
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelection and characterization of botanical natural products for research studies: a NaPDI center recommended approach†
Joshua J.
Kellogg
 a,
Mary F.
Paine
b,
Jeannine S.
McCune
c,
Nicholas H.
Oberlies
a and
Nadja B.
Cech
*a
a,
Mary F.
Paine
b,
Jeannine S.
McCune
c,
Nicholas H.
Oberlies
a and
Nadja B.
Cech
*a
aDepartment of Chemistry & Biochemistry, University of North Carolina at Greensboro, Greensboro, North Carolina, USA. E-mail: nbcech@uncg.edu
bDepartment of Pharmaceutical Sciences, College of Pharmacy, Washington State University, Spokane, Washington, USA
cDepartment of Population Sciences, City of Hope, Duarte, California, USA
First published on 25th January 2019
Abstract
Covering: up to the end of 2018
Dietary supplements, which include botanical (plant-based) natural products, constitute a multi-billion-dollar industry in the US. Regulation and quality control for this industry is an ongoing challenge. While there is general agreement that rigorous scientific studies are needed to evaluate the safety and efficacy of botanical natural products used by consumers, researchers conducting such studies face a unique set of challenges. Botanical natural products are inherently complex mixtures, with composition that differs depending on myriad factors including variability in genetics, cultivation conditions, and processing methods. Unfortunately, many studies of botanical natural products are carried out with poorly characterized study material, such that the results are irreproducible and difficult to interpret. This review provides recommended approaches for addressing the critical questions that researchers must address prior to in vitro or in vivo (including clinical) evaluation of botanical natural products. We describe selection and authentication of botanical material and identification of key biologically active compounds, and compare state-of-the-art methodologies such as untargeted metabolomics with more traditional targeted methods of characterization. The topics are chosen to be of maximal relevance to researchers, and are reviewed critically with commentary as to which approaches are most practical and useful and what common pitfalls should be avoided.
 Joshua J. Kellogg | Dr Joshua J. Kellogg obtained his PhD in natural product chemistry and ethnobotanical nutraceuticals from North Carolina State University. He is currently a researcher in the lab of Professor Nadja Cech at the University of North Carolina at Greensboro, investigating the relative contributions of natural product mixtures to antibacterial activity. Dr Kellogg is the recipient of an NRSA Postdoctoral Fellowship from the National Institutes of Health. His research is centered around the development and application of metabolomic approaches to complex mixture analyses, with a focus on endosymbiotic microorganisms to drive discovery of novel bioactive compounds. |
 Mary F. Paine | Dr Mary F. Paine is an Associate Professor in the Department of Pharmaceutical Sciences, College of Pharmacy and Pharmaceutical Sciences, Washington State University. Dr Paine received her BS in pharmacy at Oregon State University, her PhD in pharmaceutics at the University of Washington, and completed a post-doctoral fellowship in clinical pharmacology at the University of Michigan. Her longstanding research program, funded continuously by the National Institutes of Health, focuses on adverse interactions between conventional medications and natural products. Dr Paine leads the Center of Excellence for Natural Product-Drug Interaction Research, a multidisciplinary effort involving clinical pharmacologists, natural products and analytical chemists, and health informaticists to provide leadership in the study of these complex interactions. Dr Paine has coauthored more than 90 publications as original research articles, invited reviews, and book chapters. She is currently an associate editor for the journals Clinical Pharmacology and Therapeutics and Drug Metabolism and Disposition. |
 Jeannine S. McCune | Dr Jeannine S. McCune, Pharm. D. is Professor in Population Sciences at City of Hope Cancer Center. Dr McCune's NIH funded research seeks to identify clinically relevant biomarkers using pharmacokinetic and pharmacodynamic modeling of cancer prevention and treatment. Her natural project research focuses on natural product–drug interactions that can affect the efficacy of cancer prevention and treatment. She has received over $2 million in NIH funding as a Principal Investigator to support her research and has over 100 publications. She has served in various professional leadership positions, and is currently the Chair of the CSR Cancer Prevention Study Section. |
 Nicholas H. Oberlies | Dr Nicholas H. Oberlies earned his Ph.D. in Medicinal Chemistry and Pharmacognosy from Purdue University, where he studied under Professor Jerry L. McLaughlin. After a year of postdoctoral work in industry, he joined Research Triangle Institute, specifically to be mentored by Dr Mansukh Wani and the now late, Dr Monroe Wall, who are the co-discoverers of both taxol and camptothecin. He rose through the ranks of RTI and eventually directed the Natural Products Laboratory. In 2009, he moved his group to the Department of Chemistry & Biochemistry at the University of North Carolina at Greensboro, where he helped to grow their Ph.D. program in Medicinal Biochemistry. His lab largely focuses on the characterization and development of chemical entities from natural sources. This includes efforts that seek new drug leads, as well as, projects, like this, that focus on insuring the safety and quality of herbal preparations. |
 Nadja B. Cech | Dr Nadja B. Cech is the Patricia A. Sullivan Distinguished Professor of Chemistry at the University of North Carolina at Greensboro (UNCG). She has had a lifelong interest in the use of plants for medicine, stemming from her involvement as a child in establishing one of the world's largest medicinal plant and seed companies, a company still operated by her family today. Dr Cech leads a dynamic research group at UNCG, for which a major focus in the development of metabolomics as a tool to understand synergy and complexity in biologically active botanical natural products. This work has been continuously funded by the National Institutes of Health for more than 15 years, and was awarded the Jack L. Beal Award from the Journal of Natural Products in 2011. Dr Cech is a member of the Center of Excellence for Natural Product Drug Interaction Research, and Co-Director of the Medicinal Chemistry Collaborative (https://mcsquared.uncg.edu/). |
1. Introduction
More than 50% of Americans acknowledge taking dietary supplements,1,2 with sales more than tripling since passage of the Dietary Supplement Health and Education Act in 1994.3,4 In many cases, these dietary supplements are prepared from botanical (plant based) material, and as such are referred to as ‘botanical natural products.’ Countless studies have been devoted to the scientific evaluation of the safety and/or efficacy of botanical natural products. Investigators involved in such studies face a unique set of challenges (Table 1). The research methodology for evaluating safety and efficacy of conventional (pharmaceutical) drugs operates with the assumption that the product being tested is a single compound of known purity, identity, and concentration. Natural products differ from their pharmaceutical counterparts in that they are typically complex mixtures, for which the identities and quantities of components present are not fully known. The composition of these natural products can vary depending on the method of preparation or source material used.5 Such variability can impact the interpretation of in vitro, non-clinical in vivo, and/or clinical studies.| (1) Representative of what is commonly used by consumers |
| (2) Authentic (species verified) |
| (3) Well-characterized |
| (3.1) Active constituents known |
| (3.2) Concentrations of active or marker metabolites have been characterized and comply with monograph |
| (4) Free of contamination and adulteration |
| (5) Sufficient material available to conduct in vitro and/or clinical studies |
| (6) Material consistent for duration of the trial |
| (6.1) Shelf life (i.e. stability) |
| (6.2) Batch-to-batch reproducibility |
To enable studies to be as robust and as widely applicable as possible, selecting, characterizing, and ensuring continued quality and consistency of a botanical natural product is an essential task. The ideal characteristics of a botanical natural product used for research studies include that it is authenticated (of known identity), well-characterized in terms of potentially active constituents, and stable (Table 1). At present, many studies are conducted with botanical natural products that lack one or more of these characteristics. Herein, we seek to provide guidance for effective selection of botanical natural products prior to in vitro, non-clinical in vivo, or clinical evaluation. We discuss the relative merits of potential analytical approaches, including state-of-the-art metabolomics techniques, and recommend specific, effective and practical guidelines (Fig. 1). The recommendations we provide are informed by the coauthors' experience for several decades researching botanical natural products, culminating in our role as investigators participating in the Center of Excellence for Natural Product Drug Interaction Research (NaPDI Center). The National Center for Complementary and Integrative Health established the NaPDI Center to address the need for robust methods to study the potential for natural products to precipitate clinically significant pharmacokinetic interactions with conventional medications (natural product–drug interactions).6 While the focus of the NaPDI Center is specifically on the evaluation of interactions between conventional drugs and natural products, the recommendations provided herein are more broadly applicable to the selection of botanical natural products for any in vitro or in vivo investigation (including studies to evaluate efficacy or toxicity).
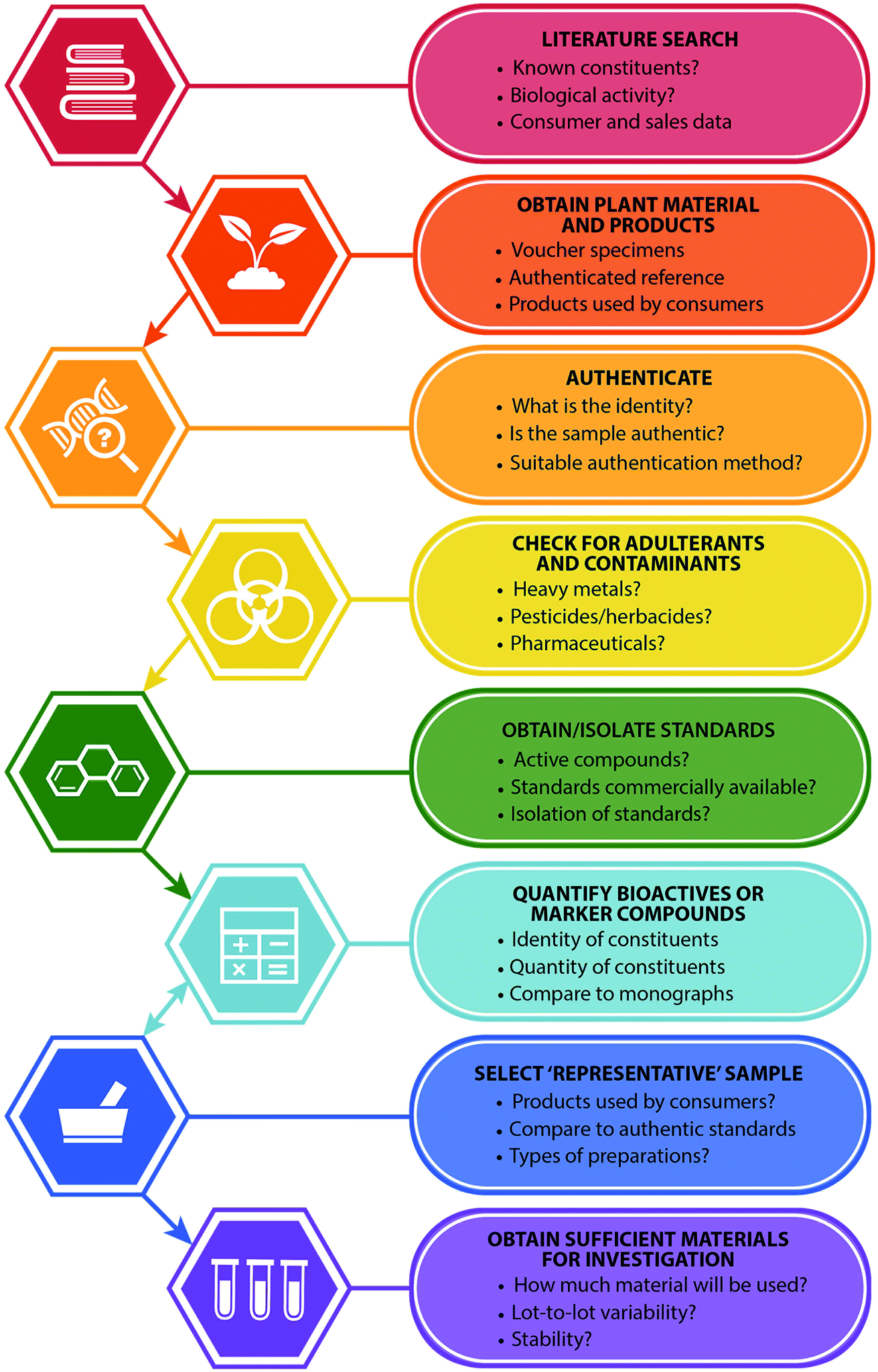 | ||
| Fig. 1 Recommended steps in the selection of botanical natural products for research purposes (in vitro and/or in vivo studies), and important questions for consideration with each step. | ||
2. Terminology relevant to botanical natural products
“Natural products” are a broad range of substances that can come from a variety of sources, including bacteria, fungi, marine organisms, and plants. The term can be used to describe both complex mixtures and single isolated compounds that come from these mixtures. For this publication, we focus specifically on botanical natural products, i.e. natural products derived from plants. Such botanical natural products are available for use by consumers in a variety of forms, including capsules containing raw or extracted material, extracts, teas (typically raw plant material that is extracted in hot water prior to use), tinctures (ethanolic extracts), and traditional formulations such as powders used in Traditional Chinese Medicine or Ayurvedic practices. In this manuscript, we use “botanical natural products” as an umbrella term to describe complex plant-based preparations. Terms used elsewhere include: supplements, herbal medicines, herbal drugs, herbs, botanical preparations, nutraceuticals, phytomedicines, and botanical medicines. Additional definitions for terms used in this manuscript are provided in Table 1S (ESI†).From a regulatory perspective, distinctions are often made between various botanical natural products depending on how they are used by consumers. The regulatory approaches and classifications of botanical natural products are complex and differ from country to country, and a detailed comparison is beyond the scope of the current work.7 The majority of botanical natural products consumed in the US (including the examples discussed in this manuscript) are regulated either as foods or dietary supplements. From a scientific perspective, the way in which the products are classified and regulated is less important than the material from which they are prepared, and much of the information presented herein is relevant to botanical natural products broadly defined, regardless of their regulatory classification.
3. Journal and government agency guidelines for characterization of natural product study material
Several agencies provide guidance for the type of information that should be obtained about a natural product prior to its use for research purposes. The United States Food and Drug Administration (FDA) recommendation regarding botanical drug clinical trials specifies that the investigational new drug application contains “a chemical identification for the active constituents or characteristic markers in the drug substance, if possible”.8 The National Center for Complementary and Integrative Health (NCCIH), a component of the National Institutes of Health, funds a large portion of the research conducted on botanical natural products in the United States. NCCIH has established a “Natural Product Integrity Policy”, which requires that researchers provide information about the identity, extraction solvent, characterization (i.e., metabolite profile), stability, standardization, and storage of all natural products that will be used in NCCIH funded studies.9 Some individual journals also provide specifications for how complex botanical natural products should be characterized before data on these products can be published.10–13 Journal editors have a great deal of influence in this regard, since their publishing requirements cross geopolitical borders.144. Literature research
Before beginning botanical natural product studies, it is useful to obtain information from the peer-reviewed literature regarding: (1) identities of major metabolites, especially those that may be biologically active; (2) potential in vitro or in vivo targets and activities; and (3) current knowledge gaps. A recent publication by the NaPDI Center addresses useful strategies for critically evaluating botanical natural product literature to identify gaps in the knowledge base.6 Due to the complexity of natural product constituents, it is important to note that reported chemical structures may contain errors or inconsistencies. Thus, the literature review by an organic or natural product chemist trained in structure elucidation is recommended.Another important topic to research during sourcing of botanical natural products is information regarding traditional and consumer usage of the product to be studied. The investigator will wish to answer questions such as: what are the most common species used medicinally? Which plant parts (for example, roots, leaves, berries) are typically used? What types of preparations (capsule, tincture, tea, compress, topical versus internal) are employed? Some information of this nature may be available in the peer-reviewed literature, but it is often necessary to seek other sources. Common products used by consumers can be evaluated via national surveys, such as the National Institute of Health's Office of Dietary Supplement's Dietary Supplement Label Database,15 National Health and Nutrition Examination Survey (NHANES),16 consumer quality reports17,18 as well as industry or retail sales reports.4,19,20
Box 1 Summary of recommendations: literature review for botanical natural products• Examine references to identify known constituents and biological targets.• Investigate botanical usage information, including common species used, physiological portions harvested, and preparation. • Utilize both peer-reviewed resources, government reports, and industry-based information. |
5. Obtaining botanical natural product study material
Prior to conducting in vitro, non-clinical in vivo, and/or clinical studies, it is important that researchers identify an authentic natural product (i.e. correct assignment of genus and species) that is available in sufficient quantity to conduct the studies. In the case of clinical studies, this product should be selected to closely resemble (or be identical to) the commercial product(s) used by the target population. Where is such a product obtained? One option is to purchase this material from a commercial distributor. Alternately, researchers may opt to obtain raw material and prepare a natural product themselves or to employ a contract laboratory for formulation.21 Finally, several suppliers provide high quality reference materials of botanicals for research purposes. Here we discuss the importance of voucher specimens and compare the relative merits of various sources of botanical material.5.1 Voucher specimens
Under optimal circumstances, a voucher specimen of any botanical natural product to be studied is collected at the same time and from the same lot that the study material is obtained. The voucher specimen consists of an intact, dried sample of the plant material, including the flower when possible, and as many parts as can reasonably be collected, i.e. roots, stem, leaves, flowers and/or seeds.22 The voucher specimen is used for taxonomic identification of the study material by a trained botanist or otherwise qualified individual. Herbaria have existed for centuries,23 and these vouchers serve as historical records of plant specimens, and modern uses of these go beyond taxonomy, including areas like conservation biology.24 Thus, a pressed and dried sample of the voucher is deposited in a regional or national herbarium, where it is catalogued and stored for future reference. Herbarium vouchers are essential for preserving a record of the original sample tested and provide lasting, public access to that material in perpetuity. Authentication and retention of voucher specimens is required by major natural product journals.10–12,25 For example, for the Journal of Natural Products, authors who “purchase dried ‘herbal remedies’ or other materials from companies must make provision for their proper deposit in a herbarium or other permanent repository, for access by future workers”.10 Another example is Phytochemistry, which mandates that samples, “must also include a reference to voucher specimen(s) and voucher number(s) of the plants or other material examined” as well as “the name and address of the authority who identified each non-cultivated plant investigated”. Similar guidelines are in place for other major natural product journals.11,12,255.2 Contracting an independent laboratory to prepare study material
One way to obtain quality study material is to contract a laboratory to prepare the botanical natural product study material from raw materials to pre-determined specifications. This approach has been employed by other research groups.21,26,27 A major advantage of employing a contract laboratory to produce study materials is that the researchers can be responsible for ensuring the quality, consistency, and rigor with which the material is prepared. However, contracting a laboratory to prepare a formulation can be expensive, and the production of such formulations is time-consuming and requires access to specialized facilities and expertise. Even more importantly, a botanical natural product prepared by a contract laboratory may not reflect commercial products being consumed by the general public. Thus, many researchers opt instead to conduct studies with commercial products (the same products used by consumers). This was the approach that we took with the NaPDI Center's studies of both green tea [Camellia sinensis (L.) Kuntze (Theaceae)]28 and goldenseal [Hydrastis canadensis L. (Ranunculaceae)].295.3 Purchasing study material from a commercial supplier
Many botanical natural products can be purchased from online distributors, health food stores, grocery stores. The advantage of purchasing commercial botanical natural products for study is that it is possible to select material that directly reflects what is being used by consumers (with the caveat that there may be a great deal of variability among commercial products). In addition, for clinical studies, commercial products have the advantage of being “food grade” meaning that they can be administered to human subjects. On the other hand, it is difficult to control quality and ensure correct identity of botanical natural products obtained from commercial sources. Commercial botanical natural products typically do not come with associated voucher specimens. Additionally, adulteration or mis-identification of commercial natural products (both intentional and unintentional) is common.30–32 For example, in the process of selecting commercial products of goldenseal for clinical studies by the NaPDI Center, we identified several preparations sold as goldenseal (Hydrastis canadensis) that were a different botanical, Chinese goldenthread (Coptis chinensis).295.4 Optimal number of samples to analyze
When a study will be conducted with commercial products, the question arises as to how many samples should be evaluated before one is selected for study material. The goal is to select a sample set that is representative of the variability in products used by the target population. How can this be accomplished? Purchasing every potential commercial botanical natural product would theoretically be an ideal strategy. However, this is not always feasible due to constraints of time and cost. If a botanical natural product has a relatively small commercial footprint, it is possible to acquire all available products at a given time. As an illustrative example, the 35 commercial goldenseal samples selected for the second NaPDI Center study represented an estimated >90% of the readily available commercial products.29 However, when a botanical natural product is found in hundreds or potentially thousands of products, the logistical and potentially financial hurdles of sampling a great number of samples must be taken into account during this planning stage.For analyses of commercial samples, there has not been a conventional minimum value set on the number of samples needed to yield a robust analysis. Previous studies on botanical natural products have sampled as few as five products,33 with many sampling 10–18 samples;34–37 while some procure over 75 commercial products.38 We elected to select a minimum of 30 commercial samples for each of the NaPDI Center's studies on green tea (34 products)28 and goldenseal (35 products).29 Importantly, these samples were selected based on sales data to represent the products most widely purchased for use by US consumers (our target population). The result was a sample set that captured variability among the products most widely consumed and could be realistically handled within constraints of processing and analysis time.
5.5 Purchasing authenticated botanical reference materials
Several sources provide authenticated botanical reference materials. The National Institute of Standards and Technology (NIST, https://www.nist.gov) offers a number of botanical natural products as verified reference materials available for testing and authentication purposes, as does the U.S. Pharmacopeia (USP, https://www.usp.org). Additionally, phytochemical reference suppliers like ChromaDex (Irvine, CA, https://chromadex.com) provide different grades of botanical reference material. A major advantage of these reference materials is that they have often been characterized already, and information is available about known genetic markers and the presence and identity of known metabolites. Some sources, such as ChromaDex, may also provide information regarding their internal voucher specimens. Even if the intention is to use a commercial product for the final studies, it is advisable, where possible, to obtain an authenticated reference material. The composition of this reference material can then be compared to that of commercial botanical natural products to verify identity and compare quality.28,295.6 Collecting cultivated or wild plant material
Certain botanicals are unavailable as authenticated references, and references must be obtained by other means. In some cases, it is possible to contract third-party vendors to obtain vouchered botanical samples. Another option is to purchase raw or minimally processed plant material from a nursery or farm, or, in the case of a botanical that grows wild, to harvest it from its native environment (sometimes termed “wild crafting”). In these cases, a voucher specimen should be collected at the time of harvest. For wild-harvested plants, permission of the landowner or overseeing agency should be sought prior to collection.39 Researchers seeking to collect botanical material for research purposes should also be aware of regulations that govern intellectual property related to that material. In 1992, the United Nations, through the U.N. Convention on Biodiversity, enacted recognition of traditional knowledge and the natural resources that go with that knowledge. The Convention on Biological Diversity codified enforcement of ecologically sound harvesting practices and sharing of any intellectual or economic benefits that may arise as a result of the utilization of that traditional knowledge.40–42 Even though the United States did not ratify this treaty, many funding agencies, particularly those of the National Institutes of Health, have chosen to adopt the principles of this convention.43 In addition, during the 10th conference of parties of the Convention on Biological Diversity,43 the Nagoya Protocol was adopted, which is a legally binding protocol for access to genetic resources and benefit sharing.44 Again, since the United States is not a party to the Convention, this creates a gray area. However, this is another example where journal editors have a great degree of influence, as some journals (for example, Planta Medica) now require verification that study materials were acquired in accordance with the Nagoya Protocol, as it is interpreted by the country from where the sample was collected. This latter point is important, as individual countries have interpreted how to handle the Nagoya Protocol differently. Ultimately, collecting a plant specimen is not a trivial matter, especially when done outside the United States, and we recommend working with local authorities and seeking guidance and consultation from individuals with relevant experience before doing so.
Box 2 Summary of recommendations: obtaining study material• When harvesting cultivated or wild botanical specimens, collect additional material for identification and accession to an herbarium.• Procure sufficient number of commercial products to be representative of the overall market population. In our studies we evaluated a minimum of 30 different products. • For commercial samples that are dried, powdered, or pre-extracted, obtain additional authenticated reference material for identification. • Either a commercial source or a contract laboratory can be used to provide botanical natural product study material. The advantage of a commercial source is that the product is representative of what is used by consumers, but care must be taken to ensure that the material is authentic, consistent, and stable. • Obtain proper permissions before harvesting; collection of samples could be restricted due to local, state, federal, or international laws. |
6. Authentication
Once a botanical sample (or series of samples) has been collected for analysis, an important step is “authentication,” i.e. the definitive determination of the species represented by the sample.13,45,46 Botanical samples can be authenticated by multiple means, including examination of voucher samples, DNA barcoding, and both untargeted and targeted characterization of chemical constituents. Whenever possible, more than one technique should be applied to confirm the identity of a botanical sample. However, the type of processing that a sample has been subjected to will determine which techniques are effective for authentication, as described in the following section.6.1 Authentication by microscopic and morphological characteristics
Traditional methods used to authenticate botanical samples rely on examining the morphological and microscopic characteristics of representative voucher specimens. For example, macroscopic observations surrounding the arrangement (e.g., alternate, opposite, whorled) and shape (e.g., elliptic, ovate, cordate) of the leaves, the shape of inflorescence (e.g., cruciform, head, spurred) as well as its type (e.g., raceme, umbel, spike), the classification of the root system (e.g., taproot, fibrous root) can provide insight into the genus and species of the plant material. Similarly, microscopic examination of the histological characteristics of plant parts (e.g., stems, roots and rhizomes, bark, leaves, flowers, seeds, wood) can also aid in positive identification. These techniques, whether independently or in combination, have formed a baseline of approaches for botanical natural product quality control, and they continue to serve as essential methods used in pharmacopeia worldwide.47 However, accurate microscopic and morphological analysis requires a trained expert, and the robustness of these procedures is also reliant on the presence (and expert knowledge) of diagnostic characteristics that enable one natural product to be separated from another. Challenges arise for closely related species and plant parts that share similar morphological structures yet could have differing biological activities.Studies conducted using natural product extracts, botanical supplements, and commercial material are subject to several obstacles to authentication. When using commercial suppliers as material sources, voucher specimens are not always readily available. Identification by morphology or microscopy is not suitable for extracted natural product materials, and it may be difficult to determine the identity to the species level. Thus, to address the need for additional, more robust methods, researchers developed complementary techniques of differentiating botanical natural products.
6.2 Authentication by DNA barcoding
DNA barcoding is a genetic technique used to identify species using specific differences in small regions of DNA.48 Barcoding is an effective tool in authentication of botanical natural products that are comprised of fresh, dried, or powdered material, where intact DNA is still present.46,49,50 However, the DNA barcoding approach is more difficult when applied to botanical extracts. The manufacturing process for botanical natural products may involve heat treatment, oxidation, extraction, distillation, filtration, pressing, encapsulation, spray drying and/or UV light exposure, and these often lead to removal or degradation of DNA. DNA barcoding is not feasible for processed botanical products where the DNA is either not present or potentially highly degraded, or where there are two or more species present (either mixtures of active botanicals or the use of botanical “filler” such as rice).50 Unfortunately, DNA barcoding is occasionally used to characterize processed botanical natural products without recognition of these limitations. For example, in 2015, the New York State Attorney General investigated potential adulteration of commercial products. This investigation employed DNA barcoding methodology to suggest that only five of 24 samples actually contained the purported botanical natural product,51 and as a result many of the products were subjected to a forced recall. However, the methods and results of the study were suspect. Among other concerns, there were reports of cross-contamination of the samples, and the investigation focused on botanical extracts, not whole plant material, such that intact DNA may not have been present even if the correct botanical natural products were used.52 This controversy may have been avoided with the use of complementary analytical approaches suitable for characterizing extracts (such as described in 6.3 and 6.4).Although beyond the scope of this review, there are other factors that make DNA barcoding of plants challenging, such as hybridization and polyploidy, slow mutation rates compared with speciation rates, as well as lineages that show rapid and recent divergence rates. In some species of plants, these factors can influence the discriminatory power of standard plant barcoding markers.53–58 Despite these limitations, the use of two core plant DNA barcode regions rbcL (ribulose-bisphosphate carboxylase) and matK (maturase K) as well as two supplementary regions, trnH–psbA (chloroplast intergeneric spacers) and internal transcribed spacer (ITS) have been used for species discrimination in identifying plants via barcoding.58 Given all these complexities and controversies, it is recommended that DNA barcoding of botanical materials be conducted in consultation with experts.
6.3 Authentication by targeted analysis of “marker compounds”
Information about the identities of known small molecule constituents (metabolites) present in a botanical extract (known as “chemotaxonomy”) can be extremely useful in the authentication process. For most botanicals sold commercially as dietary supplements, there is a broad literature base that describes known metabolites and quantities in which they are likely to be present. When these constituents are unique to the botanical of interest, they are often referred to as “marker compounds”. Information about the chemical composition of a given plant is available in the primary peer reviewed literature, and in the form of “monographs” that summarize the chemical content of a particular botanical. Monographs are published by several sources, including the German Commission E,59 US Pharmacopeia,60 and Tyler's Herbs of Choice.61 To make use of these monographs, the sample under question is analyzed by a method appropriate for detecting and quantifying the key marker compounds (see Section 9, Identification, structure elucidation, and quantitative analysis of bioactive compounds in the selected study material), and their identities and concentrations are compared with literature values. The most common analytical techniques for analyzing botanical constituents are ultraviolet/visible (UV/VIS) spectrophotometry, mass spectrometry (MS), and Nuclear Magnetic Resonance (NMR) spectroscopy. Because of the complicated nature of botanical samples, it is also typical to perform chromatographic separation prior to analysis by spectrometric approaches. These chromatographic separations are most commonly carried out using gas chromatography (GC), high-performance thin-layer chromatography (HPTLC), high-performance liquid chromatography (HPLC), or a modern advancement on HPLC, termed ultrahigh performance liquid chromatography (UHPLC) prior to analysis by spectrometric approaches. The relative merits of different separation and analysis approaches for botanical natural products have been previously described.62–65 It is advised to use multiple approaches to characterize a single sample such that orthogonal data can be collected and compared.6.4 Authentication by “chemical fingerprints”
In addition to specifically analyzing individual marker compounds, researchers seeking to authenticate botanical natural products often collect a “chemical fingerprint” that represents a more comprehensive profile of metabolites produced by the plant. This chemical fingerprint can be compared between two or more samples to assess similarity or difference (Fig. 2).66 The most common techniques used to measure chemical fingerprints are gas or liquid chromatography, coupled to either spectrophotometric or mass spectrometric detection.62,65 The resulting data can be viewed as a chromatogram, which displays detector response as a function of time (Fig. 2), such that each “peak” in the chromatogram corresponds to a chemical constituent (or multiple constituents) from the sample. Quantitative NMR (qNMR) is another technique which provides chemical fingerprinting capable of distinguishing species and authenticating botanical origins.67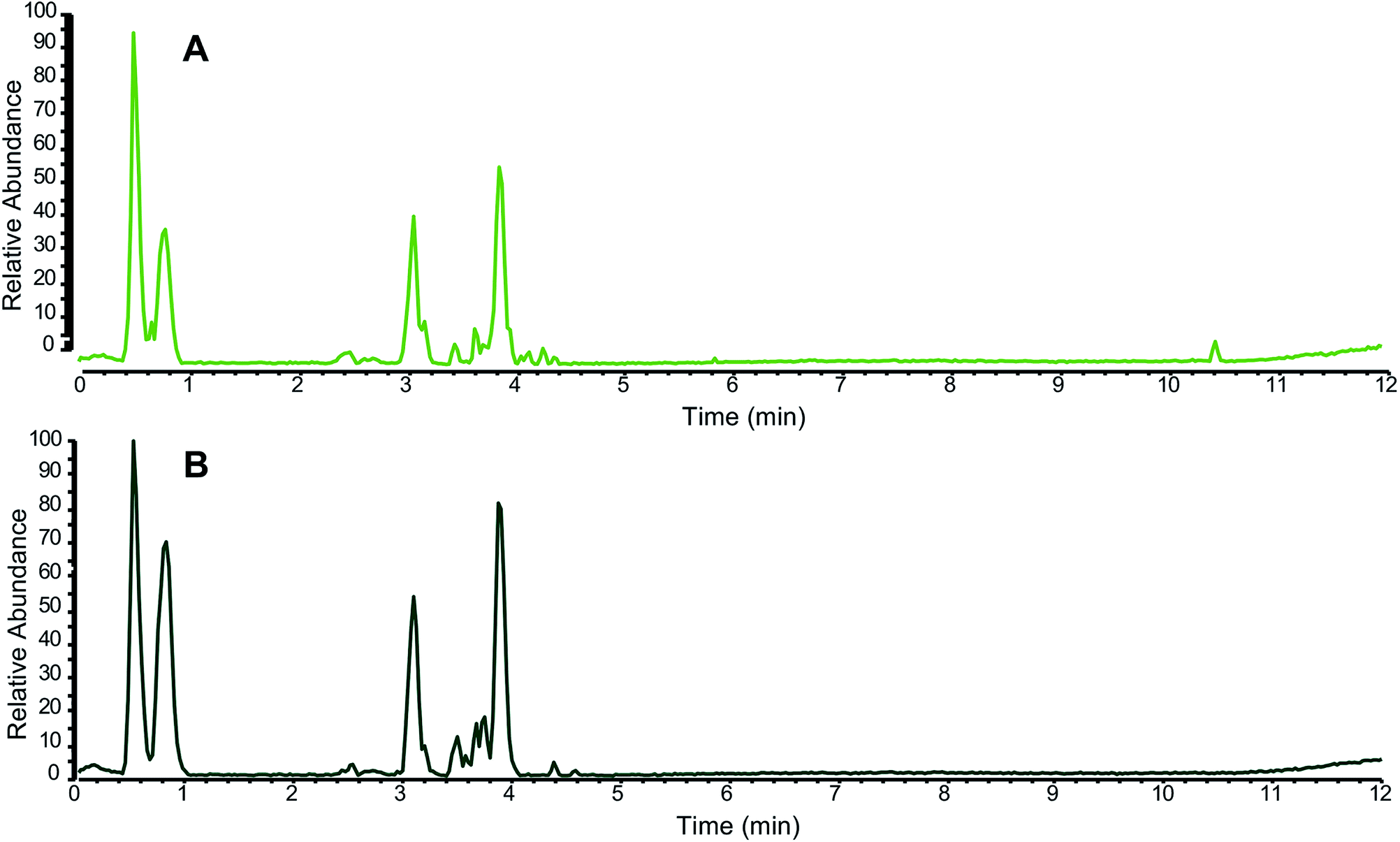 | ||
| Fig. 2 Representative liquid chromatography-mass spectrometry (LC-MS) fingerprinting of a green tea sample (A) compared against an authentic green tea (Camellia sinensis) standard from the National Institute of Standards and Technology (NIST 3254) (B). Both chromatograms were collected in the negative ion mode using ultrahigh performance liquid chromatography (UHPLC) coupled to electrospray ionization mass spectrometry; (reproduced with permission from ref. 28). Note that on balance, the majority of the peaks, both in retention time (x axis) and relative abundance (y axis), match between the two samples. | ||
The relative intensities, retention times, and spectroscopic data obtained by chromatographic analysis can be used to putatively identify a sample. Small molecules are more stable than DNA, and they typically survive the processing involved with preparing botanical natural products. Thus, it is possible to authenticate many samples based on their metabolite profiles that may not be characterizable by DNA barcoding. However, it is important to note that the “fingerprint” of which metabolites are present differs depending on the extraction procedure as well as analytical technique used to collect the data and the parameters of the specific analysis.68 Thus, it is necessary to include authenticated reference material for comparison in the same analysis with the sample under question and processed by the same extraction procedure and solvent system (see Purchasing authenticated reference materials, Section 5.5) (Fig. 2).
6.5 Authentication by untargeted metabolomics
Often, it is of interest to compare more than two botanical natural product samples to each other and assess their relative differences and similarities. The technique, currently referred to as “untargeted metabolomics,” can be very useful for this purpose. The “metabolome” is defined as the complete set of small molecules produced by a biological sample. Metabolomics techniques rely on measuring as many of these small molecules as possible (although it is never possible to measure the entire metabolome, due to limitations in analytical methods). Metabolomics has been employed to characterize the relationships between the metabolome of a given natural product and corresponding genotype, origin, quality, or other biotic or abiotic attributes.28,69–71 Two main analytical techniques are currently employed for metabolomic studies – MS and NMR. The relative advantages of these approaches have been discussed elsewhere.28,72–74 The ability of untargeted metabolomics analyses to simultaneously profile a large fraction of the phytochemical make-up of the botanical, and offer detailed characterization of the samples in question has led metabolomics to be a very useful tool in the authentication of botanical natural products.The process of data collection for a metabolomics analysis is essentially the same as that described for chemical fingerprinting, except that it is typical to analyze scores to hundreds of samples in a single analysis. This need to collect large datasets requires careful attention to experimental design, because run-times are often long, and drift in instrument response can cause artifacts in the data. Quality control checks throughout the run, as well as attention to sample order, blanks, and replication, are critically important.75,76
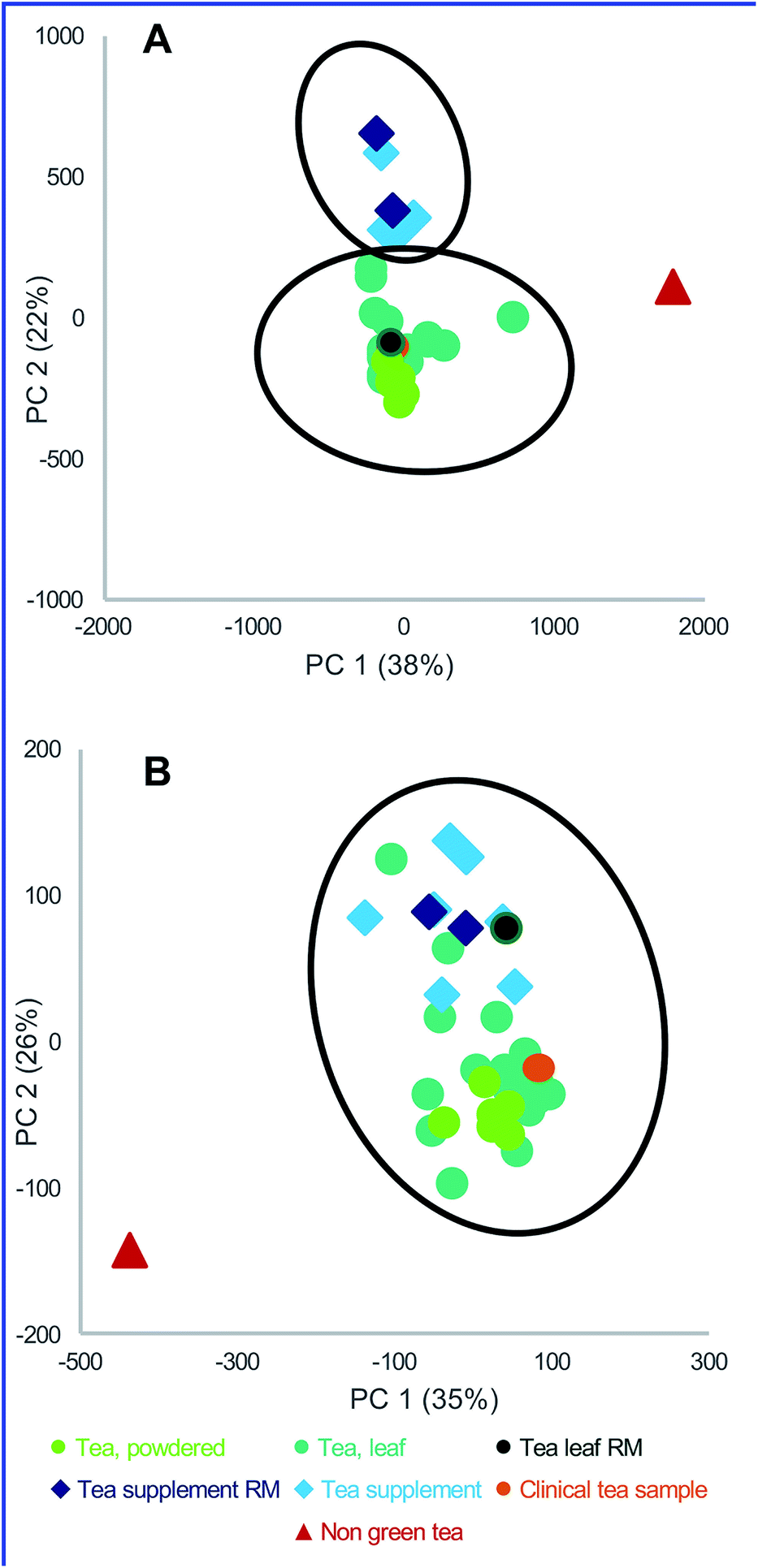 | ||
| Fig. 3 An example of principal component analysis (PCA) scores plots for green tea samples. Data points represent averaged triplicate extractions and plots are drawn with Hotelling's 95% confidence ellipses. “RM” represents green tea standard reference material from the National Institute of Standards and Technology (NIST). (A) Untargeted metabolomics analysis yielded clusters of distinct sample types (green tea supplements, green teas, and the negative control (indicated as “non-green tea”)). (B) Targeted mass spectrometry analysis, using 15 quantified standards, was employed to differentiate between green tea samples and the negative control. However, distinction between green tea supplements and green teas was not as clearly defined, and the RM samples were clustered together, irrespective of their origin. Reproduced with permission from Kellogg et al.28 | ||
Box 3 Summary of recommendations: authentication of natural products• Authenticate botanical materials to ensure the validity of in vitro, non-clinical in vivo, and/or clinical studies.• Select an authentication method that is suitable for the type of specimens being analyzed. For example, extracted botanical supplements cannot readily be identified using DNA barcoding techniques. • Authentication using only a single technique may be insufficient to ensure accurate identification of the botanical material, and the application of orthogonal methods (i.e. targeted analysis of marker compounds plus untargeted metabolomics or DNA barcoding) is recommended. |
7. Selecting a “representative” sample
Faced with a range of commercial samples purported to have been prepared from a given botanical natural product, how does the researcher select a representative product to advance to in vitro, non-clinical in vivo or clinical studies? Numerous factors influence the composition of a botanical natural product. For example, with green tea, cultivar, geography, elevation, sun exposure, climate, harvest time, harvest location (where on the plant the leaves were removed), as well as processing, fermentation, and manufacturing methodologies all led to discernable differences in final phytochemical make-up of the product.78–83 These details are not readily available to the consumer (or post-harvest researcher); thus, choosing one product out of a multitude of available options constitutes a major challenge. Researchers apply a wide variety of criteria for choosing natural products for further study, including convenience,84 consumer preference,28 or a custom prepared extract formulation contracted by the research team.85 As a result, the natural product interventions that are evaluated may or may not have a resemblance to the products that are currently on the market or used by the greatest numbers of consumers. While it is typically not possible to select a single product that represents all of the products used by consumers, it is worthwhile to capture a “snapshot” of the variability among the pool of products available, such that a desired product can be selected rationally. To conduct such a comparison, it is first necessary to obtain multiple products that represent those commercially available. For example, when the NaPDI Center prepared to conduct studies with green tea (Camellia sinensis) and goldenseal (Hydrastis canadensis), we purchased 34 and 35 commercially available samples of each botanical, respectively. We obtained fully characterized green tea samples from NIST, and several botanical reference standards of goldenseal from ChromaDex and laboratory collections.28,29 These products were then all extracted in the same fashion, and their metabolite profiles were compared to those of the reference materials to select a representative product for in vitro and clinical studies. Here we describe our selection process in more detail, using green tea as an example. A similar approach has been employed by other laboratories for other studies of botanical natural products.30,86,877.1 Selecting a formulation
Botanical natural products are available to the public in a variety of formulations. Commercially available formulations include tinctures (ethanolic or glycerol extractions of raw plant material), tea bags for making hot water extracts, raw plant material for human consumption, capsules, and tablets. A different constituent profile may be obtained by extracting in alcohol versus hot water and by using different plant material (for example, roots versus leaves) as starting material. Not all capsules are prepared in the same fashion. Some are made of raw plant material encapsulated in some coating (i.e. gelatin), some have a liquid extract directly encapsulated, whereas others are prepared by spray drying an extract on some solid support, such as cellulose. The formulation used can alter the chemical composition of the product. Spray-dried extract capsules usually are more concentrated than capsules prepared from raw plant material and may also be chemically simpler or modified by the extraction process. Because of these differences, we recommend that multiple representative formulations and, if applicable, multiple plant parts, of a given natural product be subjected to chemical evaluation. For example, for studies our group conducted with green tea, both spray-dried and raw plant material supplements and tea formulations in the 34 products were included in the initial evaluation. Metabolomics analysis demonstrated differences in profiles of the loose-leaf tea versus the encapsulated green tea supplements. The PCA loadings plot revealed several metabolites present in higher concentrations in the green tea supplements than the leaf teas: myricetin, kaempferol, and quercetin aglycones, as well as theaflavin 3-O-(3-O-methyl) gallate and the dimer epicatechin (4β→8)-epigallocatechin-3-O-gallate (Fig. 4).28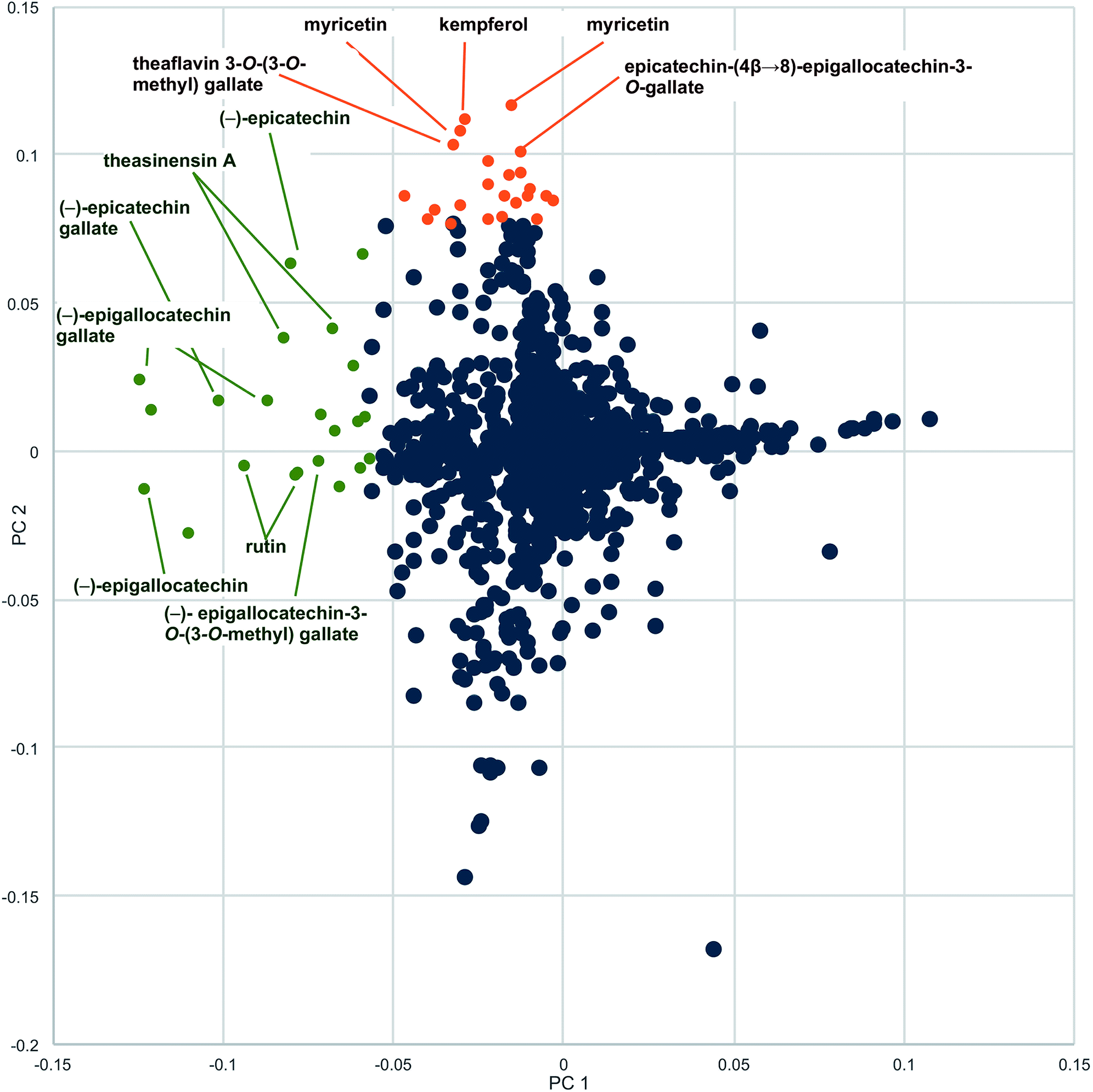 | ||
| Fig. 4 Loadings plot to identify constituents responsible for differences among commercial preparations of green tea. Metabolites with more negative values along the x-axis (PC1, green labels) were present in higher concentrations in the green tea samples versus the non-green tea (negative) control. Labeled metabolites with greater positive values along the y-axis (PC2, brown labels) were more heavily represented in green tea supplement samples compared to green tea leaf and powder samples (reproduced with permission from Kellogg et al.)28 | ||
Another consideration when selecting natural product formulation is that the dissolution and absorption of the bioactive constituents, and by extension human systemic exposure, can differ depending on formulation.88,89 These factors should be considered when designing in vitro, non-clinical in vivo, or clinical studies involving a botanical natural product.
Selection of product formulation should be guided by consumer usage. For example, we elected to conduct a clinical study with green tea as a beverage (hot tea) rather than a supplement based on popular use.16 Similarly, a goldenseal capsule containing dried botanical material was selected because this formulation is most commonly used as a botanical dietary supplement.
7.2 Standardized products
Some commercially available botanical natural products are sold with no information about chemical makeup, while others are “standardized” to (reportedly) contain a specific amount of a known constituent or constituents.27 An advantage of standardization is that researchers, health care practitioners, and consumers are provided information that may be useful for selecting and adjusting dosage.26 The standardization process may also provide consistency across products, with the caveat that methods used for standardization must be reproducible across multiple labs.90,91 Standardization can involve multiple analytical methods, including qNMR,92 UV-VIS absorbance patterns,90,93 and MS profiling.94 Even for standardized products, label claims about content and identity of a natural product should be confirmed in-house or by a contract laboratory prior to conducting in vitro, non-clinical in vivo, and clinical studies.A disadvantage of standardization is that the focus shifts from the quality of the entire mixture to a single or limited set of constituents. Standardized extracts may be chemically simpler than non-standardized counterparts, because the process of enhancing the concentration of constituents may involve extraction procedures that reduce extract complexity. Reduced extract complexity can be a disadvantage when the true “bioactive” constituent is not known, such that standardizing to one constituent may result in a lower dosage (or complete lack) of a constituent responsible for the presumed biological effect. How does a researcher decide whether to use a standardized product for an in vitro, non-clinical in vivo, or clinical study? One consideration is whether the most popular product(s) used by consumers are standardized. It is also advisable to compare metabolite profiles of standardized products to those of unstandardized counterparts to determine if components contributing to biological activity might have been lost during the standardization process (see Section 9.1).
7.3 Selection of extraction solvent
Once a series of representative products and authenticated standards has been selected, the first step in chemical analysis is to prepare extracts from these products. Typical methods of extracting botanical natural products for human consumption include the use of aqueous ethanol or aqueous glycerin (to prepared tinctures), or hot water (to prepare a tea). Applying these same extraction procedures when preparing samples for chemical analysis may seem prudent. However, water, ethanol and glycerin extracts are difficult to manipulate in a laboratory setting. Drying botanical natural product extracts (removing solvents) for long term storage is ideal because extracts are most stable and least likely to degrade when solvent is not present.95,96 Removing ethanol is a challenging process, removing water is more so, and removing glycerin is most arduous. Additionally, water extracts are unstable and contain polar constituents that confound chemical analysis and result in sticky, tar-like samples.Unlike the aforementioned solvents, methanol is an outstanding solvent for preparing botanical natural product extracts prior to analysis. Methanol has an intermediate polarity that effectively solubilizes a wide range of structurally diverse small molecules, is inexpensive, and can be rapidly and efficiently removed under vacuum or a nitrogen stream. As such, many natural product extracts are prepared in methanol or similar organic solvents.77,97,98 A disadvantage of these solvents is that they are unsafe for human consumption, raising concern that the resulting extracts are not representative of consumer use. While characterizing various green tea products prior to clinical evaluation, we addressed this concern by comparing hot water extraction (an obvious choice for tea) to methanol extraction. Methanol and hot water extracts contained similar quantities of catechins28 and showed qualitatively similar biological activity in vitro. Based on these results, methanol extraction was selected for metabolomics analysis of green tea products. Because methanol enabled rapid and reproducible extraction of samples, the extraction process did not confound comparisons of the chemical composition of the products themselves. A follow up study showed that accelerated solvent extraction improved extraction efficiency of green tea components in methanol.99 An alternative to methanol extraction includes “comprehensive” extraction techniques, with a gradient of solvents producing a variety of crude extracts with differing polarities.100 Ultimately, the ideal extraction solvent will depend on the type of material being extracted, the methods used to prepare preparations for consumer use, and the study question. Extraction solvent choice should be carefully considered and justified based both on experimental data and practical considerations. As the extracts prepared for research purposes often are not intended for human use, practical considerations of component solvation, ease of evaporation, and other physiochemical properties of the solvent may outweigh concerns about whether the extraction solvent is identical to that used to produce food grade products.
Box 4 Summary of recommendations: selecting a botanical natural product sample• Acquire multiple representative examples of different preparations and/or plant parts.• Use industry and consumer data to guide selection of products that are representative of the consumption patterns of the population of interest. • Consider whether a standardized product is appropriate for a given application; there are advantages and disadvantages of standardization. • Select an extraction solvent based on the research question, being mindful that there are many advantages to methanol extraction for stability, ease, and repeatability of laboratory work. |
8. Quality control and comparison of botanical product composition
Targeted chemical analysis of known markers and untargeted metabolomics to authenticate botanical natural product samples can be used to characterize and compare a series of candidate botanical natural products prior to selecting a product for in vitro, non-clinical in vivo, and/or clinical study. The advantage of untargeted chemical analysis for this purpose is that the analysis is relatively simple, can be highly sensitive and selective (leading to low limits of detection) and can be quantitative (when relevant reference standards are available; see Section 9.2). The disadvantage of targeted analysis for comparing samples is that the differences between samples may not be due to known “marker compounds”. For example, in green tea studies conducted by the NaPDI Center, samples of tea leaves or powder were shown to differ chemically from green tea supplements when untargeted metabolomics was applied, as indicated by the distinct grouping in a PCA plot (Fig. 3). However, when a PCA plot was generated using data from targeted analysis of 15 known green tea constituents, the differences among samples were less clear. As evidence of this lack of clear distinction, the points in the PCA plot in Fig. 3B representing supplement and tea samples overlap, suggesting (incorrectly) that the chemical makeup of the different samples is indistinguishable.8.1 Quantitative metrics for comparing similarity of natural product mixture
Untargeted metabolomics approaches comparing botanical natural products yield a tremendous quantity of data. Multivariate statistical techniques (e.g., PCA and partial least squares discriminate analysis (PLS-DA)) have become routine analytical methods used to analyze these data101 and serve as the backbone of metabolomic statistical analyses. PCA enables visualization of similarity and differences among samples by plotting them as points in 3D or 2D space (Fig. 3).102 With appropriate validation, PCA serves as an effective approach to compare qualitative differences among botanical samples.103,104 Other approaches, such as cosine calculations, measures based on vector distance, and measures based upon probabilistic difference, are used to generate quantitative metrics to compare chemometric or metabolomic spectral datasets.105–108 Recently, to facilitate selection of a green tea product for in vitro and clinical studies, we developed a new approach for quantitative comparison among samples in a metabolomics dataset, termed the composite score (CS).28 The CS is based on the scores and loadings of the totality of a multivariate PCA model, rather than pairwise comparisons of components (as in traditional PCA scores plots). The scores and loadings are integrated into a single correlation matrix, which can be used to compare similarity between multiple samples (Fig. 5). The CS ranges from −1.00 to 1.00. Values close to 1.00 indicate similarity among two samples (Fig. 5B), whereas values further from 1.00 indicate more dissimilar samples (Fig. 5C). The CS approach was used to evaluate which commercial sample was most similar to a given standard reference material.28,29,99 The PCA plot (Fig. 3A) from the green tea study yielded distinct clusterings of green tea supplements versus loose leaf green tea products (teas and powders). However, resolution was lacking to discern which specific tea was most similar to the chosen standard, NIST T26; indeed, a different answer regarding which teas are most similar would be obtained depending on which two principal components were plotted against each other (PC1 versus PC2, PC2 versus PC3, etc.). CS analysis (Fig. 5) was used to evaluate which products were most similar to the reference, as well as to select a commercial product (T21) for in vitro and clinical studies.28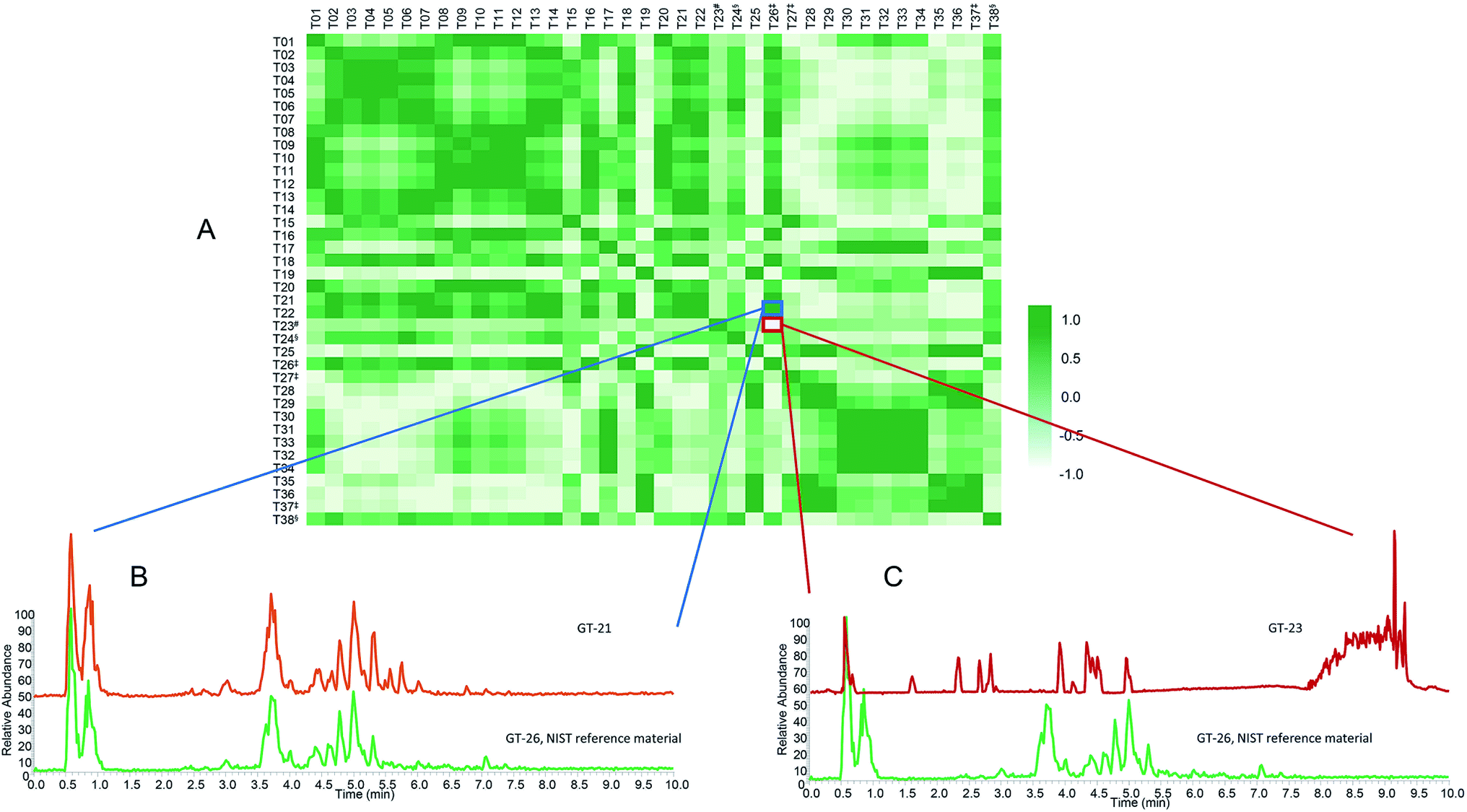 | ||
| Fig. 5 Composite score (CS) analysis for green tea samples. (A) Heat map correlation matrix of all green tea samples (indicated by codes on the horizontal and vertical axes). CS was calculated from the reproduced correlation coefficient matrix comprised of a four-component PCA model. In the heat map, darker shades (values closer to 1.00) represent stronger correlation between samples, while lighter shades (values approaching −1.00) represent decreased correlation between samples. The correlations' relationship to the metabolome are represented in two examples. (B) High correlation between two samples (CS = 0.98, dark color) indicates a strong similarity of the metabolomic profiles. (C) Low correlation (CS = −0.85 light color) indicates two samples with diverging metabolomic profiles. #Negative control (T23); ‡NIST standard reference materials (T26, T27, and T37); §green teas with botanical additives (T24 and T38). | ||
8.2 Adulteration of botanical natural products
Several reports have described suppliers of natural products selling products that are either unintentionally misidentified or intentionally adulterated (contaminated with botanical or other material different from that listed on the label).64,87,109 Adulteration can occur in a variety of ways, including spiking synthetic compounds into the plant material or adding or substituting a different (less expensive) species, representing cost-savings to a dishonest supplier.110 When studies are unknowingly conducted with adulterated or misidentified plant material, results may be irreproducible in future studies or irrelevant to the botanical supposedly under evaluation. Thus, testing botanical natural products for adulteration is critical.Several research groups have used targeted64,87,90,111–113 or untargeted methods114–116 to identify adulterants in botanical natural products. For example, we studied 35 commercial products listed as “goldenseal”. PCA of untargeted metabolomics revealed a distinct grouping of goldenseal material, including both root/rhizome- and leaf-based samples (Fig. 6), yet three samples (GS-07, GS-20, and GS-33) were distinctly separated from the other clusters.29 Separation of these samples from the others in the PCA scores plot raised suspicion that they were misidentified or adulterated with other botanical products.
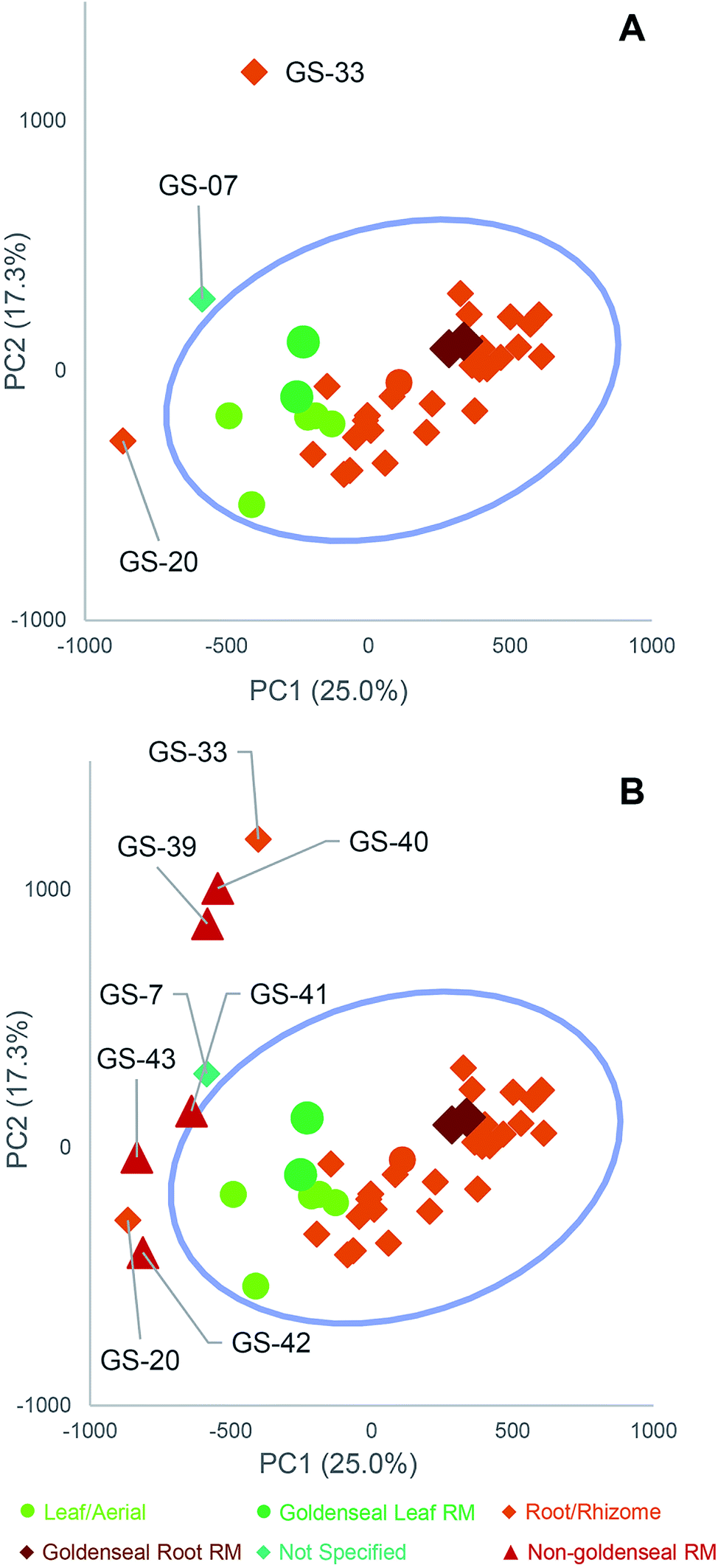 | ||
| Fig. 6 Principal component analysis (PCA) scores plots from untargeted mass spectrometry metabolomics analysis of commercial goldenseal product samples, plotted with Hotelling's 95% confidence interval (as shown by blue circle). (A) PC1 versus PC2 (25.0% and 17.3% explained variance, respectively) allowed for visualization of the samples (aerial portions and root/rhizome portions), with corresponding reference material (RM) located within the main cluster. The three samples labeled GS-07, GS-20, and GS-33 are located distinctly from other goldenseal samples and were considered outliers. (B) Follow-up metabolomics profiling and PCA scores plot including non-goldenseal reference materials (Coptis chinensis, GS-39 and GS-40; Mahonia aquifolium GS-41 and GS-42; Berberis vulgaris, GS-43) highlighted the potential sources of adulteration in the three outlier samples. Reproduced with permission from Wallace et al. (2018).29 | ||
To tentatively identify the species present in the outlier materials, reference material for three non-goldenseal species that serve as common adulterants were included in the dataset: Coptis chinensis rhizome (GS-39) and root (GS-40), Mahonia aquifolium leaf (GS-41) and root (GS-42), and Berberis vulgaris root (GS-43).110,117,118 The resulting PCA scores plot (Fig. 6B) showed close clustering of GS-33 with C. chinensis, GS-20 with M. aquifolium, and GS-07 in the middle, possibly as a blend of multiple species. Examining specific marker compounds for each of the four species (berberine, m/z 336.1229 [M]+; hydrastine, m/z 384.1440 [M + H]+; canadine, m/z 340.1545 [M + H]+; palmatine, m/z 352.1543 [M]+; coptisine m/z 320.0917 [M]+; and dihydrocoptisine, m/z 322.1074 [M]+) revealed specific concentration patterns (Fig. 7) for the putative adulterants that supported the hypotheses generated by the metabolomic analysis.29 This example illustrates the usefulness of both untargeted and targeted methods for identifying adulterated samples. PCA plots generated from untargeted metabolomics can be used to give a broad sweep analysis of the data and identify potential outliers, and follow up targeted analyses can help verify that adulteration has occurred and, if sufficient literature precedent exists, determine the identity of adulterants.29
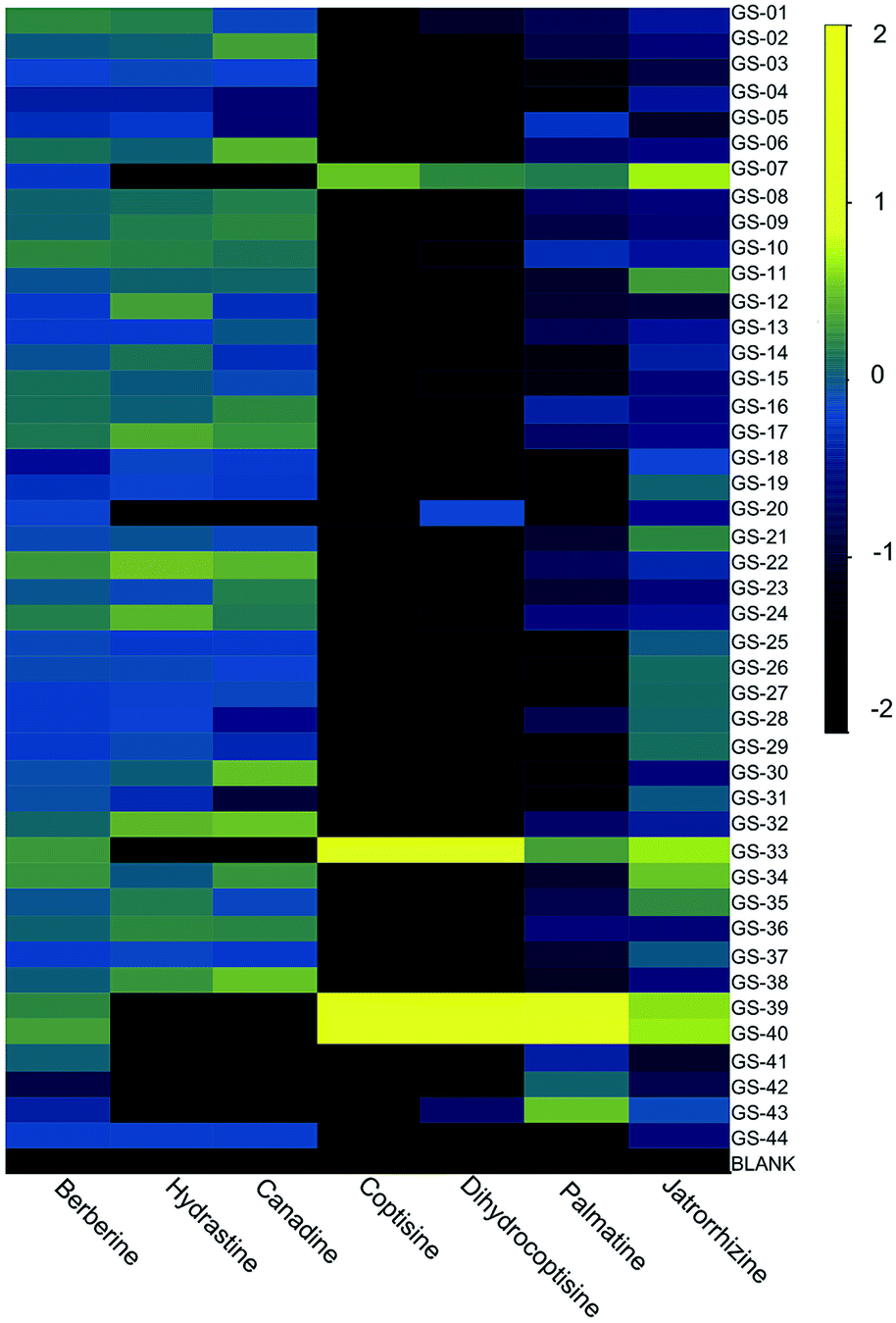 | ||
| Fig. 7 Heatmap of purported adulterants: six primary alkaloids present in H. canadensis, C. chinensis, M. aquifolium, and B. vulgaris. Heatmap was generated using the log10 of the peak area for the relevant ion detected by high resolution LC-MS. Reproduced with permission from Wallace et al.29 | ||
8.3 Consideration of contamination
A potential risk with botanical natural products is contamination by heavy metals,119 pesticides and herbicides,120 residual solvents,121 fungal spores and mycotoxins,122 or microbes.123 These concerns have received considerable attention, and recent reviews of the risks, policies, and analyses of contaminants in botanical natural products are available.27,31,112,124 In 2010, the FDA instituted the requirement that botanical natural products must be prepared using good manufacturing practices (GMP) that provide guidance for safety, consistency, and reproducibility of these products.21,125 GMP producers certify that their products are consistently produced and controlled according to quality standards. Whether the product is custom formulated21 or obtained from commercial sources, testing is required for contaminants, including “heavy metals, microbial limits, residual pesticides, adventitious toxins (e.g., aflatoxins), [and] endogenous toxins (e.g., pyrrolizidine alkaloids)” according to 21 CFR 111.70(b)(3). Product batches that do not meet the limits of contamination must be rejected.125,126 FDA guidance also specifies that at least one test must be used to verify botanical ingredients using a validated method.112 However, because the manufacturer is responsible for developing GMP procedures for each botanical natural product, there is no standardized procedure across the industry. In addition, compliance enforcement is limited and does not require evaluation or verification of authenticity or screening for contaminants by a third party.124 Given these limitations, testing botanical natural product study materials for contamination by pesticides, heavy metals, microorganisms, and residual solvents is advised. Such tests can be accomplished in house or through a contract laboratory. Screening is done in a targeted fashion, i.e. the analyst specifies a list of common or likely contaminants a priori and analyzes the sample to determine levels of these contaminants. The challenge with this approach is selecting which contaminants to test for among the almost unlimited number of potentials. Selection of contaminants can be guided by consultation with the literature or based upon knowledge regarding the environment in which the sample was grown and the processing it was subjected to post-harvest. Nonetheless, it is easy to imagine that some contaminants may be unexpected and, therefore, overlooked. Untargeted metabolomics may serve as a useful tool for identifying unexpected contaminants.29,111,115,127 However, it should be noted that untargeted methods are likely to have higher limits of detection than targeted methods; thus, the former may fail to detect low-level contaminants. Furthermore, some contaminants are detectable only with specialized analytical methods. For example, GC-MS is appropriate for the analysis of certain pesticides and residual solvents, but does not respond to many non-volatile contaminants, and a specialized system such as an inductively coupled plasma mass spectrometer (ICP-MS) is needed to detect heavy metal contaminants. With these challenges in mind, the analyst should carefully consider what the most likely contaminants are in a given sample and apply multiple analytical techniques and a combination of both targeted and untargeted methods to maximize the likelihood of detecting as many contaminants as possible.
Box 5 Summary of recommendations: quality control and comparison of botanical material• PCA is an effective method to compare qualitative differences among botanical samples.• The composite score (CS) represents a useful statistical approach for comparing the similarity or dissimilarity of multiple samples in large metabolomics datasets. • Untargeted metabolomics combined with targeted methods can effectively discern potential adulterations in botanical natural products. • Contamination testing is recommended prior to conducting studies involving botanical natural products. |
9. Identification, structure elucidation, and quantitative Analysis of bioactive compounds in the selected study material
Once a natural product has been selected for in vitro, non-clinical in vivo, or clinical studies, it should be thoroughly characterized, and concentrations and identities of key bioactive constituents (i.e., those responsible for the reported biological activity) should be determined. Such knowledge can be used to adjust dosage and conduct quality control. The challenge is that identities of bioactive constituents relevant to the activity being studied are often not known and are likely to differ depending on the biological activity evaluated.62,128,129 Assigning bioactive constituents is not trivial, as a given botanical extract may contain hundreds or thousands of constituents.130 The collective activity of the extract may be due to the combined action of these constituents acting in an additive, synergistic, or antagonistic manner.131–135 Despite these challenges, focusing solely on one or two constituents in an extract is not prudent (discussed in Sections 6.3–6.5). For some of the well characterized botanicals, nearly complete characterization of major constituents can sometimes be possible. For example, in our work with green tea, 15 commercially available pure standards were obtained that represented major and minor peaks in a LC-MS chromatogram (Fig. 2). The sections below highlight key points to consider when conducting qualitative and quantitative analysis of botanical extracts and assigning biologically active components. Based on practical experience, a summary of common pitfalls during quantitative and qualitative analysis of botanical extracts is provided (Table 2).| (1) Assuming that relative peak intensities in a chromatogram represent relative concentrations in the sample |
| Do not assume that a large peak means a high concentration of analyte in an LC-MS or LC-UV chromatogram. Instead, recognize that they reflect the responsiveness of the analyte to the detector, i.e. its ionization efficiency (for MS) or its molar absorptivity (UV). One approach to circumvent this problem is to use an evaporative light scattering detector (ELSD) or charged aerosol detector (CAD).153,154 The response of these detectors closely reflects relative abundance of analytes in the sample, but they are less sensitive than MS or UV |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| (2) Attempting to quantify analytes without reference standards |
| Because response of the detector (mass spectrometer or spectrophotometer) depends on structure, an authentic standard should be used to quantify the unknown of interest |
|
| (3) Assuming that the absence of a constituent in the data means an absence of the compound in the sample |
| Failure to observe a compound in the data means only that it was not detectable by the technique used above a specific stated limit of detection. It does not mean that the analyte was absent from the sample. Including a positive control can help address this problem. If the analyte of interest is detectable in the positive control but not in the sample, the analyte likely is absent from the sample (above the limit of detection) |
|
| (4) Overstating the accuracy to which a concentration is known |
| Absolute quantitation (knowledge of the true amount of a given compound in a botanical sample) is difficult to achieve. Factors such as inefficient extraction, lack of purity in the standard, and drift in instrument response make knowledge of true absolute concentration extremely difficult to assess. Avoid chasing perfect and absolute quantitation when relative or approximate quantitation will be sufficient. On the other hand, knowing that most quantitative measures are not as absolute as the reports may imply, avoid the pitfall of comparing samples from different runs or different laboratories when the results may be different due to factors inherent in the analysis. Include appropriate replicates, references and controls in each experiment such that comparison among experiments is possible. Report the concentration of a given analyte with its associated uncertainty and to the correct significant figures |
|
| (5) Assuming confirmation of identity with MS data |
| Even if appropriate standards are used to match retention time and fragmentation patterns of unknown compounds in a sample, mass spectrometry data does not confirm configuration of stereoisomers. NMR is needed for such confirmations. However, depending on the needs of the study, knowledge of configuration of stereoisomers may not be necessary, and MS data may be sufficient |
|
| (6) Extrapolating results beyond the linear range of the calibration curve |
| Always dilute samples so that the analyte of interest falls within the linear range of the calibration curve. Failure to do so may result in serious underestimation of analyte concentration. Errors also result from attempting to quantify analytes present at too low of a concentration, i.e. below the limit of quantification for the analytical method |
|
| (7) Failing to account for matrix interference |
| Matrix interference occurs when the matrix (everything but the analyte in the sample) alters the response of the analyte. This issue can be particularly pronounced with mass spectrometry as an analytical method. The best strategy for avoiding matrix interference is to dilute the sample as much as possible and subject it to chromatographic separation prior to analysis. A validation check that involves spiking the sample with a standard and comparing its response in matrix and solvent is necessary to check for matrix interference95 |
|
| (8) Assuming a sample is “pure” based on LC-UV data |
| When interpreting LC-UV data, it is important to remember that contaminants will not be detectable unless they absorb light in the region used for the analysis. Quantitative NMR represents an alternate method to determine purity155 |
|
| (9) Assuming that each peak in an LC-UV chromatogram represents a single compound |
| Multiple compounds in a complex sample may coelute in what appear as a single peak. Thus, one peak does not necessarily mean one compound. MS detection can be used to identify coeluting compounds if they differ in mass. Examination of UV spectra at multiple retention times across a single chromatographic peak can also aid in detecting coeluting compounds |
|
| (10) Assuming that each peak in the mass spectrum represents a different ion |
| Clustering and in-source fragmentation in mass spectrometric analysis often lead to multiple masses that represent a single ion. Identifying the true “molecular ion” can be difficult. Software packages such as RamClust156 and IntelliXtract157 can help assign identities of clusters and fragments and group associated ions |
9.1 Determining which constituents of a botanical natural product are biologically active
The gold standard approach for identifying bioactive mixture components is bioassay-guided fractionation, in which the extract is subjected to successive rounds of fractionation and purification (see Section 9.2), with each fraction prioritized for the next stage based on biological assay data. Such an approach has been used in countless studies, with perhaps the most well-known being discovery of the cancer chemotherapeutic agent taxol (aka paclitaxel) from the Pacific yew tree.136,137 An alternate approach to bioassay-guided fractionation when targets are known and available in sufficient quantity is pulsed ultrafiltration mass spectrometry,138,139 the advantages and disadvantages of which are detailed elsewhere.140 In general, we find that bioassay-guided fractionation is the most effective strategy for identifying bioactive mixture components when the specific targets and mechanism of action of a given botanical natural product are not known, and when those activities can be modeled effectively in vitro. Bioassay-guided fractionation can be enhanced using statistical approaches to integrate biological assay data with chemical metabolite (metabolomics) profiles, a process often referred to as ‘biochemometrics’.77,141In vitro assays are used to assess the potential for natural product constituents as lead compounds for a diverse array of illnesses, including cancer, bacterial infections, and diabetes.144–146 Cell-based assays can be used to guide future in vivo studies to test for safety and efficacy of natural products,147 to give insight regarding potential interactions between natural products and conventional drugs,148–150 and to predict how food matrices could affect the pharmacokinetics of natural product constituents.151 For studies evaluating interactions between natural products and conventional drugs, mechanistic insight gained from in vitro studies is imperative to guide further evaluation via mathematical (e.g., physiologically-based pharmacokinetic) modeling or clinical studies.6 However, in vitro assay data may not always translate to in vivo activity, partially due to the inherent complexity of biological systems, and the effects of other biological factors (e.g., microbiota) that are not accounted for in most in vitro models.152 Additionally, many botanical natural products are used for purported health benefits that are non-specific and difficult to model in vitro, such as “improving immune health” or “enhancing adaptability to stress”. In light of these challenges, it is important to keep in mind that the extent to which any bioassay-guided fractionation experiment is useful is fundamentally limited by the availability of a relevant, robust and translatable biological assay.
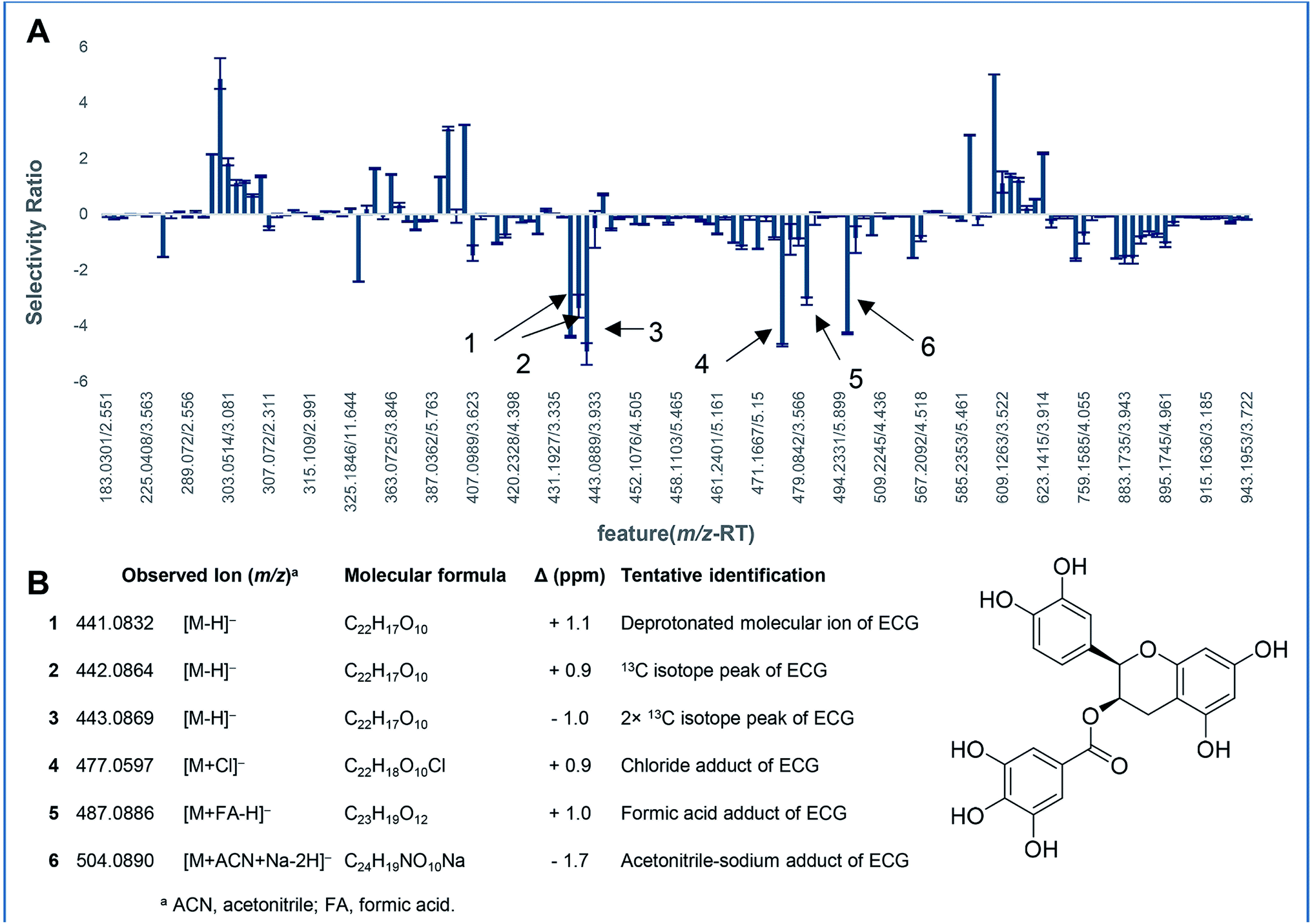 | ||
| Fig. 8 Biochemometric analysis of the inhibition of UDP-glucuronosyltransferase (UGT) activity by green tea constituents. Mass spectrometry-based metabolomics was correlated with bioactivity to generate a partial least squares (PLS) model for eight green tea subfractions. This model was used to determine the selectivity ratio for each feature detected in the green tea extract (A), the x-axis represents sample components detected as unique m/z-retention time (TR) pairs (features) in the extract. The more negative values on the y-axis represent more significant contribution to the observed UGT inhibitory activity. All six detected ions (spectral variables) represent isotope peaks and cluster ions of (−)-epicatechin gallate (ECG) (B) molecular ion, suggesting that ECG is the dominant bioactive ion in the green tea matrix. Adapted with permission from Tian et al. (2018).170 | ||
9.2 Obtaining “pure” standards
Efforts to identify and quantify constituents of botanical extracts often rely on the availability of purified standards. Several commercial suppliers provide pure samples of individual botanical compounds and include Sigma-Aldrich (St. Louis, MO, USA), ChromaDex (Irvine, CA, USA), NIST (Gaithersburg, MD, USA), and the United States Pharmacopeia (Rockville, MD, USA). The modest cost of these standards typically far outweighs the time and effort involved in obtaining standards via independent isolation methods. A standard comes with a “certificate of analysis” that reports identity and purity of the sample. Additional characterization (by LC-MS and/or NMR) is recommended to confirm accuracy of information included in the certificate of analysis.155When standards of the compounds of interest are not available commercially, isolating these compounds from the complex starting material is necessitated.171 A number of separation techniques can be used for this purpose, including solid phase extraction,172 counter current chromatography,173,174 HPLC,175 or thin layer chromatography (TLC).176 The most common strategy for isolation of constituents from natural products is a combination of liquid–liquid partitioning and column chromatography approaches. A typical fractionation scheme involves several steps, as illustrated in Fig. 9. The sample is extracted in methanol, partitioned between chloroform and water or ethyl acetate/water, and subsequently subjected to an additional partitioning step between methanol:acetonitrile and hexanes.177 These steps could be modified using acid/base conditions if the target metabolites are alkaloids. The resulting residue is dried under nitrogen and subjected to several stages of normal phase flash chromatography. Finally, pure compounds are isolated using reversed phase preparative HPLC. The solvents and stationary phases used for the separation can be varied to optimize separation of the compound of interest. Isolation of milligrams of pure material often takes weeks or months (or may be unsuccessful) depending on the difficulty of the separation and the skill of the analyst.178 As such, isolation of all constituents in a given botanical extract is typically unfeasible, and it is helpful to guide isolation efforts with biological assay data, as described in Section 9.1.
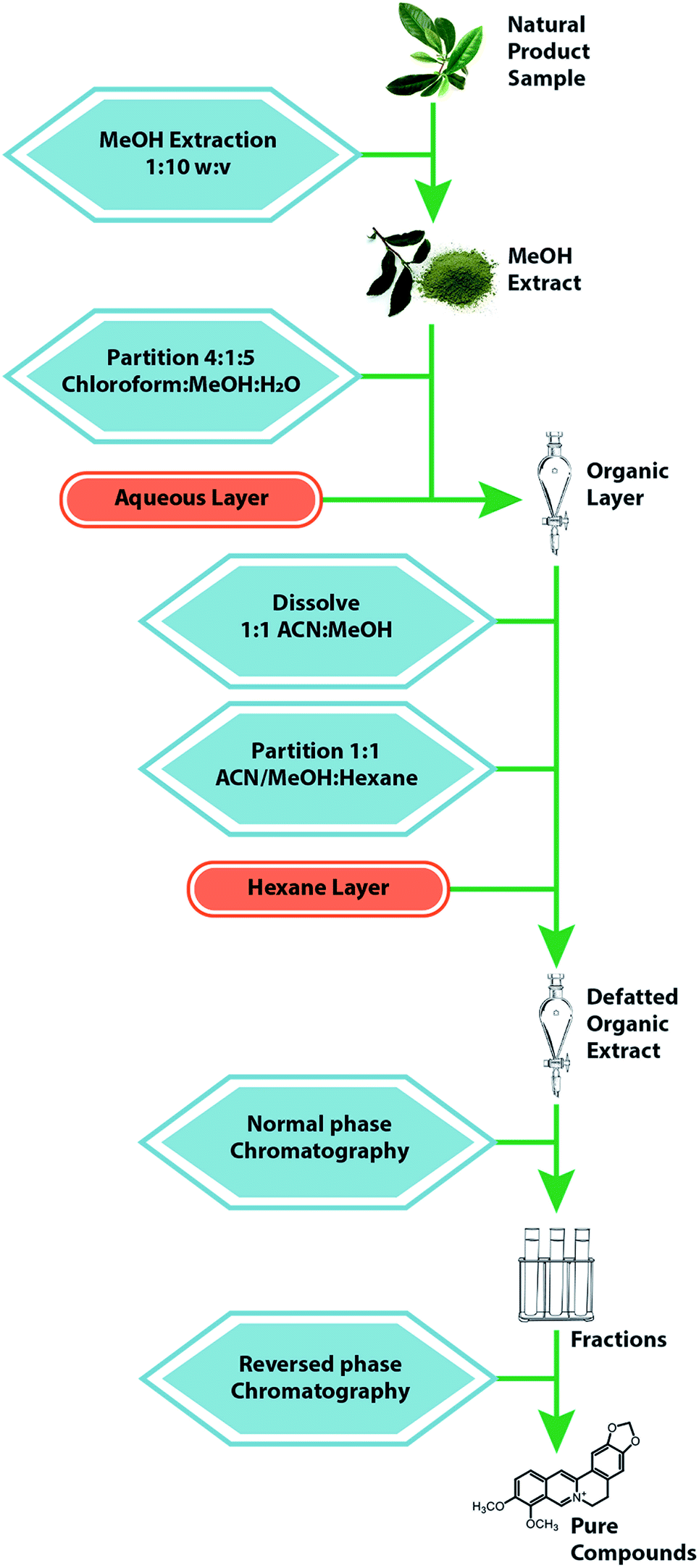 | ||
| Fig. 9 Standard extraction and fractionation scheme for a natural product. The sample material is macerated with an organic solvent, usually methanol, then subjected to repeated liquid–liquid partitions, yielding a final organic extract. This residue is fractionated with a normal-phase flash chromatography system, after which the fractions are further separated via a reverse-phase HPLC separation to yield purified compounds.77,185 | ||
9.3 Structure elucidation of constituents of botanical natural products
Identification of metabolites in botanical extracts has been reviewed previously,179,180 and structure elucidation of natural product constituents is detailed in several textbooks.181,182 Two approaches are commonly used: isolation and subsequent structure elucidation via NMR or characterization of mixture constituents without isolation via LC-MS. Conclusive assignments of structure are made via NMR and it is the preferred technique when sufficient material (typically 0.5 to 1 mg) has been isolated.183 Structure elucidation via NMR is conducted in combination with other spectroscopic approaches, notably high resolution MS, which enables determination of molecular formula.180 Accepted guidelines from peer-reviewed journals (e.g., Journal of Natural Products) recommend that assigned 1H NMR and 13C NMR spectra be presented for review, as well as molecular formulae calculated by relying on accurate mass measurements with MS, UV absorptivity, and any additional stereoscopic information as needed.10 In the very near future, we anticipate that many journals will require the deposition of raw NMR data associated with the structure elucidation of organic molecules, akin to the way DNA data are deposited in GenBank; a recent review in Natural Products Reports, co-authored by over 70 scientists, advocates for this practice.184For comprehensive characterization of mixtures without isolation, LC-MS (or GC-MS for volatile samples), is the most effective approach. Challenges, pitfalls, and strategies to identify botanical natural product metabolites by mass spectrometry have been detailed previously.62 If high resolution mass spectrometry is used, accurate measurements of mass are possible to tentatively identify extract components based on calculations of molecular formula. Mass spectrometers with the ability to measure mass with sufficient accuracy include those with Orbitrap, Fourier transform inductively coupled resonance (FT-ICR) or quadrupole time-of-flight (Q-ToF) mass analyzers. The confidence with which structure can be assigned based on mass spectrometric data is increased when retention time and fragmentation patterns for the component of the unknown sample can be compared to those of pure, isolated standards.186–188 However, mass spectrometry is not a conclusive technique for assigning configuration of stereoisomers.
9.4 Quantitative analysis
The goal of quantitative analysis is to determine the absolute concentration of a known constituent (analyte) of a complex botanical extract. Effective quantitative analysis requires an isolated standard identical to the constituent to be quantified (see Section 9.2, Obtaining “pure” standards). Dilutions of this standard are prepared and analyzed with the extract. A calibration curve of detector response (most commonly chromatographic peak area under the curve) vs. analyte concentration is plotted. The calibration curve is analyzed via regression over the linear range (range in which response increases linearly with increasing concentration). The response (peak area) for the analyte in the extract is substituted into the equation for the best fit line, and the concentration in the extract and the original plant sample is back calculated by adjusting for all necessary dilutions.189 To improve the accuracy of quantitative analyses, methods using isotopically labeled standards (when available) can also be employed.190–192The most popular techniques for quantitative analysis of botanical natural products are GC or GC-MS (for volatile sample components) and LC-UV/VIS or LC-MS (for nonvolatile sample components). The relative merits of these techniques are detailed elsewhere.62 Briefly, LC-MS often (but not always) provides lower limits of detection compared to LC-UV/VIS. An additional advantage of LC-MS is that it can be used to obtain orthogonal data about analyte mass and fragmentation pattern that is helpful for deconvoluting chromatograms and confirming identification.158 LC-UV/VIS, on the other hand, is typically less susceptible to matrix interference and for some applications demonstrates improved linearity, particularly at the high end of the calibration curve.95 Instruments with UV/VIS detectors are also less expensive and available to a broader base of analysts. Having access to instruments equipped with both UV and MS detectors, such that identity of the quantified constituents can be confirmed using more than one data type, is ideal. When both UV and MS data are available, quantitative analysis can be accomplished with whichever technique provides the most linear data in the region of interest.
When conducting quantitative analysis, analyzing the extract (botanical natural product) at multiple dilutions is recommended. A typical analysis includes a series of 2-fold or 10-fold dilutions of the extract, with the highest concentration in the range of 1.0 mg mL−1 (expressed as dry weight of extract per volume of dilution solvent). It is often desirable to quantify more than one constituent, and the ideal dilution to achieve a concentration within the linear range of the calibration curve will vary from constituent to constituent. Thus, analyzing the extract at multiple dilutions is helpful. A series of 2-fold or 10-fold dilutions of the standard is also included in the same analysis with the extract to generate a calibration curve for quantitative analysis. The resulting calibration curve should span the maximum linear range, and several dilutions above and below this range should be included to enable the full range to be determined. AOAC INTERNATIONAL (The Association of Analytical Communities) recommends including 6–8 data points within the linear range.193
One question that often arises when conducting quantitative analysis is the issue of whether (or how) a method has been validated. Validation is a process by which a given method is optimized for parameters related to accuracy, precision, limits of detection and quantitation, linear range and robustness.90 Other elements of method evaluation in the context of botanical natural product research include measurements of extraction efficiency and checking for potential matrix interference. Methods can be validated both within a single laboratory (intralaboratory validation)95,194 or across multiple laboratories (interlaboratory validation).67,90 The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) provides general guidelines for validation of analytical methods,195 and more specific guidelines and definitions related to validation of constituents of botanicals have been drafted by a subcommittee of AOAC INTERNATIONAL.13,193 When selecting a method for analysis of botanicals, we recommend reviewing multiple published methods (if available), with particular attention to those that have been validated. However, for practical reasons, including differences in available expertise, samples and instrumentation, it may be difficult to precisely replicate even a validated method for a new study. This is particularly true if the method has not already been subjected to interlaboratory validation. For this reason, it is typically useful to validate the selected method as part of the given project. One excellent strategy for checking accuracy is to obtain a botanical reference sample for which concentrations of the bioactives of interest are known, then compare the results obtained with the selected method to those reported on the certificate of analysis for the reference material. Such validation can be performed using reference standards from NIST (if available) for which marker compound concentrations have been reported. To avoid misleading results due to matrix interference, it is important to check the accuracy of the method in the botanical matrix of interest. For example, our methods for quantifying catechins as part of the NaPDI Center's green tea study were tested by analyzing a NIST green tea standard reference material, not isolated catechins.28 If no commercial reference material of the botanical is available, alternate techniques for quantifying metabolites are available. One technique is to conduct a “spike recovery” study, in which a standard is spiked into the botanical matrix at known concentrations, and the concentration is back-calculated to evaluate accuracy.95,196–198 Quantitative NMR and pre- or post-column derivatization199 also can be used to quantify metabolites without known standards.200
We end this section with a word of caution, which is that it is easy to become so focused on method validation that one is distracted from the overall research question (which typically relates to safety and/or efficacy of a botanical). It is often true that variability in biological response is so significant that it is unnecessary (and even a waste of time) to pin down concentrations in a botanical extract to the highest level achievable by a trained analytical chemist. The extent to which validation is necessary should be carefully considered in the context of the research question being evaluated.
Box 6 Summary of recommendations: identification, structure elucidation, and quantitative analysis of bioactive constituents• Knowledge of which constituents in the botanical extract are biologically active is needed to establish quality control procedures and determine dosage.• Biological activity may be due to more than one constituent, and the active constituent profile may vary depending on which biological activity is tested. • Because of the inherent complexity of botanical extracts, isolating all constituents and testing them individually for biological activity is infeasible. • Bioassay-guided fractionation, which can be enhanced with biochemometric analyses, can be used to direct isolation efforts towards identifying compounds likely to be active, provided that a relevant and robust in vitro assay is available. • Results of bioassay-guided fractionation experiments should be interpreted with consideration of the potential limitations of translatability to clinical studies. • Quantitative analysis of active compounds can be accomplished using isotopically labeled standards or calibration curves prepared from standards of the compound of interest in the relevant matrix. |
10. Conclusions
The process of selecting botanical natural products for in vitro, non-clinical in vivo, and clinical studies is complex, and the ideal approach must be tailored on a case by case basis. Nonetheless, as a result of extensive experience, and much trial and error, we have identified a few common themes, pitfalls, and questions that almost always arise. In the preceding sections, we sought to address these by presenting examples from work conducted as a part of the NaPDI Center in the context of current literature. While constructing the recommendations provided in this report, we have considered the need to balance the ideal, i.e. what would be done in a situation where time, person power, and funding were unlimited, with the practical, i.e. what is possible subject to realistic constraints. We provide Table 3 as a summary of many of the recommendations from the text, and to highlight a few additional points for consideration. It is our expectation that consideration of the topics addressed herein will enable the design of rigorous and reproducible studies with complex botanical natural products, and prevent the wasted time, funds, and effort that goes into studies where product selection has not been carefully considered.| Screen many products |
| Acquire and chemically evaluate as many different products as feasible. Most studies reviewed selected >20 products,28,29,86,94,201 although lower thresholds may be acceptable depending on limitations due to funding, logistics, and availability. Products selected for a given study or studies should be representative of consumer usage |
|
| Confirm identity claims |
| Verify all claims on labels via an independent analysis (e.g., the researcher's laboratory or a contract laboratory). This is important both for commercial botanical products and reference standards |
|
| Authenticate thoroughly |
| Authenticate using multiple orthogonal techniques and by comparing with trustworthy, vouchered samples. While this may seem obvious, avoid the wasted effort of conducting a research project starting with the wrong botanical |
|
| Obtain vouchers |
| Collect a voucher specimen and submit it to a publicly accessible collection at an herbarium. When a commercial product is used and a voucher specimen is not accessible, compare the chemical profile of the botanical to be used in the study to that of a vouchered reference material |
|
| Collect plant specimens using paper bags |
| Plant specimens should be stored in paper, rather than plastic, bags after collection. Plastic bags can contaminate the specimens with polymers that may mistakenly be attributed to the specimen. Plastic bags also prevent gas exchange, allowing moisture to accumulate rapidly, and accelerating fungal growth and potential mycotoxin contamination |
|
| Reserve samples |
| When preparing an extract, always save a labelled and catalogued sample of the starting product (extract, supplement, or dried plant material). When purifying an extract for analysis (i.e. conducting preparative chromatography or liquid–liquid partitioning), save a small sample of the extract and of each successively purified fraction. This reserved aliquot can be re-analyzed as necessary for chemical composition and biological activity. It is better to err on the side of saving too much of these materials than to save too little (or none). Relative to the cost of the future studies, the price of starting with well more than is needed is modest |
|
| Extract with methanol |
| Use methanol to prepare samples for chemical analysis and in vitro evaluation. Methanol is an effective extraction solvent because it facilitates reproducible production and stable storage of extracts. If necessary, conduct a parallel extraction with other solvents to ensure relevance to consumer usage. In the authors' experience, selection of extraction solvent typically does not change the profile of small molecule metabolites in a sample to a large extent, albeit relative amounts of these compounds may be altered. Methanol has a lower boiling point than ethanol or water, which facilitates its evaporation at lower temperatures, thus minimizing the chance of decomposition of the sample during evaporation. It is also our experience that the difficulty in working with and storing extracts prepared in other solvents introduces artefacts and unexpected complications into a research study, and some other solvents (such as DMSO) can contribute to the oxidation of extract constituents88 |
|
| Include controls in analyses |
| Botanical reference standards aid in the detection of adulterated products and enable botanical authentication, whereas chemical standards aid in quantification and provide the opportunity to monitor the performance of the analytical method |
|
| Purchase or collect more of a single lot than needed for in vitro and/or clinical studies |
| When using a commercial product, purchase double or triple the amount of product needed for the given study or studies. Where possible, purchase all of this material from a single lot to ensure consistency within and between studies |
|
| Characterize fully (not just for marker compounds) |
| Obtain the most comprehensive profile of metabolites possible when characterizing an extract. Techniques that focus on a few metabolites (or purported “marker compounds”) may produce misleading results and may unintentionally imply similarity among products that may possess different biological activities |
|
| Consider the limitations of the analytical technique |
| A few common errors in interpreting data analysis of botanical extracts are listed in Table 2. Careful data analysis and inclusion of appropriate controls and replicates is important to avoid these errors |
|
| Be cognizant of potential endotoxin contamination |
| Lipopolysaccharides (LPS, also referred to as endotoxins) are bacterial components that can profoundly influence in vitro assays, particularly those evaluating immunomodulatory activity. Because botanical extracts contain endophytic and epiphytic bacteria (and other microbes), it is virtually impossible to avoid contamination with LPS in extracts prepared from botanicals. Even ethanol extracts can contain sufficient LPS content to alter the results of biological assays202 |
|
| Store samples cool, dark, and dry |
| Never store extracts in liquid form, even in the freezer. Freeze/thaw cycles, especially in solvents such as DMSO, can result in accelerated degradation. Storing samples dry and in dark and cool conditions is the best way to prevent degradation. Cooler temperatures, such as refrigeration or freezing, are indicated to prolong the activity and chemical stability of prepared extracts.203 However, the type and extent of degradation that occurs will differ depending on the natural product being evaluated. Thus, it is advisable to monitor for potential degradation products by collecting chromatographic profiles immediately after the sample is prepared, and then at regular intervals or prior to each experiment in which it will be evaluated for biological activity |
12. Conflicts of interest
There are no conflicts to declare.13. Acknowledgements
This project was supported by the National Institutes of Health via the National Center for Complementary and Integrative Health, specifically the Center of Excellence for Natural Product Drug Interaction Research (NaPDI Center, U54 AT008909). JSM was supported in part by the National Cancer Institute [R01 CA182963]. MFP was supported in part by the National Institute of General Medical Sciences [R01 GM077482]. We thank the following individuals for insightful comments and helpful discussions: Dr D. Craig Hopp, NCCIH Program Officer; Dr Amy Roe, Principal Toxicologist at Procter and Gamble; Dr Huzefa Raja, research scientist at the University of North Carolina at Greensboro; Tyler Graf, research scientist at the University of North Carolina at Greensboro; and Dr Danny D. Shen, Professor of Pharmaceutics at the University of Washington. We would like to thank Matt Bryant and Cindy Graf for their expertise and assistance in designing the workflow figures presented here. The high-resolution mass spectrometry data were collected at the Triad Mass Spectrometry Facility at the University of North Carolina at Greensboro (http://chem.uncg.edu/triadmslab/).14. References
- C. H. Wu, C. C. Wang, M. T. Tsai, W. T. Huang and J. Kennedy, J. Evidence-Based Complementary Altern. Med., 2014, 2014, 872320 Search PubMed.
- K. M. Job, T. K. L. Kiang, J. E. Constance, C. M. T. Sherwin and E. Y. Enioutina, Expert Rev. Clin. Pharmacol., 2016, 9, 1597–1609 CrossRef CAS PubMed.
- B. T. Gufford, J. T. Barr, V. González-Pérez, M. E. Layton, J. R. White, N. H. Oberlies and M. F. Paine, CPT: Pharmacometrics Syst. Pharmacol., 2015, 4, 701–710 CAS.
- T. Smith, K. Kawa, V. Eckl, C. Morton and R. Stredney, HerbalGram, 2017, 115, 56–65 Search PubMed.
- M. A. Klein, R. L. Nahin, M. J. Messina, J. I. Rader, L. U. Thompson, T. M. Badger, J. T. Dwyer, Y. S. Kim, C. H. Pontzer, P. E. Starke-Reed and C. M. Weaver, J. Nutr., 2010, 140, 1192S–1204S CrossRef CAS PubMed.
- M. F. Paine, D. D. Shen and J. S. McCune, Drug Metab. Dispos., 2018, 46, 1041–1045 CrossRef CAS PubMed.
- J. Dwyer, P. Coates and M. Smith, Nutrients, 2018, 10, 41 CrossRef PubMed.
- Center for Drug Evaluation and Research, Botanical Drug Development: Guidance for Industry, U.S. Department of Health and Human Services, 2015 Search PubMed.
- National Center for Complementary and Integrative Health (NCCIH), NCCIH Policy: Natural Product Integrity, National Institutes of Health, 2016, http://https://nccih.nih.gov/research/policies/naturalproduct.htm (accessed February 18, 2018) Search PubMed.
- Journal of Natural Products, Preparation and Submission of Manuscripts, http://pubs.acs.org/paragonplus/submission/jnprdf/jnprdf_authguide.pdf, (accessed June 8, 2017).
- Phytochemistry Letters, Guide for Authors, https://www.elsevier.com/journals/phytochemistry-letters/1874-3900/guide-for-authors, (accessed June 8, 2017).
- Planta Medica, Guidelines for Authors, https://www.thieme.de/statics/dokumente/thieme/final/de/dokumente/zw_thieme-en/PlantaMedica-Authors-December-2016.pdf, (accessed June 8, 2017).
- AOAC International, J. AOAC Int., 2012, 95, 268–272 CrossRef.
- D. J. Kroll and N. H. Oberlies, Focus. Altern. Complement. Ther., 2003, 8, 302–306 Search PubMed.
- Office of Dietary Supplements, Dietary Supplement Label Database, https://www.dsld.nlm.nih.gov/dsld/index.jsp, (accessed September 16, 2018) Search PubMed.
- Centers for Disease Control and Prevention (CDC), National Health and Nutrition Examination Survey Data, U.S. Department of Health and Human Services, 2017, http://https://www.cdc.gov/nchs/nhanes/index.htm (accessed August 23, 2017) Search PubMed.
- Anon., Consumer Reports, 2003, vol. 68, pp. 40–43 Search PubMed.
- J. Health, Green Tea Supplement Reviews, http://www.benefitsofgreentea.info/2007/green-tea-supplement-reviews.php, (accessed November 11, 2015).
- T. Smith, K. Kawa, V. Eckl and J. Johnson, HerbalGram, 2016, 111, 67–73 Search PubMed.
- Amazon.com, Amazon Best Sellers, http://www.amazon.com/Best-Sellers-Grocery-Gourmet-Food-Green-Tea-Beverages/zgbs/grocery/16318471, (accessed November 11, 2015).
- R. B. van Breemen, J. Med. Chem., 2015, 58 CrossRef CAS PubMed.
- National Park Service (NPS), Preparing and storing herbarium specimens, U.S. Department of the Interior, 2009, http://https://www.nps.gov/museum/publications/conserveogram/11-12.pdf (accessed September 19, 2017) Search PubMed.
- W. T. Stearn, Biol. J. Linn. Soc., 1971, 3, 225–233 CrossRef.
- M. Greve, A. M. Lykke, C. W. Fagg, R. E. Gereau, G. P. Lewis, R. Marchant, A. R. Marshall, J. Ndayishimiye, J. Bogaert and J.-C. Svenning, S. Afr. J. Bot., 2016, 105, 317–323 CrossRef.
- Phytochemistry, Author Information Pack, https://www.elsevier.com/journals/phytochemistry/0031-9422?generatepdf=true, (accessed August 31, 2017).
- C. E. Piersen, N. L. Booth, Y. Sun, W. Liang, J. E. Burdette, R. B. v. Breemen, S. E. Geller, C. Gu, S. Banuvar, L. P. Shulman, J. L. Bolton and N. R. Farnsworth, Curr. Med. Chem., 2004, 11, 1361–1374 CrossRef CAS PubMed.
- R. B. van Breemen, H. H. S. Fong and N. R. Farnsworth, Chem. Res. Toxicol., 2007, 20, 577–582 Search PubMed.
- J. J. Kellogg, T. N. Graf, M. F. Paine, J. S. McCune, O. M. Kvalheiam, N. H. Oberlies and N. B. Cech, J. Nat. Prod., 2017, 80, 1457–1466 CrossRef CAS PubMed.
- E. D. Wallace, N. H. Oberlies, N. B. Cech and J. J. Kellogg, Food Chem. Toxicol., 2018, 120, 439–447 CrossRef CAS PubMed.
- J. M. Harnly, D. Luthria and P. Chen, J. AOAC Int., 2012, 95, 1579–1587 CrossRef CAS PubMed.
- P. Posadzki, L. Watson and E. Ernst, Eur. J. Clin. Pharmacol., 2013, 69, 295–307 CrossRef PubMed.
- D. J. Kroll, H. S. Shaw, J. M. Mathews and N. H. Oberlies, Acta Hortic., 2005, 720, 137–147 Search PubMed.
- S. Ding, E. Dudley, S. Plummer, J. Tang, R. P. Newton and A. G. Brenton, Phytochemistry, 2008, 69, 1555–1564 CrossRef CAS PubMed.
- P. Chen, M. Ozcan and J. Harnly, Anal. Bioanal. Chem., 2007, 389, 251–261 CrossRef CAS PubMed.
- W. Li, Y. Sun, J. F. Fitzloff and R. B. Van Breemen, Chem. Res. Toxicol., 2002, 15, 1174–1178 Search PubMed.
- E. A. Abourashed and I. A. Khan, J. Pharm. Sci., 2001, 90, 817–822 CrossRef CAS PubMed.
- S. M. Henning, C. Fajardo-Lira, H. W. Lee, A. A. Youssefian, V. L. W. Go and D. Heber, Nutr. Cancer, 2003, 45, 226–235 CrossRef CAS PubMed.
- M. Friedman, C. E. Levin, S. H. Choi, E. Kozukue and N. Kozukue, J. Food Sci., 2006, 71, C328–C337 CrossRef CAS.
- D. D. Soejarto, J. Ethnopharmacol., 1996, 51, 1–15 CrossRef CAS PubMed.
- D. D. Soejarto, H. H. S. Fong, G. T. Tan, H. J. Zhang, C. Y. Ma, S. G. Franzblau, C. Gyllenhaal, M. C. Riley, M. R. Kadushin, J. M. Pezzuto, L. T. Xuan, N. T. Hiep, N. V. Hung, B. M. Vu, P. K. Loc, L. X. Dac, L. T. Binh, N. Q. Chien, N. V. Hai, T. Q. Bich, N. M. Cuong, B. Southavong, K. Sydara, S. Bouamanivong, H. M. Ly, T. V. Thuy, W. C. Rose and G. R. Dietzman, J. Ethnopharmacol., 2005, 100, 15–22 CrossRef CAS PubMed.
- H. Zedan, Handbook of the Convention on Biological Diversity, Earthscan Publications Ltd., 2001 Search PubMed.
- T. A. Kursar, C. C. Caballero-George, T. L. Capson, L. Cubilla-Rios, W. H. Gerwick, M. P. Gupta, A. Ibañez, R. G. Linington, K. L. McPhail, E. Ortega-Barría, L. I. Romero, P. N. Solis and P. D. Coley, BioScience, 2006, 56, 1005–1012 CrossRef.
- Nagoya Protocol, Nagoya protocol on access to genetic resources and the fair and equitable sharing of benefits arising from their utilization to the convention on biological diversity, Convention on Biological Diversity, 2011, http://www.cbd.int/abs/text/default.shtml (accessed November 12, 2017) Search PubMed.
- International Institute for Environment and Development, Nagoya Protocol on Access to Genetic Resources and Benefit-Sharing, https://biocultural.iied.org/nagoya-protocol-access-genetic-resources-and-benefit-sharing, (accessed June 1, 2018).
- M. C. Leal, A. Hilario, M. H. G. Munro, J. W. Blunt and R. Calado, Nat. Prod. Rep., 2016, 33, 747–750 RSC.
- H. A. Raja, T. R. Baker, J. G. Little and N. H. Oberlies, Food Chem., 2017, 214, 383–392 CrossRef CAS PubMed.
- R. Upton, A. Graff, G. Jolliffe, R. Langer and R. Williamson, American Herbal Pharmacopoeia: Botanical Pharmacognosy - Microscopic Characterization of Botanical Medicines, CRC Press, Boca Raton, FL, 2016 Search PubMed.
- N. V. Ivanova, M. L. Kuzmina, T. W. A. Braukmann, A. V. Borisenko and E. V. Zakharov, PLoS One, 2016, 11, e0156426 CrossRef PubMed.
- D. P. Little, Genome, 2014, 57, 513–516 CrossRef CAS PubMed.
- D. F. Coutinho Moraes, D. W. Still, M. R. Lum and A. M. Hirsch, Planta Med., 2015, 81, 687–695 CrossRef CAS PubMed.
- New York State Office of the Attorney General, A. G. Schneiderman asks major retailers to halt sales of certain herbal supplements as DNA tests fail to detect plant materials listed on majority of products tested, https://ag.ny.gov/press-release/ag-schneiderman-asks-major-retailers-halt-sales-certain-herbal-supplements-dna-tests Search PubMed.
- I. Parveen, S. Gafner, N. Techen, S. J. Murch and I. A. Khan, Planta Med., 2016, 82, 1225–1235 CrossRef CAS PubMed.
- P. M. Hollingsworth, D.-Z. Li, M. van der Bank and A. D. Twyford, Philos. Trans. R. Soc., B, 2016, 371, 20150338 CrossRef PubMed.
- P. M. Hollingsworth, S. W. Graham and D. P. Little, PLoS One, 2011, 6, e19254 CrossRef CAS PubMed.
- W. L. Clement and M. J. Donoghue, BMC Evol. Biol., 2012, 12, 73 CrossRef PubMed.
- A. J. Fazekas, P. R. Kesanakurti, K. S. Burgess, D. M. Percy, S. W. Graham, S. C. Barrett, S. G. Newmaster, M. Hajibabaei and B. C. Husband, Mol. Ecol. Resour., 2009, 9, 130–139 CrossRef CAS PubMed.
- N. Pei, D. L. Erickson, B. Chen, X. Ge, X. Mi, N. G. Swenson, J.-L. Zhang, F. A. Jones, C.-L. Huang and W. Ye, Sci. Rep., 2015, 5, 15127 CrossRef CAS PubMed.
- A. J. Fazekas, M. L. Kuzmina, S. G. Newmaster and P. M. Hollingsworth, Methods Mol. Biol., 2012, 858, 223–252 CrossRef CAS PubMed.
- M. Blumenthal and W. R. Busse, The complete German Commission E monographs, American Botanical Council, Austin, Texas, 1998 Search PubMed.
- U. S. Pharmacopeia, Herbal Medicines Compendium, http://hmc.usp.org/, (accessed September 19, 2017, 2017).
- V. E. Tyler, Herbs of choice: the therapeutic use of phytomedicinals, Pharmaceutical Products Press (imprint of Haworth Press, Inc.), 1994 Search PubMed.
- N. B. Cech, LCGC North Am., 2013, 31, 938–947 CAS.
- C. Simmler, J. G. Napolitano, J. B. McAlpine, S.-N. Chen and G. F. Pauli, Curr. Opin. Biotechnol., 2014, 25, 51–59 CrossRef CAS PubMed.
- L. Vaclavik, A. J. Krynitsky and J. I. Rader, Anal. Bioanal. Chem., 2014, 406, 6767–6790 CrossRef CAS PubMed.
- J. Zhu, X. Fan, Y. Cheng, R. Agarwal, C. M. V. Moore, S. T. Chen and W. Tong, PLoS One, 2014, 9, e87462 CrossRef PubMed.
- A. Navarrete, B. Avula, Y.-W. Choi and I. A. Khan, J. AOAC Int., 2006, 89, 8–15 CAS.
- M. A. Markus, J. Ferrier, S. M. Luchsinger, J. Yuk, A. Cuerrier, M. J. Balick, J. M. Hicks, K. B. Killday, C. W. Kirby, F. Berrue, R. G. Kerr, K. Knagge, T. Gödecke, B. E. Ramirez, D. C. Lankin, G. F. Pauli, I. Burton, T. K. Karakach, J. T. Arnason and K. L. Colson, Planta Med., 2014, 80, 732–739 CrossRef CAS PubMed.
- J. Harnly, P. Chen, J. Sun, H. Huang, K. L. Colson, J. Yuk, J.-A. H. McCoy, D. T. H. Reynaud, P. B. Harrington and E. J. Fletcher, Planta Med., 2016, 82, 250–262 CAS.
- B. Avula, Y.-H. Wang, G. Isaac, J. Yuk, M. Wrona, K. Yu and I. A. Khan, Planta Med., 2017, 83, 1097–1308 CrossRef PubMed.
- D. Kumar, Crit. Rev. Anal. Chem., 2016, 46, 400–412 CrossRef CAS PubMed.
- J. Yuk, K. L. McIntyre, C. Fischer, J. Hicks, K. L. Colson, E. Lui, D. Brown and J. T. Arnason, Anal. Bioanal. Chem., 2013, 405, 4499–4509 CrossRef CAS PubMed.
- H. G. Gika, G. A. Theodoridis, R. S. Plumb and I. D. Wilson, J. Pharm. Biomed. Anal., 2014, 87, 12–25 CrossRef CAS PubMed.
- T. F. Jorge, J. A. Rodrigues, C. Caldana, R. Schmidt, J. T. van Dongen, J. Thomas-Oates and C. António, Mass Spectrom. Rev., 2016, 35, 620–649 CrossRef CAS PubMed.
- J. L. Markley, R. Brüschweiler, A. S. Edison, H. R. Eghbalnia, R. Powers, D. Raftery and D. S. Wishart, Curr. Opin. Biotechnol., 2017, 43, 34–40 CrossRef CAS PubMed.
- T. Sangster, H. Major, R. Plumb, A. J. Wilson and I. D. Wilson, Analyst, 2006, 131, 1075–1078 RSC.
- W. B. Dunn, I. D. Wilson, A. W. Nicholls and D. Broadhurst, Bioanalysis, 2012, 4, 2249–2264 CrossRef CAS PubMed.
- J. J. Kellogg, D. A. Todd, J. M. Egan, H. A. Raja, N. H. Oberlies, O. M. Kvalheiam and N. B. Cech, J. Nat. Prod., 2016, 79, 376–386 CrossRef CAS PubMed.
- J.-E. Lee, B.-J. Lee, J.-O. Chung, J.-A. Hwang, S.-J. Lee, C.-H. Lee and Y.-S. Hong, J. Agric. Food Chem., 2010, 58, 10582–10589 CrossRef CAS PubMed.
- Y. Fujimura, K. Kurihara, M. Ida, R. Kosaka, D. Miura, H. Wariishi, M. Maeda-Yamamoto, A. Nesumi, T. Saito, T. Kanda, K. Yamada and H. Tachibana, PLoS One, 2011, 6, e23426 CrossRef CAS PubMed.
- J.-E. Lee, B.-J. Lee, J.-O. Chung, H.-J. Shin, S.-J. Lee, C.-H. Lee and Y.-S. Hong, Food Res. Int., 2011, 44, 597–604 CrossRef CAS.
- J.-E. Lee, B.-J. Lee, J.-A. Hwang, K.-S. Ko, J.-O. Chung, E.-H. Kim, S.-J. Lee and Y.-S. Hong, J. Agric. Food Chem., 2011, 59, 10579–10585 CrossRef CAS PubMed.
- L.-S. Lee, J. H. Choi, N. Son, S.-H. Kim, J.-D. Park, D.-J. Jang, Y. Jeong and H.-J. Kim, J. Agric. Food Chem., 2013, 61, 332–338 CrossRef CAS PubMed.
- J.-E. Lee, B.-J. Lee, J.-O. Chung, H. N. Kim, E.-H. Kim, S. Jung, H. Lee, S. J. Lee and Y.-S. Hong, Food Chem., 2015, 174, 452–459 CrossRef CAS PubMed.
- P. K. Gupta, G. W. Barone, B. Gurley, E. K. Fifer and H. P. Hendrickson, Drug Metab. Dispos., 2015, 43, 534–552 CrossRef CAS PubMed.
- P. Bogdanski, J. Suliburska, M. Szulinska, M. Stepien, D. Pupek-Musialik and A. Jablecka, Nutr. Res., 2012, 32, 421–427 CrossRef CAS PubMed.
- C. Bergeron, J. F. Livesey, D. V. C. Awang, J. T. Arnason, J. Rana, B. R. Baum and W. Letchamo, Phytochem. Anal., 2000, 11, 207–215 CrossRef CAS.
- H. Wohlmuth, K. Savage, A. Dowell and P. Mouatt, Phytomedicine, 2014, 21, 912–918 CrossRef CAS PubMed.
- T. G. McCloud, Molecules, 2010, 15, 4526–4563 CrossRef CAS PubMed.
- B. J. Gurley, Clin. Pharmacol. Ther., 2011, 89, 915–919 CrossRef CAS PubMed.
- P. N. Brown and M. C. Roman, J. AOAC Int., 2008, 91, 694–701 CAS.
- E. M. Mudge, J. M. Betz and P. N. Brown, Adv. Nutr., 2016, 7, 390–398 CrossRef CAS PubMed.
- J. G. Napolitano, T. Gödecke, M. F. Rodríguez-Brasco, B. U. Jaki, S.-N. Chen, D. C. Lankin and G. F. Pauli, J. Nat. Prod., 2012, 75, 238–248 CrossRef CAS PubMed.
- D. L. Luthria, S. Mukhopadhyay, R. J. Robbins, J. W. Finley, G. S. Banuelos and J. M. Harnly, J. Agric. Food Chem., 2008, 56, 5457–5462 CrossRef CAS PubMed.
- M. Wang, J. Zhao, B. Avula, Y.-H. Wang, C. Avonto, A. G. Chittiboyina, P. L. Wylie, J. F. Parcher and I. A. Khan, J. Agric. Food Chem., 2014, 62, 12103–12111 CrossRef CAS PubMed.
- T. N. Graf, N. B. Cech, S. J. Polyak and N. H. Oberlies, J. Pharm. Biomed. Anal., 2016, 126, 26–33 CrossRef CAS PubMed.
- M. Coimbra, B. Isacchi, L. van Bloois, J. S. Torano, A. Ket, X. Wu, F. Broere, J. M. Metselaar, C. J. Rijcken, G. Storm, R. Bilia and R. M. Schiffelers, Int. J. Pharm., 2011, 416, 433–442 CrossRef CAS PubMed.
- B. Lapornik, M. Prošek and A. Golc Wondra, J. Food Eng., 2005, 71, 214–222 CrossRef.
- J. Azmir, I. S. M. Zaidul, M. M. Rahman, K. M. Sharif, A. Mohamed, F. Sahena, M. H. A. Jahurul, K. Ghafoor, N. A. N. Norulaini and A. K. M. Omar, J. Food Eng., 2013, 117, 426–436 CrossRef CAS.
- J. J. Kellogg, E. D. Wallace, T. N. Graf, N. H. Oberlies and N. B. Cech, J. Pharm. Biomed. Anal., 2017, 145, 604–610 CrossRef CAS PubMed.
- N. D. Yuliana, A. Khatib, R. Verpoorte and Y. H. Choi, Anal. Chem., 2011, 83, 6902–6906 CrossRef CAS PubMed.
- E. Saccenti, H. C. J. Hoefsloot, A. K. Smilde, J. A. Westerhuis and M. M. W. B. Hendriks, Metabolomics, 2014, 10, 361–374 CrossRef CAS.
- H. Abdi and L. J. Williams, Wiley Interdiscip. Rev. Comput. Stat., 2010, 2, 433–459 CrossRef.
- R. A. LaBudde and J. Harnly, J. AOAC Int., 2012, 95, 273–285 CrossRef CAS PubMed.
- M. I. López, M. P. Callao and I. Ruisánchez, Anal. Chim. Acta, 2015, 891, 62–72 CrossRef PubMed.
- D. A. Sheen, W. F. C. Wocha, K. A. Lippa and D. W. Bearden, Chemom. Intell. Lab. Syst., 2017, 162, 10–20 CrossRef CAS PubMed.
- D. Grapov, K. Wanichthanarak and O. Fiehn, Bioinformatics, 2015, 31, 2757–2760 CrossRef CAS PubMed.
- C. V. Rider, G. E. Dinse, D. M. Umbach, J. E. Simmons and R. C. Hertzberg, in Chemical Mixtures and Combined Chemical and Nonchemical Stressors, Springer, 2018, pp. 235–270 Search PubMed.
- C. V. Rider, N. J. Walker and S. Waidyanatha, Clinical Pharmacology & Therapeutics, 2018 Search PubMed.
- E. Ernst, J. Intern. Med., 2002, 252, 107–113 CrossRef CAS PubMed.
- M. Tims, Botanical Adulterants Bulletin, 2016, June, pp. 1–6 Search PubMed.
- P. Geng, J. M. Harnly, J. Sun, M. Zhang and P. Chen, Anal. Bioanal. Chem., 2017, 409, 2591–2600 CrossRef CAS PubMed.
- N. Sarma, G. Giancaspro and J. Venema, Drug Test. Anal., 2016, 8, 418–423 CrossRef CAS PubMed.
- J. M. Betz, P. N. Brown and M. C. Roman, Fitoterapia, 2011, 82, 44–52 CrossRef CAS PubMed.
- C. Simmler, J. G. Graham, S.-N. Chen and G. F. Pauli, Fitoterapia, 2018, 129, 401–414 CrossRef PubMed.
- P. Geng, J. M. Harnly and P. Chen, J. Agric. Food Chem., 2015, 63, 6189–6211 CrossRef CAS PubMed.
- N. P. Mncwangi, A. M. Viljoen, J. Zhao, I. Vermaak, W. Chen and I. A. Khan, Phytochemistry, 2014, 106, 104–115 CrossRef CAS PubMed.
- A. Pengelly, K. Bennett, K. Spelman and M. Tims, Goldenseal: An Appalachian Plant Monograph, Appalachian Center for Ethnobotanical Studies, 2012, http://https://www.frostburg.edu/fsu/assets/File/ACES/Hydrastis%20canadensis%20for%20ACES%20website.pdf (accessed October 25, 2017) Search PubMed.
- H. A. Weber, M. K. Zart, A. E. Hodges, H. M. Molloy, B. M. O'Brien, L. A. Moody, A. P. Clark, R. K. Harris, J. D. Overstreet and C. S. Smith, J. Agric. Food Chem., 2003, 51, 7352–7358 CrossRef CAS PubMed.
- A. A. Grippo, B. Hamilton, R. Hannigan and B. J. Gurley, Am. J. Health-Syst. Pharm., 2006, 63 CrossRef CAS PubMed.
- J. W. Wong, K. Zhang, K. Tech, D. G. Hayward, A. J. Krynitsky, I. Cassias, F. J. Schenck, K. Banerjee, S. Dasgupta and D. Brown, J. Agric. Food Chem., 2010, 58, 5884–5896 CrossRef CAS PubMed.
- C. Witschi and E. Doelker, Eur. J. Pharm. Biopharm., 1997, 43, 215–242 CrossRef CAS.
- Z. Veprikova, M. Zachariasova, Z. Dzuman, A. Zachariasova, M. Fenclova, P. Slavikova, M. Vaclavikova, K. Mastovska, D. Hengst and J. Hajslova, J. Agric. Food Chem., 2015, 63, 6633–6643 CrossRef CAS PubMed.
- P. Raman, L. C. Patino and M. G. Nair, J. Agric. Food Chem., 2004, 52, 7822–7827 CrossRef CAS PubMed.
- A. C. Brown, Food Chem. Toxicol., 2017, 107, 449–471 CrossRef CAS PubMed.
- Food and Drug Administration (FDA), Guidance for Industry: Current Good Manufacturing Practice in Manufacturing, Packaging, Labeling, or Holding Operations for Dietary Supplements; Small Entity Compliance Guide, U.S. Department of Health and Human Services, 2010, http://https://www.fda.gov/Food/GuidanceRegulation/GuidanceDocumentsRegulatoryInformation/DietarySupplements/ucm238182.htm (accessed October 4, 2017) Search PubMed.
- R. Danko, Testing Ingredient and Components for Identity and Quality, Natural Product Insider, 2016, vol. 21, p. 48 Search PubMed.
- R. Dowlatabadi, F. Farshidfar, Z. Zare, M. Pirali, M. Rabiei, M. R. Khoshayand and H. J. Vogel, Metabolomics, 2017, 13, 1–11 CrossRef CAS.
- R. Alvarez-Zapata, A. Sánchez-Medina, M. Chan-Bacab, K. García-Sosa, F. Escalante-Erosa, R. V. García-Rodríguez and L. M. Peña-Rodríguez, J. Chromatogr. Aa, 2015, 1422, 213–221 CrossRef CAS PubMed.
- C. Simmler, D. Kulakowski, D. C. Lankin, J. B. McAlpine, S.-N. Chen and G. F. Pauli, Adv. Nutr., 2016, 7, 179–189 CrossRef CAS PubMed.
- C. G. Enke and L. J. Nagels, Anal. Chem., 2011, 83, 2539–2546 CrossRef CAS PubMed.
- H. A. Junio, A. A. Sy-Cordero, K. A. Ettefagh, J. T. Burns, K. T. Micko, T. N. Graf, S. J. Richter, R. E. Cannon, N. H. Oberlies and N. B. Cech, J. Nat. Prod., 2011, 74, 1621–1629 CrossRef CAS PubMed.
- B. G. Chiari, J. A. Severi, D. Pauli-Credendio, P. Abackerli, C. M. d. Sylos, W. Vilegas, M. A. Correa and V. L. B. Isaac, Int. J. Pharm. Pharm. Sci., 2012, 331–336 Search PubMed.
- H. Wagner, Fitoterapia, 2011, 82, 34–37 CrossRef.
- K. Niemeyer, I. R. Bell and M. Koithan, J Herb. Med., 2013, 3, 112–119 CrossRef PubMed.
- J. Burns, L. Zhao, E. W. Taylor and K. Spelman, Toxicology, 2010, 278, 140–159 CrossRef CAS PubMed.
- N. H. Oberlies and D. J. Kroll, J. Nat. Prod., 2004, 67, 129–135 CrossRef CAS PubMed.
- M. C. Wani, H. L. Taylor, M. E. Wall, P. Coggon and A. T. McPhail, J. Am. Chem. Soc., 1971, 93, 2325–2327 CrossRef CAS PubMed.
- R. B. van Breemen, C. R. Huang, D. Nikolic, C. P. Woodbury, Y. Z. Zhao and D. L. Venton, Anal. Chem., 1997, 69, 2159–2164 CrossRef CAS PubMed.
- Z. Yang, Y. Zhang, L. Sun, Y. Wang, X. Gao and Y. Cheng, Anal. Chim. Acta, 2012, 719, 87–95 CrossRef CAS PubMed.
- L. Ciesla and R. Moaddel, Nat. Prod. Rep., 2016, 33, 1131–1145 RSC.
- H. Martens, S. W. Bruun, I. Adt, G. D. Sockalingum and A. Kohler, J. Chemom., 2006, 20, 402–417 CrossRef CAS.
- M. Balouiri, M. Sadiki and S. K. Ibnsouda, J. Pharm. Anal., 2016, 6, 71–79 CrossRef PubMed.
- G. Brusotti, I. Cesari, A. Dentamaro, G. Caccialanza and G. Massolini, J. Pharm. Biomed. Anal., 2014, 87, 218–228 CrossRef CAS PubMed.
- B. M. Dietz, A. Hajirahimkhan, T. L. Dunlap and J. L. Bolton, Pharmacol. Rev., 2016, 68, 1026–1073 CrossRef CAS PubMed.
- A. L. Harvey, R. Edrada-Ebel and R. J. Quinn, Nat. Rev. Drug Discovery, 2015, 14, 111 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS PubMed.
- W. Chun-Guang, Y. Jun-Qing, L. Bei-Zhong, J. Dan-Ting, W. Chong, Z. Liang, Z. Dan and W. Yan, Eur. J. Pharmacol., 2010, 627, 33–41 CrossRef PubMed.
- B. T. Gufford, G. Chen, A. G. Vergara, P. Lazarus, N. H. Oberlies and M. F. Paine, Drug Metab. Dispos., 2015, 43, 1353–1359 CrossRef CAS PubMed.
- G. Li, K. Huang, D. Nikolic and R. B. Van Breemen, Drug Metab. Dispos., 2015, 43, 1670–1678 CrossRef CAS PubMed.
- P. Posadzki, L. Watson and E. Ernst, Br. J. Clin. Pharmacol., 2013, 75, 603–618 CrossRef CAS PubMed.
- C. A. Van Loo-Bouwman, T. H. J. Naber, M. Minekus, R. B. van Breemen, P. J. M. Hulshof and G. Schaafsma, J. Agric. Food Chem., 2014, 62, 950–955 CrossRef CAS PubMed.
- S. Possemiers, S. Bolca, W. Verstraete and A. Heyerick, Fitoterapia, 2011, 82, 53–66 CrossRef CAS PubMed.
- J. M. Price, J. G. Little and T. R. Baker, Planta Med., 2014, 80, CL12 Search PubMed.
- K. L. Krivos, B. T. Regg, J. M. Price, D. A. McMillan and T. R. Baker, Planta Med., 2014, 80, PPL19 Search PubMed.
- J. G. Napolitano, T. Gödecke, D. C. Lankin, B. U. Jaki, J. B. McAlpine, S.-N. Chen and G. F. Pauli, J. Pharm. Biomed. Anal., 2014, 93, 59–67 CrossRef CAS PubMed.
- C. D. Broeckling, F. A. Afsar, S. Neumann, A. Ben-Hur and J. E. Prenni, Anal. Chem., 2014, 86, 6812–6817 CrossRef CAS PubMed.
- N. D. Paguigan, T. El-Elimat, D. Kao, H. A. Raja, C. J. Pearce and N. H. Oberlies, J. Antibiot., 2017, 70, 553–561 CrossRef CAS PubMed.
- T. El-Elimat, M. Figueroa, B. M. Ehrmann, N. B. Cech, C. J. Pearce and N. H. Oberlies, J. Nat. Prod., 2013, 76, 1709–1716 CrossRef CAS PubMed.
- M. Wang, J. J. Carver, V. V. Phelan, L. M. Sanchez, N. Garg, Y. Peng, D. D. Nguyen, J. Watrous, C. A. Kapono and T. Luzzatto-Knaan, Nat. Biotechnol., 2016, 34, 828 CrossRef CAS PubMed.
- T. Hoffmann, D. Krug, S. Hüttel and R. Müller, Anal. Chem., 2014, 86, 10780–10788 CrossRef CAS PubMed.
- S. P. Gaudêncio and F. Pereira, Nat. Prod. Rep., 2015, 32, 779–810 RSC.
- D. G. Cox, J. Oh, A. Keasling, K. L. Colson and M. T. Hamann, Biochim. Biophys. Acta, 2014, 1840, 3460–3474 CrossRef CAS PubMed.
- N. D. Yuliana, A. Khatib, Y. H. Choi and R. Verpoorte, Phytother. Res., 2011, 25, 157–169 CAS.
- F. Qiu, G. Cai, B. U. Jaki, D. C. Lankin, S. G. Franzblau and G. F. Pauli, J. Nat. Prod., 2013, 76, 413–419 CrossRef CAS PubMed.
- E. R. Britton, J. J. Kellogg, O. M. Kvalheim and N. B. Cech, J. Nat. Prod., 2018, 81, 484–493 CrossRef CAS PubMed.
- L. K. Caesar, J. J. Kellogg, O. M. Kvalheim, R. A. Cech and N. B. Cech, Planta Med., 2018 Search PubMed.
- M. Farrés, S. Platikanov, S. Tsakovski and R. Tauler, J. Chemom., 2015, 29, 528–536 CrossRef.
- T. Rajalahti, R. Arneberg, F. S. Berven, K.-M. Myhr, R. J. Ulvik and O. M. Kvalheim, Chemom. Intell. Lab. Syst., 2009, 95, 35–48 CrossRef CAS.
- T. Rajalahti, R. Arneberg, A. C. Kroksveen, M. Berle, K.-M. Myhr and O. M. Kvalheim, Anal. Chem., 2009, 81, 2581–2590 CrossRef CAS PubMed.
- D.-D. Tian, J. J. Kellogg, N. Okut, N. H. Oberlies, N. B. Cech, D. D. Shen, J. S. McCune and M. F. Paine, Drug Metab. Dispos., 2018, 46, 552–560 CrossRef CAS PubMed.
- F. Bucar, A. Wube and M. Schmid, Nat. Prod. Rep., 2013, 30, 525–545 RSC.
- A. Michalkiewicz, M. Biesaga and K. Pyrzynska, J. Chromatogr. A, 2008, 1187, 18–24 CrossRef CAS PubMed.
- J. B. McAlpine, J. B. Friesen and G. F. Pauli, in Natural Products Isolation, ed. S. D. Sarker and L. Nahar, Humana Press, Totowa, NJ, 2012, pp. 221–254, DOI:10.1007/978-1-61779-624-1_9.
- N.-C. Kim, N. H. Oberlies, D. R. Brine, R. W. Handy, M. C. Wani and M. E. Wall, J. Nat. Prod., 2001, 64, 251–253 CrossRef CAS.
- N. Pfoze, B. Myrboh, Y. Kumar and M. R. Rohman, Journal of Medicinal Plants Studies, 2014, 2, 48–57 Search PubMed.
- S. Agatonovic-Kustrin, D. W. Morton and A. P. Yusof, Mod. Chem. Appl., 2015 Search PubMed.
- T. El-Elimat, H. A. Raja, T. N. Graf, S. H. Faeth, N. B. Cech and N. H. Oberlies, J. Nat. Prod., 2014, 77, 193–199 CrossRef CAS PubMed.
- T. N. Graf, M. C. Wani, R. Agarwal, D. J. Kroll and N. H. Oberlies, Planta Med., 2007, 73, 1495–1501 CrossRef CAS PubMed.
- N. Bross-Walch, T. Kuhn, D. Moskau and O. Zerbe, Chem. Biodivers., 2005, 2, 147–177 CrossRef PubMed.
- A. Bouslimani, L. M. Sanchez, N. Garg and P. C. Dorrestein, Nat. Prod. Rep., 2014, 31, 718–729 RSC.
- P. Crews, J. Rodriguez and M. Jaspars, Organic Structure Analysis 2nd edn, Oxford University Press, New York, NY, USA, 2009 Search PubMed.
- R. G. Linington, P. G. Williams and J. B. MacMillan, Problems in Organic Structure Determination: A Practical Approach to NMR Spectroscopy, CRC Press, Boca Raton, FL, USA, 2015 Search PubMed.
- M. Halabalaki, K. Vougogiannopoulou, E. Mikros and A. L. Skaltsounis, Curr. Opin. Biotechnol., 2014, 25, 1–7 CrossRef CAS PubMed.
- J. B. McAlpine, S.-N. Chen, A. Kutateladze, J. B. MacMillan, G. Appendino, A. Barison, M. A. Beniddir, M. W. Biavatti, S. Bluml, A. Boufridi, M. S. Butler, R. J. Capon, Y. H. Choi, D. Coppage, P. Crews, M. T. Crimmins, M. Csete, P. Dewapriya, J. M. Egan, M. J. Garson, G. Genta-Jouve, W. H. Gerwick, H. Gross, M. K. Harper, P. Hermanto, J. M. Hook, L. Hunter, D. Jeannerat, N.-Y. Ji, T. A. Johnson, D. G. I. Kingston, H. Koshino, H.-W. Lee, G. Lewin, J. Li, R. G. Linington, M. Liu, K. L. McPhail, T. F. Molinski, B. S. Moore, J.-W. Nam, R. P. Neupane, M. Niemitz, J.-M. Nuzillard, N. H. Oberlies, F. M. M. Ocampos, G. Pan, R. J. Quinn, D. S. Reddy, J.-H. Renault, J. Rivera-Chávez, W. Robien, C. M. Saunders, T. J. Schmidt, C. Seger, B. Shen, C. Steinbeck, H. Stuppner, S. Sturm, O. Taglialatela-Scafati, D. J. Tantillo, R. Verpoorte, B.-G. Wang, C. M. Williams, P. G. Williams, J. Wist, J.-M. Yue, C. Zhang, Z. Xu, C. Simmler, D. C. Lankin, J. Bisson and G. F. Pauli, Nat. Prod. Rep., 2018 Search PubMed , in press.
- M. Figueroa, A. K. Jarmusch, H. A. Raja, T. El-Elimat, J. S. Kavanaugh, A. R. Horswill, R. G. Cooks, N. B. Cech and N. H. Oberlies, J. Nat. Prod., 2014, 77, 1351–1358 CrossRef CAS PubMed.
- H. Xu, H. Niu, B. He, C. Cui, Q. Li and K. Bi, Molecules, 2016, 21, 664 CrossRef PubMed.
- E. P. Ekanayaka, M. D. Celiz and A. D. Jones, Plant Physiol., 2015, 167, 1221–1232 CrossRef CAS PubMed.
- T. Xie, Y. Liang, H. Hao, A. Jiye, L. Xie, P. Gong, C. Dai, L. Liu, A. Kang and X. Zheng, J. Chromatogr. Aa, 2012, 1227, 234–244 CrossRef CAS PubMed.
- K. A. Reynertson and K. Mahmood, Botanicals: Methods and techniques for quality & authenticity, CRC Press, Boca Raton, FL, USA, 2015 Search PubMed.
- S. Arrivault, M. Guenther, S. C. Fry, M. M. Fuenfgeld, D. Veyel, T. Mettler-Altmann, M. Stitt and J. E. Lunn, Anal. Chem., 2015, 87, 6896–6904 CrossRef CAS PubMed.
- O. Chahrour, D. Cobice and J. Malone, J. Pharm. Biomed. Anal., 2015, 113, 2–20 CrossRef CAS PubMed.
- K. Habler and M. Rychlik, Anal. Bioanal.Chem., 2016, 408, 307–317 CrossRef CAS PubMed.
- AOAC International, AOAC Official Methods of Analysis, 2012, pp. 1–32 Search PubMed.
- K. M. VanderMolen, N. B. Cech, M. F. Paine and N. H. Oberlies, Phytochem. Anal., 2013, 24, 654–660 CrossRef CAS PubMed.
- ICH Harmonized Tripartite Guideline, Q2 (R1), 2005, 1.
- M. C. Roman, J. M. Betz and J. Hildreth, J. AOAC Int., 2007, 90, 68–81 CAS.
- A. K. Jarmusch, A. M. Musso, T. Shymanovich, S. A. Jarmusch, M. J. Weavil, M. E. Lovin, B. M. Ehrmann, S. Saari, D. E. Nichols and S. H. Faeth, J. Pharm. Biomed. Anal., 2016, 117, 11–17 CrossRef CAS PubMed.
- K. M. VanderMolen, G. R. Ainslie, M. F. Paine and N. H. Oberlies, J. Pharm. Biomed. Anal., 2014, 98, 260–265 CrossRef CAS PubMed.
- Y. Lv, X. Yang, Y. Zhao, Y. Ruan, Y. Yang and Z. Wang, Food Chem., 2009, 112, 742–746 CrossRef CAS.
- Y. Liu, S.-N. Chen, J. B. McAlpine, L. L. Klein, J. B. Friesen, D. C. Lankin and G. F. Pauli, J. Nat. Prod., 2014, 77, 611–617 CrossRef CAS PubMed.
- A. Booker, A. Suter, A. Krnjic, B. Strassel, M. Zloh, M. Said and M. Heinrich, J. Pharm. Pharmacol., 2014, 66, 811–822 CAS.
- D. A. Todd, T. V. Gulledge, E. R. Britton, M. Oberhofer, M. Leyte-Lugo, A. N. Moody, T. Shymanovich, L. F. Grubbs, M. Juzumaite, T. N. Graf, N. H. Oberlies, S. H. Faeth, S. M. Laster and N. B. Cech, PLoS One, 2015, 10, e0124276 CrossRef PubMed.
- J. Dai, A. Gupte, L. Gates and R. Mumper, Food Chem. Toxicol., 2009, 47, 837–847 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8np00065d |
| This journal is © The Royal Society of Chemistry 2019 |