
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metabolic functions of the human gut microbiota: the role of metalloenzymes
Lauren J.
Rajakovich
 and
Emily P.
Balskus
*
and
Emily P.
Balskus
*
Department of Chemistry and Chemical Biology, Harvard University, USA. E-mail: balskus@chemistry.harvard.edu
First published on 19th November 2018
Abstract
Covering: up to the end of 2017
The human body is composed of an equal number of human and microbial cells. While the microbial community inhabiting the human gastrointestinal tract plays an essential role in host health, these organisms have also been connected to various diseases. Yet, the gut microbial functions that modulate host biology are not well established. In this review, we describe metabolic functions of the human gut microbiota that involve metalloenzymes. These activities enable gut microbial colonization, mediate interactions with the host, and impact human health and disease. We highlight cases in which enzyme characterization has advanced our understanding of the gut microbiota and examples that illustrate the diverse ways in which metalloenzymes facilitate both essential and unique functions of this community. Finally, we analyze Human Microbiome Project sequencing datasets to assess the distribution of a prominent family of metalloenzymes in human-associated microbial communities, guiding future enzyme characterization efforts.
 Lauren J. Rajakovich | Lauren Rajakovich received her BS in chemistry from Wake Forest University and her PhD in biochemistry from Pennsylvania State University. Her graduate work in the joint laboratory of J. Martin Bollinger, Jr. and Carsten Krebs focused on mechanistic characterization of non-heme diiron oxidases and oxygenases involved in microbial biofuel production and organophosphonate metabolism. She is currently a Helen Hay Whitney postdoctoral fellow with Emily Balskus at Harvard University. Her research focuses on transformations of dietary components by the human gut microbiota that have been implicated in host disease. |
 Emily P. Balskus | Emily Balskus received her BA from Williams College and her PhD in synthetic organic chemistry from Harvard University, where she worked with Eric Jacobsen. Her postdoctoral studies at Harvard Medical School with Christopher T. Walsh focused on deciphering the biosynthesis of cyanobacterial sunscreens. She began her independent career at Harvard University in 2011 and is now a Professor of Chemistry and Chemical Biology. Her research program explores the intersection of organic chemistry, biochemistry, and microbiology, with a major focus on understanding metabolic activities of the human microbiota and their contributions to host health and disease. |
1 Introduction
The human gastrointestinal (GI) tract harbors a unique and complex microbial ecosystem consisting of trillions of bacteria, fungi, archaea, and viruses. This microbial community inhabits our gut from infancy and plays a critical role in the development and maintenance of healthy human physiology. It aids in developing the innate and adaptive immune systems,1,2 provides nutrients and vitamins,3 and protects against pathogen invasion.4 Counter to its vital role in normal physiology, the gut microbiota has also been linked to a wide range of human diseases.5 Unsurprisingly, many of these conditions are localized to the gastrointestinal tract, including colorectal cancer6 and inflammatory bowel diseases (IBD)7 like Crohn's disease and ulcerative colitis. However, the gut microbiota has also been connected with systemic diseases, such as obesity and diabetes,8 and with maladies of distal organs, including neurological and cardiovascular disorders.9,10The intriguing relationship between the gut microbiota and its host elicits fundamental questions about the composition of this community in both health and disease states, the gut microbial functions that influence host biology, and ultimately, the ways that this knowledge can be leveraged to improve human health. Over the last decade and a half, the application of next-generation sequencing has significantly advanced the field toward addressing the first point.11 Taxonomic profiling of gut microbial communities has shown that only two bacterial phyla, Bacteroidetes and Firmicutes, typically dominate this habitat in healthy western populations.12,13 However, the bacteria within these two phyla that constitute the community have been found to vary greatly in healthy populations depending on geography, age, and environmental factors.14,15 These discoveries have dispelled the concept of a core community of gut bacteria that are characteristic of a healthy person. Instead, the focus has shifted toward defining conserved microbial functions that promote host health or affect disease susceptibility.16
The metabolic potential of the gut microbiota greatly surpasses that of the host as its genetic content exceeds that of the human genome 150-fold.17,18 Consequently, gut microbes produce a vast set of small molecules that are often chemically distinct from those generated by host metabolism.19 The nature of these metabolites and their abundance can vary substantially between individuals depending on the composition of their gut microbiota and dietary intake.20 In many cases, metabolites produced by the host and the gut microbiota are exchanged and can be co-metabolized to generate molecules that can have unique consequences in the context of the human body.21 Indeed, many microbially-produced metabolites have been strongly correlated with human disease.22 Thus, the chemical output of microbial metabolism is becoming recognized as a critical component of human health and disease. However, we currently lack a molecular understanding of gut microbial metabolic activities. Bacteria from this habitat are often difficult to cultivate in the laboratory. In addition, bacteria of the same genus and even from the same species can have substantially divergent metabolic capabilities. Therefore, taxonomic profiling, which at present is limited to genus level assignments, is insufficient to provide an accurate assessment or prediction of the metabolic functions of the gut microbiota.
The identification of specific organisms, genes, and enzymes responsible for metabolic functions of interest will improve our understanding of the metabolic potential of the gut microbiota and its role in host health and disease. Metagenomic, metatranscriptomic and metaproteomic approaches are currently used to profile the gut microbiota on a community level. Ideally, specific genes, transcripts, or proteins could be used as indicators in complex meta'omic data to predict the presence of a metabolic function and potentially serve as a personalized read-out of health status. Microbial enzymes could even be direct targets for therapeutics.23,24 However, the present challenge to leverage this type of analysis or manipulate gut microbial functions is that only ∼50% of genes from human stool metagenomes can be assigned a broad functional annotation and less than half of that 50% can be given more descriptive annotations.25 This gap in knowledge limits our understanding of the metabolic functions of the gut microbiota and the molecular basis of the diseases linked to this community. This vast area of unexplored biochemical space creates a need, as well as great opportunity, for biochemists and chemical biologists to contribute to this exciting field.
Metalloenzymes are an important class of enzymes for all domains of life. In enzyme catalysis, metallocofactors enable unique molecular rearrangements and transformations of relatively chemically inert molecules. In particular, anaerobic microbes often use metalloenzymes to promote radical catalysis, which presents challenges in aerobic environments due to the reactivity of dioxygen with organic radical intermediates.26 In the anoxic environment of the human colon, anaerobic bacteria deploy metalloenzymes to perform many interesting metabolic functions. These metal-dependent transformations can enable gut microbes to access nutrients or energy from alternative substrates or to generate molecules that can have important consequences for the host. This review describes unique metabolic functions of the human gut microbiota that involve metalloenzymes. Rather than providing a comprehensive account of all extant metalloenzymes employed by gut microbes for housekeeping functions, we will focus on enzymatic transformations that enable gut microbes to interact with their environment, pathogens, ingested compounds (dietary molecules and xenobiotics), and the human host. Finally, we evaluate the prevalence and distribution of the major families of metalloenzymes across the healthy human microbiota, with the goal of inspiring further investigation into characterized and uncharacterized microbial enzymes that play important roles in human biology.
2 Commensal colonization of the human gut: fitness and adaptation
The dense population of microbes in the human gut creates a highly competitive environment for nutrient resources. Consequently, commensal organisms utilize a variety of energy sources for colonization and survival in this ecological niche. Carbohydrates are rich energy sources that gut microbes obtain from the diet, the colonic mucosal layer, and epithelial cell debris.27 The dietary carbohydrates that microbes access are complex polysaccharides typically derived from plant components that the human host is unable to digest.28 Whereas the availability of these exogenous carbohydrates depends on host intake, the mucosal layer of the GI tract provides a constant and accessible endogenous supply of glycans for microbial consumption. This mucosal lining is a matrix primarily composed of glycoproteins (e.g., mucins) that serves as a barrier between the colonic epithelial cells and the microbes that reside in the lumen.29 Commensal bacteria, notably strains of Bacteroides, Akkermansia, Ruminococcus and Bifidobacteria, are known to utilize mucins as energy sources,30 which contributes to their population stability in the community. Additionally, there is evidence for cooperative degradation of mucin polysaccharides by multiple bacteria, suggesting a role for this metabolism in ecology structure in the gut.31,32 However, an over-abundance of mucin-degraders has been noted in patients with inflammatory bowel diseases, suggesting that microbes can compromise this protective layer and induce an immune response.30 Enteric pathogens have also evolved to take advantage of this mucus-derived resource to breakdown this protective barrier and enable infection.33Gut microbes produce specialized enzymes for reducing both host and dietary polysaccharides into smaller units for energy.34 For example, Bacteroides thetaiotaomicron (B. theta) is one of the most prevalent and prominent members of the gut microbiota in average healthy adult humans (found in 46% of individuals from the HMP cohort at a mean abundance of 0.6%)13,35 and excels at carbohydrate metabolism.36–38 Nearly 20% of its genome is dedicated to degradation of these macromolecules,37 allowing B. theta to access carbohydrates from various sources depending on their availability. Transcriptional responses to available carbohydrates in mice colonized with B. theta revealed the ability of this bacterium to prioritize transient dietary resources when available and forage alternative resources (e.g., mucin) when ingested polysaccharides have been depleted.39 This expanded metabolic repertoire may explain why B. theta is a prominent colonizer of the healthy human gut. Indeed, deletion of only a small subset of the carbohydrate metabolism genes impaired the ability of B. theta to persist in the gut of a mouse model and to be transmitted vertically to offspring.37
The carbohydrate active enzymes of gut microbes act upon polysaccharides that are diverse in monosaccharide composition, linkages, and modifications.30,32,40 Metalloenzymes in particular help with the degradation of polysaccharides containing two common modifications: (i) sulfation and (ii) fucosylation. These modifications are especially abundant on glycans that make up the mucus layer of the GI tract.
2.1 Glycan sulfation
Glycosaminoglycans, such as chondroitin and heparin, and mucin sugars like galactose and N-acetylglucosamine found in the gut mucosa are commonly modified with sulfate groups.40–43 Removal of this modification is a prerequisite for further utilization of the mucus-derived monosaccharides and is postulated to be the rate-limiting step in mucin breakdown.44–46 Gut microbes possess exo- and endo-sulfatases that desulfate both simple and complex carbohydrates (Fig. 1a).47–51 While sulfatase activity plays an integral role in nutrient acquisition and bacterial colonization, there is accumulating evidence connecting it with intestinal inflammation. Sulfatase genes were shown to be essential for inducing B. theta triggered colitis in a susceptible mouse model,52 and increased sulfatase activity was detected in fecal samples of patients with ulcerative colitis.45,53 Thus, the potential role of this microbial activity in development of disease phenotypes suggests it could represent a target for therapeutics. | ||
| Fig. 1 Maturation of gut microbial sulfatases by the radical SAM anSMEs. (a) Sulfate removal from an N-acetyl-glucosamine-6-sulfate substrate by sulfatases harboring an formylglycine (fGly) cofactor. (b) Post-translational modification of a sulfatase L-serine or L-cysteine residue to an fGly residue that is hydrated to the gem-diol active form. (c) Depiction of the anaerobic sulfatase maturating enzyme (PDB accession code: 4K38) with SAM coordinating the radical SAM cluster and a peptide substrate analogue (dark grey sticks) bound in the active site. (d) Reductive cleavage of S-adenosylmethionine (SAM) by the radical SAM [4Fe–4S]1+ cluster to generate 5′-deoxyadenosine radical (5′dA˙) and L-methionine. (e) Proposed mechanism for fGly generation by anSMEs. | ||
Sulfatases are ubiquitous throughout all domains of life and can be delineated into three main groups: the Zn-dependent alkylsulfatases, the Fe-dependent dioxygenase sulfatases, and the formylglycine-dependent sulfatases.54,55 The latter class constitutes the largest group, and its members are frequently encoded in the genomes of gut microbes.48,55 These sulfatases employ a unique post-translational Cα-formylglycine (fGly) modification that is critical for activity (Fig. 1b).56 The hydrated gem-diol form of the fGly cofactor (Fig. 1b) has been observed in crystal structures of both eukaryotic and prokaryotic homologs,57,58 implicating it as the catalytically relevant form. The proposed mechanism involves nucleophilic attack of a deprotonated alcohol of the gem-diol onto the sulfur atom of the sulfate monoester, eliminating the attached sugar and forming a sulfated-enzyme intermediate.59,60 Deprotonation of the second hydroxyl group would then promote release of sulfate from this tetrahedral intermediate, regenerating the aldehyde cofactor.
The catalytic fGly residue originates from either an active site cysteine (in eukaryotes56,61 and prokaryotes62–64) or serine (in prokaryotes63,65,66) residue that is post-translationally modified by a separate maturase enzyme (Fig. 1b). The type of maturase found in eukaryotes and some prokaryotes, termed formylglycine-generating enzyme (FGE), is dioxygen-dependent and operates through an acid–base mechanism without the use of a cofactor.61,67,68 In prokaryotes, two other types of maturases have been identified,64,66,69,70 the best studied of which are the bacterial anaerobic sulfatase-maturating enzymes (anSMEs). The anSMEs are commonly found in the anaerobic bacteria that inhabit the human gut. This class of maturase is oxygen-sensitive and uses radical chemistry to generate the fGly cofactor.64,69,71 AnSMEs belong to the large superfamily of radical S-adenosylmethionine (SAM) enzymes, which harbor an essential [4Fe–4S] cluster within a β-barrel structural domain.72 Three iron ions of the cluster are ligated by cysteine residues in a strictly conserved CX3CX2C sequence motif, and the fourth iron ion is coordinated by the enzyme co-substrate, SAM (Fig. 1c).72 The canonical function of this metallocofactor is to reductively cleave the 5′-C–S+ bond of SAM (Fig. 1d) to generate the potent one-electron oxidant, 5′-deoxyadenosyl radical (5′-dA˙), which then initiates subsequent radical chemistry.72–75 In the anSMEs, the 5′-dA˙ abstracts the pro-S-Cβ-hydrogen76,77 of the target sulfatase Cys/Ser residue located in close proximity to the radical SAM cluster (Fig. 1c), generating a Cβ-centered radical (Fig. 1e). Deprotonation of the residue's thiol or alcohol group is proposed to result in a transient radical anion intermediate that is oxidized to the (thio)aldehyde (Fig. 1e). In the case of cysteine modification, hydrolysis of the thioaldehyde would yield the final fGly functional group.
The anSMEs are members of a subclass of the radical SAM superfamily comprised of proteins that have a C-terminal “SPASM” (subtilosin, PQQ, anSME, mycofactocin) domain.78 SPASM domains possess additional cysteine-rich sequence motifs, which coordinate at least one auxiliary [2Fe–2S] or [4Fe–4S] cluster.78 The postulated roles of these clusters differ from the canonical function of the radical SAM iron–sulfur cluster because they are not known to catalyze cleavage of SAM. Instead they are purported to mediate electron transfer during catalysis and/or aid in substrate positioning and activation. In the case of the anSMEs, Mössbauer-spectroscopic characterization and crystal structures of both Cys- and Ser-type anSMEs determined that the SPASM domain harbors two additional [4Fe–4S] clusters.77,79,80 Their positions in the protein structure, one close to the radical SAM cluster (Fig. 1c) and the other near the protein surface,80 suggest that they constitute an electron transfer relay pathway to mediate the final oxidation of the radical anion intermediate to the product (Fig. 1e). This relay ultimately shuttles the electron to acceptors in solution that can reduce the oxidized radical SAM [4Fe–4S]2+ cluster to enable multiple turnovers, as is observed in vitro.77,79
The crystal structure of the anSME AtsB in the presence of a peptide substrate analogue (Fig. 1c) demonstrated the mode of binding of the sulfatase polypeptide,80 and suggested a possible rationale for substrate promiscuity. The majority of substrate peptide contacts to the anSME protein are established through the peptide backbone, instead of the residue side chains.80 Though the sequence immediately following the target Cys/Ser has a distinct pattern [(C/S)X(P/A)XR],81 this mode of interaction would enable recognition of diverse peptide sequences for maturation. This promiscuity appears to be biologically important as analysis of bacterial genomes often reveals numerous sulfatases, but only a few or just a single anSME.82 Indeed, in B. theta, mutation of the single anSME present in its genome dramatically reduced the ability of the organism to utilize sulfated mucin and glycosaminoglycan carbohydrates as energy sources for growth in vitro.82 This deficiency manifested in vivo as reduced fitness to persist or colonize in the gut of a germ-free mouse when introduced in competition with or after colonization of the wild-type strain, respectively.82 Thus, disruption of this single, metallocofactor-containing maturase enzyme dramatically altered the metabolic capabilities of this organism that allow it to thrive in the gut environment.
2.2 Glycan fucosylation
Mucin glycans are often terminally decorated with the simple sugar monomers L-fucose and sialic acid.30 These host-derived glycan modifications are critical mediators of host–microbe interactions, influencing the composition of the intestinal microflora by enriching microbes capable of hydrolyzing and consuming these sugars.83 Interestingly, the genomes of ∼20% of the human population encode for a non-functional fucosyltransferase (Fut2) that normally adds terminal L-fucose molecules to glycans,84 and this genotype has been associated with decreased microbiota diversity and a higher risk for Crohn's disease.85–87 Conversely, gut bacteria can influence host mucin fucosylation patterns for their own benefit. The commensal microflora, or even a single bacterium (e.g., B. theta), has been observed to stimulate host glycan fucosylation during enterocyte differentiation88,89 and in response to pathogen invasion.90 This interaction allows gut bacteria to maintain a sufficient nutrient supply. For example, metabolism of L-fucose gives B. theta a growth advantage in a competitive environment89 and L-fucose scavenged by Bacteroides fragilis is used for construction of their own polysaccharide cellular components and confers a fitness advantage in vivo.91 However, the simple sugars released by glycosidases that are secreted from commensal organisms can alternatively be co-opted by pathogenic organisms when the gut microbiota has been compromised.92 Thus, although L-fucose metabolism is critical in shaping and maintaining the commensal microbial community, it can also facilitate expansion of enteric pathogens.The L-fucose sugars released from polysaccharides can have multiple fates depending on the degrading organism and the environmental conditions. One of the pathways, termed the “fucose utilization” (fuc) pathway, starts with steps similar to those of glycolysis, involving aldol cleavage to yield lactaldehyde and dihydroxyacetone phosphate.93 The lactaldehyde product is then reduced to (S)-1,2-propanediol (Fig. 2a),93 which can be further metabolized either oxidatively or reductively. In the oxidative pathway, known as the “propanediol utilization” (pdu) pathway, (S)-1,2-propanediol is converted via the intermediate propionaldehyde to the short-chain fatty acid propionate (Fig. 2a).94 Short-chain fatty acids are common products of gut microbial fermentation of carbohydrates and amino acids and have been noted for their beneficial effects on host biology.95–98 Most notably, they can serve as an energy source for colonic epithelial cells, contributing ∼10% of the human daily caloric requirement.99 In addition, short-chain fatty acids can serve as health-promoting signaling molecules in host cells. Propionate, in particular, has been connected with beneficial health effects, such as lowering lipid biosynthesis and serum cholesterol levels, and reducing carcinogenesis.100
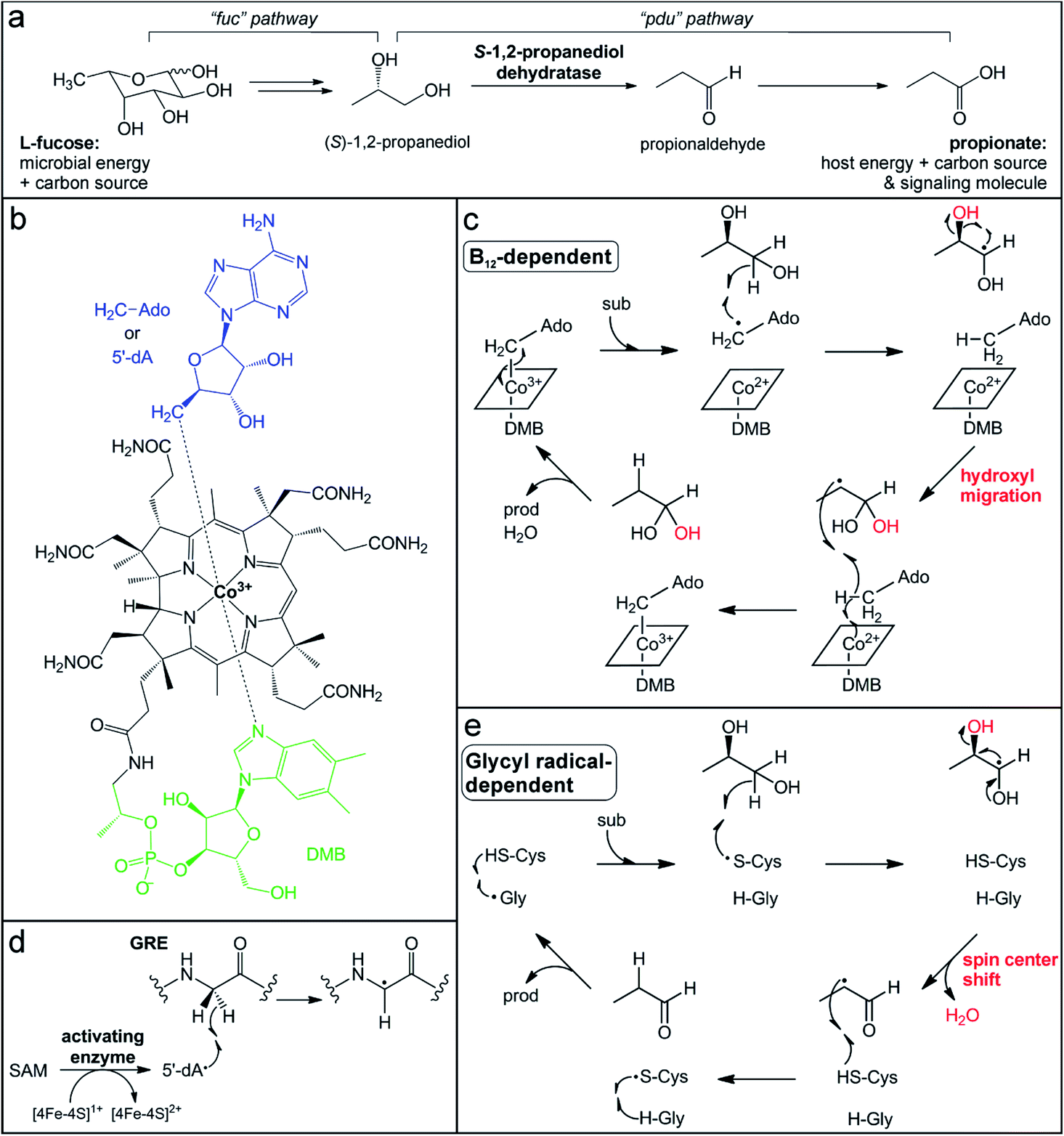 | ||
| Fig. 2 Participation of B12-dependent and glycyl radical-dependent enzymes in gut microbial L-fucose metabolism. (a) Propionate generation via the L-fucose and (S)-1,2-propanediol utilization pathways (fuc and pdu, respectively). (b) Structure of adenosylcobalamin in the “base-on” form. (c) Proposed mechanism for (S)-1,2-propanediol dehydration catalyzed by the B12-dependent enzyme PduC invoking a hydroxyl migration step. (d) Glycyl radical installation by the radical SAM activating enzyme. (e) Proposed mechanism for (S)-1,2-propanediol dehydration catalyzed by the GRE dehydratase involving a spin center shift. | ||
The penultimate step in the metabolism of L-fucose to propionate is the dehydration of (S)-1,2-propanediol to propionaldehyde (Fig. 2a). This reaction is catalyzed by two different microbial enzymes that each requires a metallocofactor for either enzyme activation or catalysis. The B12-dependent (S)-1,2-propanediol dehydratase (PduC) has been extensively characterized since its identification over 50 years ago.101–104 PduC exists as a dimer of heterotrimeric subunits in an α2β2γ2 stoichiometry with the vitamin B12 (cobalamin) cofactor located at the αβ interface of each monomer.105 The cobalamin cofactor consists of a tetrapyrrole corrin ring structure, which provides four equatorial nitrogen ligands to coordinate a cobalt ion at the center (Fig. 2b).103,106–108 Spectroscopic109,110 and structural105,111,112 characterization determined that a 5,6-dimethylbenzimidazole (DMB) moiety extending from the corrin ring coordinates the cobalt ion in the lower axial position in “base-on” form and a 5′-deoxyadenosine molecule occupies the upper axial position (Fig. 2b). In the resting state, the cobalt ion of the cofactor is in the 3+ oxidation state with a Co–C coordination bond to the 5′-deoxyadenosine (adenosylcobalamin).
The conserved first step in the mechanism of adensoylcobalamin enzymes is homolytic cleavage of this Co–C bond to generate a Co2+ center and 5′-dA˙ (Fig. 2c),104 the same radical oxidant generated by radical SAM enzymes. Many factors trigger activation of the Co–C bond, including cofactor and substrate binding to the enzyme active site, as well as kinetic coupling to the subsequent hydrogen atom abstraction step.113,114 In (S)-1,2-propanediol dehydratase, two different metal ions, a K+ ion and a Ca2+ ion, are thought to contribute to activation of the Co–C bond. An essential K+ ion binds in the protein active site near the adenine ring, inducing a protein conformational change and resulting in Co–C bond cleavage even in the absence of substrate.111,112,115 The substrate coordinates a Ca2+ ion via its two hydroxyl groups, thereby increasing its effective size and resulting in a larger energetic release upon binding,111,112,116 which could balance the energetic cost of Co–C bond cleavage.117
When the Co–C bond cleavage event occurs in the presence of substrate, the resultant 5′-dA˙ abstracts a hydrogen atom from the C1 position of (S)-1,2-propanediol to generate an α-hydroxyalkyl substrate radical (Fig. 2c).118 Based on the crystal structure, this C–H bond cleavage would necessitate rotation about the glycosidic bond to bring the 5′-dA˙ radical in close proximity to the target carbon.104,112,119 The substrate-based radical then undergoes hydroxyl group migration from the C2 position to generate a C1 gem-diol intermediate with a C2-centered radical (Fig. 2c).120 This migration is mediated by acidic and basic residues in the active site, as well as the aforementioned Ca2+ ion that interacts with the two hydroxyl groups of the substrate.111,112,116 This mechanism is consistent with the observed retention of the 18O-label in the product when using a C2-18O-isotopically labeled substrate.120,121 The C2-centered substrate radical then abstracts a hydrogen atom from the same 5′-deoxyadenosine molecule that initiated chemistry,122,123 regenerating the 5′-dA˙ that can reform the Co–C bond with concomitant oxidation of Co2+ to Co3+ (Fig. 2c). Finally, dehydration of the C1 gem-diol intermediate yields the final propionaldehyde product.
Recently, another enzyme from a different superfamily was discovered to catalyze the same (S)-1,2-propanediol dehydration reaction.124,125 Transcriptional analysis of the gut microbe Roseburia inulinivorans grown on L-fucose as a carbon source noted the absence of a B12-dependent (S)-1,2-propanediol dehydratase, PduC.126 Instead, a member of the glycyl radical enzyme (GRE) superfamily was observed in a gene cluster that was up-regulated during L-fucose growth.126 GREs utilize an active site glycyl radical to initiate radical chemistry.127 This radical is installed post-translationally by a dedicated partner activating enzyme (GRE-AE) that belongs to the radical SAM superfamily.128 The canonical [4Fe–4S]1+ cluster in the activating enzyme reductively cleaves SAM to generate the 5′-dA˙ oxidant, which abstracts a hydrogen atom from the active site glycine in the partner GRE protein (Fig. 2d).127,128 The glycyl radical is then thought to react with a nearby conserved and essential cysteine residue to generate a thiyl radical that acts on the substrate.127,129 In the (S)-1,2-propanediol dehydration reaction, the thiyl radical is proposed to abstract a hydrogen atom from the C1 position of the substrate (Fig. 2e),124 in a step analogous to the C–H bond cleavage by the 5′-dA˙ in the B12-dependent enzyme. However, the mechanisms of the GRE and the B12-dependent dehydratases are thought to diverge from this point. Instead of hydroxyl group migration, the resultant C1-based radical intermediate in the GRE mechanism is postulated to undergo a spin center shift to the C2 position. In this mechanism (Fig. 2e) supported by computational studies,126,127 deprotonation of the C1 hydroxyl group initiates elimination of the C2 hydroxyl group with concomitant formation of the C1 aldehyde and radical migration (Fig. 2e).130,131 This elimination mechanism is consistent with the observed loss of the 18O-label in the product when using a C2-18O-isotopically labeled substrate.121 Hydrogen atom abstraction by the C2 alkyl radical from the catalytic cysteine thiol regenerates the thiyl radical and forms the propionaldehyde product. At the end of each cycle, the radical migrates back to the initial conserved glycine residue.
These two enzymes represent a striking example of convergent enzyme evolution. They carry out identical dehydration reactions of (S)-1,2-propanediol, yet have different mechanisms to generate the initial radical and stabilize different substrate-based radical intermediates during catalysis. One feature they share is the ability to recycle or store the radical oxidant that activates the substrate, enabling catalytic turnover. However, the protein-based radicals of GREs and the metallocofactor of their activating enzymes are extremely sensitive to oxidation and enzyme inactivation by dioxygen or reactive oxygen species.122,128,129 Consequently, expression of the GRE (S)-1,2-propanediol dehydratase and fermentation of (S)-1,2-propanediol are induced in low-oxygenic or anaerobic conditions in the gut environment.126 In contrast, the B12-dependent enzyme is less vulnerable to oxidation,94 which may allow for gut microbes possessing this enzyme to utilize L-fucose under conditions of host intestinal inflammation when oxygen levels are higher.132,133 Interestingly, the B12-dependent dehydratases are primarily found in opportunistic enteric pathogens, including Salmonella and Klebsiella, perhaps enabling their expansion in the inflamed gut.134 Indeed, the expression of the B12-dependent enzyme in Salmonella enterica serovars Typhimurium confers a fitness advantage and is considered a genetic determinant of pathogenicity.135 Conversely, the GREs have been identified more often in commensal organisms of the Clostridia class.124,125 This observation suggests the possibility of targeting the pathogen-associated B12-dependent dehydratases to impair their ability to utilize L-fucose, without disrupting L-fucose metabolism by commensal organisms.
3 Colonization resistance to pathogenic microbes
The commensal gut microbiota acts through multiple mechanisms to protect the host from the outgrowth of pathogens, a phenomenon referred to as colonization resistance.136,137 Gut microbes prepare the host to detect and combat invasive harmful microorganisms by training and aiding in the development of the innate and adaptive immune systems.138 The importance of this interaction is highlighted by the increased susceptibility of germ-free mice to enteric pathogen infections.138 In addition, commensal microbes indirectly aid their host by creating a competitive ecosystem in which space and resources are limited, preventing colonization or expansion of pathogenic bacteria. Disruption or abolishment of the gut microbial community due to diet, disease, or antibiotic treatment can make these niches available to other microorganisms, resulting in overgrowth of opportunistic pathogens already residing in the GI tract and/or increased susceptibility to pathogenic invaders.139–141 Due to this phenomenon, restoring the commensal microbial population through fecal microbiota transplantation has proven to be an effective mechanism to treat Clostridium difficile infection.142,143 Finally, commensal microbes can directly antagonize microbial pathogens or competitors through the production of toxins and antibiotics.One large class of antibiotics produced by gut microbes is the bacteriocins, which are ribosomally synthesized and post-translationally modified peptides (RiPPs).144–147 These natural products are encoded by a precursor gene that is ribosomally translated to yield a short peptide, typically consisting of 20–100 amino acids.144 This precursor peptide consists of a core sequence and a short leader sequence that is cleaved following post-translational modifications of the core peptide to yield the final mature natural product.146 These modifications result in unique structural features that serve to classify the bacteriocin RiPPs into smaller subgroups, which have been comprehensively reviewed.144 Bioinformatic analyses of human-associated microbial genomes and metagenomes showed that they encode an array of bacteriocins representing all structural subclasses.148–152 While many putative RiPP gene clusters mapped to bacteria in the GI tract,151,152 an even higher abundance were identified from microbes residing in other sites of the human body, with the majority in the oral and vaginal cavities.152 Collectively, these analyses underscore the competitive microbial ecological niches on and in the human body and suggest that they could be untapped reservoirs of natural product antibiotics.
Sactipeptides (sacti- = sulfur-to-alpha-carbon), also known as sactibiotics, are a growing family of bacteriocins with a defining thioether peptide linkage that is installed by a metalloenzyme.144 Inhabitants of the human gut produce sactipeptides that exhibit antibiotic activity against enteric pathogens. These peptides are thought to act by creating pores in the bacterial membrane, causing an influx of ions that leads to membrane depolarization and cell death.153,154 Subtilosin A, produced by strains of Bacillus subtilis, was the first sactipeptide to be identified and characterized (Fig. 3a).155–157 Its biosynthetic gene cluster has been detected in stool metagenomic samples, as well as the genomes of microbes from various habitats.151,152 Subtilosin A demonstrates relatively broad bacteriocidal activity against Gram-positive pathogenic bacteria implicated in bacterial vaginosis, food poisoning, and hospital infections, including Gardnerella vaginalis, Listeria monocytogenes, Enterococcus faecium, and Staphylococcus aureus.153,158,159 Other sactipeptides have been isolated exclusively from gut microbes, such as thuricin CD154,160–162 (Fig. 3a) and thurincin H163,164 (Fig. 3a) that are produced by strains of Bacillus thuringiensis, and ruminococcin C made by the commensal bacterium Ruminococcus gnavus.165–167 The latter demonstrates narrow antibiotic activity toward Clostridium perfringes,168 a bacterium that causes food-borne illnesses. The thuricin CD product is a peptide heterodimer (Trnαβ) that displays antibiotic activity against the infectious gut pathogen C. difficile.160,161 Importantly, whereas other broad spectrum antibiotics commonly used in C. difficile treatment cause major disruption to the rest of the gut microflora, thuricin CD does not alter the community composition due to its narrow spectrum activity, yet is equally as effective against C. difficile.161 The use of these narrow-spectrum antibiotics against enteric pathogens could improve outcomes and mitigate adverse effects, including infection reoccurrence, caused by destroying the resident commensal gut microbiota.
 | ||
| Fig. 3 Sactipeptide RiPP natural products synthesized by commensal gut microbes contain metalloenzyme-installed thioether linkages. (a) Structures of sactipeptides produced by human-associated bacteria [PDB accession codes: subtilosin A, 1PXQ; thurincin H, 2LBZ; thuricin CD, 2L9X and 2LA0]. Leader sequences are shown in blue and brackets indicate thioether linkages. (b) Mechanistic proposals for thioether bond formation invoking an iron–sulfur cluster activated thiol (radical scheme) or a ketoimine intermediate (polar scheme). (c) The CteB (PDB accession code: 5WGG) active site depicting one of the SPASM domain auxiliary clusters located in close proximity to the SAM substrate bound to the radical SAM cluster. | ||
The unique thioether structural motif common to all sactipeptides arises from a C–S bond-forming reaction between the thiol group of a cysteine residue and the α-carbon of a variable amino acid residue in the peptide.144 While thioether bonds are also found in lantibiotics (another subclass of bacteriocin RiPPs), those linkages are formed from conjugate addition between a cysteine thiol and a dehydroalanine or dehydrobutyrine residue, which is mediated by enzymes that employ acid–base chemistry.144 In contrast, generation of the thioether linkages in sactipeptides requires radical chemistry to enable hydrogen atom abstraction from the amino acid α-carbon. Sactipeptide biosynthetic gene clusters invariantly include a gene encoding for a radical SAM enzyme, leading to its suspected involvement in thioether-bond formation.169 The first thioether bond forming radical SAM maturase to be characterized was AlbA, the enzyme that modifies the sactipeptide subtilosin A.170 This single enzyme catalyzes the crosslinking of three different cysteine residues with the respective α-carbons of one threonine and two phenylalanine residues in the subtilosin A precursor peptide (Fig. 3a). Sactipeptides commonly have multiple thioether crosslinks with either stereochemical configuration. Thus, the mechanism of C–S bond formation by radical SAM sactipeptide maturases and the basis for their regio- and stereoselectivity have been points of intrigue.
AlbA and the other identified sactipeptide maturases are members of the aforementioned SPASM-domain subgroup of radical SAM enzymes. AlbA harbors one extra [4Fe–4S] cluster in its SPASM domain,170 whereas other recently described maturases harbor two auxiliary [4Fe–4S] clusters.171,172 As expected, the radical SAM [4Fe–4S] cluster alone is competent and sufficient for reductive SAM cleavage to generate the 5′-dA˙ intermediate.170,171 This oxidant is proposed to perform the initial hydrogen atom abstraction from the α-carbon of the target amino acid. Conversely, the SPASM [4Fe–4S] cluster is not essential for SAM cleavage, but is required for thioether bond formation.170 A recent crystal structure of the sactipeptide radical SAM maturase CteB revealed that, unexpectedly, only three of the four iron ions in the SPASM cluster were ligated with cysteine residues, while the fourth site remained open.172 This unsaturated coordination sphere contrasts with the full cysteine coordination observed for the SPASM domain clusters in the anSMEs, which act as electron transfer mediators, perhaps suggesting another role for this cluster.
An additional structure of CteB solved in the presence of a truncated peptide substrate provided potential insights into the role of the SPASM cluster and its open coordination site in thioether-bond formation.172 In this structure, a cysteine from the peptide substrate (albeit not the thioether bond-forming cysteine) coordinates the open site of the SPASM auxiliary cluster, which is located <11 Å from the 5′-position of SAM (Fig. 2c).172 Prior to this structure, such a direct interaction had been postulated based on perturbations in the UV/visible absorption spectrum of the SPASM [4Fe–4S] cluster in AlbA in the presence of the peptide substrate.170 The authors proposed that the SPASM cluster could act as an electron acceptor and a Lewis acid to activate the coordinated thiol for oxidative coupling to the carbon-centered radical (Fig. 3b).170 There are mechanistic precedents for iron ion activation of a coordinated sulfur atom for oxidation and C–S bond formation. In the enzyme isopenicillin-N-synthase (IPNS), the cysteine thiol of the substrate tripeptide directly coordinates a mononuclear non-heme Fe3+ ion.173 This sulfur undergoes attack by a carbon-centered radical to form a C–S bond with concomitant one-electron reduction of the iron ion.173,174 In the case of the sulfur-inserting radical SAM enzyme BioB (described in detail in a later section), the activated sulfur atom is a sulfide bridge of an auxiliary [2Fe–2S] cluster. In this mechanism, a substrate-based alkyl radical attacks the bridging sulfide and reduces one of the Fe3+ ions through inner-sphere electron transfer (Fig. 5b).175,176 Extending this rationale to the sactipeptide maturases, radical coupling of the α-carbon radical to the iron-coordinated cysteine thiol would yield the thioether linkage with concomitant loss of an electron through inner-sphere electron transfer to the [4Fe–4S]2+ SPASM cluster (Fig. 3b).
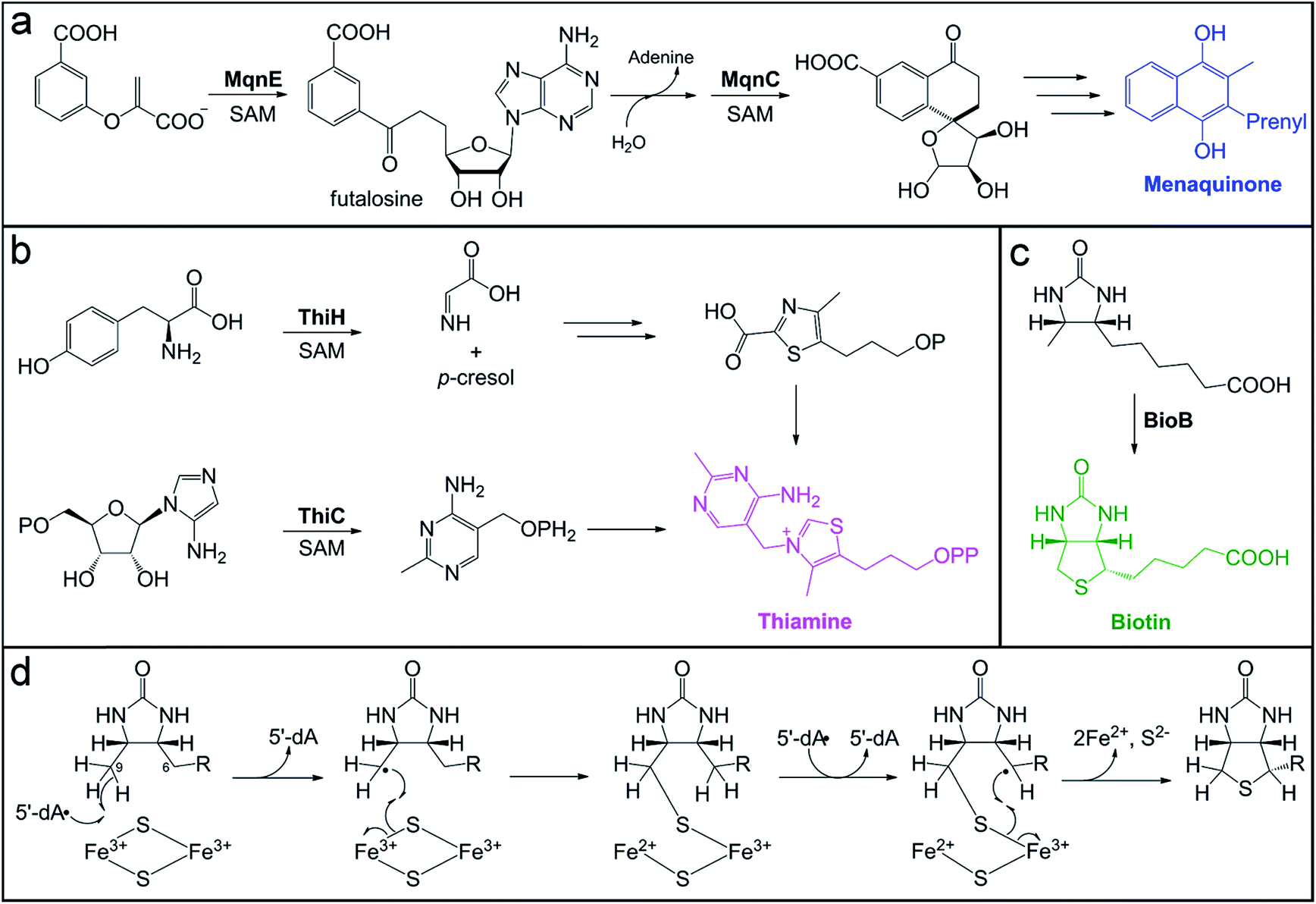 | ||
| Fig. 4 Microbial pathways for vitamin biosynthesis that involve radical SAM enzymes. (a) Futalosine biosynthetic pathway for vitamin K (menaquinone) production, highlighting the transformations catalyzed by the radical SAM enzymes, MqnE and MqnC. (b) Biosynthesis of thiamine from the pyrimidine and thiazole precursors, highlighting the reactions catalyzed by radical SAM enzymes, ThiC and ThiH, in each branch of the pathway. (c) Final step of sulfur installation in biotin biosynthesis, catalyzed by the radical SAM enzyme BioB. (d) Proposed mechanism for BioB sulfur insertion using the auxiliary [2Fe–2S] cluster as the sulfur source. | ||
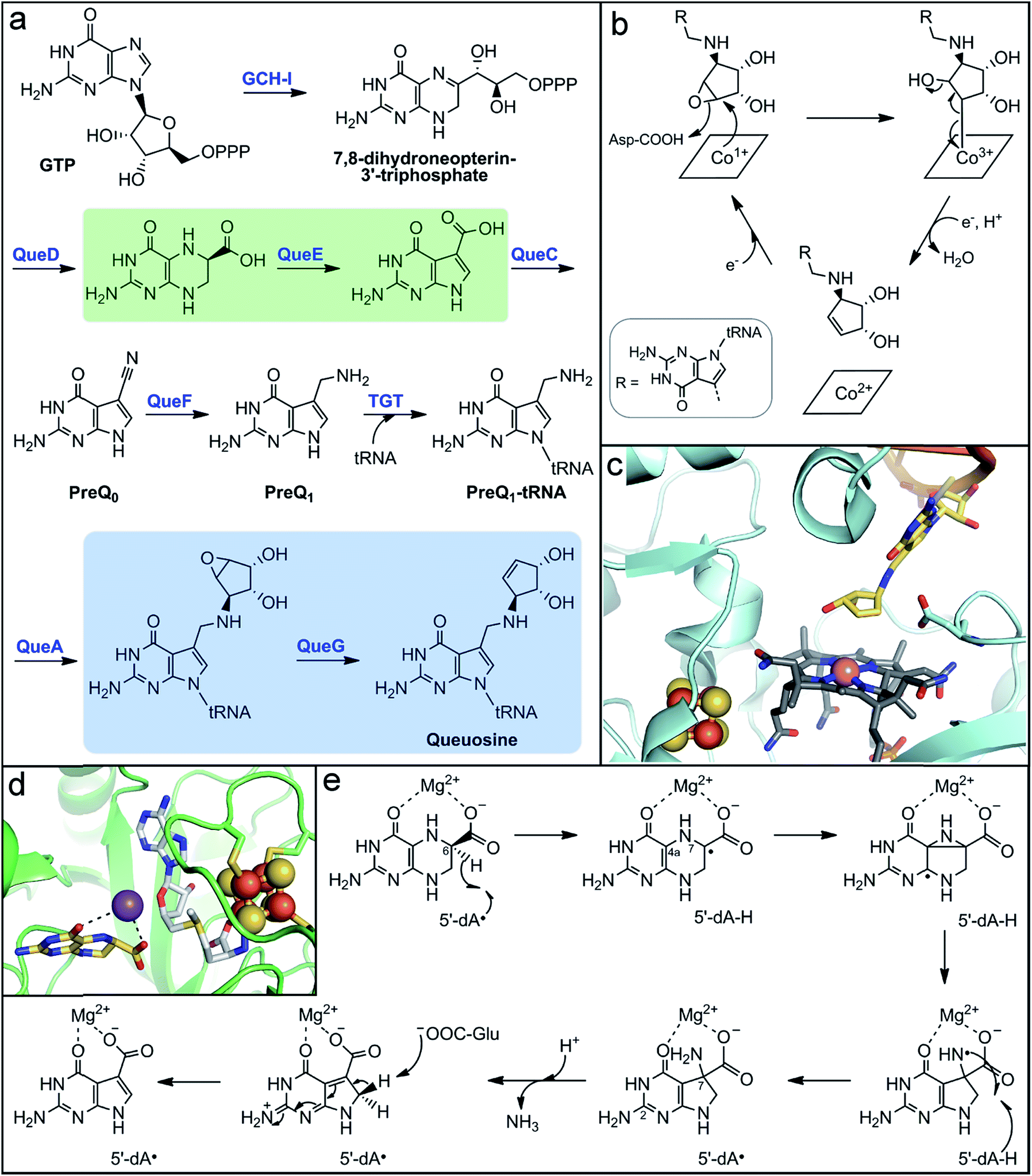 | ||
| Fig. 5 Microbial biosynthesis of the alternative tRNA base, queuosine, involves two metalloenzymes. (a) Complete biosynthetic pathway for queuosine production with the reactions catalyzed by metalloenzymes highlighted in green and blue boxes. (b) Mechanistic proposal for the final step in queuosine biosynthesis catalyzed by the Cbl-dependent epoxyqueuosine reductase, QueG. (c) Active site depiction of product-bound QueG (PDB accession code 5D0B) showing the open-form cobalamin (grey sticks) with the central cobalt ion (pink sphere), the two proximal [4Fe–4S] clusters (orange and yellow spheres), and the queuosine base (yellow sticks) of the bound tRNA mimic. (d) Active site depiction of substrate-bound QueE (PDB accession code 4NJH) showing the radical SAM cluster (orange and yellow spheres) with the co-substrate SAM (light grey sticks) bound and the substrate (yellow sticks) coordinating the essential Mg2+ ion (purple sphere). (e) Proposed mechanism for 7-deazapurine synthesis by the radical SAM enzyme, QueE. | ||
An alternative mechanism has been put forth that invokes a distinct oxidation event preceding C–S bond formation via polar chemistry.171 In this mechanism (Fig. 3b), the α-carbon-centered radical first undergoes one-electron oxidation, presumably via electron transfer to the SPASM [4Fe–4S]2+ cluster, to form a ketoimine intermediate. This electrophilic intermediate is then attacked by the deprotonated cysteine thiol to form the C–S bond.171 In this case, the observed cysteine coordination to the SPASM domain auxiliary cluster could be rationalized by a role in substrate positioning. The core sequences of sactipeptides often contain many cysteine residues, which either form crosslinks in a processive fashion or remain unmodified. Coordination of the additional, unmodified cysteines to the SPASM domain cluster could be serving to lock the substrate into the proper configuration such that both the target carbon and cysteine are positioned close to the radical SAM cluster.
Importantly, both of these mechanistic hypotheses satisfy the observation of both D- and L-stereochemistry for the thioether bonds in sactipeptide natural products. In some cases, a single peptide has both D- and L-linkages that are all installed by the action of the same maturase, as observed for subtilosin A.156,157,170 As the precursor peptides contain only L-amino acids, the lack of stereoretention at the α-carbon could support the formation of a ketoimine intermediate, which could be attacked on either planar face by the deprotonated thiol. Alternatively, this outcome could also arise from the planar character of the carbon-centered radical, which is stabilized by the captodative effect. Indeed, epimerization of amino acids in RiPP peptides is known to be promoted by radical SAM enzymes.177 These epimerases are proposed to generate a similar Cα-radical substrate intermediate that is quenched by a hydrogen atom from a protein residue on the opposite face to invert the Cα stereocenter, yielding a D-amino acid.177 Further structural and spectroscopic characterization of the auxiliary SPASM cluster in sactipeptide maturases in the presence of native peptide substrates will help to resolve these two mechanistic proposals and the precise role(s) of the SPASM clusters.
Regardless of the operant mechanism, both hypotheses imply precise positioning of the reacting cysteine residue with respect to the activated α-carbon to ensure the stereoselective outcome of each respective crosslink. The stereochemistry and location of the thioether crosslinks are expected to greatly impact the peptide structure due to its small size and therefore influence its bioactivity. The antibiotic activity of these sactipeptides, and, in particular, the promise of thuricin CD as a therapeutic agent against C. difficile infection, warrants further investigation into their structural properties and the enzymes responsible for thioether installation.
4 Biosynthesis of essential nutrients
Whereas the human body requires substantial nutrient uptake through the diet, microbes can often be more self-sufficient through de novo biosynthesis of essential small molecules. For example, many microbes can make vitamins, cofactors, and small molecules that the human host cannot synthesize. In many cases, it is unclear if the host can take advantage of the biochemical expertise of its microbes or if the host actively selects for microbes with beneficial capabilities. The extent of symbiosis between gut microbes and the human host is interesting to contemplate with respect to our co-evolutionary history. It is also an active area of exploration in the development of probiotics to supplement the nutritional needs of the human host.4.1 Vitamin biosynthesis
Vitamins are small molecules essential to basic cellular function and metabolism, from energy conversion to macromolecule biosynthesis. Despite their indispensability, humans do not have the ability to synthesize most vitamins de novo. Instead, these molecules are acquired through the diet and from gut microbial production. The potential role of the gut microbiota in vitamin provision has been recognized for decades.178–183 However, the exact contribution of gut microbes to the total pool of vitamins available to the host has not been reliably quantified. In addition, it is not well-established whether bacteria actively export vitamins for host uptake, if vitamins are accessed by the host as a result of microbial cell death, or if certain vitamins are even available to the host. In some cases, specific colonic transporters have been identified to support the notion that certain vitamins (e.g. folate and thiamine) are absorbed by the host.184,185 Mechanisms of acquisition are also important to consider in terms of vitamin exchange between gut microbes. In the case of vitamin B12, inter-microbial exchange has been identified as playing a key role in shaping the composition of the microbiota.186,187 Conversely, the availability of microbially-produced vitamin B12 to the host is still being debated.3De novo pathways for vitamin and cofactor biosynthesis in microbes involve a variety of unusual and chemically challenging transformations, many of which are catalyzed by members of the radical SAM enzyme superfamily.188 For example, the futalosine biosynthetic pathway for vitamin K2 (menaquinone) and the biosynthesis of vitamin B1 (thiamine) require the action of radical SAM enzymes that catalyze C–C bond-forming reactions (Fig. 4). We direct the reader to a recent review for more information on the mechanisms of these enzymes, including ThiC, which catalyzes the complex rearrangement of 5-aminoimidazole ribonucleotide to form the pyrimidine moiety of thiamine.189 A second thiamine biosynthetic enzyme, tyrosine lyase (ThiH), catalyzes an elimination reaction that shares mechanistic similarities with the transformations catalyzed by radical SAM enzymes HydG, CofH, and NosL.190–192 In the proposed mechanism of ThiH, the 5′-dA˙ oxidant abstracts a hydrogen atom from the amine of L-tyrosine to generate a nitrogen-centered radical.188 The Cα–Cβ bond then undergoes β-scission to yield a one-electron oxidized p-cresol intermediate that gets reduced and protonated, while the dehydroglycine co-product feeds into thiazole biosynthesis (Fig. 4b).
Thiamine acts as an enzyme cofactor in essential pathways, such as branched-chain amino acid and carbohydrate metabolism, as well as in non-coenzymatic roles.193,194 Thiamine deficiency with systemic and neurological symptoms often occurs in infants and young children from tropical and impoverished regions.194 The diet is a major source of this vitamin, but a recent study using Drosophila as a model system195 has provided evidence for the long postulated role of the gut microbiota196 in provision of this nutrient to the host. In this model, axenic offspring that were unable to develop on a thiamine-deficient diet were rescued by introduction of a microbiota and even a single organism, Acetobacter pomorum.195 Indeed, human colonocytes express a thiamine pyrophosphate-specific transport that would allow for uptake of this vitamin by the host.184,197–199 Interestingly, variants in this gene have been identified as susceptibility markers for ulcerative colitis in Northern Indian populations, which are known to exhibit thiamine deficiency.200,201 This connection between thiamine uptake and ulcerative colitis remains to be explored further.
Another example of a radical SAM enzyme that participates in vitamin biosynthesis is one of the founding superfamily members, biotin synthase (BioB). BioB is responsible for the incorporation of a sulfur atom in the thiophane ring of the cofactor biotin (Fig. 4c) and belongs to a subgroup of radical SAM enzymes that all perform sulfur insertion reactions.202,203 Each of these enzymes possesses additional auxiliary [4Fe–4S] or [2Fe–2S] cluster(s) that serve as sacrificial donors of one or two sulfur atom(s) that are incorporated into the final product.202 The auxiliary cluster in BioB is a [2Fe–2S] cluster ligated by three cysteine residues and an unusual arginine residue.204–206 The mechanism of BioB (Fig. 4d) initiates with generation of a 5′-dA˙ at the radical SAM [4Fe–4S] cluster and subsequent abstraction of a hydrogen atom from the C9 position of the desthiobiotin substrate.207 The substrate-based radical then attacks a bridging sulfide of the auxiliary [2Fe–2S]2+ cluster, forming the first C–S bond with concomitant inner-sphere electron transfer to an Fe3+ ion of the cluster.175 This reaction yields a chemically competent 9-mercaptodethiobiotin intermediate species208 that remains cross-linked to the Fe3+ ion of the [2Fe–2S]1+ cluster.176 BioB then catalyzes another SAM cleavage event, generating a second 5′-dA˙ equivalent that abstracts a hydrogen atom from the C6 position of the substrate.207 The resultant substrate alkyl radical undergoes a similar reaction with the cross-linked sulfur to cyclize the substrate, which also reduces the second Fe3+ ion of the cluster to Fe2+.
Upon reduction and loss of sulfur to the product, the auxiliary cluster degrades, resulting in an inactive enzyme in vitro.209,210 However, evidence of catalytic activity in vivo211 suggested that additional components might enable cluster reassembly to allow for multiple turnovers in the cell. A recent report reconciled the issue of cluster degradation in vitro through study of another sulfur inserting enzyme, lipoyl synthase (LipA). Scaffold proteins NfuA and IscU, associated with iron–sulfur cluster biogenesis, were demonstrated to reassemble and insert a new auxiliary cluster into lipoyl synthase, rendering it capable of multiple turnovers in vitro.212 This type of cluster repair mechanism could very well extend to other sulfur inserting enzymes, like BioB, to enable their catalytic activity in vivo.
Biotin is an essential cofactor for carboxylase enzymes in pathways such as fatty acid biosynthesis, branched-chain amino acid catabolism, and gluconeogenesis.213 Deficiency of this vitamin in humans causes alopecia and skin dermatitis.214 A recent study using murine models demonstrated the impact of the gut microbiota on systemic host biotin levels and the display of alopecia phenotypes.215 A shift in gut microbiota composition consisting of a bloom of Lactobacillus murinus, a biotin auxotroph, was identified as the cause of low biotin levels in the host.215 This example represents an interesting case in which the gut microbiota is actively depleting the vitamin pool available to the host, an aspect of vitamin homeostasis that has not been adequately explored with respect to the gut microbiota.
4.2 Queuosine biosynthesis
In addition to vitamins and cofactors, gut microbes can synthesize other small molecule products that are incorporated into host macromolecules. One such metabolite is queuine, a modified 7-deazapurine nucleobase that is found in tRNA molecules of all eukaryotic and bacterial organisms.216–219 It substitutes for guanine exclusively at the “wobble” position-34 of the 5′-GUN-3′ anticodon that is found in aspartyl-, tyrosyl-, histidinyl-, and asparginyl-tRNAs.216,218 Although the function of the queuine nucleoside (queuosine) has not been definitively established, its ubiquity in living organisms implies a critical biological role. Indeed, its absence in tRNA has been implicated in numerous, but ill-defined physiological phenomena, including cell proliferation and differentiation, cancer progression, and neurological abnormalities.220–223 Interestingly, mammals lack the biosynthetic machinery to make queuine de novo. The queuine base is scavenged by host cells and transferred into the guanine-34 position of tRNAs.224,225 A major source of queuine is the diet, but early experiments showed that conventional mice maintained on a queuine-deficient diet still possessed this modified base in their tRNA.226 Conversely, queuosine was not detected in tRNA from germ-free mice fed a queuine-deficient diet,226,227 implicating gut microbes as a significant source of this micronutrient. Queuosine biosynthesis has been well-characterized in the model organisms Escherichia coli and B. subtilis; however, the distribution of this biosynthetic pathway in gut microbes has not been evaluated.Although the structure of queuosine had been known for decades,228,229 the complete pathway for its de novo biosynthesis in bacteria (Fig. 5a) has only been recently established.223,230 The pathway begins with conversion of GTP to 7,8-dihydroneopterin-3′-triphosphate by a GTP cyclohydrolase, analogous to the first steps in folate and biopterin biosynthesis. The unique 7-deazaguanine ring is synthesized from this intermediate through the action of the enzymes QueD and QueE, the latter of which is a radical SAM enzyme. The ATP-dependent enzyme QueC transforms the carboxyl group of the 7-deazaguanine to a nitrile in the PreQ0 intermediate. The NADH-dependent enzyme QueF then reduces the nitrile to an aminomethyl group, generating the PreQ1 intermediate. The enzyme tRNA guanine transglycosylase (TGT) then inserts the PreQ1 intermediate into the target tRNA, replacing guanine at position 34. The final two enzymes act on the PreQ1–tRNA complex to yield the queuosine–tRNA final product. The SAM-dependent enzyme QueA catalyzes transfer and isomerization of the ribose group from SAM to the aminomethyl of PreQ1 to form epoxyqueuosine–tRNA. Lastly, the enzyme QueG reduces the epoxide to generate the cyclopentene ring of queuosine.
The enzyme responsible for this final transformation remained elusive for many years, even following the elucidation of the rest of the pathway. It was finally discovered by screening a strain library of E. coli single gene knockout mutants for the accumulation of epoxyqueuosine and the absence of queuosine in isolated tRNA nucleotides.231 The protein identified, QueG, shares sequence homology to reductive dehalogenases, which utilize a cobalamin (vitamin B12) cofactor and multiple iron–sulfur clusters for catalysis. These enzymes belong to the class III group of cobalamin-dependent enzymes, which to date remain grossly under-characterized despite the amazing chemistry attributed to the few known members.232 Cofactor analysis of QueG by EPR spectroscopy and structural characterization confirmed the presence of a cobalamin cofactor with square-pyramidal geometry, existing in the “free-base” configuration (i.e., no lower axial ligand) with an upper axial water ligand.233–235 However, in the presence of substrate, this water ligand is displaced (Fig. 5c).235 The lack of an upper ligand is unique to enzymes of this class, in contrast to the other classes of cobalamin-dependent enzymes which have either a 5′-deoxyadenosyl or methyl axial ligand, and is proposed to have a key function in their mode of action.232,236
In the case of QueG, the “open-Cbl” form is predicted to enable formation of a covalent substrate adduct intermediate during catalysis.224,225 The proposed mechanism for QueG234,235 (Fig. 5b)230,231 begins with nucleophilic attack by the reduced Co1+ on the substrate epoxide to form a Co3+–C adduct and open the epoxide ring, which is facilitated by protonation of the oxygen atom. Single electron reduction of the alkyl–Co3+ species induces homolytic cleavage of the metal–carbon bond, formation of the alkene, and concomitant elimination of the hydroxyl group. Reduction of the resultant Co2+ center to regenerate the Co1+ state for another turnover is mediated by two [4Fe–4S] clusters positioned between the protein surface and the cobalamin cofactor (Fig. 5c).234,235 These clusters were determined to have sufficiently low reduction potential to reduce the low redox potential Co2+/1+ couple.235
Further insight into the mechanisms of substrate recognition and binding, as well as support for the proposed catalytic mechanism, were obtained from structural comparison of substrate-free QueG and a co-crystal structure with a short oligonucleotide tRNA mimic containing the queuosine product.235 QueG is composed of three structural domains: the N-terminal Cbl-binding domain, a ferredoxin-like fold that harbors the iron–sulfur clusters, and a triple HEAT-repeat domain that interacts with the tRNA substrate. The cyclopentenediol ring of the queuosine product is observed in the structure directly above the cobalamin cofactor with the target carbon at a distance of ∼4 Å (Fig. 5c),235 supporting the proposal of a metal–carbon adduct intermediate at the open axial position. In addition, a conserved aspartate residue is positioned in the active site approximately 3 Å from the double bound of the product cyclopentene (Fig. 5c), suggesting it could aid in substrate positioning through interaction with the epoxide oxygen and could serve as a catalytic acid. The unique mechanism of QueG formulated based on structural, mechanistic, and spectroscopic studies highlights the key role of the potent nucleophilic Co1+ reactant species. This reactivity could be exploited in the design of inhibitors for use as experimental tools to probe the role of queuosine in host biology.
Another metalloenzyme involved in the biosynthesis of this alternative nucleobase is the radical SAM enzyme QueE. This enzyme and its homologs catalyze an unusual pterin ring contraction/rearrangement reaction that generates the 7-deazapurine ring found in the tRNA bases queuosine and archeosine (found exclusively in archaea), as well as a number of natural products.230,237 The structure of QueE consists of a minimal TIM barrel fold that harbors the radical SAM [4Fe–4S] cluster via either the traditional CX3CX2C or an atypical CX14CX2C sequence motif.238 The 6-carboxy-5,6,7,8-tetrahydropterin substrate is bound in close proximity to the radical SAM cluster and is stabilized through coordination of an essential Mg2+ ion via its 4- and 6-carboxylate oxygen atoms (Fig. 5d).238,239 In the proposed mechanism (Fig. 5e), the 5′-dA˙ oxidant generated from reductive SAM cleavage initiates the reaction through abstraction of the substrate C6 hydrogen atom.239 The resulting substrate radical then rearranges to form a strained aziridine intermediate or transition state, which is supported by computational studies.240 This mechanism is reminiscent of the migration reactions catalyzed by radical SAM aminomutases. Alternatively, the pyrazine ring could open through β-scission of the N7–C4a bond to form an imine intermediate. In the next step, formation of the 5-membered deazapyrrole ring is proposed to yield an exocyclic amine with a nitrogen-centered radical. This amine-based radical is quenched by hydrogen abstraction from the original 5′-dA molecule to regenerate the 5′-dA˙, which rationalizes the observed catalytic nature of SAM in QueE catalysis.239 Deprotonation of either the C2 pyrimidine exocyclic amine or the pyrrole nitrogen by a basic amino acid residue238 promotes elimination of ammonia from the C7-gem-aminocarboxylate pyrrole intermediate. Finally, a basic residue abstracts the pro-R-proton from the activated C8 position to rearomatize the final pyrrolopyrimide structure.238,239
Although the physiological role of queuine remains enigmatic, preliminary findings suggest it may regulate cell proliferation.220,241 Queuosine has been reported to influence mitotic signaling pathways that rely on protein phosphorylation patterns.242 In cancer cells, protein tyrosine phosphorylation levels are abnormally high and tRNA molecules are hypomodified with queuosine.243 Exogenous queuine administration reverses both of these phenotypes in cancer cells,242,243 suggesting that queuine plays a role in regulation of tyrosine kinases that are critical in cell proliferation processes. However, these studies have not been able to differentiate the potential effects of the free queuine base from the queuosine-containing tRNA. The connection between queuine, cell proliferation, and cancer malignancy warrants further investigation.
5 Production of host immune-modulatory metabolites
The gut microbiota is a critical component in human immune system development and maintenance. Gut microbes produce small molecules that can modulate the immune response of the host. In particular, fermentation products of tryptophan metabolism, including indole-3-aldehyde, indole-3-acetic acid, indolelactic acid, indolepropionic acid, and indoleacrylic acid, promote fortification of the intestinal epithelial barrier and influence immune cell differentiation and function. Their ability to act as ligands for pregnane X receptor (PXR) and arylhydrocarbon receptor (AhR)244–247 has been suggested as the mechanism by which these metabolites protect against chemically-induced colitis in mice. Specifically, indoleacrylic acid was shown to stimulate IL-10 production, suppressing production of TNF and IL-6, and up-regulate expression of anti-oxidant pathways through NRF2 activation.248 In contrast, some metabolites derived from aromatic amino acid fermentation can also be further transformed by gut microbes to disease-associated molecules (see following section).The acrylate and propionate derivatives of tryptophan are produced via a reductive fermentation pathway consisting of a promiscuous set of enzymes encoded by the aromatic amino acid metabolism gene operon, fldAIBC.249 In this pathway (Fig. 6), the aromatic amino acid first undergoes transamination to form the 2-oxo-acid, which is reduced via an NADH-dependent enzyme to the corresponding 2-hydroxy-acid. A CoA-ligase (FldA) appends CoA via a thioester linkage to activate the 2-hydroxy-acid, which is a prerequisite for the next enzymatic transformation.250 The 2-hydroxyacyl-CoA molecule is then dehydrated (FldIBC) to the acrylate-CoA and reduced in the final step to the fully saturated derivative.249 While most of these steps can be achieved through acid–base chemistry, the dehydration of a 2-hydroxy-acid is chemically challenging due to the high pKa of the protons at the β-carbon.251,252 Instead of acid–base chemistry, 2-hydroxyacyl-CoA dehydratases utilize reductive radical chemistry to perform this reaction, with the CoA thioester playing a key role in stabilizing reaction intermediates. The proposed mechanism of the 2-hydroxyacyl-CoA dehydratases (Fig. 6)251,252 initiates with a one-electron reduction of the substrate to generate a ketyl radical anion. Next, the carbanion eliminates the hydroxyl group to form an enoxy radical intermediate. The pKa (∼14) of the β-proton of this intermediate is markedly lower than that of the 2-hydroxy-acid (by >25 pK units),253 facilitating its deprotonation to yield another ketyl radical anion that undergoes one-electron oxidation to form the final enoyl-CoA product.
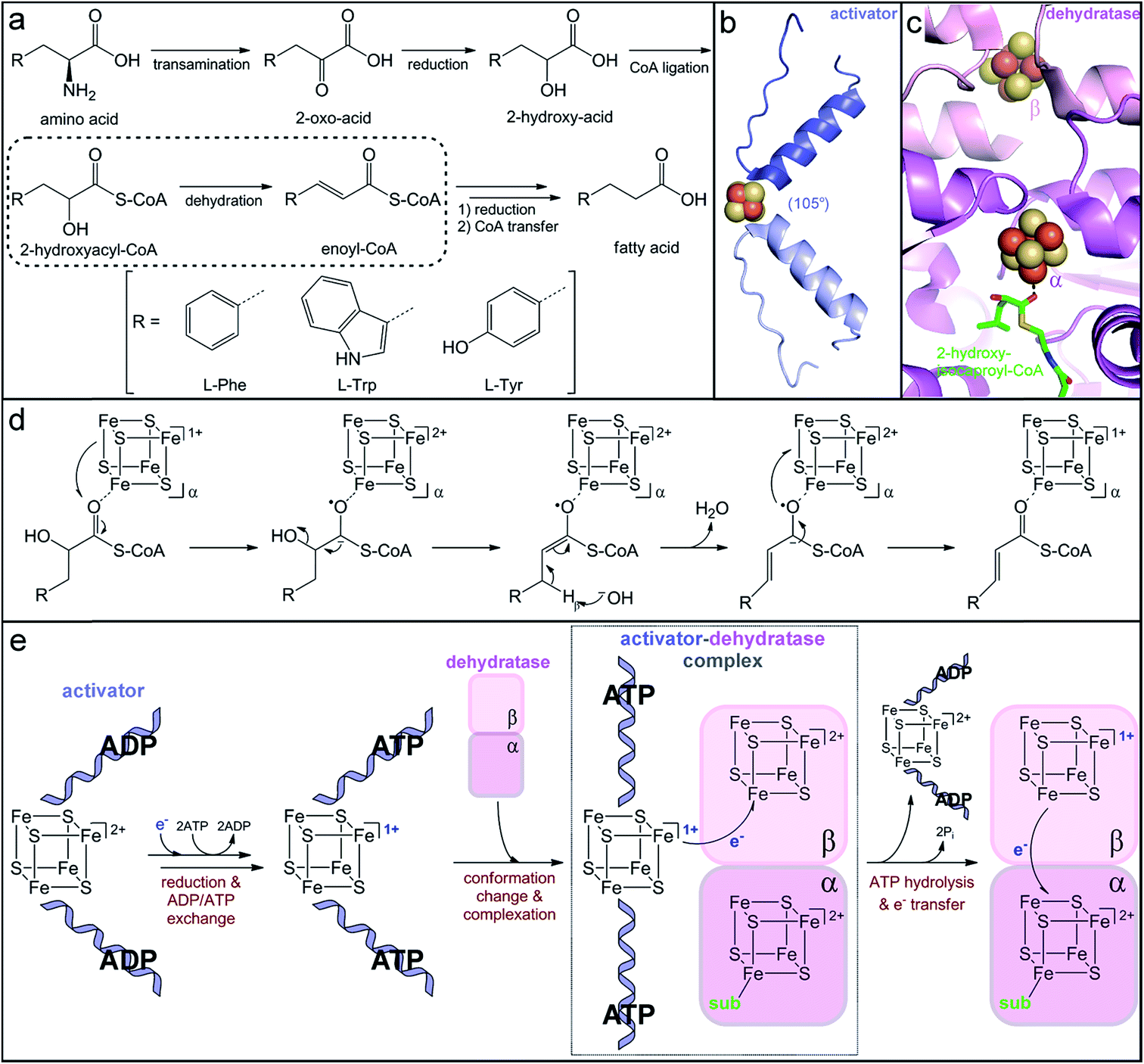 | ||
| Fig. 6 Production of immune-modulatory indole derivatives by gut-associated Clostridia. (a) Reductive amino acid fermentation pathway involving dehydration of a 2-hydroxy-acid. (b) Homodimer interface of the dehydratase activator (2-hydroxyisocaproyl-CoA dehydratase activator, PDB accession code: 4EHT) highlighting the helix–cluster–helix motif in the ADP-bound 105° angle conformation. (c) Heterodimer interface of the dehydratase component (2-hydroxyisocaproyl-CoA dehydratase, PDB accession code: 3O3N) depicting the [4Fe–4S] clusters in each subunit and the direct substrate coordination to the α-cluster. (d) Umpolung charge reversal mechanism of 2-hydroxyacyl-CoA dehydration. (e) Proposed ATP-dependent electron transfer mechanism of dehydratase activation. | ||
The critical single electron transfer that initiates dehydratase catalysis is promoted by an essential partner activating enzyme, a [4Fe–4S] cluster-dependent electron transfer protein with ATPase activity.249,254 Structural characterization of homologs that activate other 2-hydroxyacyl-CoA dehydratases has provided insight into the conserved mechanism of activation (Fig. 6e). The activating partner exists as a homodimer with a single ATP/ADP bound per monomeric subunit and a [4Fe–4S] cluster at the interface of the dimer (Fig. 6b).255,256 The cluster is highly solvent exposed and thus extremely sensitive to oxidation;249 it is ligated by two cysteine residues contributed by a helix of each monomer to form an interesting helix–cluster–helix structural motif (Fig. 6b).255,256 In the ADP-bound form, the helix–cluster–helix has an angle of 105° (Fig. 6b),255,256 which inspired the epithet for these “Archerase” enzymes.252 Reduction of the cluster and substitution of the ADP molecules for ATP is proposed to induce a conformational change to form a 180° angle at the helix–cluster–helix dimer juncture that promotes complex formation with the dehydratase (Fig. 6e).256,257 ATP hydrolysis then drives the electron transfer from the [4Fe–4S]1+ cluster of the activating partner to an oxidized [4Fe–4S]2+ cluster in the dehydratase (Fig. 6e).254,256,257 This dehydratase cluster then transfers the electron via a second [4Fe–4S] cluster to ultimately reduce the substrate (Fig. 6e).
This mechanism of enzyme activation has been largely devised based on analogy to the activation of the nitrogen-fixation metalloenzyme, nitrogenase.254,258 Although the activating partners of 2-hydroxyacyl-CoA dehydratases and nitrogenases both have the helix–cluster–helix structural motif, their structures do not otherwise share apparent homology.259 The nitrogenase activator is most similar to the family of G-proteins, while the dehydratase activator resembles the ASKHA (acetate and sugar kinases, heat shock protein 70 and actin) proteins,259 implying that they independently evolved similar ATP-dependent strategies to promote one-electron reduction. As expected from their analogous roles in electron transfer, the [4Fe–4S] clusters of the dehydratase and the nitrogenase activator proteins share distinctive electronic properties. Spectroscopic characterization of the [4Fe–4S]1+ cluster in the phenyllactyl-CoA dehydratase activator (FldI) revealed that it has an unusual S = 3/2 ground state.249 Studies of the homologous 2-hydroxyglutaryl-CoA dehydratase activator demonstrated that, when treated with the strong reductant titanium(III) citrate, the [4Fe–4S]1+ cluster can be further reduced to the superreduced [4Fe–4S]0 oxidation state.260 To our knowledge, the nitrogenase and dehydratase activators are the only examples of superreduced clusters in biology. However, at least in the case of the dehydratase activator protein, only the more traditional [4Fe–4S]2+/1+ redox couple is likely to be biologically relevant for electron transfer, as the superreduced cluster cannot activate the 2-hydroxyglutaryl-CoA dehydratase in vitro.260
After one-electron reduction by the activating protein, the dehydratase is catalytically active and capable of thousands of turnovers through storage of the reducing equivalent in its own iron–sulfur clusters.249,261 The crystal structure of the 2-hydroxyisocaproyl-CoA dehydratase provided insight into the electron transfer cycle during catalysis. The dehydratase is a heterodimer composed of two structurally similar subunits that share low sequence homology.262 Each subunit harbors a [4Fe–4S] cluster positioned at the interface of the αβ dimer (Fig. 6c).262 The β-subunit cluster is ligated by three protein cysteine residues and another thiolate ligand and is believed to be the initial acceptor of the electron provided by the activating protein.262 The α-subunit cluster has three cysteine ligands and an open coordination site that is occupied by a water molecule in the absence of substrate.262 Upon substrate binding, this water ligand is displaced by the carbonyl oxygen of the substrate thioester (Fig. 6c),262 indicating that the α-subunit harbors the catalytic active site. The direct, monodentate coordination of the substrate to the iron–sulfur cluster enables facile inner-sphere electron transfer and stabilization of the ketyl radical anion intermediate. Interestingly, neither dehydratase cluster can be reduced by any tested chemical reductants,263 suggesting that they have very low reduction potentials. Either the reduction potential of the activator cluster is lowered upon complex formation due to the large postulated 105° to 180° conformational change and likely desolvation of the cluster256 or the reduction potentials of the dehydratase clusters are altered upon complexation and/or as a result of desolvation observed upon substrate binding.262 Once the dehydratase is reduced, the electron can continuously cycle between the two dehydratase clusters and the substrate. Overall, this elaborate and elegant mechanism of ATP-dependent electron transfer to a CoA-activated substrate is used to achieve a charge reversal (Umpolung effect) for water elimination from an unactivated 2-hydroxy-acid substrate.
Reductive aromatic amino acid metabolism has only been demonstrated for a handful of gut bacteria, all belonging to the phylum Firmicutes, but it has important implications for human health and disease. The fldAIBC gene operon responsible for this metabolism was shown to be less prevalent and less abundant in metagenomes of patients with inflammatory bowel diseases (both Crohn's and ulcerative colitis) compared to healthy populations.244,248 As expected, the abundance of the gene operon correlated with observed levels of tryptophan metabolites in biological samples of these patients,248 suggesting that the lack of this operon could be a good genetic marker for inflammatory disease. Interestingly, reduction of tryptophan metabolism is correlated with reduced mucin degradation, specifically of L-fucosylated glycans.248 In the healthy gut, the host presents fucosylated mucins, which promote colonization of fucose-degrading microbes that are known to produce tryptophan-derived metabolites with immune suppression activity. The host immune response to these microbial metabolites then results in increased mucin fucosylation. This co-metabolism thus perpetuates a positive feedback cycle between the gut microbiota and the host to suppress inflammation. Conversely in IBD, the observed reduction of these beneficial microbes could reflect disruption of this cycle as either a cause or a consequence of host inflammation and could present a target for microbiota-based therapeutics.
6 Gut microbial metabolism linked to human disease
The metabolism of gut microbes is centered around nutrient and energy extraction for their own benefit. These functions can be detrimental to the host by consuming metabolites that it may need. In addition, because of the physical proximity between microbes and their host, the host is exposed to microbial metabolic waste products. These compounds can vary in distribution and abundance depending on the composition of the individual's gut microbiota. Importantly, many microbial metabolites have been associated with risk for disease development in the host. In addition, unique metabolic functions can allow for pathogenic bacteria or pathobionts to colonize or expand in the GI tract, resulting in host infection.6.1 Trimethylamine production
The quaternary amine choline is an essential nutrient for humans and major dietary component of red meat, eggs, dairy, and soy. Gut microbes metabolize this diet-derived molecule under anaerobic conditions to generate acetaldehyde and trimethylamine (TMA) (Fig. 7a).264,265 Whereas acetaldehyde can be utilized by the microbe as a source of carbon and energy, TMA is not consumed by the producing organism. Instead, it is absorbed by host cells and circulated in the bloodstream to the liver where it is converted by a host flavin monooxygenase (FMO3) to trimethylamine N-oxide (TMAO) (Fig. 7a).266 This final metabolite plays a causative role in the development of atherosclerosis and cardiac disease in mice,267–269 and has been associated with numerous other diseases, including diabetes, kidney disease, and non-alcoholic fatty liver disease.270–272 Thus, TMAO production is currently viewed as a therapeutic target. A priori, the FMO3 enzyme presents as an attractive candidate for inhibition; however, individuals with genetic mutations in the fmo3 gene have the disease trimethylaminuria that results in accumulation and excretion of TMA causing an undesired fishy malodor.273 Conversely, targeting the microbial component of this pathway could have additional benefits beyond decreased TMAO production. Choline plays essential roles in biology as a methyl donor, a precursor to the neurotransmitter acetylcholine, and a component of lipid biomolecules. The depletion of available choline due to gut microbial choline metabolism has been recently shown to have other effects on host physiology.274 The decreased abundance of choline in a mouse model of metabolic disease led to altered lipid metabolism and one-carbon metabolism that manifested as changes in DNA methylation.274 This host-microbial metabolic pathway is a clear example by which the microbial transformation of a dietary molecule can promote disease and thus represents a specific target for manipulation of the gut microbiota to influence host health.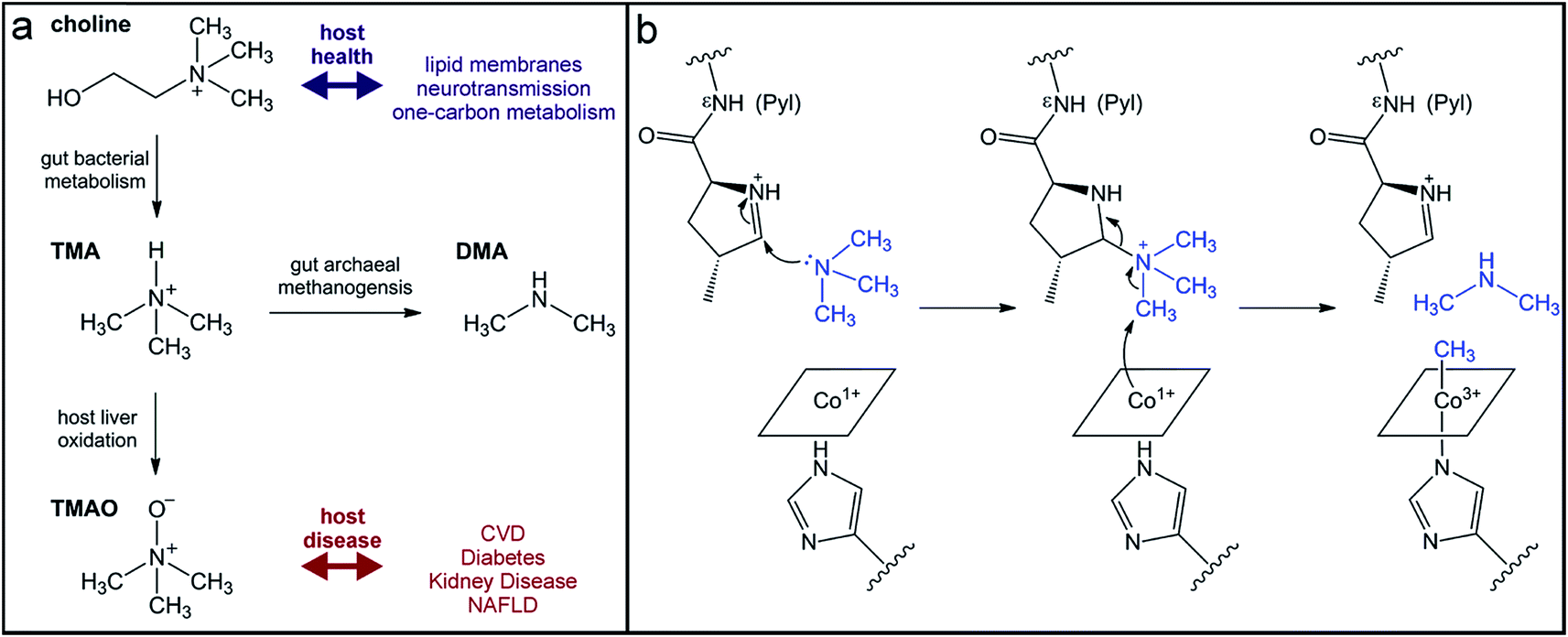 | ||
| Fig. 7 Production of the TMAO precursor TMA results from gut microbial metabolism. (a) Choline metabolism by gut microbes produce trimethylamine that can be oxidized by the host liver enzyme FMO3 or demethylated by human-associated archaea for methanogenesis. (b) Mechanism for TMA demethylation by archaeal pyrrolysine (Pyl)-containing, coronoid-dependent methyltransferases. | ||
The enzyme responsible for anaerobic microbial choline metabolism, choline TMA-lyase (CutC), was recently identified through a genome mining approach and is widely distributed in gut microbial genomes and human gut metagenomes.275–277 CutC is another member of the GRE family, but prior to its characterization, C–N bond cleavage was not a known transformation for these enzymes. As with all GREs, a partner radical SAM activating enzyme generates the catalytic glycyl radical on CutC.275 The mechanism of C–N bond cleavage is proposed to initiate with hydrogen atom abstraction by the thiyl radical from the C1 position of choline to generate an α-hydroxyalkyl radical intermediate. Deprotonation of the hydroxyl group and formation of a transient ketyl radical intermediate is then proposed to promote heterolytic cleavage of the C–N bond to directly eliminate TMA. This proposal is supported by the substrate-bound crystal structure of CutC.278 The substrate is positioned in the active site pocket with a gauche conformation that is imposed by CH–O hydrogen bonding interactions between the partial positively charged, polarized N-methyl groups and the oxygen atoms of active site residues.278 This conformation allows for hyperconjugation between the p-orbital of the carbon-centered radical and the σ* anti-bonding orbital of the C–N bond and is expected to facilitate elimination of TMA.278 Following this step, the resultant acetaldehyde radical can abstract the hydrogen atom from the active site cysteine to regenerate the thiyl radical.
Instead of uptake and oxidation of TMA by the host, other gut microbes may further metabolize this molecule. Microbes from different environments have been shown to demethylate methylamines, such as TMA, by the action of corrinoid-dependent methyltransferases. In the first step, the donor amine forms a covalent bond with a post-translationally modified pyrrolysine residue in the active site (Fig. 7b).279 Once activated, the supernucleophilic Co1+ state of the vitamin B12 cofactor attacks the donor methyl group in an SN2-like reaction (Fig. 7b), cleaving the C–N bond of TMA.279 The resultant CH3–Co3+ cofactor can then be utilized as a methyl donor in one-carbon metabolism. Certain archaea possess N-methyltransferases that are able to initiate methanogenesis through the specific demethylation of TMA, dimethylamine, and monomethylamine.280 Interestingly, this activity has been demonstrated in human-associated methanogenic archaea and has been proposed as a route to lowering TMA levels and consequently TMAO production by the host.281,282 This “archaebiotic” proposal is an intriguing example of targeting a microbially-produced metabolite to treat its associated human diseases, such as trimethylaminuria and cardiovascular disease.
6.2 p-Cresol production
In addition to the reductive pathway for amino acid fermentation described above, Clostridia also commonly oxidize aromatic amino acids to their corresponding arylacetate derivatives. Whereas the tryptophan-derived indole-3-acetate produced via this pathway has beneficial properties, the tyrosine-derived metabolite can be further metabolized to molecules linked to human disease. The enteric pathogen C. difficile metabolizes the tyrosine-derived oxidation product p-hydroxyphenylacetate to p-cresol (Fig. 8a). This metabolite has both antioxidant and antimicrobial properties.283,284 Thus, production of p-cresol has been proposed as a mechanism for this pathogen to eliminate microbial competition in the gut during invasion.285 Importantly, p-cresol production appears to be predictive of C. difficile virulence. So-called ‘hypervirulent’ strains of C. difficile have been shown to produce higher levels of p-cresol in culture and can tolerate higher concentrations of this molecule than less virulent strains.285,286 The production of p-cresol is an inducible activity in C. difficile; however, the mechanisms of regulation have not been elucidated. Comparative genomics of C. difficile strains identified an additional transcriptional regulator of phenolic acid metabolism in more virulent strains.287 Thus, differential regulation could be a possible explanation for the higher production of and/or tolerance to p-cresol exhibited in hypervirulent strains. Beyond the bactericidal effects of p-cresol, this metabolite can have other negative consequences on host biology. For example, p-cresol can be sulfated or glucuronidated by human enzymes to generate metabolites that have been implicated in chronic kidney disease.288,289 In fact, p-cresol competes with other substrates, including pharmaceuticals (e.g., acetaminophen), for sulfate conjugation by host enzymes, reducing the host's capacity to detoxify them.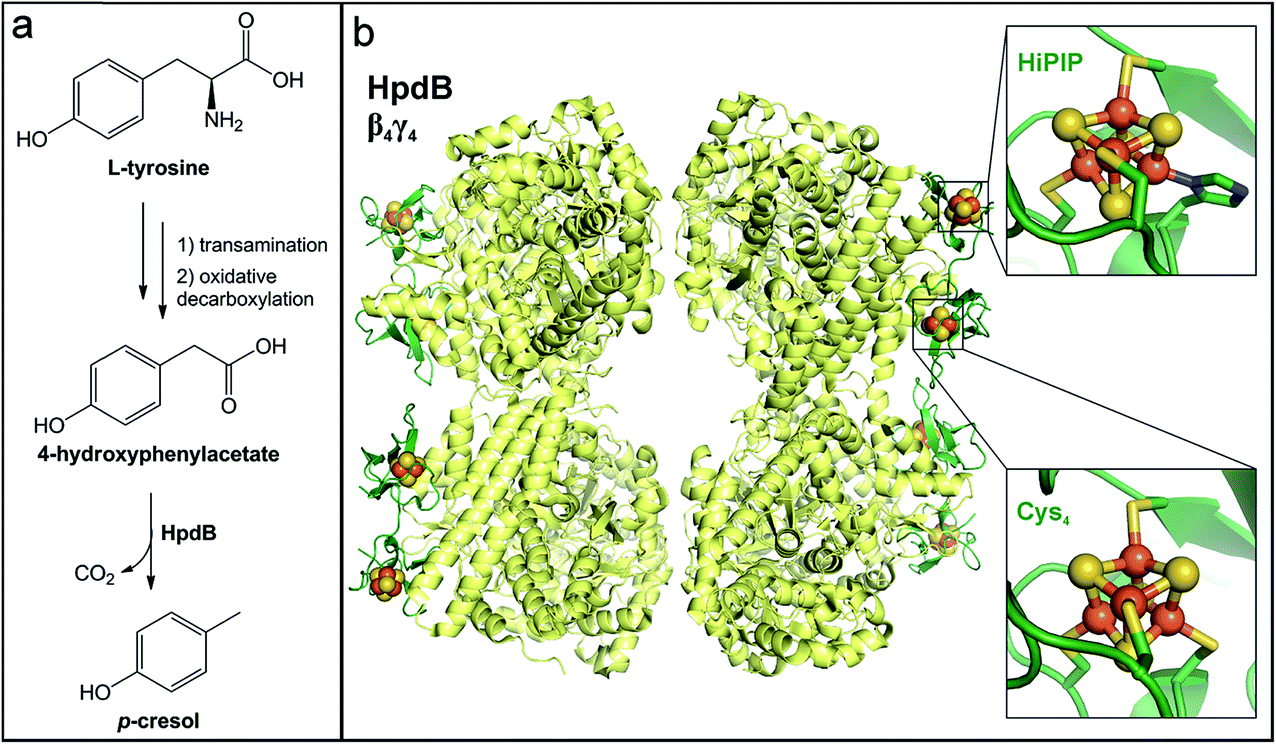 | ||
| Fig. 8 Generation of p-cresol by a gut microbial glycyl radical enzyme. (a) Oxidative metabolic pathway in Clostridia for L-tyrosine conversion to p-cresol via a 4-hydroxyphenylacetate intermediate. (b) Hetero-octameric structure of the glycyl radical enzyme 4-hydroxyphenylacetate decarboxylase (PDB accession code: 2YAJ) with the β-subunit shown in yellow and the γ-subunit shown in dark green. Insets show the two iron–sulfur clusters harbored in the γ-subunit. | ||
The microbial enzyme that catalyzes production of p-cresol, p-hydroxyphenylacetate decarboxylase (HpdB), is a member of the GRE family.290 While it shares the common mechanistic features of protein-based radical-mediated chemistry that define this family, HpdB belongs to a separate subclass of the GRE family together with the enzyme benzylsuccinate synthase.291 These GREs are unique in their requirement for an accessory scaffold protein (γ). This small γ-subunit is composed of two homologous domains that each harbor a [4Fe–4S] cluster (Fig. 8b).291,292 The C-terminal domain cluster has complete cysteine ligation, whereas the N-terminal domain contains a HiPIP (high-potential iron–sulfur proteins)-like cluster coordinated by one histidine and three cysteine ligands (Fig. 8b).292 Redox titrations revealed only a single reduction potential of −287 mV, suggesting that only one cluster, likely that with full cysteine ligation, is able to redox cycle, whereas the HiPIP cluster might play a structural role.293 Although the precise roles of these iron–sulfur clusters in GRE activity remain unclear, the γ-subunit is essential for decarboxylase activity.294 The active form of the HpdB decarboxylase protein (β) is a β4γ4 hetero-octamer that associates only upon phosphorylation of a serine residue in the β-subunit.291 Conversely, when the serine is dephosphorylated, the γ-subunits dissociate and the β-subunits form inactive homodimers.294 The active hetero-octamer only has a single glycyl radical equivalent at any given time,294 and it is unusually short-lived in the absence of substrate relative to the lifetimes of glycyl radicals in other GREs.291 It has been postulated that dissipation of the glycyl radical is facilitated by one-electron reduction via the iron–sulfur cluster(s) in the γ-subunit.291 Glycyl radical quenching could be a regulatory mechanism to limit enzymatic production of the toxic molecule, p-cresol.
The radical SAM activating enzyme of the HpdB protein possesses, in addition to the canonical N-terminal radical SAM [4Fe–4S] cluster, extra [4Fe–4S] cluster(s) that are located in a C-terminal ferredoxin-like domain.291 With the exceptions of the archetypal GRE-AEs of pyruvate formate-lyase and anaerobic Class III ribonucleotide reductase, many other GRE-AEs possess a homologous ferredoxin-like C-terminal domain with conserved cysteine sequence motifs, suggesting that they also harbor one or more additional iron–sulfur clusters. However, the purpose of these clusters is not well established. In the case of the HpdB activating enzyme, elimination of the domain harboring the auxiliary [4Fe–4S] cluster did not abolish 5′-dA˙ production and the enzyme was still able to activate the decarboxylase.295 Yet this truncated variant reduced the stability of the glycyl radical in the decarboxylase enzyme, decreasing its half-life 10-fold.295 The observed radical lability has led to speculation that this domain could protect the glycyl radical from solvent during dissociation of the decarboxylase from the activating enzyme.295 Yet structural and spectroscopic characterization of GREs in complex with their activating enzymes is lacking to support this hypothesis.
Once activated, the mechanism of HpdB diverges from that of traditional glycyl radical enzymes. In the canonical mechanism, the first step in substrate activation is hydrogen atom abstraction by the active site cysteine-based thiyl radical. This type of mechanism was originally postulated for 4-hydroxyphenylacetate decarboxylation. In this proposal, the phenolic O–H bond would be homolytically cleaved, generating an intermediate that acts as an electron sink for subsequent decarboxylation.290 However, the structure of the decarboxylase solved in the presence of substrate did not support that hypothesis, as it showed the carboxylate end of 4-hydroxyphenylacetate positioned near the cysteine.292 Thus, a mechanism was proposed in which the thiyl radical initiates a concerted proton-coupled electron transfer, abstracting an electron from the substrate carboxylate and a proton from a nearby glutamate residue. The resultant substrate radical undergoes a Kolbe-type decarboxylation, facilitated by parallel orbital alignment of the carboxylate-methylene C–C bond with the benzene ring, to generate CO2 and a 4-benzoquinone methide radical anion upon deprotonation of the phenolic hydroxyl. Hydrogen atom abstraction from the catalytic cysteine would yield the p-cresol product and regenerate the thiyl radical. Quantum mechanical calculations support this proposed mechanism,296 postulating a transition state that is already induced by the protein in substrate binding to promote the Kolbe-type decarboxylation.292
p-Cresol is a metabolite biomarker for autism spectrum disorder.297 High urinary levels of p-cresol, its sulfated-derivative, and a similar molecule, 4-ethylphenylsulfate, have been detected in young children with autism and are correlated with disorder severity.298 A higher abundance of C. difficile, in addition to other community shifts in the gut microflora, has been noted in children with autism,298 which could explain the increased levels of p-cresol. However, this metabolic activity has also been attributed to Clostridium sporogenes as well as other gut microbes299,300 and has not been extensively profiled across other commensal microbes. Despite the observed accumulation of p-cresol in this patient population, its causative effect in autism, if any, has not yet been elucidated. Conversely, administration of 4-ethylphenylsulfate to conventional mice has been shown to induce anxiety phenotypes associated with the disorder.301 Interestingly, a single health-promoting bacterium, Bacteroides fragilis, reduced levels of 4-ethylphenylsulfate and other elevated metabolites in a mouse model of autism spectrum disorder.301 Both p-cresol-sulfate and 4-ethylphenylsulfate are thought to derive from tyrosine and are speculated to have similar bioactivities based on their chemical structures; however, it is worth noting that their enzymatic production likely differs. The Kolbe-type decarboxylation that results in p-cresol production relies on the orbital overlap between the C–C bond to be cleaved and the π-system of the benzene ring, as well as resonance stabilization of the 4-benzoquinone methide radical anion intermediate. These molecular features are not conserved when the carbon chain is extended by a methylene group in the putative 4-ethylphenylsulfate precursor, and thus, an alternative enzymatic mechanism is likely used to achieve production of this metabolite. The enzyme(s) responsible for 4-ethylphenylsulfate production have not yet been identified.
6.3 Ammonia production
Urea is the major circulating pool and waste-product of nitrogen in humans.302 It is primarily generated in the liver, transferred through the kidneys, and excreted in urine.303 However, ∼20–30% of this metabolite reaches the intestinal organs,304 where it can be used by gut bacteria as a source of nitrogen. Gut microbes capable of metabolizing urea possess the enzyme urease, which hydrolyzes urea to generate two equivalents of ammonia from every molecule of substrate.305 Some of the ammonia produced is metabolically assimilated by the urease-positive microbe or by other gut inhabitants. Interestingly, the introduction of a urease-positive strain of E. coli into a mouse model reshaped the gut community composition and exacerbated the colitis phenotype in this model.306 The free ammonia that is not microbially assimilated becomes protonated, leading to high concentrations of positively-charged ammonium ions in the host intestine. Some ammonium ions can recirculate to the liver to be reused by hepatic cells.303 However, in individuals with liver dysfunction, ammonium ions accumulate throughout the body, leading to a condition termed hyperammonemia that can cause dysfunction of the central nervous system.307,308 Under normal physiological conditions, the urea-derived ammonium ions that do not get recirculated to the liver remain in the colon. These ions cause an increase in pH that helps to offset the acidity arising from short-chain fatty acids produced by gut microbes. While the shift in pH resulting from urease activity is beneficial in the context of the colon, it also enables the pathogen Helicobacter pylori to colonize in the stomach. The low pH of the stomach normally precludes bacterial colonization, but the urease activity of H. pylori creates a microenvironment with a more neutral pH.309 In addition, ammonium ions have been shown to have direct cytotoxic effects on gastric epithelial cells, contributing to the pathology of H. pylori infection.310 For these reasons, urease is considered a virulence factor of H. pylori and other pathogens, and is a primary antigen for the host immune response to H. pylori.309,311The decomposition of urea by urease occurs in two steps. The first is enzymatic hydrolysis to produce ammonia and carbamate, followed by non-enzymatic hydrolysis of carbamate in water to yield the second molecule of ammonia and carbonic acid (Fig. 9a).312 The enzymatic reaction carried out by urease is facilitated by a rare dinickel cofactor, which acts as a Lewis acid to activate the substrate and water. The dinickel cofactor enhances the electrophilicity of the urea carbonyl and lowers the pKa of water, generating the more nucleophilic hydroxide ion.305 Urease was the first enzyme to be crystallized, an experiment which confirmed that enzymes are proteins and garnered a Nobel Prize.305 Numerous crystal structures have been solved of this enzyme from bacterial and plant sources in its resting state and in the presence of small molecule inhibitors that illustrate key features of the proposed mechanism for urea hydrolysis.305 The protein exists as a trimer of trimeric subunits (αβγ)3 with the dinickel cofactor in the α subunit, giving three active sites per trimeric complex.313,314 The two Ni2+ ions in each active site are located within ∼3 Å from one another in a weakly antiferromagnetically-coupled cluster.305,313,314 The coupling is achieved through two bridging ligands – a hydroxide and a post-translationally modified carbamylated lysine residue (Fig. 9b).314 The former acts as the nucleophile in urea hydrolysis and the latter is essential for nickel ion binding.315 The dinickel cofactor is asymmetrically coordinated with two histidine ligands at the Ni2+ ion in site 1 and two histidine residues and a monodentate aspartate residue ligating the Ni2+ ion in site 2 (Fig. 9b).313,314 In the resting state, the first coordination sphere of both sites includes a water ligand, resulting in a six-coordinate metal at site 2 and a five-coordinate metal at site 1 (Fig. 9b).314
 | ||
| Fig. 9 Microbial urea metabolism catalyzed by the dinickel enzyme urease. (a) Two-step breakdown of urea. (b) Proposed catalytic cycle of urease mediated by its dinickel (green spheres) cofactor. Active site depictions of inhibitor-bound structures that support the mechanism are shown as insets (PDB accession codes: H2O = 2UBP, acetohydroxamic acid, AHA = 4UBP, BO33− = 1S3T, diamidophosphate, DAP = 3UBP). | ||
In the presence of substrate, both water ligands are displaced and urea is proposed to bind directly via its carboxyl oxygen to the open coordination site of the nickel ion in site 1. This coordination is supported by the structure solved in the presence of the urease inhibitor, acetohydroxamic acid (AHA),316 which effectively mimics the bridging hydroxyl group and the urea structure (Fig. 9b). A conformational change is then proposed to induce coordination of the urea amine to the Ni2+ at site 2, poising the molecule as a μ-1,3-bridging ligand for attack by the bridging hydroxide. This bridging mode is analogous to that observed in the borate-inhibited structure (Fig. 9b).317 The structure with the inhibitor diamidophosphate (DAP) (Fig. 9b)314 has led to the proposal of a tetrahedral intermediate (or transition state) upon nucleophilic attack of the bridging hydroxide that is stabilized through an extensive hydrogen-bonding network. Finally, protonation of the coordinated amine by a nearby histidine residue is postulated to facilitate its elimination.305
Urease is one of just a handful of enzymes that use a nickel-containing metallocofactor.305,318,319 Most metal-dependent hydrolases utilize the non-redox active Zn2+ ion in mono-, di- and tri-nuclear clusters for catalysis. However, urease is highly specific for its dinuclear Ni2+ cofactor, as it is inactive with Zn2+ and other divalent metals.320,321 Both Ni2+ and Zn2+ have large positive charge density that can facilitate polarization of the hydroxide nucleophile and the substrate. However, the protein active site could favor intrinsic stereoelectronic properties of Ni2+ over Zn2+ ions.305,322 The d8 electronic configuration of Ni2+ favors an octahedral coordination sphere, whereas Zn2+ ion complexes in biology typically have tetrahedral geometry. Thus, the active site geometry could rearrange with Zn2+ ions to potentially preclude bridging of the water nucleophile and of the substrate. Conversely, the preference for an octahedral coordination sphere results in open Ni2+ coordination sites occupied by labile water molecules that can be displaced for proper positioning of the activated substrate with respect to the bridging hydroxide ligand to facilitate ureolysis.
The importance of the dinickel cofactor is also underscored by the suite of accessory proteins dedicated to its incorporation into the urease apo-protein.323 Four accessory proteins, UreDEFG, are often encoded in the same operon with the urease gene and are minimally essential for urease activity in most bacteria.323,324 UreD is postulated to act as a protein scaffold for a multicomponent complex of the apo-urease enzyme with the other accessory proteins.325–327 UreF binding to the apo-urease–UreD complex induces a conformational change that exposes the urease active site, providing access for the nickel ions and CO2 for lysine carbamylation.325,328 Lastly, UreG catalyzes GTP hydrolysis that is proposed to drive Ni2+ transfer from the metallochaperone UreE to the urease active site.329 Nickel binding sites have been identified in the UreE metallochaperone,330–332 as well as UreG and UreF proteins,333,334 suggesting a metal shuttling mechanism from UreE via UreG and UreF to the final destination in the urease active site.327 The UreDEFG proteins are sufficient to activate urease that is heterologously expressed in E. coli.335 However, in some native bacteria, additional proteins can play essential roles in urease nickel-activation, including nickel ion permeases, transporters, and binding proteins.324,336 Furthermore, there is an interesting, yet not fully understood, connection between the proteins responsible for biogenesis of the [NiFe]-hydrogenase cofactor (HydA and HydB) and the UreG and UreE proteins that is essential for urease activation in H. pylori.336–338
In addition to the urease enzyme itself, the importance of these accessory proteins in urease activity renders them each unique targets for inhibition of this metabolism. Inhibitors of urease activity have been studied for more than 50 years,339,340 but primarily have been applied to combat H. pylori gastric and Proteus urinary infections.341 More recently, efforts to minimize gut microbial urease activity via microbiota transplant have been directed toward the treatment of hyperammonemia.342 Introduction of a model gut community with minimal urease activity into mice promoted development of a more complex microflora with reduced urease activity342,343 and was shown to improve phenotypes associated with hyperammonemia.342 In a similar fashion, manipulation of the gut microbiota, or specific targeting of urease and its cofactor biosynthetic machinery, could be applied to the treatment of Crohn's disease, which is exacerbated by high urease activity.306
6.4 Hydrogen sulfide production
The typical gut microbiota in a healthy adult human is dominated by two phyla, Firmicutes and Bacteroides, representing up to 90% of all microbes.12,13 However, environmental factors, such as diet,344 can enable blooms of residents that are typically only present in low abundance. For example, the Deltaproteobacterium Bilophila wadsworthia is barely detectable in the average healthy human gut; however, in mice fed a typical high-fat Western diet, the population of B. wadsworthia expands.345 Importantly, this shift in community composition has been correlated with ulcerative colitis and colon cancer.345–347The genus name of this organism means “bile-loving”, hinting at the underlying cause of its expansion. Bile acids are produced exclusively by vertebrates to emulsify dietary lipids and fat-soluble vitamins, which facilitates their absorption in the intestines.348 They are also signaling molecules, acting as ligands to nuclear and G-protein coupled receptors that regulate lipid, glucose, and energy metabolism.348,349 These biomolecules are synthesized from cholesterol in the liver and conjugated with glycine and taurine to form salts that can then enter the digestive system.350 While some microbes are sensitive to the moderate antibacterial activity of bile acids,351 other gut microbes and pathogens transform these molecules into secondary bile acids through deconjugation, oxidation, and dehydroxylation reactions.352 Some of these secondary bile acids can be reabsorbed through enterohepatic circulation to accumulate in the liver, whereas others are excreted.352 High levels of these microbially-derived molecules in bile, blood, and feces have been correlated with gallstone disease and colon cancer.352 Although the purposes of bile acid oxidation and dehydroxylation are not entirely understood, the deconjugation of bile salts liberates free glycine and taurine, which can be sources of carbon, nitrogen, and sulfur, as well as substrates for energy production.
In the case of B. wadsworthia, the released taurine is metabolized to generate sulfite (Fig. 10a), which can be used as an electron acceptor for anaerobic respiration.353 Indeed, only taurine-conjugated bile salts induced a bloom of B. wadsworthia in IL10−/− deficient mice.345 This expansion can be recapitulated by a milk, saturated fat-based diet, as it stimulates increased production of taurine-conjugated bile salts.345 Notably, this diet and the associated expansion of B. wadsworthia have been connected with development of colitis in IL10−/− deficient mice.345 Although genetic susceptibility is essential in this model, the colonization of this pathobiont and its metabolic functions are causative in development of the observed disease phenotypes.
 | ||
| Fig. 10 Microbial taurine metabolism linked to human disease. (a) Deconjugation of taurine-bile salts by microbes releases free taurine, followed by reduction of the downstream metabolite sulfite to hydrogen sulfide as a means of anaerobic respiration. (b) Quaternary structure of the dissimilatory sulfite reductase (PDB accession code: 2V4J), composed of a dimer of trimeric subunits: DsrA (light green), DsrB (blue), and DsrC (red). Iron–sulfur clusters are depicted as spheres. (c) DsrB active site located at the interface with the DsrA subunit, depicting the siroheme cofactor bridged to one iron–sulfur cluster and a sulfite ligand. The C-terminal tail of the DsrC subunit is shown in red. (d) Proposed mechanism for the first two two-electron reduction steps mediated by the DsrABC complex that yields the protein-based trisulfide substrate for anaerobic respiration. | ||
The final product of taurine metabolism and sulfite respiration, hydrogen sulfide (Fig. 10a), has been detected at higher levels in patients with ulcerative colitis.354,355 While this observation might suggest a role of this gaseous molecule in inducing the disease, contradictory properties of hydrogen sulfide have complicated efforts to establish its causality in ulcerative colitis.356 Hydrogen sulfide is an inhibitor of host β-oxidation of microbially-produced short-chain fatty acids in colonocytes,357 preventing energy acquisition and compromising the epithelial barrier. It has also been shown to stimulate T-cell activation,358 which could induce antigen production against commensal microbes. Conversely, various reports have shown hydrogen sulfide to be anti-inflammatory and ameliorate colitis in animal models.359–361 Thus, the role of hydrogen sulfide in the development or mediation of inflammatory bowel disease remains to be definitively established.
Hydrogen sulfide is generated from the six-electron reduction of sulfite. In B. wadsworthia, this reaction is catalyzed by the metalloenzyme dissimilatory sulfite reductase (dSiR).362 Its structure consists of three subunits with α2β2γ2 stoichiometry (Fig. 10b), encoded by the genes dsrA, dsrB and dsrC, respectively. The DsrA and DsrB proteins are evolutionarily related as paralogs that likely resulted from gene duplication and divergent evolution to only maintain ∼20% sequence identity in contemporary bacteria. Thus, these two subunits form a structurally symmetric heterodimer, but only the DsrB subunit harbors a functional active site capable of sulfite reduction.363 The active site in the DsrB subunit, situated at the interface of the αβ dimer, contains a siroheme cofactor that is bridged via the axial cysteine ligand to a [4Fe–4S] cluster (Fig. 10c). In contrast, the corresponding site in the DsrA subunit contains a demetallated siroheme (termed sirohydrochlorin), lacks critical conserved residues, and has an occluded substrate channel. The role of the third subunit DsrC (γ) was contentious for some time; however, recent biochemical characterization of this protein, described below, revealed its essential role in sulfite reduction.364
The overall six-electron reduction of sulfite to hydrogen sulfide was originally proposed to occur by three consecutive two-electron reduction steps.365–367 However, at present the data suggest that the three steps occur by different mechanisms and with different electron sources. In the current proposal, the siroheme cofactor in DsrB activates the sulfite substrate and mediates the first two-electron reduction (Fig. 10d). This type of cofactor is found in all types of sulfite reductases (assimilatory and dissimilatory), as well as some nitrite reductases, underscoring its ability to mediate multi-electron reduction of inorganic anions.367 The two inorganic components, the iron ion of the siroheme and the [4Fe–4S] cluster, are electronically coupled through the bridging cysteine ligand,368 which enables the cofactor to effectively store two electron equivalents. The two reducing equivalents are provided by an unknown exogenous source and are thought to be transferred via an additional [4Fe–4S] cluster that is located close to the protein surface in a ferredoxin-like domain of the DsrB subunit.363 In the reduced state, the coupled iron–sulfur cluster is in the 1+ oxidation state and the siroheme has a high-spin Fe2+ ion with an open axial coordination site. The structure of the dSiR complex solved in the presence of substrate demonstrated direct sulfite ligation via the sulfur atom to the open coordination site of the DsrB siroheme with tetrahedral geometry (Fig. 10c).363 The substrate oxygen atoms are stabilized in the active site by positively-charged lysine and arginine residues. Coordination of the π-acceptor sulfite substrate to the electron-rich siroheme-Fe2+ weakens the S–O bond through π-backbonding, facilitating its cleavage upon reduction.365,366 This bond breaking step is likely also facilitated by protonation of the departing oxygen atom.
The next step in sulfite reduction is primarily mediated by the DsrC subunit. DsrC possesses two conserved cysteine residues at the C-terminal tail that are critical for conversion of sulfite to sulfide with fidelity.364 This tail is disordered in solution, but the structure of DsrAB in complex with DsrC showed the C-terminal tail positioned in a crevice between the αβ dimer in close proximity to the siroheme cofactor (Fig. 10c).363In vitro characterization revealed that this cysteine thiol forms a trisulfide with the sulfur atom derived from sulfite and the other conserved cysteine residue.369 This intermediate was rationalized to arise from nucleophilic attack of the deprotonated cysteine thiol on the two-electron reduced sulfite intermediate ligated to the siroheme (Fig. 10d). This step would result in dehydration and reduction of the intermediate to generate sulfur in the 1+ oxidation state, which would weaken the S–Fe3+ bond and induce dissociation. Attack by the other cysteine thiol would eliminate the final hydroxyl group to form the trisulfide intermediate (Fig. 10d).
The elucidation of this intermediate illuminated the key to sulfite respiration. The neutral oxidation state of the sulfite-derived sulfur atom in the trisulfide intermediate implies that the dissimilatory sulfite reductase system (DsrABC) only reduces sulfite by four electrons. The final two electrons needed to reduce the trisulfide are predicted to derive from the membrane-bound DsrMKJOP complex369 that had been previously implicated in sulfite reduction.364 The DsrK subunit is homologous to the heterodisulfide reductase HdrD in methanogens and, like HdrD, possesses a [4Fe–4S] cluster that could reduce the trisulfide intermediate.370,371 Involvement of this complex enables coupling of electron transfer and subsequent substrate reduction to the transfer of protons across the membrane, generating a proton motive force to drive energy production.369,371 This concept could not be rationalized previously invoking a six-electron reduction by the cytoplasmic DsrABC components alone because no membrane components were involved. In this model, DsrABC are responsible for the four-electron reduction of sulfite to the protein-based trisulfide, which then serves as the terminal electron acceptor for anaerobic respiration by the DsrMKJOP complex.
The complexity of this multi-component protein system is interesting in light of the observation that siroheme itself is competent for sulfite reduction to hydrogen sulfide in isolation with exogenous reductants.372,373 In this instance, Nature has evolved a mechanism to limit the involvement of the siroheme cofactor in order to generate energy from the final two-electron reduction step. Ultimately, the ability to anaerobically respire on taurine-derived sulfite gives B. wadsworthia a competitive advantage in the gut. However, this microbial adaptation can have inadvertent negative consequences for the host and provides a clear example of the complex interactions between external factors (i.e., diet), host biology, microbial metabolism, and human disease.
7 Microbial metabolism of xenobiotics
The human host and gut microbes are continually exposed to foreign substances, or xenobiotics, including pharmaceuticals, dietary bioactive molecules, and environmental chemicals. Transformation of pharmaceuticals by human enzymes is well-established, but the gut microbiota also plays a role in drug metabolism and can affect both drug availability and efficacy.374,375 However, in the vast majority of cases these transformations have not been connected to specific microbes and/or enzymes. Human enzymes that process xenobiotics typically catalyze oxidative and conjugative reactions to increase the polarity and molecular weight of these substances, thus facilitating their excretion.375 In contrast, microbial transformations of xenobiotics are most often hydrolytic and reductive, allowing the bacteria to access sources of nutrients and energy.374,375 The cancer drug irinotecan is a well-studied example of a pharmaceutical small molecule drug that undergoes a hydrolytic transformation by gut microbes to provide a source of carbon and energy. The active form of the drug is detoxified through glucuronidation by human liver enzymes, leading to its inactivation and excretion.376 However, as the glucuronide form traverses the GI tract, microbes in the large intestine cleave the sugar appendage through the action of β-glucuronidases, releasing it for further metabolism.376 This reaction reactivates the drug in the gut, causing severe side effects that limit its pharmacological use.376 Whereas this transformation is an example of an undesirable gut microbe–drug interaction, in other cases microbial metabolism is required for drug activation. For instance, the rheumatoid arthritis and ulcerative colitis drug sulfasalazine (Fig. 11a) is delivered as a prodrug with an azo moiety that is converted by microbial azoreductases to the biologically active agent.377–380 The metabolizing bacteria may be using sulfasalazine and other azo-containing drugs as terminal electron acceptors for anaerobic respiration to increase their energy production. However, the specific azoreductases for many of these prodrugs have not been discovered or characterized.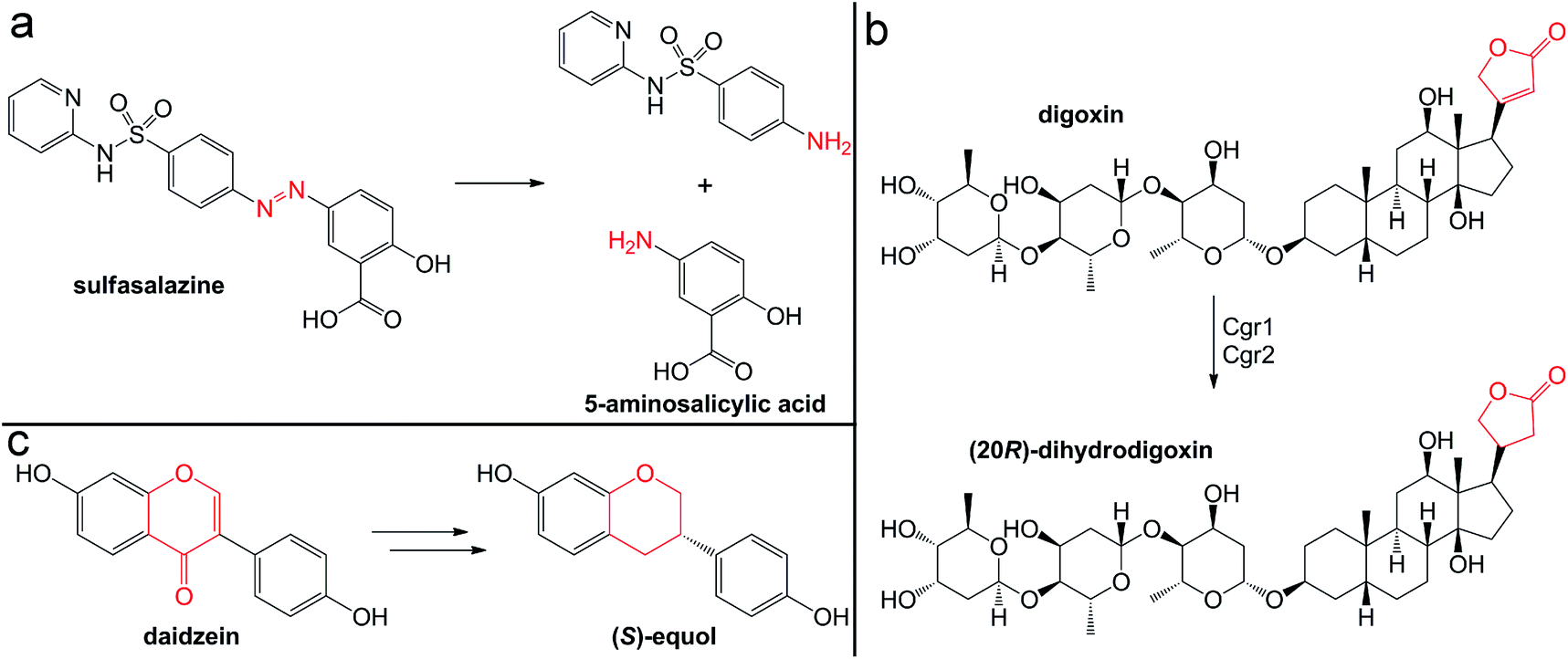 | ||
| Fig. 11 Reductive transformations of the xenobiotics (a) sulfasalazine, (b) digoxin, and (c) daidzein by gut microbes. | ||
The nature of reductive chemistry necessitates electron transfer, a common function of metallocofactors. Digoxin is a cardiac drug that is metabolized by strains of the gut microbe Eggerthella lenta, rendering it inactive.381,382 The reduction of digoxin to (20R)-dihydrodigoxin (Fig. 11b) is catalyzed by two proteins, termed Cgr1 and Cgr2.381,382 Cgr1 shares sequence homology with membrane-associated cytochrome proteins and is predicted to harbor multiple cytochrome c-type hemes.382 This protein is postulated to participate in electron delivery to Cgr2, which catalyzes the reduction of the α,β-unsaturated lactone moiety in digoxin.382 Cgr2 shares homology with flavin-dependent reductases and requires FAD for activity, but also harbors a [4Fe–4S] cluster that can potentially mediate electron transfer from the Cgr1 partner to the flavin cofactor for digoxin reduction.383 The presence of a [4Fe–4S] cluster in Cgr2 was not predicted bioinformatically from the protein sequence, and thus Cgr2 likely uses a novel fold to accommodate this metallocofactor. Interestingly, the reduction activity of Cgr2 is specific for substrates of the cardenolide structural family, which originate from plant sources.383 The lack of enzymatic activity toward additional endogenous host and dietary substrates suggests that this activity may not have arisen from promiscuous enzyme activity. One possible explanation could be that Cgr2 evolved to protect the host from the toxicity of these plant-derived compounds.383 This discovery portends that there may be other novel enzymes to uncover from the gut environment that have evolved specifically in response to xenobiotic exposure.
Similar reductive strategies are predicted in the transformation of dietary molecules, such as phytochemicals that are associated with altered disease risk. The soy-derived isoflavonoid, daidzein, is sequentially reduced by gut microbes to the estrogenic molecule, equol (Fig. 11c).384 Equol has been linked to reduced risk for breast and prostate cancers, likely due to its ability to act as a ligand for the human estrogen receptor.384 However, there is substantial variation among individuals in the ability of their gut microbiota to metabolize daidzein to equol.385 Equol-producing organisms possess a gene cluster that encodes for three essential enzymes that convert daidzein to equol.386–389 The first enzyme, daidzein reductase, catalyzes the initial flavin-dependent reduction to generate dihydrodaidzein.387 In addition, this enzyme is predicted to harbor a [4Fe–4S] cluster based on conservation of a cysteine-rich sequence motif.387 Although this hypothesis remains to be experimentally confirmed, an electron transfer role for this metallocofactor is easily envisioned in isoflavanoid reduction.
These examples are some of the few enzymes that have been connected to xenobiotic metabolism. Discovery of the organisms and enzymes responsible for other transformations will enable the identification of gene markers for metabolism that could be applied to predict an individual's response to different diets or drug therapies. It could also lead to development of approaches to enhance production of beneficial bioactive compounds and mitigate negative effects that influence drug bioavailability and efficacy. The metabolic capabilities of the gut microbiota, together with the variability in drug-metabolizing enzymes in the human genome, can have dramatic impacts on inter-individual response to medications and on development of precision medicine.
8 Metalloenzyme abundance in human microbiomes
Each of the isolated cases discussed in this review demonstrates the relevance of metalloenzymes in human gut microbial metabolism. However, the characterized enzymes from the gut microbiota represent a very small portion of the genetic potential associated with this community. To evaluate the prevalence and distribution of different classes of metalloenzymes in the human gut microbiota, we analyzed the stool metagenomes originating from the first phase of the NIH Human Microbiome Project (HMP), a study of the microbiotas of healthy adult individuals from the United States.13 We first categorized the genes identified in the assembled stool metagenomes into functional protein families using criteria from the Pfam protein family database (Fig. 12b).390 Only ∼40% of these genes can be assigned to a protein family (Fig. 12b). Table 1 shows the most highly represented families that use a metallocofactor (redox or non-redox) for their function (among proteins with representation greater than 1 out of every 1000 genes). An analogous analysis using criteria from the enzyme commission (EC) classification system, which is substantially more stringent, resulted in fewer annotations. Less than 6% of the genes in the assembled stool metagenomes could be assigned a level 4 EC function (Fig. 12b), which is the level needed to evaluate the use of a metallocofactor in catalysis. Therefore, this classification system was not suitable for assessing metalloenzyme prevalence and distribution. | ||
| Fig. 12 Analysis of metalloenzyme family prevalence and distribution in HMP metagenomes using the Pfam or EC classification systems. (a) Overview of analysis workflow. (b) Percentage of genes assigned a Pfam or EC number. (c) Pfam classes with representation in HMP stool metagenomes above a threshold of 1 hit/5000 genes and 1 hit/1000 genes (inset). The radical SAM superfamily, highlighted in red, is the eleventh highest represented family overall. (d) Prevalence of radical SAM enzyme encoding genes in the HMP assembled metagenomes from different body sites. Box plots are standard Tukey plots representing the median, 25th and 75th quartile values for radical SAM representation in the metagenomes of various human body sites. [oral (1) = buccal mucosa; oral (2) = supragingival plaque; oral (3) = tongue dorsum]. | ||
| Pfam | Pfam description | Mean representation in stool metagenomes (hits/1000 genes) |
|---|---|---|
| PF04055 | Radical SAM superfamily | 2.6 |
| PF00149 | Calcineurin-like phosphoesterase | 1.5 |
| PF01609 | Transposase DDE domain | 1.4 |
| PF00293 | NUDIX domain | 0.76 |
| PF01546 | Peptidase family M20/M25/M40 | 0.66 |
| PF13408 | Recombinase Zn β-ribbon domain | 0.65 |
| PF00383 | CMP/dCMP deaminase Zn-binding domain | 0.63 |
| PF01807 | CHC2-type Zn finger domain | 0.61 |
| PF01966 | HD domain | 0.61 |
| PF00557 | Metallopeptidase family M24 | 0.60 |
Using the Pfam classification analysis, we found that the radical SAM enzyme superfamily is the most prevalent metalloenzyme family in the human gut microbiome. This observation is perhaps unsurprising, as this family is highly represented in anaerobic bacteria, which dominate the healthy human GI tract. Radical SAM enzymes are found in every subject sampled with a mean representation of 2.6 hits per 1000 genes and a range of 1.5–3.5 hits per 1000 genes in metagenome assemblies (Fig. 12c and d). This high representation of radical SAM enzymes is not universal, as similar analyses of additional HMP metagenomes revealed the skin and vaginal microbiotas have a very low prevalence of radical SAM enzymes (Fig. 12d). While this observation might be expected because of the oxygen present in these habitats, the oral cavity, which includes both aerobic and anaerobic sites, has a high representation of radical SAM enzymes. Interestingly, this trend was also noted when assessing the abundance of genes encoding GREs, which are typically co-localized with radical SAM activating enzymes, in these same data sets.125 The prevalence of GREs in both the oral and gut cavities led the authors to posit that the oral cavity could be a reservoir for bacteria that can colonize the colon and have unique impacts in that niche.125 As demonstrated throughout this review, radical SAM enzymes are already known to play a wide range of roles in gut microbial metabolism. However, our analysis reveals that the majority of the radical SAM genes present in the stool metagenomes remain uncharacterized, as less than 40% have an assigned EC number. While this likely reflects the overall lack of characterization of this extensive superfamily (>300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 members), the biological context of this particular analysis could offer leads for uncharacterized enzymes to prioritize for further study.
000 members), the biological context of this particular analysis could offer leads for uncharacterized enzymes to prioritize for further study.
Inclusive of and beyond metalloenzyme families, the vast amount of genetic data from individual organisms and microbial communities creates a challenge for identifying and prioritizing enzymes for study. With respect to the gut microbiota, the enzymes and metabolic functions that have an impact on human health and disease are obvious candidates of interest. One approach to uncover potentially disease-associated activities is to compare metagenomic, metatranscriptomic, metaproteomic, and metabolomic data sets from healthy and patient populations. This type of analysis could identify differentially regulated genes, expressed proteins, and microbial metabolites that may contribute to disease risk or development as well as provide support for proposed associations between gut microbial metabolism and host health and disease. Other experimental approaches such as activity-based functional profiling can be impactful tools for the discovery of novel enzymes by leveraging their chemical properties. Although not widely applied to gut microbial communities as of yet, this technique was used to enhance a comparative metaproteomic study of a mouse model of IBD using a cysteine nucleophile-reactive probe to assess host and microbial peptidase profiles.391 The quantification of gene abundance may be a critical component for assessing relevance in the gut microbiota. An approach termed chemically-guided functional profiling, which uses the computational tool ShortBRED392 in conjunction with Sequence Similarity Network analysis,393 has been applied to assess the abundance of GREs in the HMP metagenomes.125 This analysis identified a highly abundant, previously uncharacterized enzyme that was found to catalyze the dehydration of 4-hydroxy-L-proline.125 Another avenue is to apply well-established computational tools for identifying biosynthetic gene clusters to discover novel secondary metabolites produced by gut microbes. For example, the bioinformatics tool ClusterFinder394 has been successfully deployed to analyze the HMP metagenomes for multiple types of biosynthetic gene clusters.149 This work identified a diverse set of over 3000 putative biosynthetic gene clusters that are highly represented in the gut and oral microbiotas and are largely uncharacterized. Further studies of these cryptic gene clusters could reveal novel natural products with potential therapeutic applications, as well as improve our fundamental understanding of host–microbe or microbe–microbe interactions.
9 Conclusions
Recognition of the complex relationship between the human host and its gut microbial residents has redefined our perception of human health. In order to progress the field beyond the present correlative connections between the microbiota and human health and disease, the specific metabolic functions of these organisms must be uncovered. Integrating diverse and innovative experimental and computational approaches will enable the discovery of microbial chemical transformations and products that influence host biology. Biochemists and chemical biologists are therefore uniquely poised to increase our mechanistic understanding of the human gut microbiota and devise approaches for therapeutically targeting these organisms to improve human health.10 Conflicts of interest
There are no conflicts of interest to declare.11 Acknowledgements
The authors would like to acknowledge Abraham Waldman for helpful comments during writing and Eric Franzosa for assistance with the HMP metagenomic analysis. We acknowledge funding from the Bill and Melinda Gates Foundation (OPP1158186) (HMMI-Gates Faculty Scholar Award to E. P. B.). L. J. R. is the recipient of a Helen Hay Whitney postdoctoral research fellowship.12 References
- C. A. Thaiss, N. Zmora, M. Levy and E. Elinav, Nature, 2016, 535, 65–74 CrossRef CAS PubMed.
- K. Honda and D. R. Littman, Nature, 2016, 535, 75–84 CrossRef CAS PubMed.
- H. K. Biesalski, Ann. N. Y. Acad. Sci., 2016, 1372, 53–64 CrossRef PubMed.
- A. J. Bäumler and V. Sperandio, Nature, 2016, 535, 85–93 CrossRef PubMed.
- J. A. Gilbert, R. A. Quinn, J. Debelius, Z. Z. Xu, J. Morton, N. Garg, J. K. Jansson, P. C. Dorrestein and R. Knight, Nature, 2016, 535, 94–103 CrossRef CAS PubMed.
- C. L. Sears and W. S. Garrett, Cell Host Microbe, 2014, 15, 317–328 CrossRef CAS PubMed.
- A. D. Kostic, R. J. Xavier and D. Gevers, Gastroenterology, 2014, 146, 1489–1499 CrossRef CAS PubMed.
- P. D. Cani and N. M. Delzenne, Curr. Pharm. Des., 2009, 15, 1546–1558 CrossRef CAS PubMed.
- H. Tremlett, K. C. Bauer, S. Appel-Cresswell, B. B. Finlay and E. Waubant, Ann. Neurol., 2017, 81, 369–382 CrossRef PubMed.
- W. H. Tang, T. Kitai and S. L. Hazen, Circ. Res., 2017, 120, 1183–1196 CrossRef CAS PubMed.
- J. W. Arnold, J. Roach and M. A. Azcarate-Peril, Trends Microbiol., 2016, 24, 887–901 CrossRef CAS PubMed.
- P. B. Eckburg, E. M. Bik, C. N. Bernstein, E. Purdom, L. Dethlefsen, M. Sargent, S. R. Gill, K. E. Nelson and D. A. Relman, Science, 2005, 308, 1635–1638 CrossRef PubMed.
- The Human Microbiome Project Consortium, Nature, 2012, 486, 207–214 CrossRef PubMed.
- T. Yatsunenko, F. E. Rey, M. J. Manary, I. Trehan, M. G. Dominguez-Bello, M. Contreras, M. Magris, G. Hidalgo, R. N. Baldassano, A. P. Anokhin, A. C. Heath, B. Warner, J. Reeder, J. Kuczynski, J. G. Caporaso, C. A. Lozupone, C. Lauber, J. C. Clemente, D. Knights, R. Knight and J. I. Gordon, Nature, 2012, 486, 222–227 CrossRef CAS PubMed.
- C. A. Lozupone, J. I. Stombaugh, J. I. Gordon, J. K. Jansson and R. Knight, Nature, 2012, 489, 220–230 CrossRef CAS PubMed.
- A. Heintz-Buschart and P. Wilmes, Trends Microbiol., 2017, 26(7), 563–574 CrossRef PubMed.
- B. Zhu, X. Wang and L. Li, Protein Cell, 2010, 1, 718–725 CrossRef PubMed.
- J. Qin, R. Li, J. Raes, M. Arumugam, K. S. Burgdorf, C. Manichanh, T. Nielsen, N. Pons, F. Levenez, T. Yamada, D. R. Mende, J. Li, J. Xu, S. Li, D. Li, J. Cao, B. Wang, H. Liang, H. Zheng, Y. Xie, J. Tap, P. Lepage, M. Bertalan, J. M. Batto, T. Hansen, D. Le Paslier, A. Linneberg, H. B. Nielsen, E. Pelletier, P. Renault, T. Sicheritz-Ponten, K. Turner, H. Zhu, C. Yu, S. Li, M. Jian, Y. Zhou, Y. Li, X. Zhang, S. Li, N. Qin, H. Yang, J. Wang, S. Brunak, J. Dore, F. Guarner, K. Kristiansen, O. Pedersen, J. Parkhill, J. Weissenbach, H. I. T. C. Meta, P. Bork, S. D. Ehrlich and J. Wang, Nature, 2010, 464, 59–65 CrossRef CAS PubMed.
- M. S. Donia and M. A. Fischbach, Science, 2015, 349, 1254766 CrossRef PubMed.
- L. K. Ursell, H. J. Haiser, W. Van Treuren, N. Garg, L. Reddivari, J. Vanamala, P. C. Dorrestein, P. J. Turnbaugh and R. Knight, Gastroenterology, 2014, 146, 1470–1476 CrossRef CAS PubMed.
- J. K. Nicholson, E. Holmes, J. Kinross, R. Burcelin, G. R. Gibson, W. Jia and S. Pettersson, Science, 2012, 336, 1262–1267 CrossRef CAS PubMed.
- E. Holmes, J. V. Li, T. Athanasiou, H. Ashrafian and J. K. Nicholson, Trends Microbiol., 2011, 19, 349–359 CrossRef CAS PubMed.
- N. Koppel and E. P. Balskus, Cell Chem Biol, 2016, 23, 18–30 CrossRef CAS PubMed.
- B. D. Wallace, H. Wang, K. T. Lane, J. E. Scott, J. Orans, J. S. Koo, M. Venkatesh, C. Jobin, L. A. Yeh, S. Mani and M. R. Redinbo, Science, 2010, 330, 831–835 CrossRef CAS PubMed.
- R. Joice, K. Yasuda, A. Shafquat, X. C. Morgan and C. Huttenhower, Cell Metab., 2014, 20, 731–741 CrossRef CAS PubMed.
- W. Buckel and B. T. Golding, Annu. Rev. Microbiol., 2006, 60, 27–49 CrossRef CAS PubMed.
- N. M. Koropatkin, E. A. Cameron and E. C. Martens, Nat. Rev. Microbiol., 2012, 10, 323–335 CrossRef CAS PubMed.
- E. C. Martens, E. C. Lowe, H. Chiang, N. A. Pudlo, M. Wu, N. P. McNulty, D. W. Abbott, B. Henrissat, H. J. Gilbert, D. N. Bolam and J. I. Gordon, PLoS Biol., 2011, 9, e1001221 CrossRef CAS PubMed.
- M. E. Johansson, H. Sjovall and G. C. Hansson, Nat. Rev. Gastroenterol. Hepatol., 2013, 10, 352–361 CrossRef CAS PubMed.
- L. E. Tailford, E. H. Crost, D. Kavanaugh and N. Juge, Front. Genet., 2015, 6, 81 Search PubMed.
- D. A. Ravcheev and I. Thiele, Front. Genet., 2017, 8, 111 CrossRef PubMed.
- M. M. Grondin, K. Tamura, G. Déjean, D. W. Abbott and H. Brumer, J. Bacteriol., 2017, 199, e00860-16 CrossRef PubMed.
- M. A. McGuckin, S. K. Linden, P. Sutton and T. H. Florin, Nat. Rev. Microbiol., 2011, 9, 265–278 CrossRef CAS PubMed.
- T. Bhattacharya, T. S. Ghosh and S. S. Mande, PLoS One, 2015, 10, e0142038 CrossRef PubMed.
- W. E. C. Moore and L. V. Holdeman, Appl. Microbiol., 1974, 27, 961–979 CAS.
- A. A. Salyers, J. R. Vercellotti, S. E. H. West and T. D. Wilkins, Appl. Environ. Microbiol., 1977, 33, 319–322 CAS.
- E. C. Martens, H. C. Chiang and J. I. Gordon, Cell Host Microbe, 2008, 4, 447–457 CrossRef CAS PubMed.
- E. C. Martens, N. M. Koropatkin, T. J. Smith and J. I. Gordon, J. Biol. Chem., 2009, 284, 24673–24677 CrossRef CAS PubMed.
- J. L. Sonnenburg, J. Xu, D. D. Leip, C.-H. Chen, B. P. Westover, J. Weatherford, J. D. Buhler and J. I. Gordon, Science, 2005, 307, 1955–1959 CrossRef CAS PubMed.
- D. K. Podolsky, J. Biol. Chem., 1985, 260, 8262–8271 CAS.
- S. Inoue and Z. Yosizawa, Arch. Biochem. Biophys., 1966, 117, 257–265 CrossRef CAS PubMed.
- M. Filipe, Invest. Cell Pathol., 1979, 2, 195–216 CAS.
- A. Nieuw Amerongen, J. Bolscher, E. Bloemena and E. Veerman, Biol. Chem., 1998, 379, 1–18 CAS.
- N. Mian, C. E. Anderson and P. W. Kent, Biochem. J., 1979, 181, 387–399 CrossRef CAS PubMed.
- A. P. Corfield, S. A. Wagner, L. J. D. O'Donnell, P. Durdey, R. A. Mountford and J. R. Clamp, Glycoconjugate J., 1993, 10, 72–81 CrossRef CAS PubMed.
- H. H. Tsai, D. Sunderland, G. R. Gibson, C. A. Hart and J. M. Rhodes, Clin. Sci., 1992, 82, 447–454 CrossRef CAS PubMed.
- D. P. Wright, C. G. Knight, S. G. Parkar, D. L. Christie and A. M. Roberton, J. Bacteriol., 2000, 182, 3002–3007 CrossRef CAS PubMed.
- J. Xu, M. A. Mahowald, R. E. Ley, C. A. Lozupone, M. Hamady, E. C. Martens, B. Henrissat, P. M. Coutinho, P. Minx, P. Latrelle, H. Cordum, A. V. Brunt, K. Kim, R. S. Fulton, L. A. Fulton, S. W. Clifton, R. K. Wilson, R. D. Knight and J. I. Gordon, PLoS Biol., 2007, 5, e156 CrossRef PubMed.
- J. E. Ulmer, E. M. Vilen, R. B. Namburi, A. Benjdia, J. Beneteau, A. Malleron, D. Bonnaffe, P. A. Driguez, K. Descroix, G. Lassalle, C. Le Narvor, C. Sandstrom, D. Spillmann and O. Berteau, J. Biol. Chem., 2014, 289, 24289–24303 CrossRef CAS PubMed.
- M. Egan, H. Jiang, M. O'Connell Motherway, S. Oscarson and D. van Sinderen, Appl. Environ. Microbiol., 2016, 82, 6611–6623 CrossRef CAS PubMed.
- A. Cartmell, E. C. Lowe, A. Basle, S. J. Firbank, D. A. Ndeh, H. Murray, N. Terrapon, V. Lombard, B. Henrissat, J. E. Turnbull, M. Czjzek, H. J. Gilbert and D. N. Bolam, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 7037–7042 CrossRef CAS PubMed.
- C. A. Hickey, K. A. Kuhn, D. L. Donermeyer, N. T. Porter, C. Jin, E. A. Cameron, H. Jung, G. E. Kaiko, M. Wegorzewska, N. P. Malvin, R. W. Glowacki, G. C. Hansson, P. M. Allen, E. C. Martens and T. S. Stappenbeck, Cell Host Microbe, 2015, 17, 672–680 CrossRef CAS PubMed.
- H. H. Tsai, A. D. Dwarakanath, C. A. Hart, J. D. Milton and J. M. Rhodes, Gut, 1995, 36, 570–576 CrossRef CAS PubMed.
- T. Barbeyron, L. Brillet-Gueguen, W. Carre, C. Carriere, C. Caron, M. Czjzek, M. Hoebeke and G. Michel, PLoS One, 2016, 11, e0164846 CrossRef PubMed.
- A. Benjdia and O. Berteau, Biochem. Soc. Trans., 2016, 44, 109–115 CrossRef CAS PubMed.
- M. J. Appel and C. R. Bertozzi, ACS Chem. Biol., 2015, 10, 72–84 CrossRef CAS PubMed.
- I. Boltes, H. Czapinska, A. Kahner, R. v. Bülow, T. Dierks, B. Schmidt, K. v. Figura, M. A. Kertesz and I. Usón, Structure, 2001, 9, 483–491 CrossRef CAS PubMed.
- R. v. Bülow, B. Schmidt, T. Dierks, K. v. Figura and I. Usón, J. Mol. Biol., 2001, 305, 269–277 CrossRef PubMed.
- C. S. Bond, P. R. Clements, S. J. Ashby, C. A. Collyer, S. J. Harrop, J. J. Hopwood and J. M. Guss, Structure, 1996, 5, 277–289 CrossRef.
- F. G. Hernandez-Guzman, T. Higashiyama, W. Pangborn, Y. Osawa and D. Ghosh, J. Biol. Chem., 2003, 278, 22989–22997 CrossRef CAS PubMed.
- T. Dierks, A. Dickmanns, A. Preusser-Kunze, B. Schmidt, M. Mariappan, K. von Figura, R. Ficner and M. G. Rudolph, Cell, 2005, 121, 541–552 CrossRef CAS PubMed.
- A. Schirmer and R. Kolter, Chem. Biol., 1998, 5, R181–R186 CrossRef CAS PubMed.
- T. Dierks, C. Miech, J. Hummerjohann, B. Schmidt, M. A. Kertesz and K. von Figura, J. Biol. Chem., 1998, 273, 25560–25564 CrossRef CAS PubMed.
- O. Berteau, A. Guillot, A. Benjdia and S. Rabot, J. Biol. Chem., 2006, 281, 22464–22470 CrossRef CAS PubMed.
- C. Miech, T. Dierks, T. Selmer, K. v. Figura and B. Schmidt, J. Biol. Chem., 1998, 273, 4835–4837 CrossRef CAS PubMed.
- C. Marquordt, Q. Fang, E. Will, J. Peng, K. von Figura and T. Dierks, J. Biol. Chem., 2003, 278, 2212–2218 CrossRef CAS PubMed.
- D. Roeser, A. Preusser-Kunze, B. Schmidt, K. Gasow, J. G. Wittmann, T. Dierks, K. von Figura and M. G. Rudolph, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 81–86 CrossRef CAS PubMed.
- B. L. Carlson, E. R. Ballister, E. Skordalakes, D. S. King, M. A. Breidenbach, S. A. Gilmore, J. M. Berger and C. R. Bertozzi, J. Biol. Chem., 2008, 283, 20117–20125 CrossRef CAS PubMed.
- C. Szameit, C. Miech, M. Balleininger, B. Schmidt, K. von Figura and T. Dierks, J. Biol. Chem., 1999, 274, 15375–15381 CrossRef CAS PubMed.
- A. Benjdia, G. Deho, S. Rabot and O. Berteau, FEBS Lett., 2007, 581, 1009–1014 CrossRef CAS PubMed.
- Q. Fang, J. Peng and T. Dierks, J. Biol. Chem., 2004, 279, 14570–14578 CrossRef CAS PubMed.
- J. B. Broderick, B. R. Duffus, K. S. Duschene and E. M. Shepard, Chem. Rev., 2014, 114, 4229–4317 CrossRef CAS PubMed.
- P. A. Frey, A. D. Hegeman and F. J. Ruzicka, Crit. Rev. Biochem. Mol. Biol., 2008, 43, 63–88 CrossRef CAS PubMed.
- S. C. Wang and P. A. Frey, Trends Biochem. Sci., 2007, 32, 101–110 CrossRef CAS PubMed.
- S. J. Booker and T. L. Grove, F1000 Biology Reports, 2010, 2, 52 Search PubMed.
- A. Benjdia, J. Leprince, C. Sandstrom, H. Vaudry and O. Berteau, J. Am. Chem. Soc., 2009, 131, 8348–8349 CrossRef CAS PubMed.
- T. L. Grove, J. H. Ahlum, R. M. Qin, N. D. Lanz, M. I. Radle, C. Krebs and S. J. Booker, Biochemistry, 2013, 52, 2874–2887 CrossRef CAS PubMed.
- T. A. Grell, P. J. Goldman and C. L. Drennan, J. Biol. Chem., 2015, 290, 3964–3971 CrossRef CAS PubMed.
- T. L. Grove, K.-H. Lee, J. S. Clair, C. Krebs and S. J. Booker, Biochemistry, 2008, 47, 7523–7538 CrossRef CAS PubMed.
- P. J. Goldman, T. L. Grove, L. A. Sites, M. I. McLaughlin, S. J. Booker and C. L. Drennan, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8519–8524 CrossRef CAS PubMed.
- T. Dierks, M. R. Lecca, P. Schlotterhose, B. Schmidt and K. von Figura, EMBO J., 1999, 18, 2084–2091 CrossRef CAS PubMed.
- A. Benjdia, E. C. Martens, J. I. Gordon and O. Berteau, J. Biol. Chem., 2011, 286, 25973–25982 CrossRef CAS PubMed.
- P. C. Kashyap, A. Marcobal, L. K. Ursell, S. A. Smits, E. D. Sonnenburg, E. K. Costello, S. K. Higginbottom, S. E. Domino, S. P. Holmes, D. A. Relman, R. Knight, J. I. Gordon and J. L. Sonnenburg, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17059–17064 CrossRef CAS PubMed.
- R. J. Kelly, S. Rouquier, D. Giorgi, G. G. Lennon and J. B. Lowe, J. Biol. Chem., 1995, 270, 4640–4649 CrossRef CAS PubMed.
- D. P. McGovern, M. R. Jones, K. D. Taylor, K. Marciante, X. Yan, M. Dubinsky, A. Ippoliti, E. Vasiliauskas, D. Berel, C. Derkowski, D. Dutridge, P. Fleshner, D. Q. Shih, G. Melmed, E. Mengesha, L. King, S. Pressman, T. Haritunians, X. Guo, S. R. Targan, J. I. Rotter and I. B. D. G. C. International, Hum. Mol. Genet., 2010, 19, 3468–3476 CrossRef CAS PubMed.
- P. Rausch, A. Rehman, S. Künzel, R. Häsler, S. J. Ott, S. Schreiber, P. Rosenstiel, A. Franke and J. F. Baines, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 19030–19035 CrossRef CAS PubMed.
- L. Maroni, S. F. van de Graaf, S. D. Hohenester, R. P. Oude Elferink and U. Beuers, Clin. Rev. Allergy Immunol., 2015, 48, 182–191 CrossRef CAS PubMed.
- L. Bry, P. G. Falk, T. Midtvedt and J. I. Gordon, Science, 1996, 273, 1380–1383 CrossRef CAS PubMed.
- L. V. Hooper, J. Xu, P. G. Falk, T. Midtvedt and J. I. Gordon, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9833–9838 CrossRef CAS.
- J. M. Pickard, C. F. Maurice, M. A. Kinnebrew, M. C. Abt, D. Schenten, T. V. Golovkina, S. R. Bogatyrev, R. F. Ismagilov, E. G. Pamer, P. J. Turnbaugh and A. V. Chervonsky, Nature, 2014, 514, 638–641 CrossRef CAS PubMed.
- M. J. Coyne, B. Reinap, M. M. Lee and L. E. Comstock, Science, 2005, 307, 1778–1781 CrossRef CAS PubMed.
- K. M. Ng, J. A. Ferreyra, S. K. Higginbottom, J. B. Lynch, P. C. Kashyap, S. Gopinath, N. Naidu, B. Choudhury, B. C. Weimer, D. M. Monack and J. L. Sonnenburg, Nature, 2013, 502, 96–99 CrossRef CAS PubMed.
- J. Badía, J. Ros and J. Aguilar, J. Bacteriol., 1985, 161, 435–437 Search PubMed.
- R. M. Jeter, J. Gen. Microbiol., 1990, 136, 887–896 CrossRef CAS PubMed.
- M. A. Vinolo, H. G. Rodrigues, R. T. Nachbar and R. Curi, Nutrients, 2011, 3, 858–876 CrossRef CAS PubMed.
- J. Tan, C. McKenzie, M. Potamitis, A. N. Thorburn, C. R. Mackay and L. Macia, Adv. Immunol., 2014, 121, 91–119 CAS.
- M. Kasubuchi, S. Hasegawa, T. Hiramatsu, A. Ichimura and I. Kimura, Nutrients, 2015, 7, 2839–2849 CrossRef CAS PubMed.
- D. J. Morrison and T. Preston, Gut Microbes, 2016, 7, 189–200 CrossRef PubMed.
- G. den Besten, K. van Eunen, A. K. Groen, K. Venema, D. J. Reijngoud and B. M. Bakker, J. Lipid Res., 2013, 54, 2325–2340 CrossRef CAS PubMed.
- E. Hosseini, C. Grootaert, W. Verstraete and T. Van de Wiele, Nutr. Rev., 2011, 69, 245–258 CrossRef PubMed.
- H. A. Lee, Jr. and R. H. Abeles, J. Biol. Chem., 1963, 238, 2367–2373 Search PubMed.
- H. A. Barker, Annu. Rev. Biochem., 1972, 784, 55–90 CrossRef PubMed.
- R. H. Abeles and D. Dolphin, Acc. Chem. Res., 1975, 9, 114–120 CrossRef.
- T. Toraya, Chem. Rev., 2003, 103, 2095–2127 CrossRef CAS PubMed.
- M. Yamanishi, M. Yunoki, T. Tobimatsu, H. Sato, J. Matsui, A. Dokiya, Y. Iuchi, K. Oe, K. Suto, N. Shibata, Y. Morimoto, N. Yasuoka and T. Toraya, Eur. J. Biochem., 2002, 269, 4484–4494 CrossRef CAS PubMed.
- D. C. Hodgkin, J. Kamper, M. Mackay and J. Pickworth, Nature, 1956, 178, 64–66 CrossRef CAS PubMed.
- R. Bonnnett, J. R. Cannon, V. M. Clark, A. W. Johnson, L. F. J. Parker, E. L. Smith and S. A. Todd, J. Chem. Soc., 1957, 227, 1158–1168 RSC.
- J. Pilbrow, EPR of B12-dependent enzyme reactions and related systems, John Wiley & Sons, Inc., New York, 1982 Search PubMed.
- M. Yamanishi, S. Yamada, H. Muguruma, Y. Murakami, T. Tobimatsu, A. Ishida, J. Yamauchi and T. Toraya, Biochemistry, 1998, 37, 4799–4803 CrossRef CAS PubMed.
- A. Abend, R. Nitsche, V. Bandarian, E. Stupperich and J. Rétey, Angew. Chem., Int. Ed., 1998, 37, 625–627 CrossRef CAS PubMed.
- N. Shibata, J. Masuda, T. Tobimatsu, T. Toraya, K. Suto, Y. Morimoto and N. Yasuoka, Structure, 1999, 7, 997–1008 CrossRef CAS PubMed.
- J. Masuda, N. Shibata, Y. Morimoto, T. Toraya and N. Yasuoka, Structure, 2000, 8, 775–788 CrossRef CAS PubMed.
- E. N. G. Marsh and G. D. Melendez, Biochim. Biophys. Acta, 2012, 1824, 1154–1164 CrossRef CAS PubMed.
- T. Toraya, Cell. Mol. Life Sci., 2000, 57, 106–127 CrossRef CAS PubMed.
- P. A. Schwartz and P. A. Frey, Biochemistry, 2007, 46, 7293–7301 CrossRef CAS PubMed.
- T. Toraya, S. Honda and K. Mori, Biochemistry, 2010, 49, 7210–7217 CrossRef CAS PubMed.
- T. Toraya, K. Yoshizawa, M. Eds and T. Yamabe, J. Biochem., 1999, 126, 650–654 CrossRef CAS PubMed.
- M. Yamanishi, H. Ide, Y. Murakami and T. Toraya, Biochemistry, 2005, 44, 2113–2118 CrossRef CAS PubMed.
- V. B. Pett, M. N. Liebman, P. Murray-Rust, K. Prasad and J. P. Glusker, J. Am. Chem. Soc., 1987, 109, 3207–3215 CrossRef CAS.
- J. Rétey, A. Umani-Ronchi, J. Seible and D. Arigoni, Experientia, 1966, 22, 502–503 CrossRef.
- B. J. Levin and E. P. Balskus, Biochemistry, 2018, 57(23), 3222–3226 CrossRef CAS PubMed.
- O. W. Wagner, H. A. Lee, Jr., P. A. Frey and R. H. Abeles, J. Biol. Chem., 1966, 241, 1751–1762 CAS.
- P. A. Frey, M. K. Essenberg and R. H. Abeles, J. Biol. Chem., 1967, 242, 5369–5377 CAS.
- J. W. LaMattina, N. D. Keul, P. Reitzer, S. Kapoor, F. Galzerani, D. J. Koch, I. E. Gouvea and W. N. Lanzilotta, J. Biol. Chem., 2016, 291, 15515–15526 CrossRef CAS PubMed.
- B. J. Levin, Y. Y. Huang, S. C. Peck, Y. Wei, A. Martinez-Del Campo, J. A. Marks, E. A. Franzosa, C. Huttenhower and E. P. Balskus, Science, 2017, 355(6325), eaai8386 CrossRef PubMed.
- K. P. Scott, J. C. Martin, G. Campbell, C. D. Mayer and H. J. Flint, J. Bacteriol., 2006, 188, 4340–4349 CrossRef CAS PubMed.
- T. Selmer, A. J. Pierik and J. Heider, Biol. Chem., 2005, 386, 981–988 CAS.
- K. A. Shisler and J. B. Broderick, Arch. Biochem. Biophys., 2014, 546, 64–71 CrossRef CAS PubMed.
- S. G. Reddy, K. K. Wong, C. V. Parast, J. Peisach, R. S. Magliozzo and J. W. Kozarich, Biochemistry, 1998, 37, 558–563 CrossRef CAS PubMed.
- M. Feliks and G. M. Ullmann, J. Phys. Chem. B, 2012, 116, 7076–7087 CrossRef CAS PubMed.
- B. Kovačević, D. Barić, D. Babić, L. Bilić, M. Hanževački, G. M. Sandala, L. Radom and D. M. Smith, J. Am. Chem. Soc., 2018, 140, 8487–8496 CrossRef PubMed.
- F. Rivera-Chávez, L. F. Zhang, F. Faber, C. A. Lopez, M. X. Byndloss, E. E. Olsan, G. Xu, E. M. Velazquez, C. B. Lebrilla, S. E. Winter and A. J. Bäumler, Cell Host Microbe, 2016, 19, 443–454 CrossRef PubMed.
- C. A. Lopez, B. M. MIller, F. Rivera-Chávez, E. M. Velazquez, M. X. Byndloss, A. Chávez-Arroyo, K. L. Lokken, R. M. Tsolis, S. E. Winter and A. J. Bäumler, Science, 2016, 353, 1249–1253 CrossRef CAS PubMed.
- L. Staib and T. M. Fuchs, Front. Microbiol., 2015, 6, 1116 Search PubMed.
- F. Faber, P. Thiennimitr, L. Spiga, M. X. Byndloss, Y. Litvak, S. Lawhon, H. L. Andrews-Polymenis, S. E. Winter and A. J. Baumler, PLoS Pathog., 2017, 13, e1006129 CrossRef PubMed.
- C. G. Buffie and E. G. Pamer, Nat. Rev. Immunol., 2013, 13, 790–801 CrossRef CAS PubMed.
- N. Kamada, G. Y. Chen, N. Inohara and G. Nunez, Nat. Immunol., 2013, 14, 685–690 CrossRef CAS PubMed.
- M. G. Rooks and W. S. Garrett, Nat. Rev. Immunol., 2016, 16, 341–352 CrossRef CAS PubMed.
- A. Agus, J. Denizot, J. Thevenot, M. Martinez-Medina, S. Massier, P. Sauvanet, A. Bernalier-Donadille, S. Denis, P. Hofman, R. Bonnet, E. Billard and N. Barnich, Sci. Rep., 2016, 6, 19032 CrossRef CAS PubMed.
- L. K. Ursell, W. Van Treuren, J. L. Metcalf, M. Pirrung, A. Gewirtz and R. Knight, BioEssays, 2013, 35, 810–817 CrossRef PubMed.
- S. R. Modi, J. J. Collins and D. A. Relman, J. Clin. Invest., 2014, 124, 4212–4218 CrossRef CAS PubMed.
- G. Cammarota, G. Ianiro and A. Gasbarrini, J. Clin. Gastroenterol., 2014, 48, 693–702 CrossRef PubMed.
- T. J. Borody and A. Khoruts, Nat. Rev. Gastroenterol. Hepatol., 2011, 9, 88–96 CrossRef PubMed.
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K. D. Entian, M. A. Fischbach, J. S. Garavelli, U. Goransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Muller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. Reaney, S. Rebuffat, R. P. Ross, H. G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Sussmuth, J. R. Tagg, G. L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Nat. Prod. Rep., 2013, 30, 108–160 RSC.
- S. Kommineni, D. J. Bretl, V. Lam, R. Chakraborty, M. Hayward, P. Simpson, Y. Cao, P. Bousounis, C. J. Kristich and N. H. Salzman, Nature, 2015, 526, 719–722 CrossRef CAS PubMed.
- M. A. Ortega and W. A. van der Donk, Cell Chem. Biol., 2016, 23, 31–44 CrossRef CAS PubMed.
- K. J. Hetrick and W. A. van der Donk, Curr. Opin. Chem. Biol., 2017, 38, 36–44 CrossRef CAS PubMed.
- A. J. Marsh, O. O'Sullivan, R. R. Paul, P. D. Cotter and C. Hill, BMC Genomics, 2010, 11, 679 CrossRef CAS PubMed.
- M. S. Donia, P. Cimermancic, C. J. Schulze, L. C. Wieland Brown, J. Martin, M. Mitreva, J. Clardy, R. G. Linington and M. A. Fischbach, Cell, 2014, 158, 1402–1414 CrossRef CAS PubMed.
- A.-C. Letzel, S. J. Pidot and C. Hertweck, BMC Genomics, 2014, 15, 983 CrossRef PubMed.
- C. J. Walsh, C. M. Guinane, C. Hill, R. P. Ross, P. W. O'Toole and P. D. Cotter, BMC Microbiol., 2015, 15, 183 CrossRef PubMed.
- J. Zheng, M. G. Ganzle, X. B. Lin, L. Ruan and M. Sun, Environ. Microbiol., 2015, 17, 2133–2143 CrossRef CAS PubMed.
- S. van Kuijk, K. S. Noll and M. L. Chikindas, Lett. Appl. Microbiol., 2012, 54, 52–58 CrossRef CAS PubMed.
- H. Mathur, V. Fallico, P. M. O'Connor, M. C. Rea, P. D. Cotter, C. Hill and R. P. Ross, Front. Microbiol., 2017, 8, 696 CrossRef PubMed.
- K. Babasaki, T. Takao, Y. Shimonishi and K. Kurahashi, J. Biochem., 1985, 98, 585–603 CrossRef CAS PubMed.
- K. Kawulka, T. Sprules, R. T. McKay, P. Mercier, C. M. Diaper, P. Zuber and J. C. Vederas, J. Am. Chem. Soc., 2003, 125, 4726–4727 CrossRef CAS PubMed.
- K. Kawulka, T. Sprules, C. M. Diaper, R. M. Whittal, R. T. McKay, P. Mercier, P. Zuber and J. C. Vederas, Biochemistry, 2004, 43, 3385–3395 CrossRef CAS PubMed.
- C. E. Shelburne, F. Y. An, V. Dholpe, A. Ramamoorthy, D. E. Lopatin and M. S. Lantz, J. Antimicrob. Chemother., 2007, 59, 297–300 CrossRef CAS PubMed.
- K. S. Noll, P. J. Sinko and M. L. Chikindas, Probiotics Antimicrob. Proteins, 2011, 3, 41–47 CrossRef CAS PubMed.
- M. C. Rea, C. S. Sit, E. Clayton, P. M. O'Connor, R. M. Whittal, J. Zheng, J. C. Vederas, R. P. Ross and C. Hill, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9852–9857 CrossRef PubMed.
- M. C. Rea, A. Dobson, O. O'Sullivan, F. Crispie, F. Fouhy, P. D. Cotter, F. Shanahan, B. Kiely, C. Hill and R. P. Ross, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(Suppl. 1), 4639–4644 CrossRef CAS PubMed.
- C. S. Sit, R. T. McKay, C. Hill, R. P. Ross and J. C. Vederas, J. Am. Chem. Soc., 2011, 133, 7680–7683 CrossRef CAS PubMed.
- C. S. Sit, M. J. van Belkum, R. T. McKay, R. W. Worobo and J. C. Vederas, Angew. Chem., Int. Ed. Engl., 2011, 50, 8718–8721 CrossRef CAS PubMed.
- G. Wang, G. Feng, A. B. Snyder, D. C. Manns, J. J. Churey and R. W. Worobo, FEMS Microbiol. Lett., 2014, 357, 69–76 CrossRef CAS PubMed.
- J. Dabard, C. Bridonneau, C. Phillipe, P. Anglade, D. Molle, M. Nardi, M. Ladire, H. Girardin, F. Marcille, A. Gomez and M. Fons, Appl. Environ. Microbiol., 2001, 67, 4111–4118 CrossRef CAS PubMed.
- A. Gomez, M. Ladire, F. Marcille and M. Fons, J. Bacteriol., 2002, 184, 18–28 CrossRef CAS PubMed.
- F. Marcille, A. Gomez, P. Joubert, M. Ladire, G. Veau, A. Clara, F. Gavini, A. Willems and M. Fons, Appl. Environ. Microbiol., 2002, 68, 3424–3431 CrossRef CAS PubMed.
- A. Pujol, E. H. Crost, G. Simon, V. Barbe, D. Vallenet, A. Gomez and M. Fons, FEMS Microbiol. Ecol., 2011, 78, 405–415 CrossRef CAS PubMed.
- L. Flühe and M. A. Marahiel, Curr. Opin. Chem. Biol., 2013, 17, 605–612 CrossRef PubMed.
- L. Flühe, T. A. Knappe, M. J. Gattner, A. Schäfer, O. Burghaus, U. Linne and M. A. Marahiel, Nat. Chem. Biol., 2012, 8, 350–357 CrossRef PubMed.
- N. A. Bruender, J. Wilcoxen, R. D. Britt and V. Bandarian, Biochemistry, 2016, 55, 2122–2134 CrossRef CAS PubMed.
- T. L. Grove, P. M. Himes, S. Hwang, H. Yumerefendi, J. B. Bonanno, B. Kuhlman, S. C. Almo and A. A. Bowers, J. Am. Chem. Soc., 2017, 139, 11734–11744 CrossRef CAS PubMed.
- N. I. Burzlaff, P. J. Rutledge, I. J. Clifton, C. M. H. Hensgens, M. Pcikford, R. M. Adlington, P. L. Roach and J. E. Baldwin, Nature, 1999, 401, 721–724 CrossRef CAS PubMed.
- E. Tamanaha, B. Zhang, Y. Guo, W. C. Chang, E. W. Barr, G. Xing, J. St Clair, S. Ye, F. Neese, J. M. Bollinger, Jr. and C. Krebs, J. Am. Chem. Soc., 2016, 138, 8862–8874 CrossRef CAS PubMed.
- A. M. Taylor, S. Stoll, R. D. Britt and J. T. Jarrett, Biochemistry, 2011, 50, 7953–7963 CrossRef CAS PubMed.
- C. J. Fugate, T. A. Stich, E. G. Kim, W. K. Myers, R. D. Britt and J. T. Jarrett, J. Am. Chem. Soc., 2012, 134, 9042–9045 CrossRef CAS PubMed.
- A. Benjdia, A. Guillot, P. Ruffié, J. Leprince and O. Berteau, Nat. Chem., 2017, 9, 698–707 CrossRef CAS PubMed.
- P. R. Burkholder and I. McVeigh, Proc. Natl. Acad. Sci. U. S. A., 1942, 28, 285–289 CrossRef CAS.
- V. A. Najjar and R. Barrett, The Synthesis of B Vitamins by Intestinal Bacteria, Vitamins & Hormones, ed. R. S. Harris and K. V. Thimann, Academic Press, 1945, vol. 3, pp. 23–48 Search PubMed.
- M. E. Coates, Proc. Nutr. Soc., 1973, 32, 53–58 CrossRef CAS PubMed.
- M. J. Albert, V. I. Mathan and S. J. Baker, Nature, 1980, 283, 781–782 CrossRef CAS PubMed.
- B. S. Wostmann, Annu. Rev. Nutr., 1981, 1, 257–279 CrossRef CAS PubMed.
- M. J. Hill, Eur. J. Cancer Prev., 1997, 6, S43–S45 CrossRef PubMed.
- S. M. Nabokina, K. Inoue, V. S. Subramanian, J. E. Valle, H. Yuasa and H. M. Said, J. Biol. Chem., 2014, 289, 4405–4416 CrossRef CAS PubMed.
- A. Qiu, M. Jansen, A. Sakaris, S. H. Min, S. Chattopadhyay, E. Tsai, C. Sandoval, R. Zhao, M. H. Akabas and I. D. Goldman, Cell, 2006, 127, 917–928 CrossRef CAS PubMed.
- P. H. Degnan, N. A. Barry, K. C. Mok, M. E. Taga and A. L. Goodman, Cell Host Microbe, 2014, 15, 47–57 CrossRef CAS PubMed.
- P. H. Degnan, M. E. Taga and A. L. Goodman, Cell Metab., 2014, 20, 769–778 CrossRef CAS PubMed.
- A. P. Mehta, S. H. Abdelwahed, N. Mahanta, D. Fedoseyenko, B. Philmus, L. E. Cooper, Y. Liu, I. Jhulki, S. E. Ealick and T. P. Begley, J. Biol. Chem., 2015, 290, 3980–3986 CrossRef CAS PubMed.
- K. Yokoyama and E. A. Lilla, Nat. Prod. Rep., 2018, 35, 660–694 RSC.
- M. R. Challand, F. T. Martins and P. L. Roach, J. Biol. Chem., 2010, 285, 5240–5248 CrossRef CAS PubMed.
- M. Kriek, F. Martins, M. R. Challand, A. Croft and P. L. Roach, Angew. Chem., Int. Ed. Engl., 2007, 46, 9223–9226 CrossRef CAS PubMed.
- M. Kriek, F. Martins, R. Leonardi, S. A. Fairhurst, D. J. Lowe and P. L. Roach, J. Biol. Chem., 2007, 282, 17413–17423 CrossRef CAS PubMed.
- G. Mkrtchyan, V. Aleshin, Y. Parkhomenko, T. Kaehne, M. L. Di Salvo, A. Parroni, R. Contestabile, A. Vovk, L. Bettendorff and V. Bunik, Sci. Rep., 2015, 5, 12583 CrossRef CAS PubMed.
- L. Hiffler, B. Rakotoambinina, N. Lafferty and D. Martinez Garcia, Front. Nutr., 2016, 3, 16 Search PubMed.
- D. R. Sannino, A. J. Dobson, K. Edwards, E. R. Angert and N. Buchon, mBio, 2018, 9 CrossRef PubMed.
- V. A. Najjar and L. E. Holt, Jr., JAMA, J. Am. Med. Assoc., 1943, 123, 683–684 CrossRef CAS.
- S. M. Nabokina, M. B. Ramos, J. E. Valle and H. M. Said, Am. J. Physiol.: Cell Physiol., 2015, 308, C750–C757 CrossRef PubMed.
- S. M. Nabokina, V. S. Subramanian and H. M. Said, Biochim. Biophys. Acta, 2016, 1858, 866–871 CrossRef CAS PubMed.
- S. M. Nabokina, M. B. Ramos and H. M. Said, PLoS One, 2016, 11, e0149255 CrossRef PubMed.
- G. Juyal, S. Negi, A. Sood, A. Gupta, P. Prasad, S. Senapati, J. Zaneveld, S. Singh, V. Midha, S. van Sommeren, R. K. Weersma, J. Ott, S. Jain, R. C. Juyal and B. K. Thelma, Gut, 2015, 64, 571–579 CrossRef CAS PubMed.
- A. Gupta and B. K. Thelma, Genes Immun., 2016, 17, 105–109 CrossRef CAS PubMed.
- S. J. Booker, R. M. Cicchillo and T. L. Grove, Curr. Opin. Chem. Biol., 2007, 11, 543–552 CrossRef CAS PubMed.
- N. D. Lanz and S. J. Booker, Biochim. Biophys. Acta, 2015, 1853, 1316–1334 CrossRef CAS PubMed.
- M. M. Cosper, G. N. L. Jameson, H. L. Hernández, C. Krebs, B. H. Huynh and M. K. Johnson, Biochemistry, 2004, 43, 2007–2021 CrossRef PubMed.
- G. N. L. Jameson, M. M. Cosper, H. L. Hernández, M. K. Johnson and B. H. Huynh, Biochemistry, 2004, 43, 2022–2031 CrossRef CAS PubMed.
- F. Berkovitch, Y. Nicolet, J. T. Wan, J. T. Jarrett and C. L. Drennan, Science, 2004, 303, 76–79 CrossRef CAS PubMed.
- F. Escalettes, D. Florentin, B. Tse Sum Bui, D. Lesage and A. Marquet, J. Am. Chem. Soc., 1999, 121, 3571–3578 CrossRef CAS.
- A. M. Taylor, C. E. Farrar and J. T. Jarrett, Biochemistry, 2008, 47, 9309–9317 CrossRef CAS PubMed.
- N. B. Ugulava, C. J. Sacanell and J. T. Jarrett, Biochemistry, 2001, 40, 8352–8358 CrossRef CAS PubMed.
- M. R. Reyda, R. Dippold, M. E. Dotson and J. T. Jarrett, Arch. Biochem. Biophys., 2008, 471, 32–41 CrossRef CAS PubMed.
- M. R. Reyda, C. J. Fugate and J. T. Jarrett, Biochemistry, 2009, 48, 10782–10792 CrossRef CAS PubMed.
- E. L. McCarthy and S. J. Booker, Science, 2017, 358, 373–377 CrossRef CAS PubMed.
- L. Tong, Cell. Mol. Life Sci., 2013, 70, 863–891 CrossRef CAS PubMed.
- D. Mock, Seminars in Dermatology, 1991, 10, 296–302 CAS.
- A. Hayashi, Y. Mikami, K. Miyamoto, N. Kamada, T. Sato, S. Mizuno, M. Naganuma, T. Teratani, R. Aoki, S. Fukuda, W. Suda, M. Hattori, M. Amagai, M. Ohyama and T. Kanai, Cell Rep., 2017, 20, 1513–1524 CrossRef CAS PubMed.
- C. Fergus, D. Barnes, M. A. Alqasem and V. P. Kelly, Nutrients, 2015, 7, 2897–2929 CrossRef CAS PubMed.
- H. Kasai, Y. Kuchino, K. Nihei and S. Nishimura, Nucleic Acids Res., 1975, 2, 1931–1939 CrossRef CAS PubMed.
- F. Harada and S. Nishimura, Biochemistry, 1972, 11, 301–308 CrossRef CAS PubMed.
- J. R. Katze, B. Basile and J. A. McCloskey, Science, 1982, 216, 55–56 CrossRef CAS PubMed.
- M. Vinayak and C. Pathak, Biosci. Rep., 2009, 30, 135–148 CrossRef PubMed.
- T. Marks and W. R. Farkas, Biochem. Biophys. Res. Commun., 1997, 230, 233–237 CrossRef CAS PubMed.
- C. Pathak, Y. K. Jaiswal and M. Vinayak, BioFactors, 2007, 29, 159–173 CrossRef CAS PubMed.
- D. Iwata-Reuyl, Bioorg. Chem., 2003, 31, 24–43 CrossRef CAS PubMed.
- J. R. Katze, U. Gündüz, D. L. Smith, C. S. Cheng and J. A. McCloskey, Biochemistry, 1984, 23, 1171–1176 CrossRef CAS PubMed.
- U. Gündüz and J. R. Katze, J. Biol. Chem., 1984, 259, 1110–1113 Search PubMed.
- W. R. Farkas, J. Biol. Chem., 1980, 255, 6832–6835 CAS.
- J. P. Reyniers, J. R. Pleasants, B. S. Wostmann, J. R. Katze and W. R. Farkas, J. Biol. Chem., 1981, 256, 11591–11594 CAS.
- S. Yokoyama, T. Miyazawa, Y. Iitaka, Z. Yamaizuma, H. Kasai and S. Nishimura, Nature, 1975, 282, 107–109 CrossRef.
- H. Kasai, Z. Ohashi, F. Harada, S. Nishimura, N. J. Oppenheimer, P. F. Crain, J. G. Liehr, D. L. von Minden and J. A. McCloskey, Biochemistry, 1975, 14, 4198–4208 CrossRef CAS PubMed.
- R. M. McCarty and V. Bandarian, Bioorg. Chem., 2012, 43, 15–25 CrossRef CAS PubMed.
- Z. D. Miles, R. M. McCarty, G. Molnar and V. Bandarian, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 7368–7372 CrossRef CAS PubMed.
- J. Bridwell-Rabb and C. L. Drennan, Curr. Opin. Chem. Biol., 2017, 37, 63–70 CrossRef CAS PubMed.
- Z. D. Miles, W. K. Myers, W. M. Kincannon, R. D. Britt and V. Bandarian, Biochemistry, 2015, 54, 4927–4935 CrossRef CAS PubMed.
- K. A. Payne, K. Fisher, H. Sjuts, M. S. Dunstan, B. Bellina, L. Johannissen, P. Barran, S. Hay, S. E. Rigby and D. Leys, J. Biol. Chem., 2015, 290, 27572–27581 CrossRef CAS PubMed.
- D. P. Dowling, Z. D. Miles, C. Kohrer, S. J. Maiocco, S. J. Elliott, V. Bandarian and C. L. Drennan, Nucleic Acids Res., 2016, 44, 9965–9976 CAS.
- R. Banerjee and S. W. Ragsdale, Annu. Rev. Biochem., 2003, 72, 209–247 CrossRef CAS PubMed.
- V. Bandarian and C. L. Drennan, Curr. Opin. Struct. Biol., 2015, 35, 116–124 CrossRef CAS PubMed.
- D. P. Dowling, N. A. Bruender, A. P. Young, R. M. McCarty, V. Bandarian and C. L. Drennan, Nat. Chem. Biol., 2013, 10, 106–112 CrossRef PubMed.
- R. M. McCarty, C. Krebs and V. Bandarian, Biochemistry, 2013, 52, 188–198 CrossRef CAS PubMed.
- W. Zhu and Y. Liu, ACS Catal., 2015, 5, 3953–3965 CrossRef CAS.
- P. Close, D. Bose, A. Chariot and S. A. Leidel, in Cancer and Noncoding RNAs, ed. D. S. Mitra, Academic Press, Boston, 2018, vol. 1 Search PubMed.
- C. Pathak, Y. K. Jaiswal and M. Vinayak, Mol. Biol. Rep., 2008, 35, 369–374 CrossRef CAS PubMed.
- C. Pathak, Y. K. Jaiswal and M. Vinayak, RNA Biol., 2005, 2, 143–148 CrossRef CAS PubMed.
- B. Lamas, M. L. Richard, V. Leducq, H. P. Pham, M. L. Michel, G. Da Costa, C. Bridonneau, S. Jegou, T. W. Hoffmann, J. M. Natividad, L. Brot, S. Taleb, A. Couturier-Maillard, I. Nion-Larmurier, F. Merabtene, P. Seksik, A. Bourrier, J. Cosnes, B. Ryffel, L. Beaugerie, J. M. Launay, P. Langella, R. J. Xavier and H. Sokol, Nat. Med., 2016, 22, 598–605 CrossRef CAS PubMed.
- T. Zelante, R. G. Iannitti, C. Cunha, A. De Luca, G. Giovannini, G. Pieraccini, R. Zecchi, C. D'Angelo, C. Massi-Benedetti, F. Fallarino, A. Carvalho, P. Puccetti and L. Romani, Immunity, 2013, 39, 372–385 CrossRef CAS PubMed.
- M. Venkatesh, S. Mukherjee, H. Wang, H. Li, K. Sun, A. P. Benechet, Z. Qiu, L. Maher, M. R. Redinbo, R. S. Phillips, J. C. Fleet, S. Kortagere, P. Mukherjee, A. Fasano, J. Le Ven, J. K. Nicholson, M. E. Dumas, K. M. Khanna and S. Mani, Immunity, 2014, 41, 296–310 CrossRef CAS PubMed.
- L. Cervantes-Barragan, J. N. Chai, M. D. Tianero, B. Di Luccia, P. P. Ahern, J. Merriman, V. S. Cortez, M. G. Caparon, M. S. Donia, S. Gilfillan, M. Cella, J. I. Gordon, C.-S. Hsieh and M. Colonna, Science, 2017, 357, 806–810 CrossRef CAS PubMed.
- M. Wlodarska, C. Luo, R. Kolde, E. d'Hennezel, J. W. Annand, C. E. Heim, P. Krastel, E. K. Schmitt, A. S. Omar, E. A. Creasey, A. L. Garner, S. Mohammadi, D. J. O'Connell, S. Abubucker, T. D. Arthur, E. A. Franzosa, C. Huttenhower, L. O. Murphy, H. J. Haiser, H. Vlamakis, J. A. Porter and R. J. Xavier, Cell Host Microbe, 2017, 22, 25–37 CrossRef CAS PubMed.
- S. Dickert, A. J. Pierik and W. Buckel, Mol. Microbiol., 2002, 44, 49–60 CrossRef CAS PubMed.
- S. Dickert, A. J. Pierik, D. Linder and W. Buckel, Eur. J. Biochem., 2000, 267, 3874–3884 CrossRef CAS PubMed.
- W. Buckel, B. M. Martins, A. Messerschmidt and B. T. Golding, Biol. Chem., 2005, 386, 951–959 CAS.
- W. Buckel, J. Zhang, P. Friedrich, A. Parthasarathy, H. Li, I. Djurdjevic, H. Dobbek and B. M. Martins, Biochim. Biophys. Acta, 2012, 1824, 1278–1290 CrossRef CAS PubMed.
- D. M. Smith, W. Buckel and H. Zipse, Angew. Chem., Int. Ed. Engl., 2003, 42, 1867–1870 CrossRef CAS PubMed.
- W. Buckel, M. Hetzel and J. Kim, Curr. Opin. Chem. Biol., 2004, 8, 462–467 CrossRef CAS PubMed.
- K. P. Locher, M. Hans, A. P. Yeh, B. Schmidt, W. Buckel and D. C. Rees, J. Mol. Biol., 2001, 307, 297–308 CrossRef CAS PubMed.
- S. H. Knauer, W. Buckel and H. Dobbek, Biochemistry, 2012, 51, 6609–6622 CrossRef CAS PubMed.
- J. Kim, Y. Lu and W. Buckel, C. R. Chim., 2007, 10, 742–747 CrossRef CAS.
- J. Kim, M. Hetzel, C. D. Boiangiu and W. Buckel, FEMS Microbiol. Rev., 2004, 28, 455–468 CrossRef CAS PubMed.
- H. Schindelin, C. Kisker, J. L. Schlessman, J. B. Howard and D. C. Rees, Nature, 1997, 387(6631), 370–376 CrossRef CAS PubMed.
- M. Hans, W. Buckel and E. Bill, J. Biol. Inorg Chem., 2008, 13, 563–574 CrossRef CAS PubMed.
- J. Kim, D. Darley and W. Buckel, FEBS J., 2005, 272, 550–561 CrossRef CAS PubMed.
- S. H. Knauer, W. Buckel and H. Dobbek, J. Am. Chem. Soc., 2011, 133, 4342–4347 CrossRef CAS PubMed.
- M. Hans, W. Buckel and E. Bill, Eur. J. Biochem., 2000, 267, 7082–7093 CrossRef CAS PubMed.
- H. R. Hayward and T. C. Stadtman, J. Bacteriol., 1959, 78, 557–561 CAS.
- C. Bradbeer, J. Biol. Chem., 1965, 240, 4669–4674 CAS.
- D. H. Lang, C. K. Yeung, R. M. Peter, C. Ibarra, R. Gasser, K. Itagaki, R. M. Philpot and A. E. Rettie, Biochem. Pharmacol., 1998, 56, 1005–1012 CrossRef CAS PubMed.
- Z. Wang, E. Klipfell, B. J. Bennett, R. Koeth, B. S. Levison, B. Dugar, A. E. Feldstein, E. B. Britt, X. Fu, Y. M. Chung, Y. Wu, P. Schauer, J. D. Smith, H. Allayee, W. H. Tang, J. A. DiDonato, A. J. Lusis and S. L. Hazen, Nature, 2011, 472, 57–63 CrossRef CAS PubMed.
- W. H. Tang, Z. Wang, B. S. Levison, R. A. Koeth, E. B. Britt, X. Fu, Y. Wu and S. L. Hazen, N. Engl. J. Med., 2013, 368, 1575–1584 CrossRef CAS PubMed.
- Z. Wang, W. H. Tang, J. A. Buffa, X. Fu, E. B. Britt, R. A. Koeth, B. S. Levison, Y. Fan, Y. Wu and S. L. Hazen, Eur. Heart J., 2014, 35, 904–910 CrossRef CAS PubMed.
- M. Dambrova, G. Latkovskis, J. Kuka, I. Strele, I. Konrade, S. Grinberga, D. Hartmane, O. Pugovics, A. Erglis and E. Liepinsh, Exp. Clin. Endocrinol. Diabetes, 2016, 124, 251–256 CrossRef CAS PubMed.
- S. H. Zeisel and M. Warrier, Annu. Rev. Nutr., 2017, 37, 157–181 CrossRef CAS PubMed.
- Y. M. Chen, Y. Liu, R. F. Zhou, X. L. Chen, C. Wang, X. Y. Tan, L. J. Wang, R. D. Zheng, H. W. Zhang, W. H. Ling and H. L. Zhu, Sci. Rep., 2016, 6, 19076 CrossRef CAS PubMed.
- S. C. Mitchell and R. L. Smith, Drug Metab. Dispos., 2001, 29, 517–521 CAS.
- K. A. Romano, A. Martinez-Del Campo, K. Kasahara, C. L. Chittim, E. I. Vivas, D. Amador-Noguez, E. P. Balskus and F. E. Rey, Cell Host Microbe, 2017, 22, 279–290 CrossRef CAS PubMed.
- S. Craciun and E. P. Balskus, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 21307–21312 CrossRef CAS PubMed.
- S. Craciun, J. A. Marks and E. P. Balskus, ACS Chem. Biol., 2014, 9, 1408–1413 CrossRef CAS PubMed.
- A. Martinez-del Campo, S. Bodea, H. A. Hamer, J. A. Marks, H. J. Haiser, P. J. Turnbaugh and E. P. Balskus, mBio, 2015, 6(2), e00042-15 CrossRef PubMed.
- S. Bodea, M. A. Funk, E. P. Balskus and C. L. Drennan, Cell Chem. Biol., 2016, 23, 1206–1216 CrossRef CAS PubMed.
- J. A. Krzycki, Curr. Opin. Chem. Biol., 2004, 8, 484–491 CrossRef CAS PubMed.
- L. Paul, D. J. Ferguson and J. A. Krzycki, J. Bacteriol., 2000, 182, 2520–2529 CrossRef CAS PubMed.
- J. F. Brugère, G. Borrel, N. Gaci, W. Tottey, P. W. O'Toole and C. Malpuech-Brugère, Gut Microbes, 2014, 5, 5–10 CrossRef PubMed.
- J. F. Brugère, G. Borrel, P. W. O'Toole, C. Malpuech-Brugère and M. Alric, US Pat., US20160074440A1, 2016.
- S. Hafiz and C. L. Oakley, J. Med. Microbiol., 1976, 9, 129–136 CrossRef CAS PubMed.
- L. D'Ari and H. A. Barker, Arch. Microbiol., 1985, 143, 311–312 CrossRef.
- L. F. Dawson, R. A. Stabler and B. W. Wren, J. Med. Microbiol., 2008, 57, 745–749 CrossRef PubMed.
- L. F. Dawson, E. H. Donahue, S. T. Cartman, R. H. Barton, J. Bundy, R. McNerney, N. P. Mintn and B. W. Wren, BMC Microbiol., 2011, 11, 86 CrossRef CAS PubMed.
- R. A. Stabler, M. He, L. Dawson, M. Martin, E. Valiente, C. Corton, T. D. Lawley, M. Sebaihia, M. A. Quail, G. Rose, D. N. Gerding, M. Gibert, M. R. Popoff, J. Parkhill, G. Dougan and B. W. Wren, Genome Biol., 2009, 10, R102 CrossRef PubMed.
- B. Bammens, P. Evenepoel, H. Keuleers, K. Verbeke and Y. Vanrenterghem, Kidney Int., 2006, 69, 1081–1087 CrossRef CAS PubMed.
- J. Poveda, M. D. Sanchez-Nino, G. Glorieux, A. B. Sanz, J. Egido, R. Vanholder and A. Ortiz, Nephrol., Dial., Transplant., 2014, 29, 56–64 CrossRef CAS PubMed.
- T. Selmer and P. I. Andrei, Eur. J. Biochem., 2001, 268, 1363–1372 CrossRef CAS PubMed.
- L. Yu, M. Blaser, P. I. Andrei, A. J. Pierik and T. Selmer, Biochemistry, 2006, 45, 9584–9592 CrossRef CAS PubMed.
- B. M. Martins, M. Blaser, M. Feliks, G. M. Ullmann, W. Buckel and T. Selmer, J. Am. Chem. Soc., 2011, 133, 14666–14674 CrossRef CAS PubMed.
- M. Blaser, Activation and Regulation of the 4-hydroxyphenylacetate decarboxylase system from Clostridium difficile, Ph.D. thesis, Philipps-Universit at Marburg, Marburg, Germany, 2006.
- P. I. Andrei, A. J. Pierik, S. Zauner, L. C. Andrei-Selmer and T. Selmer, Eur. J. Biochem., 2004, 271, 2225–2230 CrossRef CAS PubMed.
- B. Selvaraj, A. J. Pierik, E. Bill and B. M. Martins, J. Biol. Inorg Chem., 2014, 19, 1317–1326 CrossRef CAS PubMed.
- M. Feliks, B. M. Martins and G. M. Ullmann, J. Am. Chem. Soc., 2013, 135, 14574–14585 CrossRef CAS PubMed.
- L. Altieri, C. Neri, R. Sacco, P. Curatolo, A. Benvenuto, F. Muratori, E. Santocchi, C. Bravaccio, C. Lenti, M. Saccani, R. Rigardetto, M. Gandione, A. Urbani and A. M. Persico, Biomarkers, 2011, 16, 252–260 Search PubMed.
- A. M. Persico and V. Napolioni, Neurotoxicol. Teratol., 2012, 36, 82–90 CrossRef PubMed.
- E. Bone, A. Tamm and M. Hill, Am. J. Clin. Nutr., 1976, 29, 1448–1454 CrossRef CAS PubMed.
- M. T. Yokoyama and J. R. Carslon, Appl. Environ. Microbiol., 1981, 41, 71–76 CAS.
- E. Y. Hsiao, S. W. McBride, S. Hsien, G. Sharon, E. R. Hyde, T. McCue, J. A. Codelli, J. Chow, S. E. Reisman, J. F. Petrosino, P. H. Patterson and S. K. Mazmanian, Cell, 2013, 155, 1451–1463 CrossRef CAS PubMed.
- H. A. Krebs and K. Henseleit, Hoppe-Seyler's Z. Physiol. Chem., 1932, 210, 33–66 CrossRef CAS.
- I. D. Weiner, W. E. Mitch and J. M. Sands, Clin. J. Am. Soc. Nephrol., 2014, 10, 1444–1458 CrossRef PubMed.
- W. J. Visek, Fed. Proc., 1972, 31, 1178–1193 CAS.
- M. J. Maroney and S. Ciurli, Chem. Rev., 2014, 114, 4206–4228 CrossRef CAS PubMed.
- J. Ni, T.-C. D. Shen, E. Z. Chen, K. Bittinger, A. Bailey, M. Roggiani, A. Sirota-Madi, E. S. Friedman, L. Chau, A. Lin, I. Nissim, J. Scott, A. Lauder, C. Hoffman, G. Rivas, L. Albenberg, R. N. Baldassano, J. Braun, R. J. Xavier, C. B. Clish, M. Yudkoff, H. Li, M. Goulian, F. D. Bushman, J. D. Lewis and G. D. Wu, Sci. Transl. Med., 2017, 9, eaah6888 CrossRef PubMed.
- M. Romero-Gomez, M. Jover, J. J. Galan and A. Ruiz, Metab. Brain Dis., 2009, 24, 147–157 CrossRef CAS PubMed.
- D. J. Kang, N. S. Betrapally, S. A. Ghosh, R. B. Sartor, P. B. Hylemon, P. M. Gillvet, A. J. Sanyal, D. M. Heuman, D. Carl, H. Zhou, R. Liu, X. Wang, J. Yang, C. Jiao, J. Herzog, H. R. Lippman, M. Sikaroodi, R. R. Brown and J. S. Bajaj, Hepatology, 2016, 64, 1232–1248 CrossRef CAS PubMed.
- R. A. Burne and Y.-Y. M. CHen, Microbes Infect., 2000, 2, 533–542 CrossRef CAS PubMed.
- D. T. Smoot, H. L. T. Mobley, G. R. Chippendale, J. F. Lewison and J. H. Resau, Infect. Immun., 1990, 58, 1992–1994 CAS.
- I. Konieczna, P. Zarnowiec, M. Kwinkowski, B. Kolesińska, J. Frączyk, Z. Kamiński and W. Kaca, Curr. Protein Pept. Sci., 2012, 13, 789–806 CrossRef CAS PubMed.
- R. L. Blakeley, J. A. Hinds, H. E. Kunze, E. C. Webb and B. Zerner, Biochemistry, 1969, 8, 1991–2000 CrossRef CAS PubMed.
- E. Jabri, M. B. Carr, R. P. Hausinger and P. A. Karplus, Science, 1995, 268, 998–1004 CrossRef CAS PubMed.
- S. Benini, W. R. Rypniewski, K. S. Wilson, S. Miletti, S. Ciurli and S. Mangani, Structure, 1999, 7, 205–216 CrossRef CAS PubMed.
- M. A. Pearson, R. A. Schaller, L. O. Michel, P. A. Karplus and R. P. Hausinger, Biochemistry, 1998, 37, 6214–6220 CrossRef CAS PubMed.
- S. Benini, W. R. Rypniewski, K. S. Wilson, S. Miletti, S. Ciurli and S. Mangani, J. Biol. Inorg Chem., 2000, 5, 110–118 CrossRef CAS PubMed.
- S. Benini, W. R. Rypniewski, K. S. Wilson, S. Mangani and S. Ciurli, J. Am. Chem. Soc., 2004, 126, 3714–3715 CrossRef CAS PubMed.
- J. L. Boer, S. B. Mulrooney and R. P. Hausinger, Arch. Biochem. Biophys., 2014, 544, 142–152 CrossRef CAS PubMed.
- N. E. Dixon, C. Gazzola, R. L. Blakeley and B. Zerner, J. Am. Chem. Soc., 1975, 97, 4131–4133 CrossRef CAS PubMed.
- G. J. King and B. Zerner, Inorg. Chim. Acta, 1997, 255, 381–388 CrossRef CAS.
- I.-S. Park and R. P. Hausinger, Biochemistry, 1996, 35, 5345–5352 CrossRef CAS PubMed.
- B. Zambelli, F. Musiani, S. Benini and S. Ciurli, Acc. Chem. Res., 2011, 44, 520–530 CrossRef CAS PubMed.
- M. A. Farrugia, L. Macomber and R. P. Hausinger, J. Biol. Chem., 2013, 288, 13178–13185 CrossRef CAS PubMed.
- E. L. Carter, N. Flugga, J. L. Boer, S. B. Mulrooney and R. P. Hausinger, Metallomics, 2009, 1, 207–221 RSC.
- Z. Chang, J. Kuchar and R. P. Hausinger, J. Biol. Chem., 2004, 279, 15305–15313 CrossRef CAS PubMed.
- M. A. Farrugia, L. Han, Y. Zhong, J. L. Boer, B. T. Ruotolo and R. P. Hausinger, J. Am. Soc. Mass Spectrom., 2013, 24, 1328–1337 CrossRef CAS PubMed.
- Y. H. Fong, H. C. Wong, M. H. Yuen, P. H. Lau, Y. W. Chen and K. B. Wong, PLoS Biol., 2013, 11, e1001678 CrossRef CAS PubMed.
- Y. H. Fong, H. C. Wong, C. P. Chuck, Y. W. Chen, H. Sun and K. B. Wong, J. Biol. Chem., 2011, 286, 43241–43249 CrossRef CAS PubMed.
- X. Yang, H. Li, T. P. Lai and H. Sun, J. Biol. Chem., 2015, 290, 12474–12485 CrossRef CAS PubMed.
- R. Shi, C. Munger, A. Asinas, S. L. Benoit, E. Miller, A. Matte, R. J. Maier and M. Cygler, Biochemistry, 2010, 49, 7080–7088 CrossRef CAS PubMed.
- M. Stola, F. Musiani, S. Marngani, P. Turano, N. Safarov, B. Zambelli and S. Ciurli, Biochemistry, 2006, 45, 6495–6509 CrossRef CAS PubMed.
- K. Banaszak, V. Martin-Diaconescu, M. Bellucci, B. Zambelli, W. Rypniewski, M. J. Maroney and S. Ciurli, Biochem. J., 2012, 441, 1017–1026 CrossRef CAS PubMed.
- B. Zambelli, A. Berardi, V. Martin-Diaconescu, L. Mazzei, F. Musiani, M. J. Maroney and S. Ciurli, J. Biol. Inorg Chem., 2014, 19, 319–334 CrossRef CAS PubMed.
- V. Martin-Diaconescu, C. A. Joseph, J. L. Boer, S. B. Mulrooney, R. P. Hausinger and M. J. Maroney, J. Biol. Inorg Chem., 2017, 22, 497–503 CrossRef CAS PubMed.
- L.-T. Hu and H. L. T. Mobley, Infect. Immun., 1993, 61, 2563–2569 CAS.
- K. A. Higgins, C. E. Carr and M. J. Maroney, Biochemistry, 2012, 51, 7816–7832 CrossRef CAS PubMed.
- H. de Reuse, D. Vinella and C. Cavazza, Front. Cell. Infect. Microbiol., 2013, 3, 94 Search PubMed.
- H. Q. Hu, R. C. Johnson, D. S. Merrell and M. J. Maroney, Biochemistry, 2017, 56, 1105–1116 CrossRef CAS PubMed.
- W. N. Fishbein and P. P. Carbone, J. Biol. Chem., 1965, 240, 2407–2414 CAS.
- Z. Amtul, A. Rahman, R. A. Siddiqui and M. I. Choudhary, Curr. Med. Chem., 2002, 9, 1323–1348 CrossRef CAS PubMed.
- C. Follmer, J. Clin. Pathol., 2010, 63, 424–430 CrossRef CAS PubMed.
- T.-C. D. Shen, L. Albenberg, K. Bittinger, C. Chehoud, Y.-Y. Chen, C. A. Judge, L. Chau, J. Ni, M. Sheng, A. Lin, B. J. Wilkins, E. L. Buza, J. D. Lewis, Y. Daikhin, I. Nissim, M. Yudkoff, F. D. Bushman and G. D. Wu, J. Clin. Invest., 2015, 125, 2841–2850 CrossRef PubMed.
- T.-C. D. Shen, C. Chehoud, J. Ni, E. Hsu, Y.-Y. Chen, A. Bailey, A. Laughlin, K. Bittinger, F. D. Bushman and G. D. Wu, PLoS One, 2016, 11, e0155620 CrossRef PubMed.
- L. A. David, C. F. Maurice, R. N. Carmody, D. B. Gootenberg, J. E. Button, B. E. Wolfe, A. V. Ling, A. S. Devlin, Y. Varma, M. A. Fischbach, S. B. Biddinger, R. J. Dutton and P. J. Turnbaugh, Nature, 2014, 505, 559–563 CrossRef CAS PubMed.
- S. Devkota, Y. Wang, M. W. Musch, V. Leone, H. Fehlner-Peach, A. Nadimpalli, D. A. Antonopoulos, B. Jabri and E. B. Chang, Nature, 2012, 487, 104–108 CrossRef CAS PubMed.
- S. Devkota and E. B. Chang, Digestive diseases, 2015, 33, 351–356 CrossRef PubMed.
- J. M. Ridlon, P. G. Wolf and H. R. Gaskins, Gut Microbes, 2016, 7, 201–215 CrossRef CAS PubMed.
- S. M. Houten, M. Watanabe and J. Auwerx, EMBO J., 2006, 25, 1419–1425 CrossRef CAS PubMed.
- P. B. Hylemon, H. Zhou, W. M. Pandak, S. Ren, G. Gil and P. Dent, J. Lipid Res., 2009, 50, 1509–1520 CrossRef CAS PubMed.
- S. Mukhopadhyay and U. Maitra, Curr. Sci., 2004, 87, 1666–1683 CAS.
- M. Begley, C. G. Gahan and C. Hill, FEMS Microbiol. Rev., 2005, 29, 625–651 CrossRef CAS PubMed.
- J. M. Ridlon, D. J. Kang and P. B. Hylemon, J. Lipid Res., 2006, 47, 241–259 CrossRef CAS PubMed.
- H. Laue, K. Denger and A. M. Cook, Appl. Environ. Microbiol., 1997, 63, 2016–2021 CAS.
- J. Levine, C. J. Ellis, J. K. Furne, J. Springfield and M. D. Levitt, Am. J. Gastroenterol., 1998, 93, 83–87 CrossRef CAS PubMed.
- G. R. Gibson, J. H. Cummings and G. T. Macfarlane, FEMS Microbiol. Ecol., 1991, 86, 103–132 CrossRef CAS.
- M. C. L. Pitcher and J. H. Cummings, Gut, 1996, 39, 1–4 CrossRef CAS PubMed.
- W. Babidge, S. Millard and W. Roediger, Mol. Cell. Biochem., 1998, 118, 117–124 CrossRef.
- T. W. Miller, E. A. Wang, S. Gould, E. V. Stein, S. Kaur, L. Lim, S. Amarnath, D. H. Fowler and D. D. Roberts, J. Biol. Chem., 2012, 287, 4211–4221 CrossRef CAS PubMed.
- J. L. Wallace, L. Vong, W. McKnight, M. Dicay and G. R. Martin, Gastroenterology, 2009, 137, 569–578 CrossRef CAS PubMed.
- I. Fasolino, A. A. Izzo, T. Clavel, B. Romano, D. Haller and F. Borrelli, Mol. Nutr. Food Res., 2015, 59, 434–442 CrossRef CAS PubMed.
- J. P. Motta, K. L. Flannigan, T. A. Agbor, J. K. Beatty, R. W. Blackler, M. L. Workentine, G. J. Da Silva, R. Wang, A. G. Buret and J. L. Wallace, Inflammatory Bowel Dis., 2015, 21, 1006–1017 CrossRef PubMed.
- H. Laue, M. Friedrich, J. Ruff and A. M. Cook, J. Bacteriol., 2001, 183, 1727–1733 CrossRef CAS PubMed.
- T. F. Oliveira, C. Vonrhein, P. M. Matias, S. S. Venceslau, I. A. Pereira and M. Archer, J. Biol. Chem., 2008, 283, 34141–34149 CrossRef CAS PubMed.
- S. S. Venceslau, Y. Stockdreher, C. Dahl and I. A. Pereira, Biochim. Biophys. Acta, 2014, 1837, 1148–1164 CrossRef CAS PubMed.
- S. M. Lui, A. Soriano and J. A. Cowan, J. Am. Chem. Soc., 1993, 115, 10483–10486 CrossRef CAS.
- S. M. Lui, W. Liang, A. Soriano and J. A. Cowan, J. Am. Chem. Soc., 1994, 116, 4531–4536 CrossRef CAS.
- B. R. Crane and E. D. Getzoff, Curr. Opin. Struct. Biol., 1996, 6, 744–756 CrossRef CAS PubMed.
- I. Moura, J. LeGall, A. R. Lino, J. H. D. Peck, G. Fauque, A. V. Xavier, D. V. DerVartanian, J. J. G. Moura and B. H. Huynh, J. Am. Chem. Soc., 1988, 110, 1075–1082 CrossRef CAS.
- A. A. Santos, S. S. Venceslau, F. Grein, W. D. Leavitt, C. Dahl, D. T. Johnston and I. A. C. Pereira, Science, 2015, 350, 1541–1545 CrossRef CAS PubMed.
- F. Grein, I. A. Pereira and C. Dahl, J. Bacteriol., 2010, 192, 6369–6377 CrossRef CAS PubMed.
- F. Grein, A. R. Ramos, S. S. Venceslau and I. A. Pereira, Biochim. Biophys. Acta, 2013, 1827, 145–160 CrossRef CAS PubMed.
- Y. Seki and M. Ishimoto, J. Biochem., 1979, 86, 273–276 CAS.
- Y. Seki, N. Sogawa and M. Ishimoto, J. Biochem., 1981, 90, 1487–1492 CrossRef CAS PubMed.
- P. Spanogiannopoulos, E. N. Bess, R. N. Carmody and P. J. Turnbaugh, Nat. Rev. Microbiol., 2016, 14, 273–287 CrossRef CAS PubMed.
- N. Koppel, V. Maini Rekdal and E. P. Balskus, Science, 2017, 356(6344), eaag2770 CrossRef PubMed.
- M. K. Ma and H. K. McLeod, Curr. Med. Chem., 2003, 10, 41–49 CrossRef CAS PubMed.
- E. M. Cooke, Gut, 1969, 10, 565–568 CrossRef CAS PubMed.
- M. A. Peppercorn, Ann. Intern. Med., 1984, 3, 377–386 CrossRef.
- G. L. Plosker and K. F. Croom, Drugs, 2005, 65, 1825–1849 CrossRef CAS PubMed.
- H. Zheng, M. Chen, Y. Li, Y. Wang, L. Wei, Z. Liao, M. Wang, F. Ma, Q. Liao and Z. Xie, Front. Microbiol., 2017, 8, 1703 CrossRef PubMed.
- H. J. Haiser, D. B. Gootenberg, K. Chatman, G. Sirasani, E. P. Balskus and P. J. Turnbaugh, Science, 2013, 341, 295–298 CrossRef CAS PubMed.
- H. J. Haiser, K. L. Seim, E. P. Balskus and P. J. Turnbaugh, Gut Microbes, 2014, 5, 233–238 CrossRef PubMed.
- N. Koppel, J. E. Bisanz, M.-E. Pandelia, P. J. Turnbaugh and E. P. Balskus, eLife, 2018, 7, e33953 CrossRef PubMed.
- J. P. Yuan, J. H. Wang and X. Liu, Mol. Nutr. Food Res., 2007, 51, 765–781 CrossRef CAS PubMed.
- I. R. Rowland, H. Wiseman, T. A. Sanders, H. Adlercreutz and E. A. Bowey, Nutr. Cancer, 2000, 36, 27–32 CrossRef CAS PubMed.
- Y. Shimada, M. Takahashi, N. Miyazawa, T. Ohtani, Y. Abiru, S. Uchiyama and H. Hishigaki, J. Mol. Microbiol. Biotechnol., 2011, 21, 160–172 CrossRef CAS PubMed.
- H. Tsuji, K. Moriyama, K. Nomoto and H. Akaza, Appl. Environ. Microbiol., 2012, 78, 1228–1236 CrossRef CAS PubMed.
- C. Schröder, A. Matthies, W. Engst, M. Blaut and A. Braune, Appl. Environ. Microbiol., 2013, 79, 3494–3502 CrossRef PubMed.
- Y. Kawada, S. Yokoyama, E. Yanase, T. Niwa and T. Suzuki, Biosci. Microbiota, Food Health, 2016, 35, 113–121 CrossRef CAS PubMed.
- R. D. Finn, P. Coggill, R. Y. Eberhardt, S. R. Eddy, J. Mistry, A. L. Mitchell, S. C. Potter, M. Punta, M. Qureshi, A. Sangrador-Vegas, G. A. Salazar, J. Tate and A. Bateman, Nucleic Acids Res., 2016, 44, D279–D285 CrossRef CAS PubMed.
- M. D. Mayers, C. Moon, G. S. Stupp, A. I. Su and D. W. Wolan, J. Proteome Res., 2017, 16, 1014–1026 CrossRef CAS PubMed.
- J. Kaminski, M. K. Gibson, E. A. Franzosa, N. Segata, G. Dantas and C. Huttenhower, PLoS Comput. Biol., 2015, 11, e1004557 CrossRef PubMed.
- J. A. Gerlt, J. T. Bouvier, D. B. Davidson, H. J. Imker, B. Sadkhin, D. R. Slater and K. L. Whalen, Biochim. Biophys. Acta, 2015, 1854, 1019–1037 CrossRef CAS PubMed.
- P. Cimermancic, M. H. Medema, J. Claesen, K. Kurita, L. C. Wieland Brown, K. Mavrommatis, A. Pati, P. A. Godfrey, M. Koehrsen, J. Clardy, B. W. Birren, E. Takano, A. Sali, R. G. Linington and M. A. Fischbach, Cell, 2014, 158, 412–421 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |