
The verticillenes. Pivotal intermediates in the biosynthesis of the taxanes and the phomactins
Matthew J.
Palframan
 and
Gerald
Pattenden
*
and
Gerald
Pattenden
*
School of Chemistry, The University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: gp@nottingham.ac.uk
First published on 6th July 2018
Abstract
Covering: up to May 2018
The verticillene family of 6,12-membered ring-fused diterpenes are found in plants, liverworts, corals and insects. Carbocations derived from verticillene hydrocarbons are central intermediates in the biosynthesis of the taxane and the phomactin families of polycyclic natural products. This perspective delineates these unique biosynthetic interrelationships, which are reinforced by recent biomimetic synthesis investigations, alongside quantum chemical calculations and targeted engineering studies of the taxadiene synthase (TXS) cascade.
 Matthew J. Palframan | Matthew Palframan graduated from Oxford University with an MChem degree in 2006, and then carried out research with Professor Andy Parsons at the University of York on the synthesis of alkaloids leading to a PhD in 2010. After post-doctoral research exploiting the products of microbial oxidations in synthesis with Dr Simon Lewis at the University of Bath, Matthew joined the research group of Professor Pattenden in Nottingham where he developed biomimetic intramolecular (4 + 2) and (4 + 3) type cycloaddition reactions involving furanoxonium ion intermediate leading to the complex ring systems in the diterpene structures rameswaralide and plumarellide found in corals. Furthermore, with others, he demonstrated that (4 + 3) cycloaddition reactions between furanoxonium ions and 1,3-dienes, leading to concise syntheses of substituted cycloheptenes, proceed via two-step carbocation ion cyclisation reactions rather than concerted [4 + 3] processes. Matthew is presently working as a medicinal chemist with Prof. Chris Hayes in The School of Chemistry at Nottingham University. |
 Gerald Pattenden | Gerry Pattenden is Emeritus Professor in the School of Chemistry at the University of Nottingham. He was appointed Professor at Nottingham in 1980, and was Head and Sir Jesse Boot Professor of Organic Chemistry until 2005. He then held the title of Research Professor in Nottingham until his supposed retirement in 2009. However, Gerry continued to carry out research in synthetic organic chemistry, publish and act as a consultant in chemistry, and to give lectures until very recently. His latest publication, describing the structure of a novel verticillene shunt metabolite of the taxadiene synthase cascade, produced by males of the Brazilian sand fly was published in 2018, and is included in this Report. Gerry's research has been driven largely by his curiosity of the biosynthesis and the biosynthetic interrelationships within families of natural products. His research, described in nearly 500 publications, has involved developing new synthetic methods and strategies for organic synthesis, especially biomimetic synthesis, and the total synthesis of biologically important target natural products and their congeners. |
1. Introduction
The verticillenes are a family of 6,12-membered ring-fused diterpenes which have structures based on a bicyclo[9.3.1] pentadeca-E-3,E-7-triene framework. The tertiary alcohol verticillol 1, found in the evergreen wood of the conifer Sciandopitys verticillate (Taxodiaceae) was the first of their number to be described in 1964.1 Contemporaneously, Lythgoe et al.2 postulated that the hydrocarbon 2 corresponding to 1, called “verticillene”, might be a key intermediate in the biosynthesis of the tricyclo[6,9,3,1,03,8] pentadeca-4,11-diene, i.e., taxadiene, ring system 3 of the taxane alkaloids (Fig. 1). Although this postulate is repeated in graduate texts,3 and several naturally occurring alkene isomers of verticillene 2 have been discovered since the 1960s, at this time the particular 3,7,11-triene isomer 2, has not been found “free” in Nature. However, in 1985 our research group published a total synthesis of racemic 2 in connection with a program we had designed to examine its possible biomimetic transannular cyclisation into the ring system in taxadiene 3.4 These synthesis and biomimetic studies, alongside later investigations by others, are summarised in Sections 7 and 8.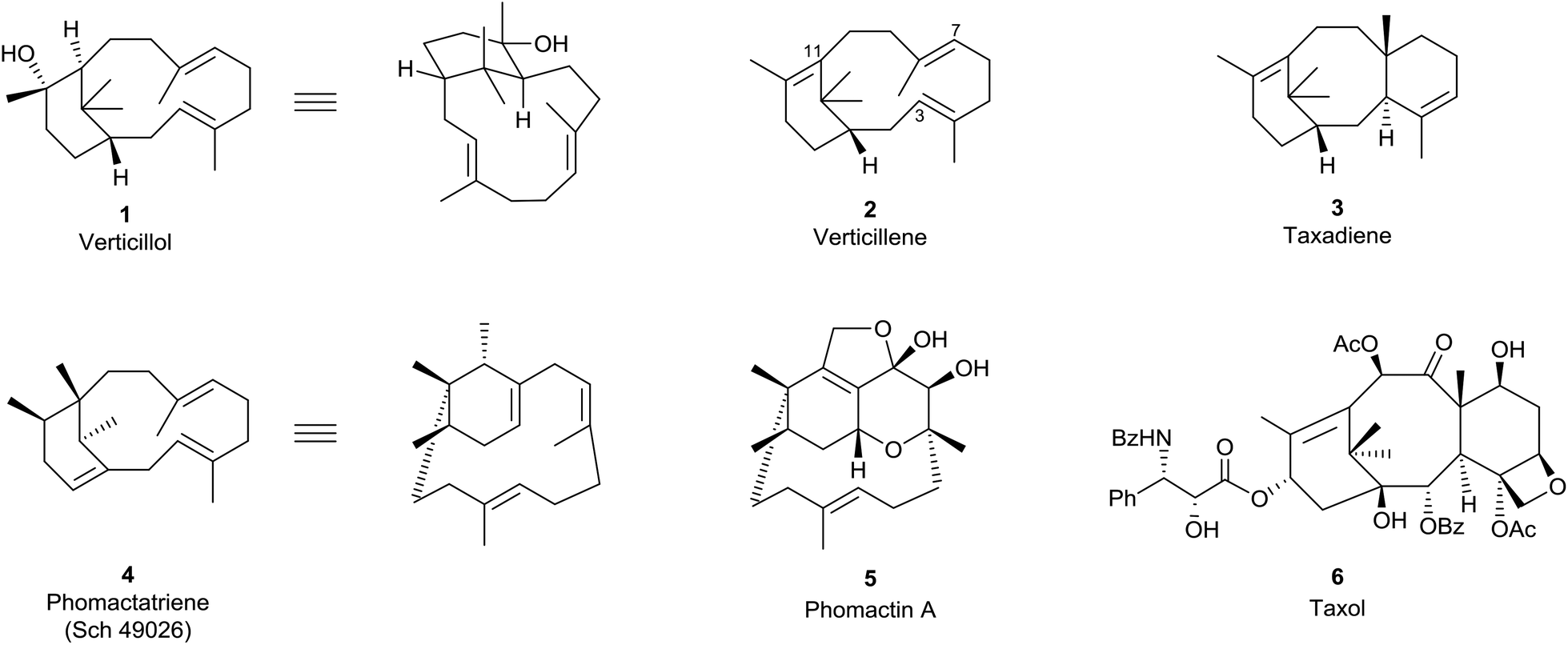 | ||
| Fig. 1 Verticillenes, taxanes and phomactins.7 | ||
The absolute stereochemistry of (+)-verticillol isolated from S. verticillate was assigned as the enantiomer of 1 in 1978,5 but this was revised some twenty five years later to 1 following anomalous dispersion X-ray crystallographic analysis of its p-iodobenzoate.6 Unfortunately, the absolute configurations of most verticillene derivatives reported in the literature between 1978 and 2005 were assigned on the basis of the incorrect ent-stereochemistry originally assigned to natural (+)-verticillol. It follows therefore that most of the absolute stereochemistries given to the verticillenes over the period 1978–2005 have needed to be reversed. Hence, throughout this report, the verticillene structures in the schemes and figures are all drawn with their correct stereochemistries.
A variety of verticillene hydrocarbons and their oxy-derivatives have been found in plants of the genus Bursera, endemic to Mexico, and in Boswellia species which are trees native to Ethiopia. They have also been isolated from liverworts collected in Japan, from a wide variety of soft corals, and especially from Taxus (yew) species. In 2018, a novel verticillene hydrocarbon was reported, for the first time, as a sex-aggregation pheromone in insects. Although some of the plant sources of natural verticillenes have shown biological properties, interest in these compounds has remained largely focussed on the implication of the hydrocarbon verticillene 2, and carbocation intermediates derived from it, in the biosynthesis of the taxadiene ring system 3, precursor to the potent antitumor taxoids, e.g. taxol (paclitaxel) 6. The same verticillene hydrocarbon 2 has also been recognised as a key intermediate in the biosynthesis of the closely related bicyclic phomactatriene ring system 4, precursor to the phomactin family of platelet activating factor (PAF) antagonists, e.g. phomactin A (5). Here we summarise the evidence for the biogenetic interrelationships between the verticillenes, the taxanes and phomactins, and show how recent targeted engineering of the taxadiene synthase (TXS) enzyme alongside biomimetic synthesis and biosynthesis investigations have added credence to earlier conclusions.
2. Verticillene vis-à-vis cembrene hydrocarbons
Although the hydrocarbon verticillene 2 has not been reported “free” in Nature, over the past 25 years several research groups have described the presence of a number of alkene regioisomers of (S)-verticillene 2, and its (R)-enantiomer, in the essential oil of Boswellia carterii olibanum, from various species of the tree Bursera, and from the Japanese liverwort Jackiella javanica. Thus, Asakawa et al.8 were the first to describe the presence of the isomeric ent-isomers 7 and 8 of exo- and endo-verticillene respectively in the liverwort J. javanica in 1995, and a few years later König et al.,9 reported the C4 exo-isomer 9 in the essential oil of Boswellia carterii (Fig. 2). In a comprehensive investigation of verticellene derivatives in Bursera species Hernández et al.10 later established the presence of the two alkene isomers 10 (ent-7) and 11 (ent-8) of verticillene, alongside 9, in Bursera suntui and B. kerberi, and Marcotullio et al.11 reported the presence of 10 in B. microphylla. During studies of the chemistry of naturally derived (+)-verticillol 1, it was shown that the isomeric verticillenes 10 and 11 could be produced following dehydration of 1 under acid conditions.4,5 The unusual and interesting Z-8,9-isomer 12 of verticillene 2, called sobralene, was recently reported to be a sex-aggregation pheromone produced by the sand fly Lutzomylia longipalpis from Sobral (Ceará State) in Brazil.12L. longipalpis is the main vector of the parasite Leischmania infantum, the causative agent of American visceral leishmaniasis (AVL) a potentially fatal human and canine disease in South and Central America.13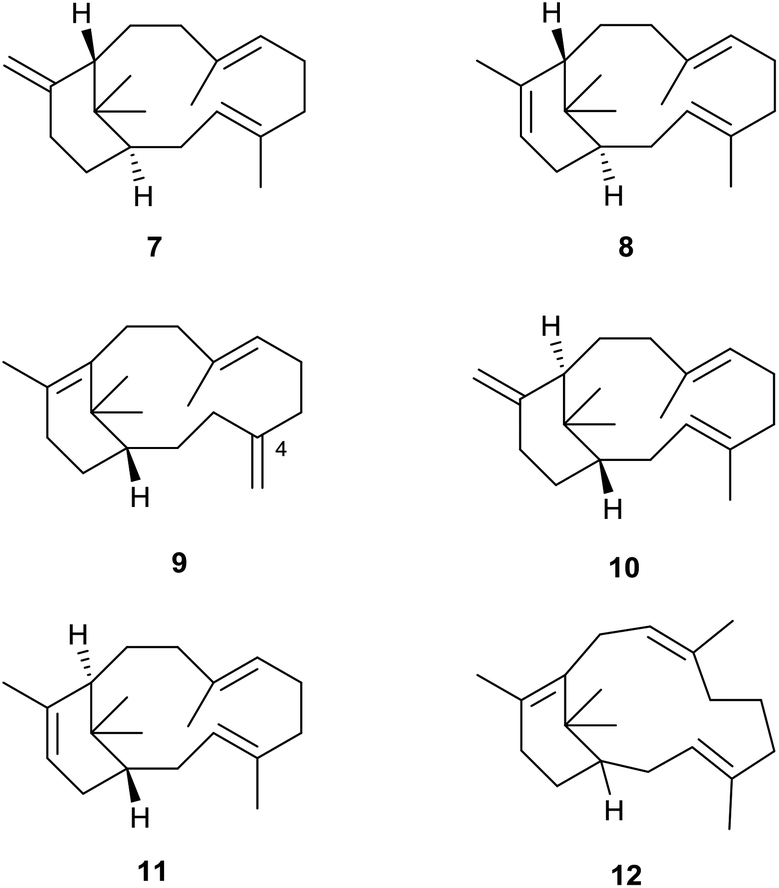 | ||
| Fig. 2 Naturally occurring isomeric verticillene hydrocarbons. | ||
The verticillene alkene isomers 7–11 have their origin in electrophilic cyclisation of geranylgeranyl diphosphate (GGPP) 13, which leads initially to the 14-membered macrocyclic carbocation intermediate 15 (Scheme 1). Quantum chemical calculations on the TXS pathway from GGPP to taxadiene 3, published by Hong and Tantillo in 2011,14 reinforced an earlier view that the GGPP carbocation species 14 in the cyclisation leading to 15a is preorganised and that the cyclisation is stereospecific with re-face attack of the π-bond and anti-displacement of the diphosphate (OPP) group.15 Correspondingly, si-face attack within 14 would lead to the enantiomeric carbocation 15b. Calculations also showed that the cyclisation process leading to 15a is likely to be concerted (albeit asynchronous) with loss of OPP. The “cembrenyl” carbocation 15a in the taxadiene synthase (TXS) cascade is a discrete intermediate, yet it rapidly undergoes transannular cyclisation to the C12 verticillyl carbocation intermediate 16a. Loss of protons from C13 and C18 in 16a/16b then lead to the enantiomeric verticillene alkene isomers 7, 8, 10 and 11. The C4 exo-isomer 9 of verticillene is most likely produced from the C12 verticillyl carbocation 16a in a two-step process, involving capture of the proton at C11 by the C3, C4 double bond leading to the C4 verticillyl carbocation 17, followed by elimination of a proton from C20 (Scheme 1).
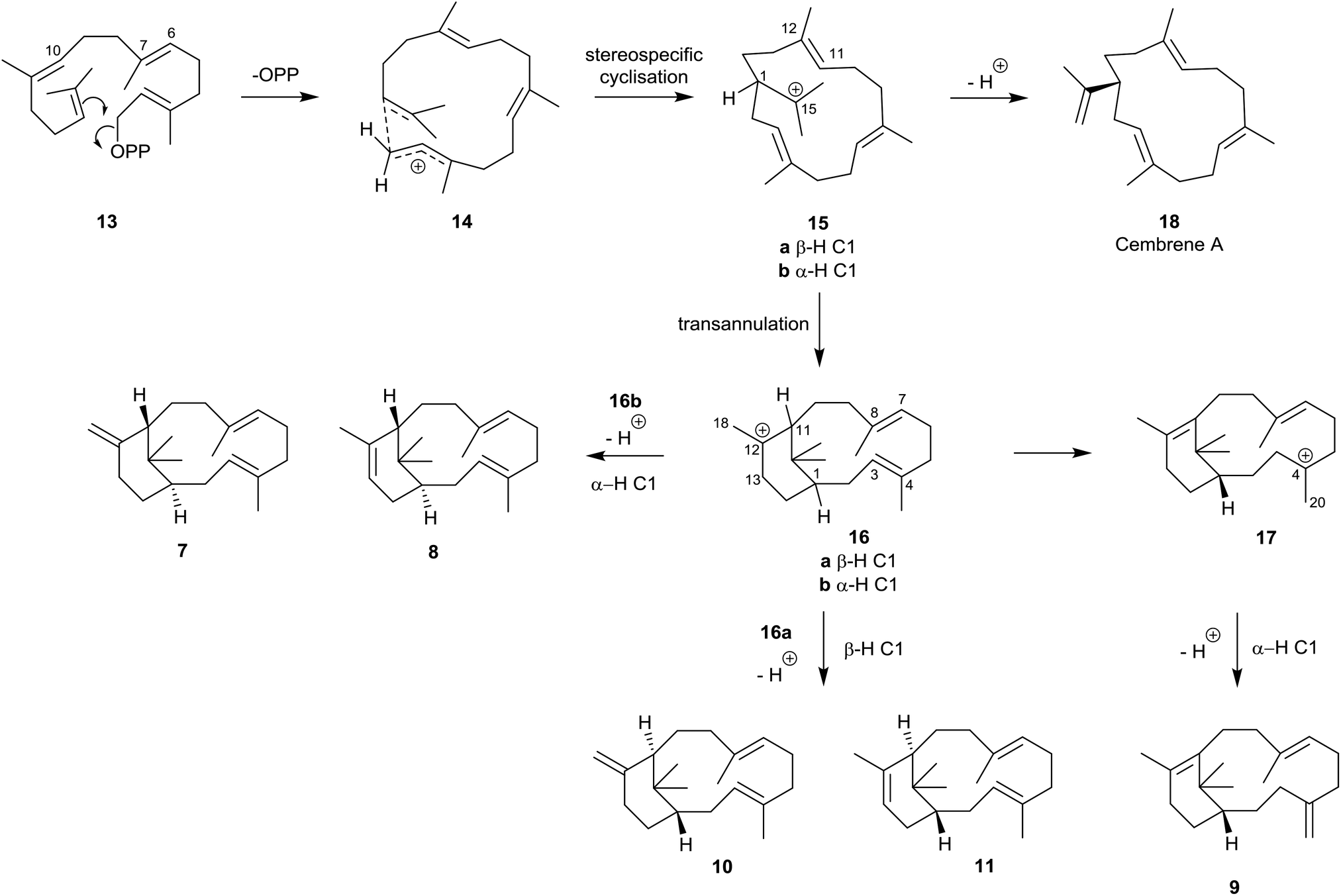 | ||
| Scheme 1 Origin of the verticillene hydrocarbons 7–11 and the cembrenes 18–23 from GGPP 13.16 | ||
Loss of a proton from one of the methyl groups at C15 in the macrocyclic carbocation intermediate 15 instead delivers cembrene A (18), a member of the “cembrene” family of natural products, e.g. hydrocarbons 20–25 (Fig. 3), which often occur naturally in both the R and S enantiomeric series. It is interesting that cembrene A (18),17 as well as the cyclopropane-based diterpene macrocycle casbene 19,18 were both once considered to be conceivable intermediates in the biosynthesis of taxadiene from GGPP. Furthermore, in some studies of the cyclisations of analogues of GGPP catalysed by TXS, Scott et al.19 established that 14-membered cembrene hydrocarbons could be major products. Later, during targeted engineering studies of the TXS pathway, Brück et al. engineered a TXS mutant incorporating histidine as a specific proton acceptor, and observed the formation of R-cembrene A (18); they then went on to develop a fermentation system which produced 18 in small quantities.20 Earlier, Kuzuyama et al.21 had described a diterpene synthase produced from Streptomyces which also led to the synthesis of cembrene A (18) alongside its isopropylidene isomer 20, and Keasling et al.22 had cloned cembrene A (18) and casbene 19 synthases from Euphorbiaceae plant genes expressed in Saccharomyces cerevisiae. Cembrene 21, which is also called thunbergene or thumbelene, was the first cembrene to be isolated and characterised from pine oleoresins in 1965.23
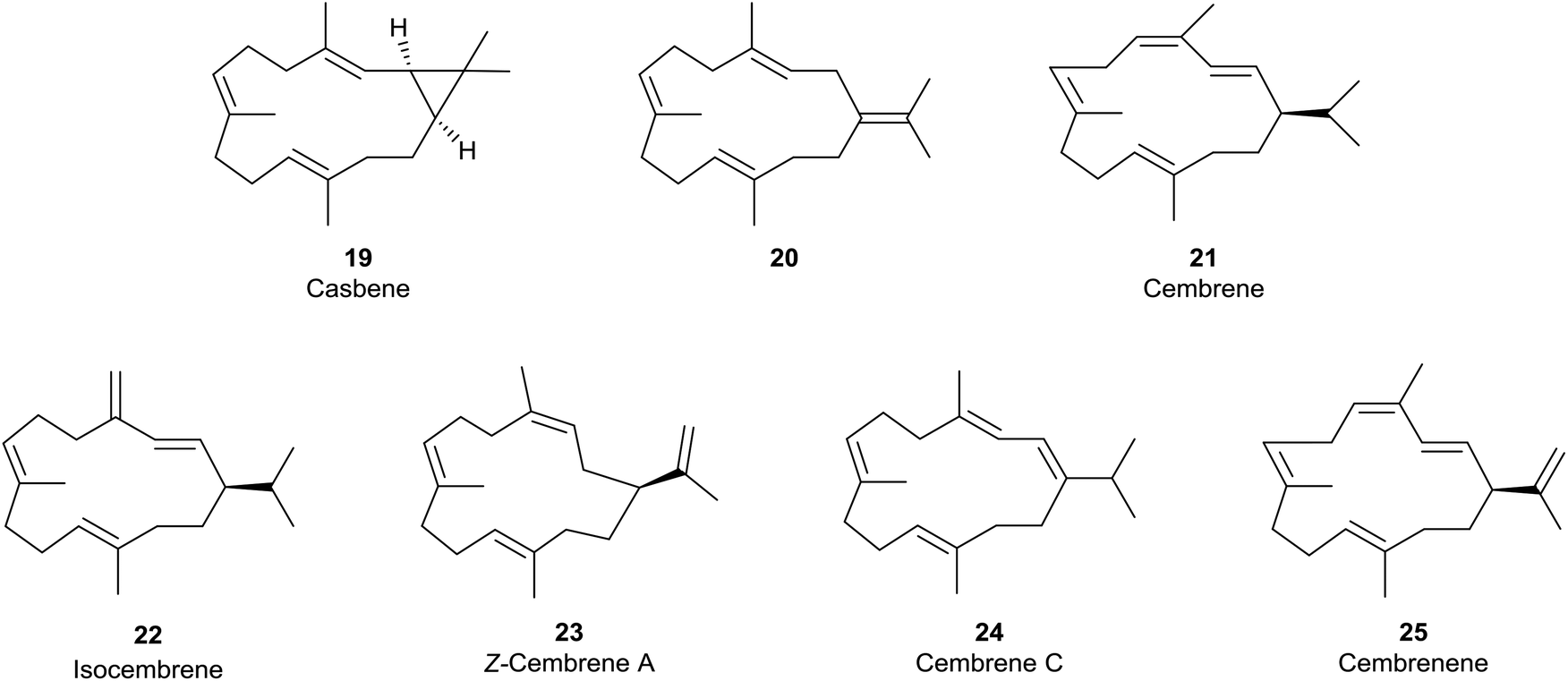 | ||
| Fig. 3 Casbene 19 and cembrene diterpene hydrocarbons 20–25. | ||
Contemporaneously, a number of isomeric cembrene hydrocarbons, including isocembrene 22,24 were found in other higher plants, e.g. the tobacco plant.25 However, cembrene A (18) (also known as neocembrene and neocembrene A) is the most widely distributed cembrene hydrocarbon, and has been isolated, for example, from the Pharaohs ant, Monomorium pharaonis,26 and from the Australian subterranean termite, Nasutitermes exitiosus,27 where it exhibits pheromone-like activity. It has also been found in paracloacal glands of the Chinese alligator, Alligator sinensis,28 and its 3Z-isomer 23 has been isolated from the heads of soldier termites Cubitermes umbratus.29 Of some relevance to this discussion, the cembrenes 18, 22 and 24 have been found to co-occur with the 4-exo verticillene 9 in Boswellia carteri.9
The majority of cembrenes that have been characterised over the past few decades, however, are oxygenated variants (e.g. OH, epoxy, cyclic ether, carbonyl and combinations thereof) of the hydrocarbons 18 and 21–23, also of cembrene C (24) and cembrenene 25.30 Furthermore, the overwhelming majority of these oxygenated cembrenes are produced by soft corals and gorgonians,31,32 where many are precursors to a variety of structurally novel and biologically interesting polycyclic cembranoids.33
3. Biosynthetic interrelationship between verticillene and taxadiene
The structures and stereochemistries of the taxane family of tricyclic diterpenes (see structures 3 and 6) were established independently by Lythgoe34 and by Nakanishi35 and their respective collaborators during the 1960s. The later revision of the absolute configuration of natural (+)-verticillol to 1 with the β-configuration at C1 in 2005![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6 was therefore consistent with the taxane stereochemistry 3. In 1966, Lythgoe et al.2 had postulated that the 6,8,6-ring-fused system in taxadiene 3 could arise in Nature from GGPP (13) via verticillene 2 (presumably via the verticillyl carbocation 16a) followed by a transannulation reaction, implicating the carbocation intermediate 26 (Scheme 2).
6 was therefore consistent with the taxane stereochemistry 3. In 1966, Lythgoe et al.2 had postulated that the 6,8,6-ring-fused system in taxadiene 3 could arise in Nature from GGPP (13) via verticillene 2 (presumably via the verticillyl carbocation 16a) followed by a transannulation reaction, implicating the carbocation intermediate 26 (Scheme 2).
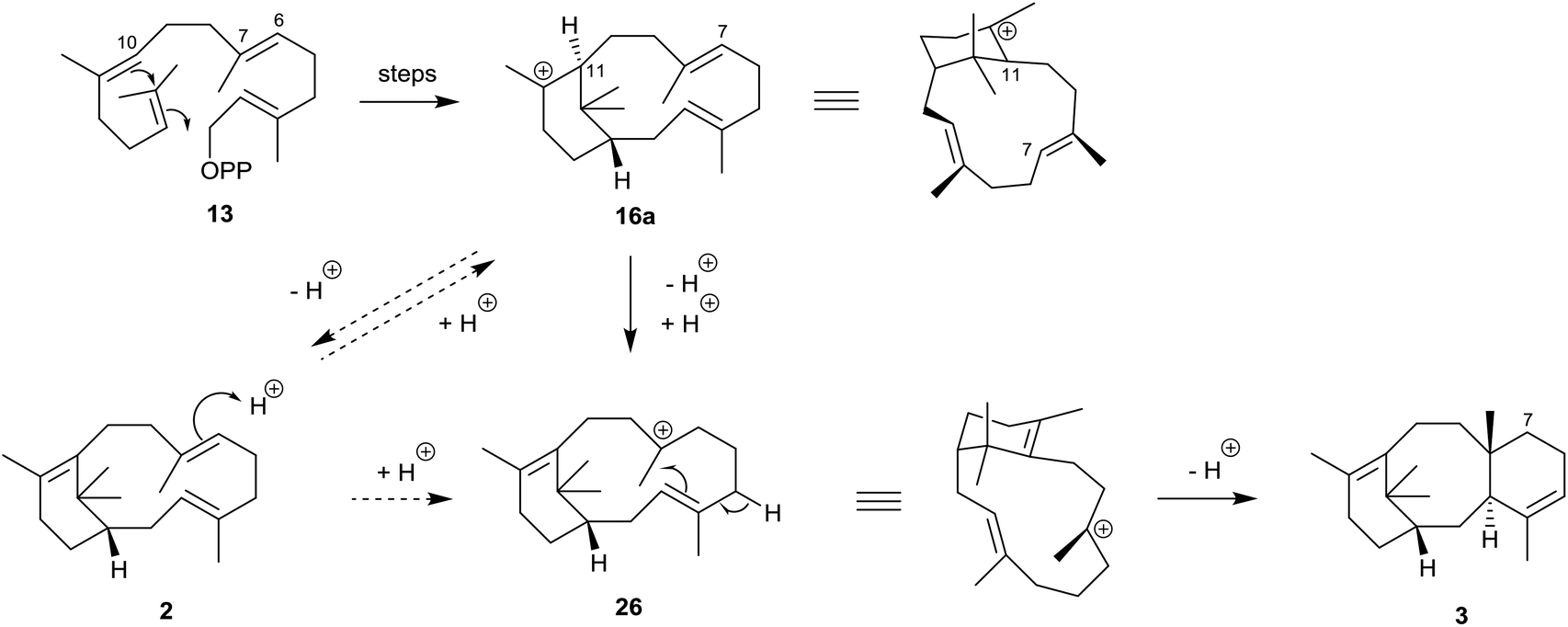 | ||
| Scheme 2 Early outline proposal for the biosynthesis of taxadiene 3 from GGPP (13) via verticillene 2 and the verticillyl carbocation intermediates 16a and 26. | ||
Lythgoe's proposal was essentially vindicated some years later during studies of the mechanism of the TXS-catalysed cyclisation of GGPP (13) by several groups of researchers during 1995–2005. Thus, early studies by Croteau et al.36 showed that when GGPP labelled with deuterium at C10 (see structure 27) was incubated with taxadiene synthase, the taxadiene isolated contained deuterium located at C7 (see structure 30). This observation implicated capture of the C11 deuterium in the first-formed verticillyl carbocation 28, by the C7,8 alkene bond, and its transfer to C7, leading firstly to the isomeric verticillyl carbocation intermediate 29 prior to its cyclisation to the taxadiene 30 (Scheme 3).
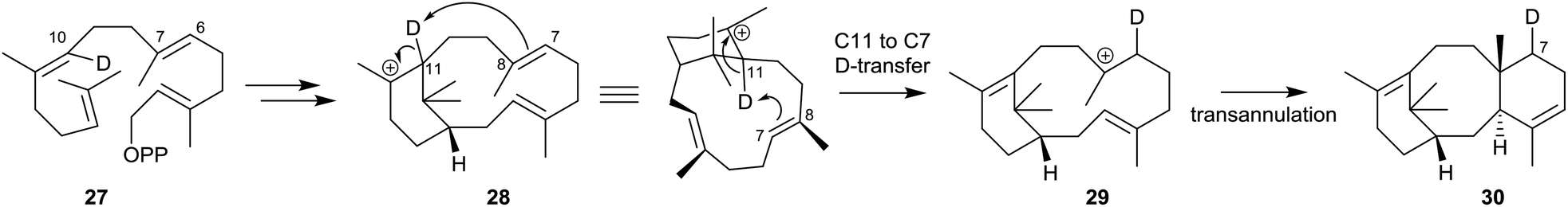 | ||
| Scheme 3 Incubation of C10 deuterium labelled GGPP (27) with taxadiene synthase leading to the C7 deuterium labelled taxadiene 30. | ||
In other investigations, Coates et al.6 showed that when the 6-fluoro analogue 31 of GGPP was similarly incubated with recombinant TXS, only the C12-exo34, the C12-endo35 and, significantly, the C4-exo36 isomeric 7-fluoroverticillenes were produced (88% yield; ratio 3:2:2) (Scheme 4). None of the C7 fluoro analogue 33 of taxadiene was produced in this incubation. This outcome demonstrated how the inductive effect of the fluoro-substituent at C7 in the fluoroverticillyl carbocation intermediate 32 inhibited the crucial C11 to C7 proton transfer, thereby blocking the cyclisation which would have led to the fluorinated taxadiene 33 and allowing the carbocation 32 to undergo alternative reactions leading to the fluoro-verticillenes 34–36.
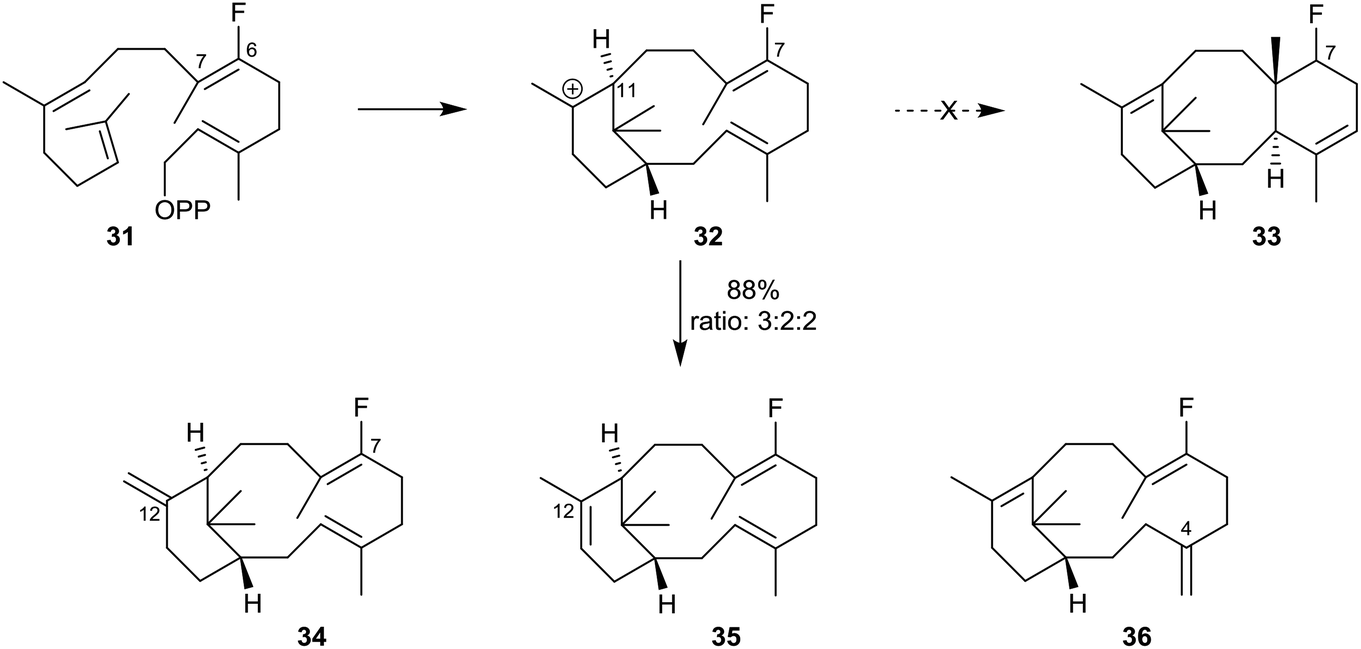 | ||
| Scheme 4 Incubation of the fluoro-analogue 31 of GGPP with recombinant TXS leading to the isomeric fluoroverticillenes 34–36 at the expense of the fluorotaxadiene 33. | ||
Interestingly, the formation of the C4 exo-fluoroverticillene 36 in this incubation, suggested that a two-step proton-transfer process is involved in the conversion of the verticillyl C12 carbocation 16a into taxadiene 3 (see Scheme 2). Thus, transfer of the proton at C11 to C3 in 16a, first leads to the verticillyl C4 carbocation 37, which, following the transfer of a proton from C3 to C7 then leads to the C8 verticillyl carbocation 39 (Scheme 5). Lastly, transannular cyclisation of 39 produces the tricyclic carbocation intermediate 38 which then loses a proton leading to taxadiene 3 (cf.Scheme 2). Indeed, additional quantum chemical calculations by Hong and Tantillo,14 suggest that the two-step route (16a → 37 → 39) is energetically more favourable that the single step route (16a → 39) for transfer of the proton at C11 in 16a to C7 in 39. The same calculations also supported an earlier proposal by Coates et al.36b that there needs to be a change in conformation of the carbocation 39, i.e. from 40 to 41, prior to its cyclisation to 38 (energy difference ca. 4 kcal mol−1; Fig. 4). Significantly, although the aforementioned early studies ruled out verticillene 2 as a “free” intermediate they consolidated the view that the verticillyl carbocation 16a was the central intermediate in the TXS cascade between GGPP (13) and taxadiene 3.
 | ||
| Scheme 5 Stepwise conversion of the verticillyl C12 carbocation 16a into taxadiene 3via the C4 and C8 verticillyl carbocations 37 and 39 and the taxane carbocation 38. | ||
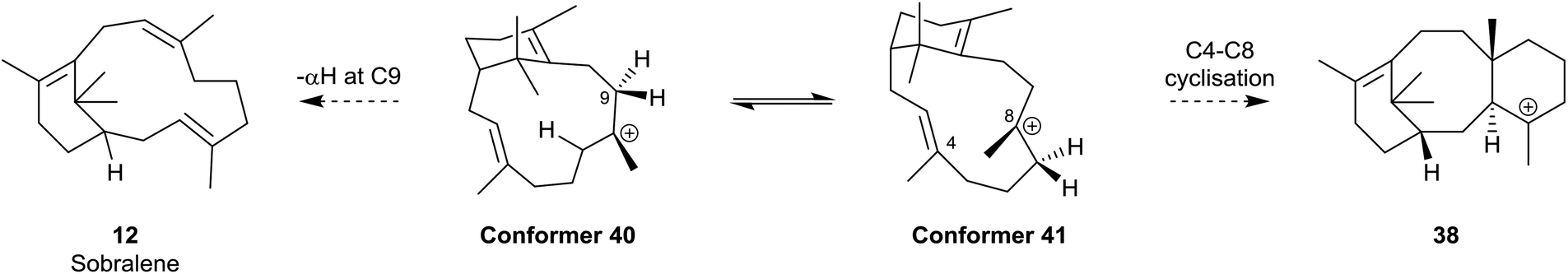 | ||
| Fig. 4 Different conformations, 40 and 41, of the verticillyl C8 carbocation 39 leading to the formation of sobralene 12 and taxadiene 3 respectively. | ||
Interestingly, it has been suggested that the biosynthesis of the novel Z-8,9 verticillene isomer sobralene 12, takes place by elimination of the α-proton at C9 in the high energy conformation 40 of the verticillyl carbocation 39 thereby leading to the Z-configuration of the C8,9 alkene bond in the natural product.12
4. Targeted engineering of TXS leading to the production of verticillene intermediates
The crystal structure of a shortened/truncated variant of the TXS enzyme was reported in 2011,37 and this exciting disclosure was a catalyst for others to pursue targeted engineering of TXS to probe further mechanistic details, as well as accessing and characterising key intermediates, on the pathway from GGPP to taxadiene.38 Although earlier mechanistic studies using deuterium and fluorine-labelled GGPP analogues, and quantum chemical calculations, had clearly established the presence of several verticillyl carbocation intermediates in the TXS cascade (see Section 3) the nature and location of the deprotonating basic sites within the enzyme leading to these reactive species remained a mystery.Brück et al.,19 constructed a closed TXS-GGPP complex with both TXS and GGPP in their productive conformations. They then used an integrated approach of docking and MM optimisation to model the catalytic sites of TXS and GGPP, and the carbocation intermediates suggested in the cascade mechanism. In this manner Brück and colleagues were able to accurately trace the reaction pathway followed by TXS which, in turn, allowed them to target and then engineer enzymes to generate important verticillene intermediates in the overall TXS cascade. Significantly, Brück et al., demonstrated that particular mutants of TXS gave rise to the production of the verticillenes 2, 9, and 11, as well as the isomer 42 of taxadiene 3 (Scheme 6). Following experiments carried out in vitro, NMR and chiroptical measurements on the isolated verticillene 11 showed that it was identical with, and had the same (+)-R,R-configuration as, the same compound isolated earlier from species of Bursera and from Boswellia carterii.9,10
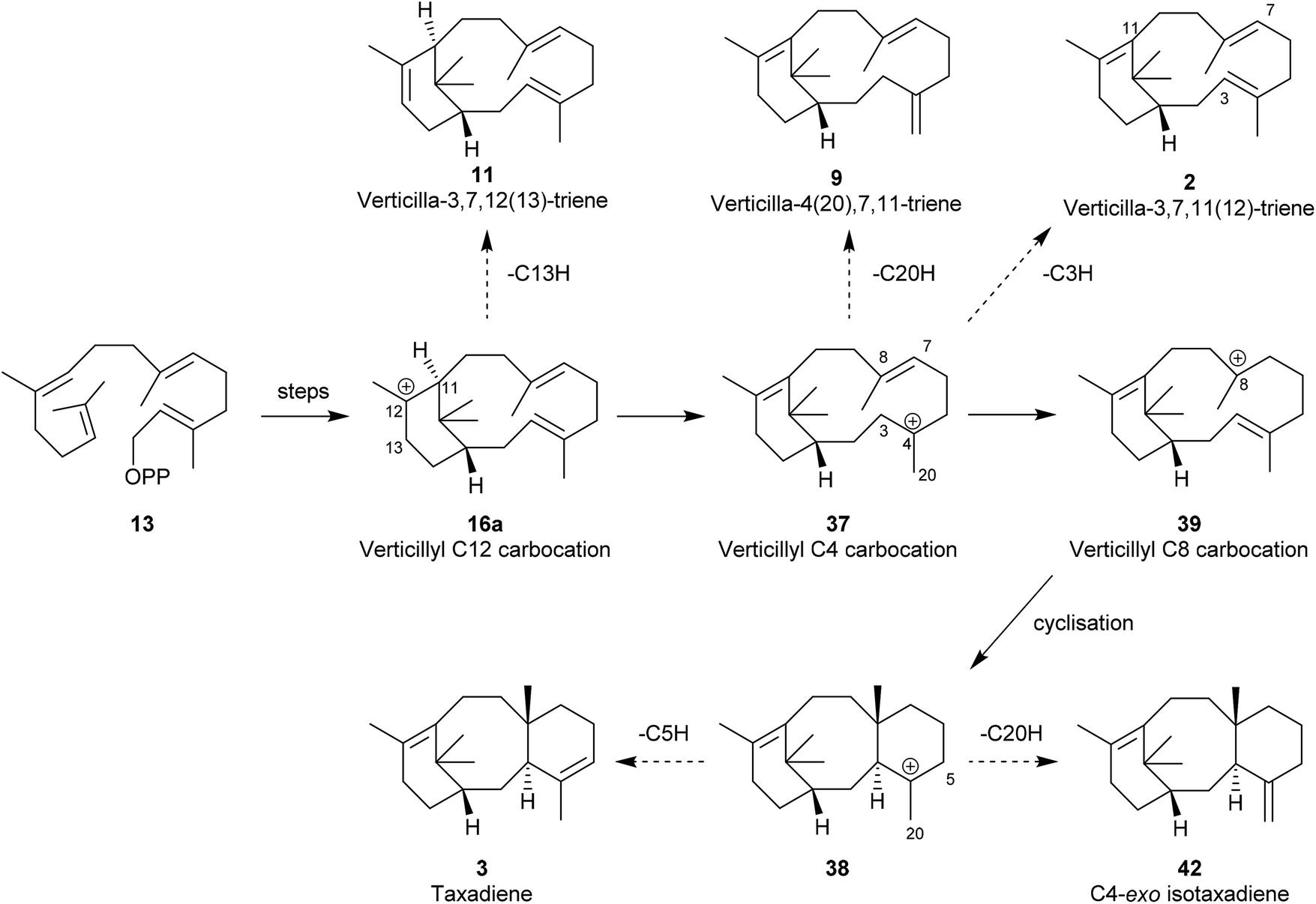 | ||
| Scheme 6 Overall picture of the TXS cascade from GGPP (13) to taxadiene 3, with the postulated verticillyl carbocation intermediates 16a, 37 and 39, the deprotonated verticillene intermediates 2, 9 and 11, and the C4-exo iso-taxadiene 42. | ||
In contemporaneous investigations, Edgar et al.,39 established a TXS mutant enzyme, expressed in an engineered E. coli, and showed that the verticillene 11 was its sole product. The verticillene isomer 11 results from premature deprotonation at C13 in the C12 carbocation intermediate 16a presumably as a result of the closer orientation of the C13 proton to a basic site in the mutant in comparison with native TXS. The formation of the C4 exo-verticillene 9 confirmed the presence of the C4 verticillyl carbocation 37 in the TXS cascade; furthermore, the carbocation 37 is also linked to the verticillene 2via deprotonation at C3. Overall, the studies of Brück and colleagues supported the earlier proposed two step route, i.e.16a → 37 → 39 as being more energetically favourable over the alternative single step process 16a → 39,40 en route to taxadiene.
5. Biosynthetic interrelationship between verticillene and the phomactins
The phomactin family of PAF antagonists have a core bicyclo[9.3.1] pentadecane ring system, and they were first reported in the marine fungus, Phoma sp., in 1991.41e.g. phomactin A (5), Sch 49026 (4), phomactin B (43), phomactin G (44) and phomactin K (45) (Fig. 5). Following their isolation it was recognised immediately that the phomactin ring system 4 exhibited a striking structural relationship with that found in the taxanes, i.e.3 which suggested that they probably shared the same verticillene 2 (or verticillyl carbocation intermediate 16a) in their biosynthesis.42 Thus, whereas an enzyme-mediated process featuring transannulation from 16a produces the C-ring in taxadiene 3, an alternative sequence of carbocation initiated 1,2-hydrogen and 1,2-methyl group shifts in 16a leads to the carbocation intermediate 46 instead (Scheme 7). Elimination of a proton in 46 then delivers the natural phomactatriene hydrocarbon 47 which, together with its isomer 4, are logical precursors to the oxygenated phomactins, i.e.5, 43, 44 and 45. Indeed, several years later the essential ingredients of this biosynthesis proposal were given credence by Oikawa et al.43 based on their feeding experiments with 13C-labelled acetates in fermentations of Phoma sp. Furthermore, during a study of the hydrocarbon constituents of the phomactin-producing fungus Phoma sp. the same authors also identified the verticillenes 9, 10 and 11 together with the phomactins 4 and 47. These observations reinforced the pivotal role of the verticillyl carbocation 16a in the biosynthesis of the phomactins and the special relationship they share with the taxanes 3 (see Scheme 7).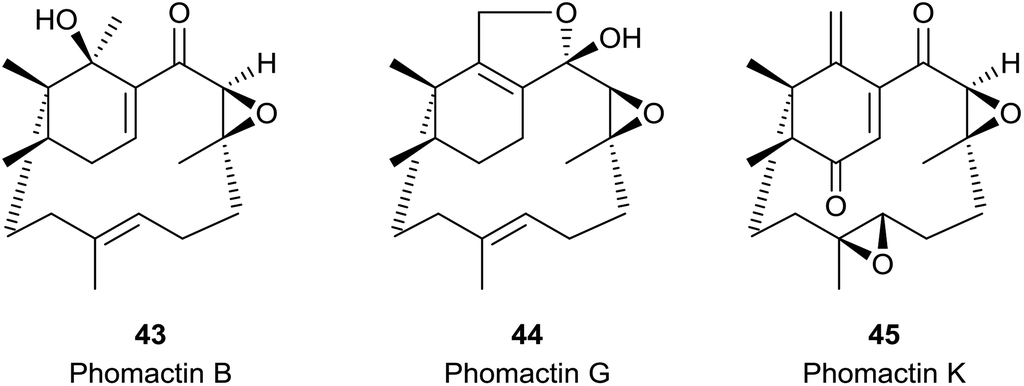 | ||
| Fig. 5 Some naturally occurring phomactins. | ||
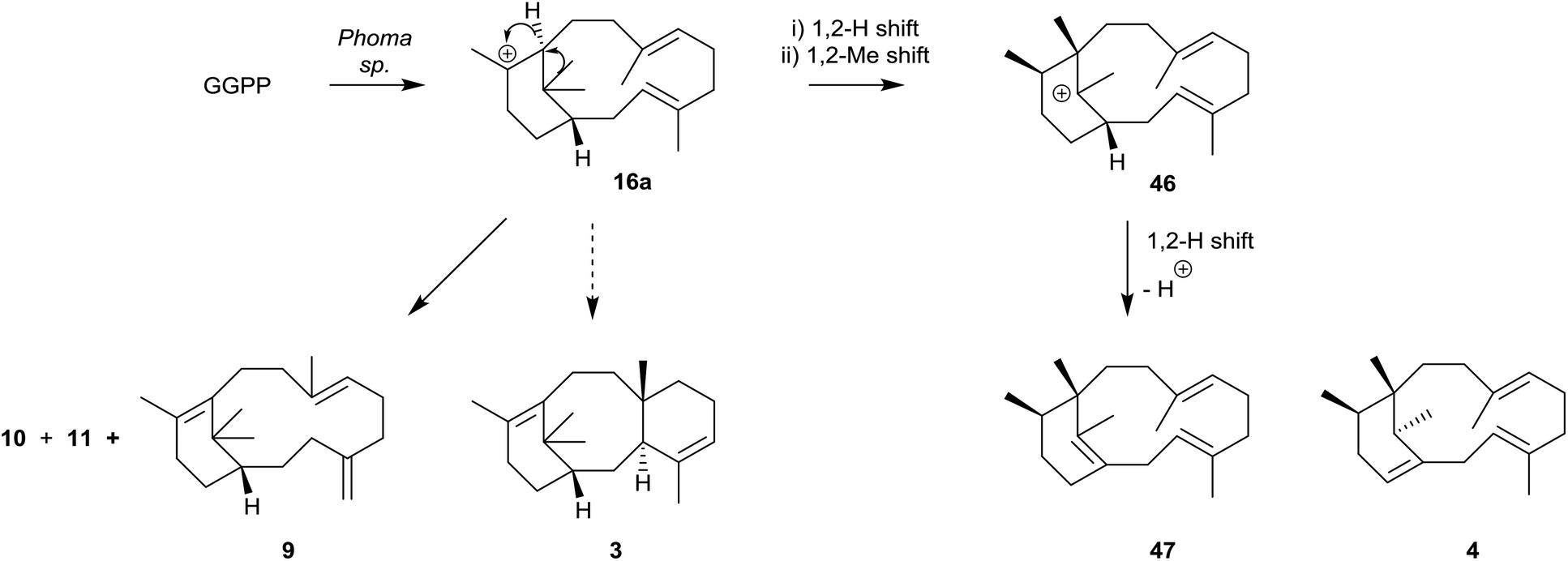 | ||
| Scheme 7 Proposed biosynthesis of the phomactatriene 47 from the verticillyl carbocation 16avia the rearranged carbocation 46. | ||
Furthermore, in contemporaneous studies Scott et al.44 demonstrated that when des-7-methylGGPP (48) was incubated with the enzyme of recombinant E. coli the major products were the isomeric des-8-methylphomactatrienes 51 and 52 (Scheme 8). None of the des-8-methyl taxadiene 50 was produced in this incubation. This outcome was rationalised on the basis that the absence of a carbocation stabilising methyl group at C8 in the verticillyl intermediate 49 diverts the pathway away from that leading to the des-methyltaxadiene 50, allowing time for the carbocation 49 to undergo rearrangement to the desmethylphomactatrienes 51 and 52 instead.
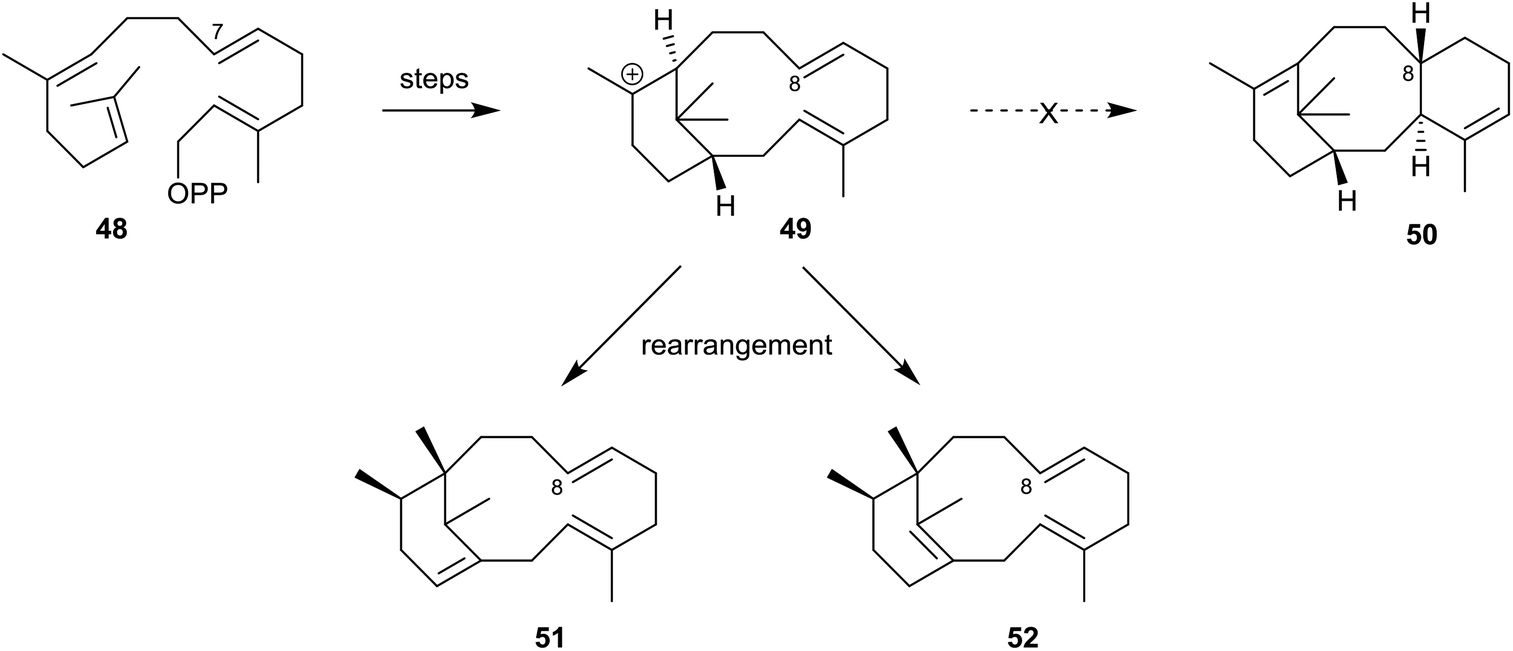 | ||
| Scheme 8 Incubation of the enzyme of recombinant E. coli with des-7-methylGGPP (48) leading to the des-8-methylphomactatrienes 51 and 52 at the expense of des-8-methyltaxadiene 50. | ||
6. Natural oxy-substituted verticillenes
The overwhelming majority of verticillenes that have been described accommodate various oxygen substituents, i.e. OH, OAc, carbonyl, epoxy, and the major portion of these metabolites have been isolated from soft corals and from Taxus species. As a result of their association and structural relationship with taxol several of these oxy-substituted verticillenes have been variously named as bicyclic taxanes,45 seco-taxanes,46 taxachitrienes,47 taxuspines,48 and canadensenes,49 amongst others. Shi and colleagues have earlier listed the structures of many oxy-substituted verticillenes that have been described in the literature up to about 2005,50 and we will not duplicate this listing here. Nevertheless, because it was one of the first oxygenated verticillenes to be characterised in the early 1980s we make special mention of the poly-oxygeneated verticillene hypoestoxide 53 (Fig. 6).51 This highly oxygenated verticillene was isolated from the tropical shrub Hypoestes rosea, found in the Nigerian rain forests, and extracts of the shrub have been used in folk medicine for generations to treat various skin rashes and fungal infection conditions. Here however, we will focus on the structures of some more recently characterised oxy-substituted verticillenes and particularly those that relate to, or co-occur with, the verticillene hydrocarbons 7–11 of importance in the biosynthesis of the taxanes and the phomactins. Thus, in investigations of the verticillols produced by plants of the genus Bursera which grow in parts of Mexico, Hernández et al.10 showed that although (+)-verticillol 1 is found in B. suntui, surprisingly its C12 epimer 54 is produced by B. kerberi. Earlier, Asakawa et al.52 had reported the presence of the enantiomers of 1 and 54 in the liverwort Jackiella javanica, and in 2017 the same verticillol 54 was found in B. microphylla.11 Hence a total of four isomeric verticillols have now been characterised in Nature i.e.1, ent-1, 54 and ent-54. The same liverwort J. javanica from which the verticillene hydrocarbons 7 and 8 were isolated is also a source of the interesting hydroxyl-substituted verticellenes, verticillenol 55, verticillenediol 56a and 12-epi-verticillenediol 56b.52 Likewise, the shrub B. suntui that provides the verticillene hydrocarbons 9, 10, and 11 also metabolises the C20 hydroxy substituted verticillol 57a and its acetate 57b,10 together with the C7–C8 epoxides and C7 hydroxy derivatives of 57a and 57b.53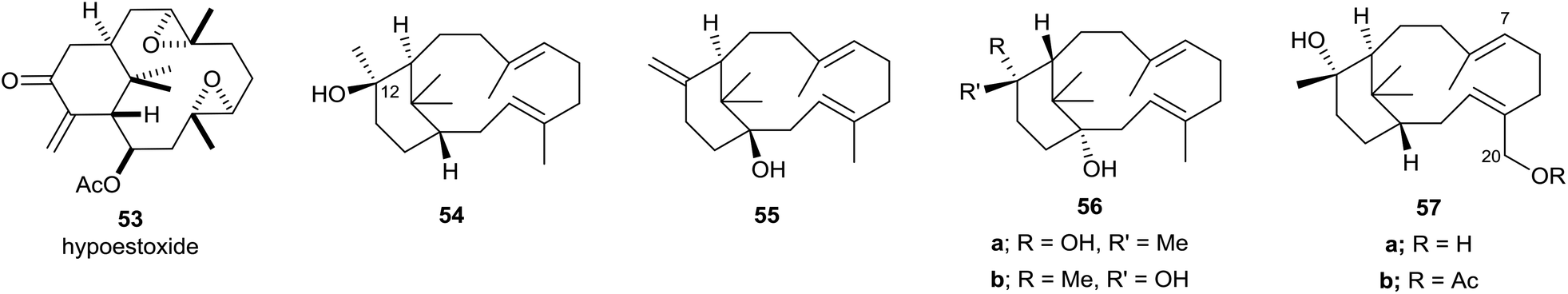 | ||
| Fig. 6 Some natural hydroxyverticillenes and their derivatives. | ||
7. Some biomimetic conversions of verticillenes into taxadiene and phomactatrienes
We have mentioned earlier that our motivation for synthesising verticillene 2 in the mid-1980s was to establish whether it could be encouraged to undergo a (biomimetic) transannular cyclisation across its 12-membered ring leading to the 6,8,6-tricyclic ring system 3 in the taxanes.4 Unfortunately we were frustrated in all of the attempts we made to convert 2 into 3 in the presence of boron trifluoride at 0–50 °C, the preferred reagent and conditions for carrying out these types of transannulations at the time; instead verticillene 2 underwent extensive decomposition. We also prepared the C7,8-epoxide 58 of our synthetic (±)-verticillene but this compound only underwent rearrangement to a mixture of the secondary alcohols 59 and 60 on treatment with boron trifluoride etherate. We next prepared the epoxide 61 of natural (+)-verticillol 1, and also the epoxide 62 produced from the isomeric verticillene 11 which we obtained by dehydration of (+)-verticillol 1 (Scheme 9). Treatment of the epoxide 61 with boron trifluoride etherate or trimethylsilyl triflate under a range of conditions produced largely the corresponding ketone 63 accompanied by small amounts of the product 62 from dehydration of 61.4 In separate experiments and under the same boron trifluoride etherate reaction conditions the epoxide 62 underwent extensive decomposition.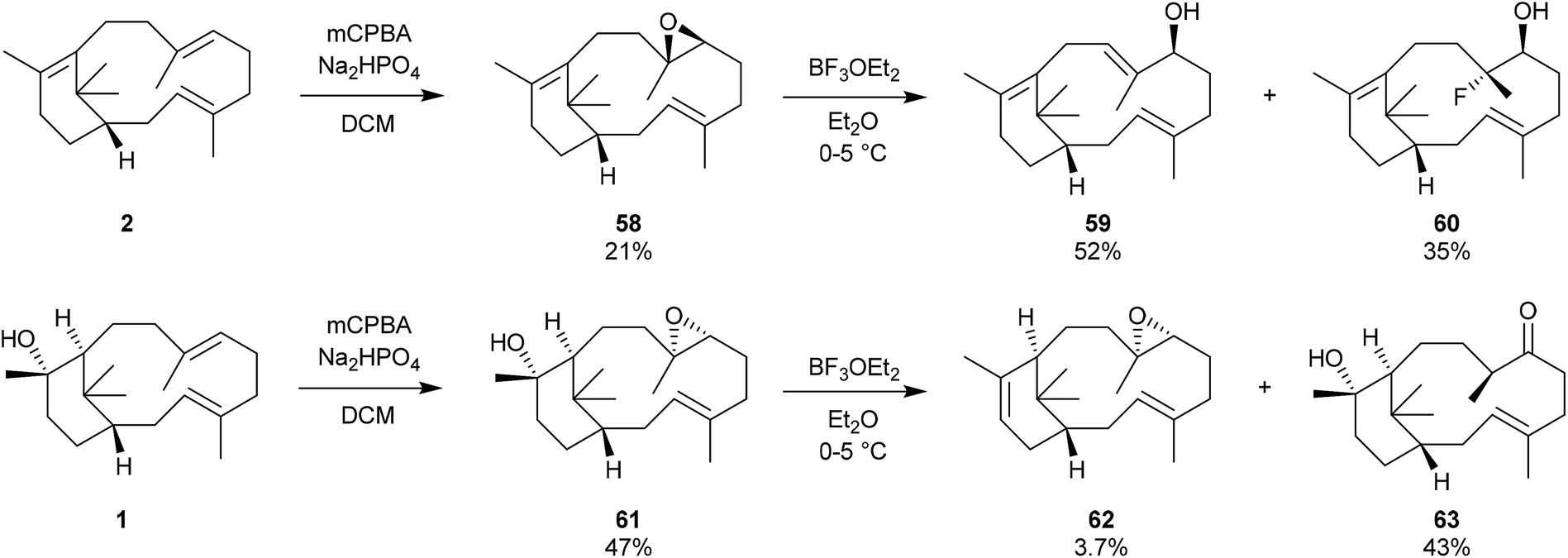 | ||
| Scheme 9 Attempted biomimetic conversions of verticillene epoxides. | ||
Some twenty years after our own work, and following the discovery of the phomactins in the fungus Phoma sp. in 1991, Oikawa et al.43 carried out a more systematic study of the rearrangement of natural verticillol 1 in the presence of boron trifluoride etherate but at various lower temperatures. Thus, they found that when verticillol was treated with boron trifluoride etherate at −78 °C almost equal amounts (i.e. 31, 32, and 37% based on peak areas in GC analysis) of the isomeric verticillene hydrocarbons 10 and 11, and the phomactatriene 47 were obtained (Scheme 10). However, at −30 °C the major product was the phomactatrine 47 (62%) together with the verticillene 11 (36%), and at −20 °C the unusual C4,5/C8,9-positional isomer 64 of 47 predominated (46%). The same phomactatriene positional isomer 64 was also the major product, i.e. 65%, when the exo-verticillene 10 was separately treated with boron trifluoride etherate at −30 °C and when verticillol was treated with La(OTf)3 at 40 °C. Interestingly, the same authors also reported the presence of taxadiene 3, estimated to the extent of 0.004% yield (after repeated HPLC, and detected by co-injection with an authentic sample), when verticillol 1 was treated with boron trifluoride etherate at room temperature. We have already pointed out that, Oikawa et al. had simultaneously identified the isomeric verticillenes 9–11 and the phomactatriene 47 in mycelium extracts of the fungus. These studies clearly reinforced the central role that the verticillyl carbocation 16a plays in linking the biosynthetic interrelationships within the verticillene, the taxane and the phomactane families of diterpenes.
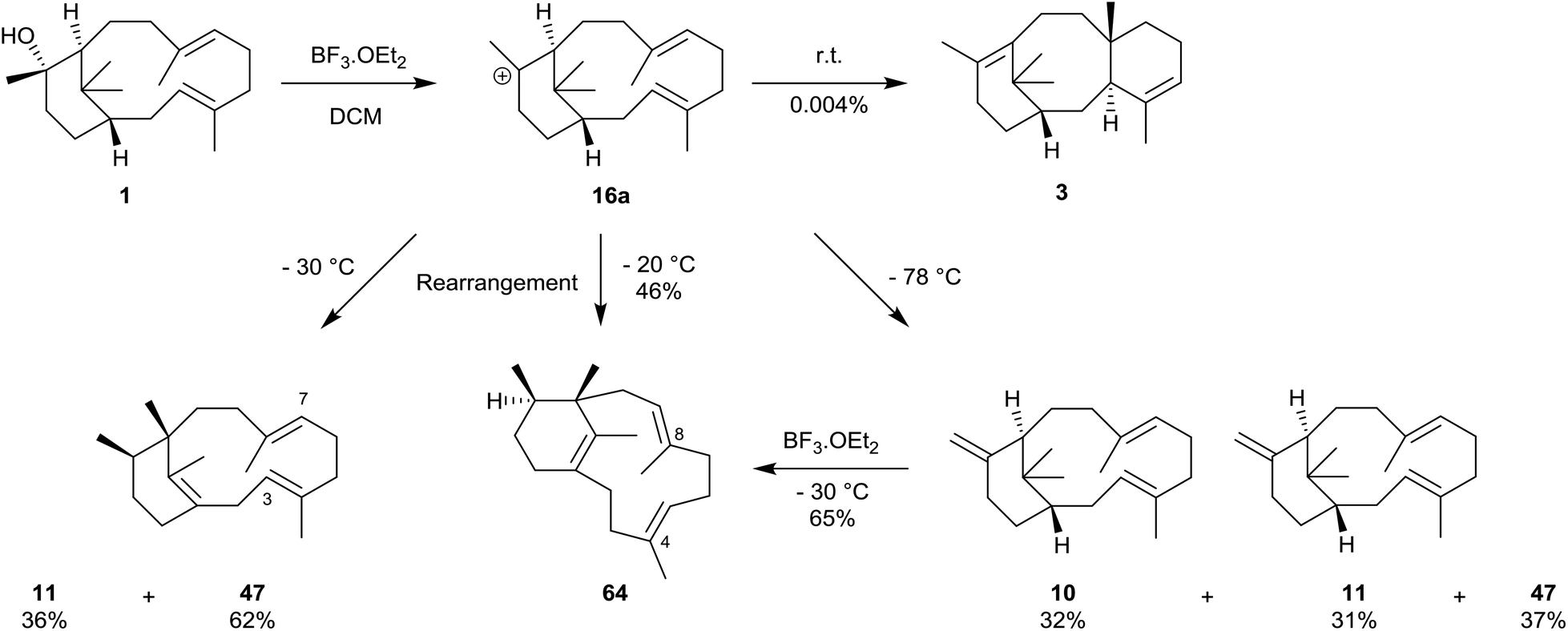 | ||
| Scheme 10 Biomimetic conversions of (+)-verticillol 1 into the phomactin 47, taxadiene 3 and the phomactin isomer 64 (note: yields were calculated from GC analysis). | ||
8. Synthetic studies towards the verticillenes
Synthetic studies within the verticillene family of natural products have been relatively sparse, although two reports of the total synthesis of verticillene 2 have been presented. The first synthesis of racemic verticillene 2 was described by ourselves in 1985,4 and was based on an intramolecular McMurray reductive coupling reaction with the bis-aldehyde 72 leading to the macrocycle 73 as the key step (Scheme 11).4 The synthesis of 72 started from the 3-tert-butoxycyclohexenone 65a which was first alkylated with geranyl bromide leading to 65b. Addition of methyllithium to 65b followed by hydrolysis next gave the cyclohexenone 66, which was then added to lithium dimethylcuprate leading to an enolate which could be trapped with TMS chloride producing the silyl enol ether 67. The silyl enol ether 67 was next alkylated with trimethyl orthoformate in the presence of TiCl4, and chromatography separated the cis-ketoacetal 68 in 58% yield. The ketoacetal 68 was next converted into the unsaturated aldehyde 70 in two steps, following treatment with methylmagnesium iodide, producing 69, and concomitant hydrolysis and dehydration of 60, using POCl3 in pyridine. Regioselective allylic oxidation of 70, using catalytic selenium dioxide/tert-butyl hydroperoxide next gave the E-allylic alcohol 71a which underwent smooth oxidation using MnO2 producing the central intermediate bis-aldehyde 72. In one of the first reported demonstrations of the scope of the intramolecular McMurray reductive coupling reaction, at the time, treatment of 72 with TiCl4 and Zn–Cu couple gave a mixture of the macrocyclic tetraene 73 and the isomer 74 which resulted from concurrent 1,5-sigmatropic rearrangement in 73. 1,4-Reduction of the conjugated 1,3-diene unit in 74 using sodium in liquid ammonia then led to E,E-verticillene 2.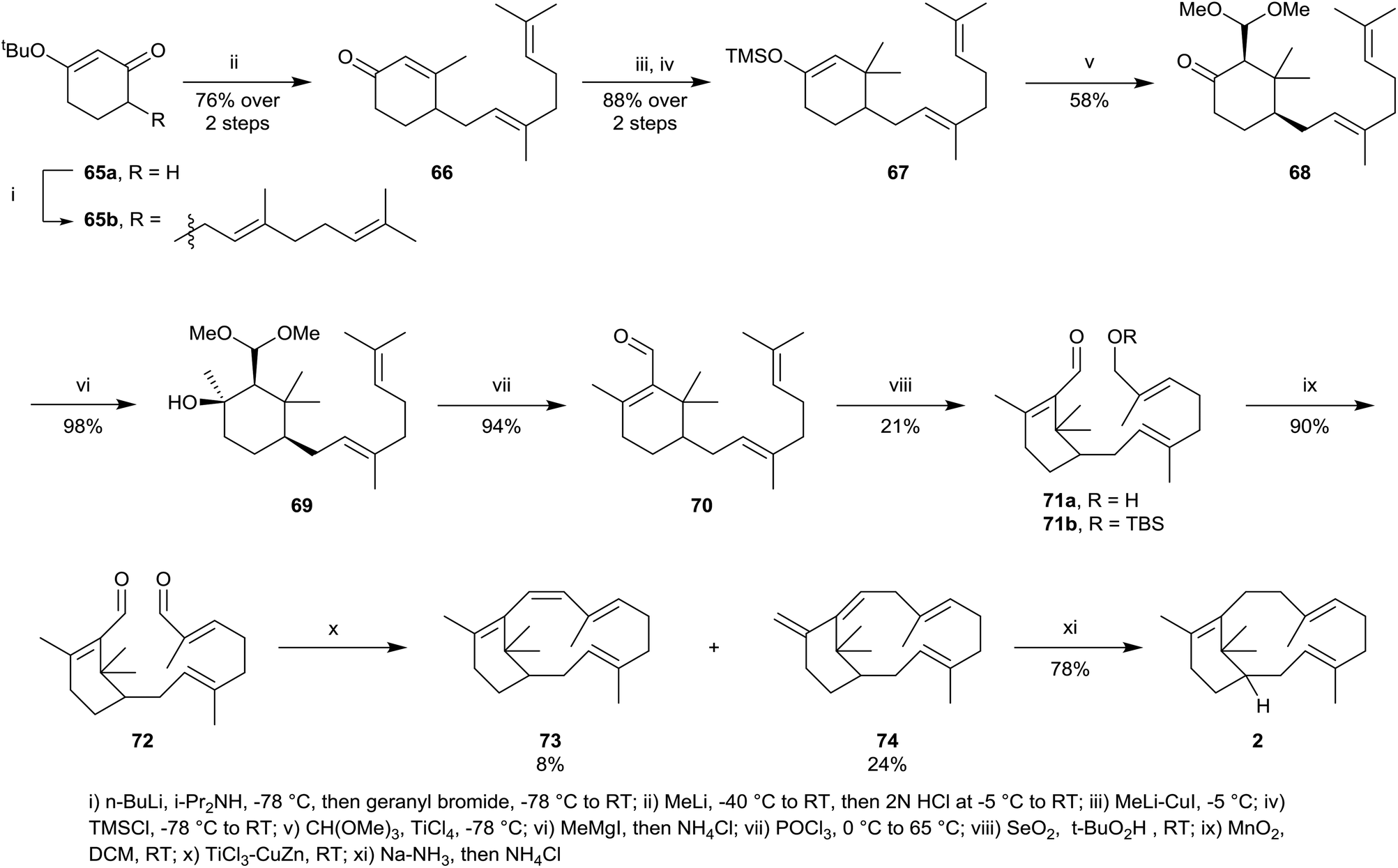 | ||
| Scheme 11 The first total synthesis of (±)-verticillene 2. | ||
Interestingly, a few years later, Croteau et al.36 used a modification of the above synthetic approach to prepare C10 deuterium-labelled verticillene 77 in connection with their investigations of the mechanism of taxadiene synthase (cf.Scheme 3). Thus, reduction of the aldehyde group in 71b using NaBD4 in the presence of CeCl3, followed by deprotection of the silyl ether protecting group in the D-labelled product, first gave the corresponding bis-allylic alcohol 75 (Scheme 12). Bromination of 75 using PBr3, followed by intramolecular reductive coupling of the resulting bis-allylic bromide 76 in the presence of Ni(CO)4 then gave C10 deuterium-labelled verticillene 77.
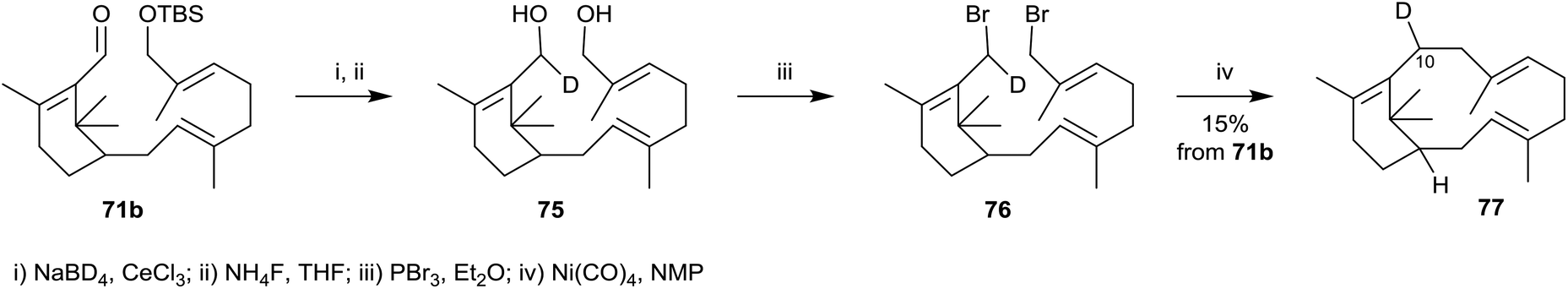 | ||
| Scheme 12 Synthesis of C10-deuterium labelled verticillene 77. | ||
A different synthetic approach towards verticillene 2 was used by Kato et al.54 whereby the cyclohexene ring in the substituted verticillene intermediate 82 was produced via an intramolecular alkylation and elimination process, reminiscent of a Friedel–Crafts reaction, from the chloride intermediate 80. Thus, the substituted C10-geranyl units 78 and 79 were first coupled in the presence of SnCl4 at −90 °C leading to 80 which on further reaction with SnCl4 at −30 °C led to a mixture of the diastereoisomeric substituted cyclohexanes 81a and 81b (Scheme 13). Then diastereoisomer 81a was treated with LiCl at 100 °C which resulted in dehydrochlorination leading to the substituted cyclohexene 82, while diastereoisomer 81b was treated with ZnCl2 leading to cyclohexene 82. In several steps the ester group in 82 was then converted into the corresponding alkyl chloride 83. An intramolecular alkylation in 83 in the presence of lithium bis-(trimethylsilyl) amide at 60 °C next gave the macrocycle 84 as a single diastereoisomer in 73% yield. Reduction of the nitrile group in 84 followed by decarbonylation of the resulting aldehyde 85 using Wilkinson's catalyst finally gave racemic verticillene 2.
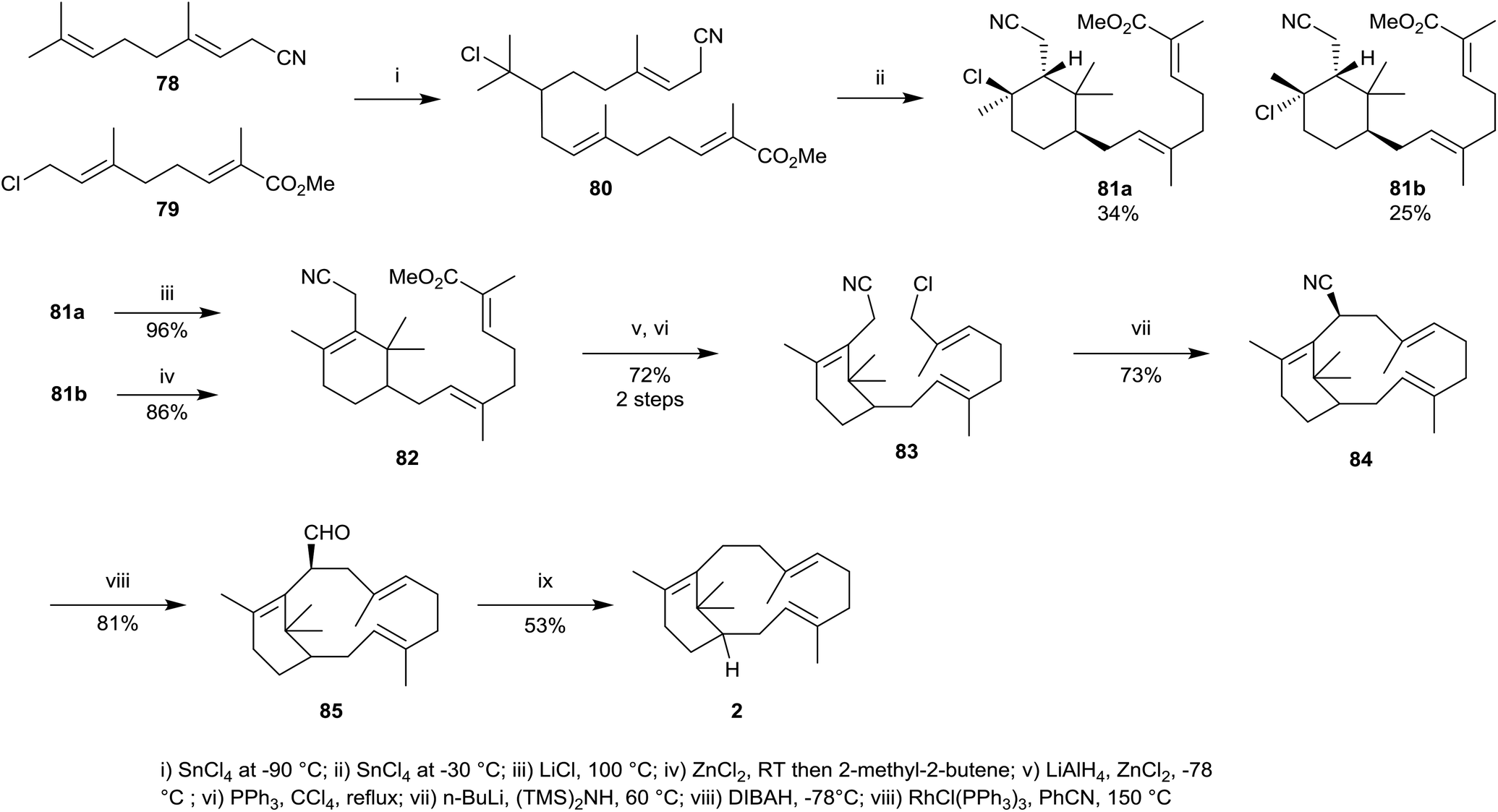 | ||
| Scheme 13 Kato's synthesis of (±)-verticillene 2. | ||
More recently, during their synthetic efforts directed towards hypoestoxide 53 Njardarson et al.,55 succeeded in preparing the Z-4,5-isomer of verticillol 1. Their synthesis started with the tert-butoxycyclohex-2-enone 65a which was first converted into the substituted cyclohexanone 87 in five straightforward steps (Scheme 14) via86, using essentially the same strategy used earlier to convert 65a into the cyclohexanone 69 in Scheme 10. Treatment of 87 with methylmagnesium bromide next led to the corresponding 1,3-diol which was protected as the cyclic carbonate 88. Ring-closure metathesis of 88 using Grubb's II catalyst in toluene under reflux, followed by carbonate deprotection resulted in the formation of the verticillane diol 89 in 40% yield, but unfortunately it had the undesired Z-3,4-stereochemistry and was also the incorrect atropisomer. Mesylation of the secondary hydroxyl group in 89 followed by reduction of the resulting mesylate using sodium naphthalenide then gave the atropisomer 90 of the Z-4,5-isomer of verticillol (cf. natural verticillol 1).
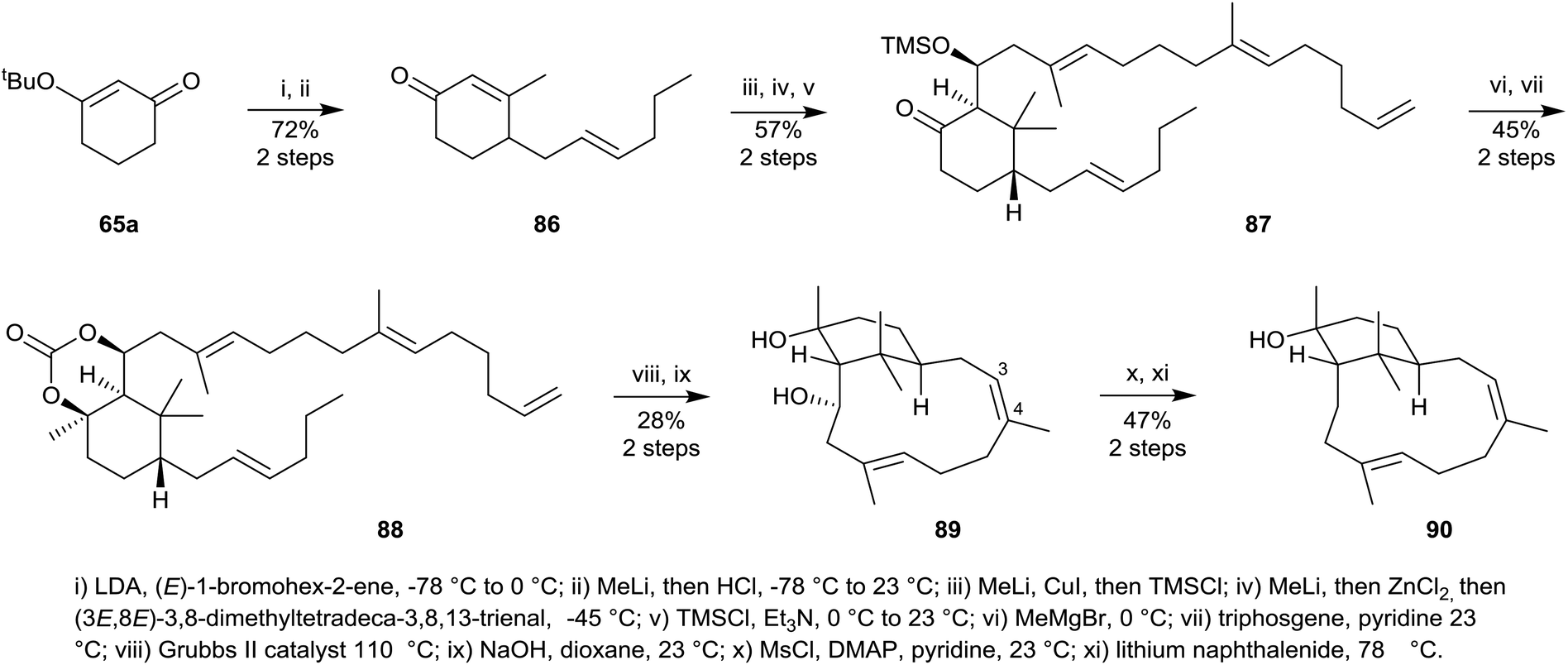 | ||
| Scheme 14 Synthesis of the incorrect atropisomer of Z-3,4-verticillol 90. | ||
9. Concluding remarks
We now have a clear insight into the origins and biosynthetic interrelationships within the taxane and phomactin families of natural products. This insight has emerged through concise studies of taxadiene synthase (TXS)-catalysed cyclisations with deuterium and fluorine-labelled GGPP and its analogues, alongside feeding experiments with 13C-labelled acetates in fermentations of Phoma sp., and detailed biomimetic interconversions with key verticillyl carbocation intermediates. These relationships, (summarised in Scheme 15), have been significantly reinforced following recent quantum chemical calculations and targeted engineering of the TXS enzyme.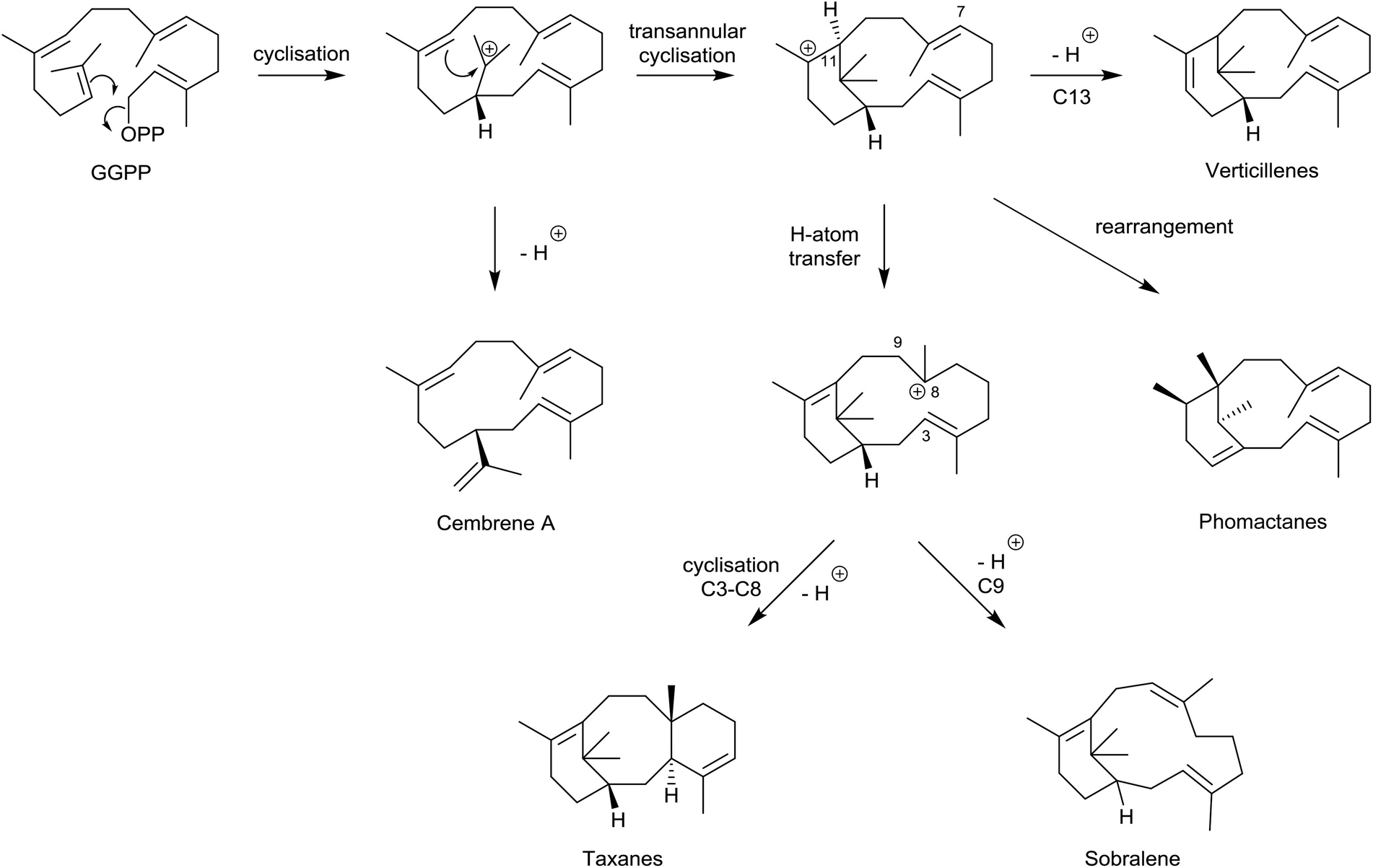 | ||
| Scheme 15 Relationships between cembrenes, phomactanes, taxanes, verticillenes and sobralene derived from GGPP. | ||
10. Conflicts of interest
There are no conflicts to declare.11. Acknowledgements
We thank the School of Chemistry at The University of Nottingham for support and for access to facilities.12. Notes and references
- H. Erdtman, T. Norin, M. Sumimoto and A. Morrison, Tetrahedron Lett., 1964, 5, 3879–3886 CrossRef.
- J. W. Harrison, R. M. Scrowston and B. Lythgoe, J. Chem. Soc. C, 1966, 1933–1945 RSC.
- See for example: P. M. Dewick, Medicinal Natural Products, A Biosynthetic Approach, J. Wiley and Sons Ltd, 3rd edn, 2009 Search PubMed.
- (a) C. B. Jackson and G. Pattenden, Tetrahedron Lett., 1985, 26, 3393–3396 CrossRef CAS; (b) M. J. Begley, C. B. Jackson and G. Pattenden, Tetrahedron, 1990, 46, 4907–4924 CrossRef CAS.
- B. Karlsson, A. M. Pilotti, A. C. Söderholm, T. Norin, S. Sundin and M. Sumimoto, Tetrahedron, 1978, 34, 2349–2354 CrossRef CAS.
- Y. Jin, D. C. Williams, R. Croteau and R. M. Coates, J. Am. Chem. Soc., 2005, 127, 7834–7842 CrossRef CAS.
- Throughout the literature authors have presented the ring systems in verticillenes and phomactins in a range of differing projections, e.g. compare the differing orientations for verticillol 1 and phomactatriene 4. We have used these differing orientations throughout this Report, largely to maximise clarity wherever possible.
- Y. Asakawa, Progress in the Chemistry of Organic Natural Products, ed. W. Herz, G. W. Kirby, R. E. Moore, W. Steglich and C. Tamm, Springer-Verlag, Vienna, 1995, vol. 65, pp. 279–280 Search PubMed.
- S. Basar, A. Koch and W. A. König, Flavour Fragrance J., 2001, 16, 315–318 CrossRef CAS.
- J. D. Hernández-Hernández, L. U. Román-Marín, C. M. Cerda-García-Rojas and P. Joseph-Nathan, J. Nat. Prod., 2005, 68, 1598–1602 CrossRef.
- F. Messina, M. Curini, C. D. Sano, C. Zadra, G. Gigliarelli, L. A. Rascón-Valenzuela, R. E. R. Zepeda and M. C. Marcotullio, J. Nat. Prod., 2015, 78, 1184–1188 CrossRef CAS.
- M. J. Palframan, K. K. Bandi, J. G. C. Hamilton and G. Pattenden, Tetrahedron Lett., 2018, 59(20), 1921–1923 CrossRef CAS.
- C. N. Spiegel, D. B. S. Dias, A. S. Araki, J. G. C. Hamilton, R. P. Brazil and T. M. Jones, Parasites Vectors, 2016, 9(580), 1–15 Search PubMed.
- Y. J. Hong and D. J. Tantillo, J. Am. Chem. Soc., 2011, 133(45), 18249–18256 CrossRef CAS.
- D. C. Williams, B. J. Carroll, Q. Jin, C. D. Rithner, S. R. Lenger, H. G. Floss, R. M. Coates, R. M. Williams and R. Croteau, Chem. Biol., 2000, 7, 969–977 CrossRef CAS ; also see ref. 6 and 36b.
- The numbering system used here for verticillene and verticillyl carbocations is the same as that used in the literature for taxadiene. Note, this numbering system differs from that used for GGPP.
- W. G. Dauben, J. P. Hubbell, P. Oberhansli and W. E. Thiessen, J. Org. Chem., 1979, 44, 669–673 CrossRef CAS.
- (a) L. Crombie, G. Kneen and G. Pattenden, Chem. Commun., 1976, 66–68 RSC; (b) L. Crombie, G. Kneen, G. Pattenden and D. Whybrow, J. Chem. Soc., Perkin Trans. 1, 1980, 1711–1777 RSC; (c) G. Pattenden and A. J. Smithies, J. Chem. Soc., Perkin Trans. 1, 1996, 57–61 RSC.
- S. Y. Chow, H. J. Williams, Q. Huang, S. Nanda and A. I. Scott, J. Org. Chem., 2005, 70(24), 9997–10003 CrossRef CAS.
- P. Schrepfer, A. Buettner, C. Goerner, M. Hertel, J. van Rijn, F. Wallrapp, W. Eisenreich, V. Sieber, R. Kourist and T. Brück, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E958–E967 CrossRef CAS.
- A. Meguro, T. Tomito, M. Nishiyama and T. Kuzuyama, Chembiochem, 2013, 14, 316–321 CrossRef CAS.
- J. Kirby, M. Nishimoto, J. G. Park, S. T. Withers, F. Nowroozi, D. Behrendt, E. J. Garcia Rutledge, J. L. Fortman, H. E. Johnson, J. V. Anderson and J. D. Keasling, Phytochemistry, 2010, 71, 1466–1473 CrossRef CAS.
- W. G. Dauben, W. E. Thiessen and P. R. Resnick, J. Org. Chem., 1965, 30, 1693–1698 CrossRef CAS.
- N. K. Kashtanova, A. I. Lisina and V. A. Pentegova, Khim. Prir. Soedin., 1968, 4, 52–53 CAS.
- I. Wahlberg and C. R. Enzell, Beikrage zur Tabakforschung International, 1984, 12(3), 93–104 CAS.
- J. P. Edwards and J. Chambers, J. Chem. Ecol., 1984, 10, 1731–1747 CrossRef CAS.
- A. J. Birch, W. V. Brown, J. E. T. Corrie and B. P. Moore, J. Chem. Soc., Perkin Trans. 1, 1972, 2653–2658 RSC.
- D. L. Mattern, W. D. Scott, C. A. McDaniel, P. J. Weldon and D. E. Graves, J. Nat. Prod., 1997, 60, 828–831 CrossRef CAS.
- D. F. Wiemer, J. Meinwald, G. D. Prestwich and I. Miura, J. Org. Chem., 1979, 44, 3950–3952 CrossRef CAS.
- Y. Uchio, H. Nabeya, M. Nakayama, S. Hayashi and T. Hase, Tetrahedron Lett., 1981, 22, 1689–1690 CrossRef CAS.
- D. J. Vanderah, N. Rutledge, F. J. Schmitz and L. S. Ciereszko, J. Org. Chem., 1978, 43, 1614–1616 CrossRef CAS.
- C.-Y. Duh, S.-K. Wang, Y.-L. Weng, M. Y. Chiang and C.-F. Dai, J. Nat. Prod., 1999, 62, 1518–1521 CrossRef CAS.
- (a) Y. Li and G. Pattenden, Nat. Prod. Rep., 2011, 28, 1269–1310 RSC; (b) Y. Li and G. Pattenden, Nat. Prod. Rep., 2011, 28, 429–440 RSC.
- (a) J. W. Harrison and B. Lythgoe, J. Chem. Soc. C, 1966, 1932–1933 RSC; (b) B. Lythgoe, Taxus Alkaloids, in The Alkaloids, Chemistry and Physiology, ed. R. H. F. Manske, Academic Press, New York, 1968, vol. 10, pp. 547–627; and earlier references cited therein Search PubMed.
- (a) M. Shiro, T. Sato, H. Koyama, Y. Maki, K. Nakanishi and S. Uyeo, Chem. Commun., 1966, 97–98 RSC; (b) M. Shiro and H. Koyama, J. Chem. Soc. B, 1971, 1342–1346 RSC; (c) N. Harada, M. Ohashi and K. Nakanishi, J. Am. Chem. Soc., 1968, 90, 7349–7351 CrossRef CAS; (d) N. Harada and K. Nakanishi, J. Am. Chem. Soc., 1969, 91, 3989–3991 CrossRef CAS.
- (a) X. Lin, M. Hezari, A. E. Koepp, H. G. Floss and R. Croteau, Biochemistry, 1996, 35, 2968–2977 CrossRef CAS; (b) Q. Jin, D. C. Williams, M. Hezari, R. Croteau and R. M. Coates, J. Org. Chem., 2005, 70, 4667–4675 CrossRef CAS ; and full list of earlier references cited therein.
- M. Köksal, Y. Jin, R. M. Coates, R. Croteau and D. W. Christianson, Nature, 2011, 469, 116–120 CrossRef.
- S. Soliman and Y. Tang, Biotechnol. Bioeng., 2015, 112, 229–235 CrossRef CAS.
- S. Edgar, F.-S. Li, K. Qiao, J. K. Weng and G. Stephanopoulos, ACS Synth. Biol., 2017, 6, 201–205 CrossRef CAS.
- (a) A. M. Escorcia, J. P. M. van Rijn, G.-J. Cheng, P. Schrepfer, T. B. Brück and W. Thiel, Comput. Chem., 2018, 39, 1215–1225 CrossRef CAS PubMed; (b) Y. Freud, T. Ansbacher and D. T. Major, ACS Catal., 2017, 7, 7653–7657 CrossRef CAS.
- M. Sugano, A. Sato, Y. Iijima, T. Oshima, K. Furuya, H. Kuwano, T. Hata and H. Hanzawa, J. Am. Chem. Soc., 1991, 113, 5463–5464 CrossRef CAS.
- (a) K. M. Foote, C. J. Hayes, M. P. John and G. Pattenden, Org. Biomol. Chem., 2003, 1, 3917–3948 RSC; (b) J. Ciesielskia and A. Frontiera, Org. Prep. Proced. Int., 2014, 46, 214–251 CrossRef; (c) W. P. D. Goldring and G. Pattenden, Acc. Chem. Res., 2006, 39, 354–361 CrossRef CAS.
- T. Tokiwano, T. Endo, T. Tsukagoshi, H. Goto, E. Fukushic and H. Oikawa, Org. Biomol. Chem., 2005, 3, 2713–2722 RSC.
- S. Y. Chow, H. J. Williams, J. D. Pennington, S. Nanda, J. Reibenspies and A. I. Scott, Tetrahedron, 2007, 63, 6204–6209 CrossRef CAS.
- Q.-W. Shi, T. Oritani and T. Sugiyama, Planta Med., 1999, 65, 356–359 CrossRef CAS.
- Q.-W. Shi, T. Oritani, T. Sugiyama and H. Kiyota, J. Nat. Prod., 1998, 61, 1437–1440 CrossRef CAS.
- W.-S. Fang, Q.-C. Fang, X.-T. Liang, Y. Lu and Q.-T. Zheng, Tetrahedron, 1995, 51, 8483–8490 CrossRef CAS.
- J. Kobayashi and H. Shigemori, Heterocycles, 1998, 47, 1111–1133 CrossRef CAS.
- L. O. Zamir, J. Zhang, K. Kutterer, F. Sauriol, O. Mamer, A. Khiat and Y. Boulanger, Tetrahedron, 1998, 54, 15845–15860 CrossRef CAS.
- Y.-F. Wang, X.-H. Su, L.-G. Li, W. Wang, M.-L. Zhang, C.-H. Huo and Q.-W. Shi, Chem. Biodiversity, 2009, 6, 1661–1673 CrossRef CAS.
- A. A. Adesomoju, J. I. Okogun, M. P. Cava and P. J. Carroll, Heterocycles, 1983, 20, 2125–2128 CrossRef CAS.
- (a) F. Nagashima, A. Tamada, N. Fujii and Y. Asakawa, Phytochemistry, 1997, 46, 1203–1208 CrossRef CAS; (b) F. Nagashima, K. Kishi, Y. Hamada, S. Takaoka and Y. Asakawa, Phytochemistry, 2005, 66, 1662–1670 CrossRef CAS.
- H. A. García-Gutiérrez, C. M. Cerda-García-Rojas, J. D. Hernández-Hernández, L. U. Román-Marín and P. Joseph-Nathan, Phytochemistry, 2008, 69, 2844–2848 CrossRef.
- (a) T. Kato, T. Hirano, M. Hoshikawa and T. Uyehara, Bull. Chem. Soc. Jpn., 1996, 69, 221–228 CrossRef CAS; (b) T. Kato, M. Hoshikawa, Y. Yoshihiro, K. Izumi, Y. Uotsu and K. Sakai, Tetrahedron, 2002, 58, 9213 CrossRef CAS.
- N. A. McGrath, C. A. Lee, H. Araki, M. Brichacek and J. T. Njardarson, Angew. Chem., Int. Ed., 2008, 47, 9450–9453 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |