J. Am. Chem. Soc., 2000, 122, 4982–4983 CrossRef CAS.
- K. C. Nicolaou, Y.-P. Sun, H. Korman and D. Sarlah, Angew. Chem., Int. Ed., 2010, 49, 5875–5878 CrossRef CAS PubMed.
- A. B. Smith, S. A. Kozmin and D. V. Paone, J. Am. Chem. Soc., 1999, 121, 7423–7424 CrossRef CAS.
- A. B. Smith, C. M. Adams, S. A. Kozmin and D. V. Paone, J. Am. Chem. Soc., 2001, 123, 5925–5937 CrossRef CAS PubMed.
- A. B. Smith, C. M. Adams and S. A. Kozmin, J. Am. Chem. Soc., 2001, 123, 990–991 CrossRef CAS PubMed.
- M. Razzak and J. K. D. Brabander, Nat. Chem. Biol., 2011, 7, 865–875 CrossRef CAS PubMed.
- M. E. Layton, C. A. Morales and M. D. Shair, J. Am. Chem. Soc., 2002, 124, 773–775 CrossRef CAS PubMed.
- R. S. Nett, M. Montanares, A. Marcassa, X. Lu, R. Nagel, T. C. Charles, P. Hedden, M. C. Rojas and R. J. Peters, Nat. Chem. Biol., 2017, 13, 69–74 CrossRef CAS PubMed.
- A. C. Huang, S. A. Kautsar, Y. J. Hong, M. H. Medema, A. D. Bond, D. J. Tantillo and A. Osbourn, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E6005–E6014 CrossRef CAS PubMed.
- S. Brameyer, D. Kresovic, H. B. Bode and R. Heermann, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 572–577 CrossRef CAS PubMed.
Footnotes |
| † Current address: Univ. de Bordeaux, UFR des Sciences Pharmaceutiques, Unité de recherche Œnologie, EA 4577, USC 1366 INRA, ISVV, Villenave d'Ornon, France. |
| ‡ MEGA X software (URL: www.megasoftware.net/) was used to create amino acid sequence alignments (MUSCLE), phylogenetic model evaluation and ML phylogenetic tree construction. |
| This journal is © The Royal Society of Chemistry 2019 |