
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigating the isolation and interconversion of two diastereoisomers in an octahedral 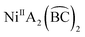 system†
system†
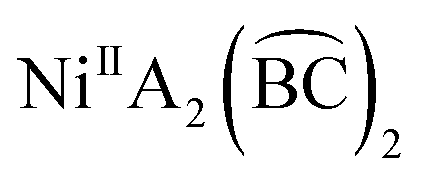
Panagiota
Danelli
a,
Sokratis T.
Tsantis
 a,
Alexandros
Kitos
a,
Vassilis
Psycharis
b,
Catherine P.
Raptopoulou
*b,
Athanassios
Tsipis
*c and
Spyros P.
Perlepes
*a
a,
Alexandros
Kitos
a,
Vassilis
Psycharis
b,
Catherine P.
Raptopoulou
*b,
Athanassios
Tsipis
*c and
Spyros P.
Perlepes
*a
aDepartment of Chemistry, University of Patras, 26504 Patras, Greece. E-mail: erlepes@patreas.upatras.gr; Tel: +30 2610 996730
bInstitute of Nanoscience and Nanotechnology, NCSR “Demokritos”, 15310 Aghia Paraskevi Attikis, Greece. E-mail: c.raptopoulou@inn.demokritos.gr; Tel: +30 210 6503346
cDepartment of Chemistry, University of Ioannina, 45110 Ioannina, Greece. E-mail: attsipis@uoi.gr; Tel: +30 26510 08333
First published on 28th October 2019
Abstract
Two diastereoisomers of bis(2-benzoylpyridine-O,N)dichloridonickel(II) have been isolated from the reactions of nickel(II) chloride hexahydrate and the organic ligand depending on the reaction solvent (ethanol, acetonitrile) used. DFT studies indicate that one of the isomers is more stable in both solvents, but nevertheless different diastereoisomers are crystallized from the two solvents. The two complexes represent the first pair of well characterized diastereoisomers in the coordination chemistry of 2-pyridyl ketones, providing a new example in the chemistry of octahedral 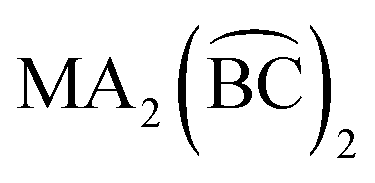 complexes (A = a monodentate ligand,
complexes (A = a monodentate ligand,  = an unsymmetrical bidentate chelate).
= an unsymmetrical bidentate chelate).
Diastereoisomeric and enantiomeric metal complexes (especially those of the d-block elements) have played a central role in the development of modern inorganic chemistry.1–5 An isomer is one of several chemical species that have the same atom composition (molecular formula), but have different constitutional formulae (atom connectivities) or different stereochemical formulae.1 Isomers exhibit different physical and/or chemical properties. If two complex molecules or ions have the same molecular formula and the same atom connectivity, but differ in the spatial arrangement of different atoms or groups about a central metal ion, then the coordination compounds are stereoisomers.1 Diastereoisomers (which are the focus of the present work) are stereoisomers that are not mirror-images of one another, whereas enantiomers are stereoisomers that are mirror-images of one another. A famous example is the diastereoisomers in the pair of the square planar complexes [PtCl2(NH3)2]; the cis isomer is a well-known antitumor drug, but the trans isomer is pharmacologically inactive.6 Diastereoisomerism continues to attract the intense interest of synthetic and physical inorganic chemists. For instance, recent examples include cis–trans interconversion in octahedral bis(2,2′-bipyridine) Ru(II) complexes;7 this results in specific synthetic applications of the trans species, potentially useful as a platform for designing highly efficient visible light-activated caged compounds. Single-crystal X-ray crystallography is the state-of-the-art method for identifying diastereoisomers in the solid state, while advanced theoretical methods can explain the preference of a given system for one diastereoisomer and predict/explain interconversions.
The introduction of chelating ligands in six-coordinate octahedral complexes usually acts to limit the number of diastereoisomers (or geometrical isomers), or at least not to extend the number.2 For example, the coordination of three symmetrical bidentate chelates  can take place in only one way, in the same sense that six identical monodentate ligands can coordinate in only one way. Likewise,
can take place in only one way, in the same sense that six identical monodentate ligands can coordinate in only one way. Likewise, 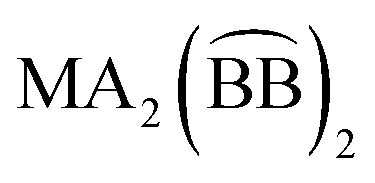 compounds (where A refers to a monodentate group) have precisely the same number and type of diastereoisomers as the all-monodentate analogue MA2X4. The same holds true for the
compounds (where A refers to a monodentate group) have precisely the same number and type of diastereoisomers as the all-monodentate analogue MA2X4. The same holds true for the  and the all-monodentate MA2X2Y2 complexes, where
and the all-monodentate MA2X2Y2 complexes, where  refers to an unsymmetrical bidentate chelate (X and Y are monodentate ligands bearing the same donor atoms that are present in the B and C parts, respectively, of
refers to an unsymmetrical bidentate chelate (X and Y are monodentate ligands bearing the same donor atoms that are present in the B and C parts, respectively, of  ); for these compounds there are now five diastereoisomers (Scheme 1). Contrary to
); for these compounds there are now five diastereoisomers (Scheme 1). Contrary to 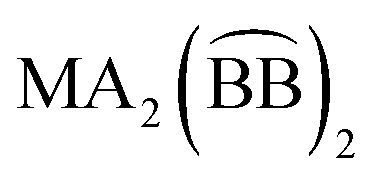 diastereoisomers,1–5 studies in
diastereoisomers,1–5 studies in  diastereoisomers are generally lacking. As a continuation of our interest in the chemistry of diastereoisomers possessing asymmetrical bidentate chelating ligands,8 we report here experimental (synthetic, structural, spectroscopic) and theoretical studies on two nickel(II) diastereoisomers that belong to the
diastereoisomers are generally lacking. As a continuation of our interest in the chemistry of diastereoisomers possessing asymmetrical bidentate chelating ligands,8 we report here experimental (synthetic, structural, spectroscopic) and theoretical studies on two nickel(II) diastereoisomers that belong to the 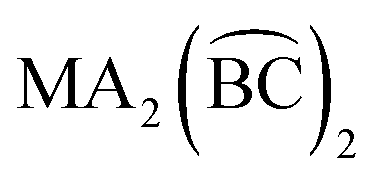 class. We selected 2-benzoylpyridine (or phenyl 2-pyridyl ketone), (py)(ph)CO, for two reasons. First, this molecule normally behaves as a neutral, O,N-bidentate chelating ligand,9,10 (exceptions are known,11–14Scheme 2) and, second, it can undergo impressive metal ion-assisted transformations,15,16 a topic that is currently one of the central themes in our research efforts.17–19
class. We selected 2-benzoylpyridine (or phenyl 2-pyridyl ketone), (py)(ph)CO, for two reasons. First, this molecule normally behaves as a neutral, O,N-bidentate chelating ligand,9,10 (exceptions are known,11–14Scheme 2) and, second, it can undergo impressive metal ion-assisted transformations,15,16 a topic that is currently one of the central themes in our research efforts.17–19
 | ||
Scheme 1 Diastereoisomers (with indication of point symmetry) possible for octahedral complexes  (in the present work M = Ni, A = Cl and (in the present work M = Ni, A = Cl and  = 2-benzoylpyridine). B (O in the present case) and C (N in the present case) denote the donor atoms of the unsymmetrical (heterotopic) bidentate chelating ligand 2-benzoylpyridine; b corresponds to complex 2 of this work and e corresponds to 1·0.4H2O·0.6EtOH. = 2-benzoylpyridine). B (O in the present case) and C (N in the present case) denote the donor atoms of the unsymmetrical (heterotopic) bidentate chelating ligand 2-benzoylpyridine; b corresponds to complex 2 of this work and e corresponds to 1·0.4H2O·0.6EtOH. | ||
 | ||
| Scheme 2 The to-date crystallographically established coordination modes of (py)(ph)CO. The ligand is neutral in I (the most common ligation mode) and II, and anionic organometallic in III. | ||
Treatment of NiCl2·6H2O with (py)(ph)CO (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) in EtOH gave an olive green solution from which were subsequently isolated dark orange-red crystals of trans,trans,trans-[NiCl2{(py)(ph)CO}2]·0.4H2O·0.6EtOH (1·0.4H2O·0.6EtOH) in moderate yields (50–55%) upon vapour diffusion with Et2O. The 1:2 reaction between NiCl2·6H2O and (py)(ph)CO (1:2) in MeCN gave a green solution from which were subsequently isolated green crystals of cis,cis,trans-[NiCl2{(py)(ph)CO}2] (2) in rather high yields (∼70%) upon liquid diffusion with Et2O. The first prefix in the formulae refers to the chlorido groups, the second to the O and the third refers to the N donor atoms (vide infra). Complex 1 is readily transformed into 2 upon dissolution in MeCN and either slow solvent evaporation or precipitation with Et2O. Solid 2 reverts to 1 in EtOH under heating and precipitation with Et2O. The IR v(C
:2) in EtOH gave an olive green solution from which were subsequently isolated dark orange-red crystals of trans,trans,trans-[NiCl2{(py)(ph)CO}2]·0.4H2O·0.6EtOH (1·0.4H2O·0.6EtOH) in moderate yields (50–55%) upon vapour diffusion with Et2O. The 1:2 reaction between NiCl2·6H2O and (py)(ph)CO (1:2) in MeCN gave a green solution from which were subsequently isolated green crystals of cis,cis,trans-[NiCl2{(py)(ph)CO}2] (2) in rather high yields (∼70%) upon liquid diffusion with Et2O. The first prefix in the formulae refers to the chlorido groups, the second to the O and the third refers to the N donor atoms (vide infra). Complex 1 is readily transformed into 2 upon dissolution in MeCN and either slow solvent evaporation or precipitation with Et2O. Solid 2 reverts to 1 in EtOH under heating and precipitation with Et2O. The IR v(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) mode appears at ∼1625 cm−1 in the spectra of the complexes (which are similar but not identical) and is shifted to lower wavenumbers by ∼45 cm−1 (compared with the free ligand) due to coordination.20
O) mode appears at ∼1625 cm−1 in the spectra of the complexes (which are similar but not identical) and is shifted to lower wavenumbers by ∼45 cm−1 (compared with the free ligand) due to coordination.20
The mononuclear octahedral complexes 1 and 2 (Fig. 1, 2 and Fig. S4, S8a, ESI†) are diastereoisomers. In the former, all the chemically similar donor atoms are in trans positions. The latter possesses a crystallographic 2-fold axis which is parallel to the a axis and passes through Ni1 bisecting the Cl1–Ni1–Cl1′ angle; the molecule is the cis,cis,trans isomer considering the positions of the chlorido ligands, the carbonyl oxygen atoms and the 2-pyridyl nitrogen atoms. In both complexes, the (py)(ph)CO molecules behave as O,N-bidentate chelating ligands (I in Scheme 2). The distortion from the regular octahedral geometry in the two complexes is primarily attributed to the somewhat small bite angles of the two 5-membered chelating rings (the N–Ni–O angles for a given ligand are in the range 77.2(1)–78.9(1)°). There is one crystallographically independent (and its symmetry equivalent) intramolecular H bond in 2 with the aromatic carbon atom C1 as donor and the chlorido group as acceptor (Fig. S8a and Table S5, ESI†). Intermolecular H-bonding interactions (1·0.4H2O·0.6EtOH), and both intermolecular H bonds and π–π stacking interactions (2) are responsible for the formation of interesting 3D architectures in the two structures (see the ESI† for full details).
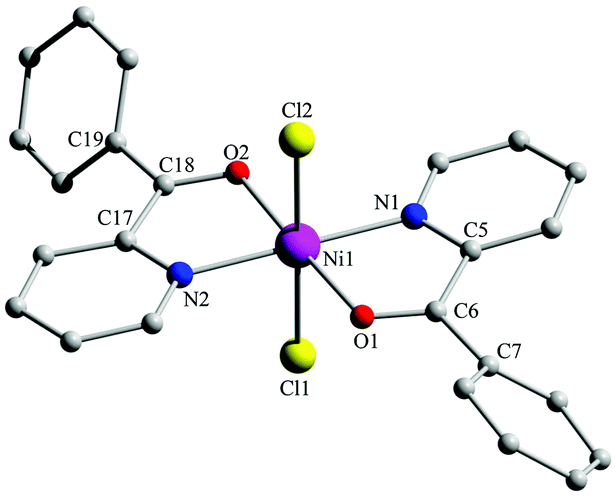 | ||
| Fig. 1 The structure of the molecule trans,trans,trans-[NiCl2{(py)(ph)CO}2] that is present in the crystal of 1·0.4H2O·0.6EtOH. Bond lengths (Å) in the coordination sphere of NiII: Ni1–Cl1 2.417(1), Ni1–Cl2 2.397(1), Ni1–O1 2.095(2), Ni1–O2 2.088(2), Ni1–N1 2.046(2), Ni–N2 2.049(3). | ||
 | ||
| Fig. 2 The structure of the molecule cis,cis,trans-[NiCl2{(py)(ph)CO}2] that is present in the crystal of 2. Bond lengths (Å) in the coordination sphere of NiII: Ni1–Cl1/Cl1′ 2.376(1), Ni1–O1/O1′ 2.160(1), Ni1–N1/N1′ 2.054(2). The N1–Ni1–N1′ angle is 169.9(1)°. Symmetry operation: (′) x, 1 − y, 3/2 − z. | ||
Complexes 1 and 2 are the third and fourth examples of structurally characterized Ni(II) complexes with (py)(ph)CO as N,O-bidentate chelating ligand.20–22 Only one of them is of the type [ ], namely [Ni(
], namely [Ni(![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif) CS)2{(py)(ph)CO}2];20,22 this compound is the cis,cis,trans diastereoisomer considering the positions of the isothiocyanato ligands, the oxygen and the 2-pyridyl nitrogen atoms, i.e. like 2. The only to-date characterized [MIICl2{(py)(ph)CO}2] (M = any divalent metal) complexes are [CuCl2{(py)(ph)CO}2],20 [CdCl2{(py)(ph)CO}2]9 and [RuCl2{(py)(ph)CO}2].23 The Cu(II) complex is trans square planar with a N-monodentate ligation of the two (py)(ph)CO molecules; the Cd(II) and Ru(II) complexes are the cis,cis,trans (like 2) and cis,cis,cis diastereoisomers, respectively. Complexes 1 and 2 represent the first authentic pair of well characterized diastereoisomers in the coordination chemistry of (py)(ph)CO.
CS)2{(py)(ph)CO}2];20,22 this compound is the cis,cis,trans diastereoisomer considering the positions of the isothiocyanato ligands, the oxygen and the 2-pyridyl nitrogen atoms, i.e. like 2. The only to-date characterized [MIICl2{(py)(ph)CO}2] (M = any divalent metal) complexes are [CuCl2{(py)(ph)CO}2],20 [CdCl2{(py)(ph)CO}2]9 and [RuCl2{(py)(ph)CO}2].23 The Cu(II) complex is trans square planar with a N-monodentate ligation of the two (py)(ph)CO molecules; the Cd(II) and Ru(II) complexes are the cis,cis,trans (like 2) and cis,cis,cis diastereoisomers, respectively. Complexes 1 and 2 represent the first authentic pair of well characterized diastereoisomers in the coordination chemistry of (py)(ph)CO.
The different spatial arrangement of the donor atoms in the two complexes is reflected in their different solid-state (diffuse reflectance) electronic spectra (Fig. S1 and S2, ESI†). The 10 Dq values, calculated directly from the wavenumber of the lowest energy 3A2g → 3T2g transition (under a simplified Oh model) are 9260 cm−1 for 1 and 9900 cm−1 for 2. The appearance of a distinct shoulder at 810 nm in the middle visible band of 1 at ∼700 nm (assigned to the 3A2g → 3T1g(F) transition) is due5 to spin–orbit coupling that mixes the 3T1g(F) and 1Eg states which are very close in energy at the 10 Dq value given by the all-trans disposition of the donor atoms, whereas in the stronger field of 2 they are so far apart that no significant mixing occurs.
We studied in detail by DFT methods (see the ESI†) the model complexes cis-[NiCl2(H2O)4] and trans-[NiCl2(H2O)4] in solution (MeCN and EtOH), from which 2 and 1, respectively, are assumed to derive. Then the equilibrium geometries of cis,cis,trans-[NiCl2{(py)(ph)CO}2] and trans,trans,trans-[NiCl2{(py)(ph)CO}2] in MeCN and EtOH solutions were calculated at the PBE0/SDD(Ni)∪6-31G(d,p)(E) level of theory; the results of the calculations are presented in Fig. 3.
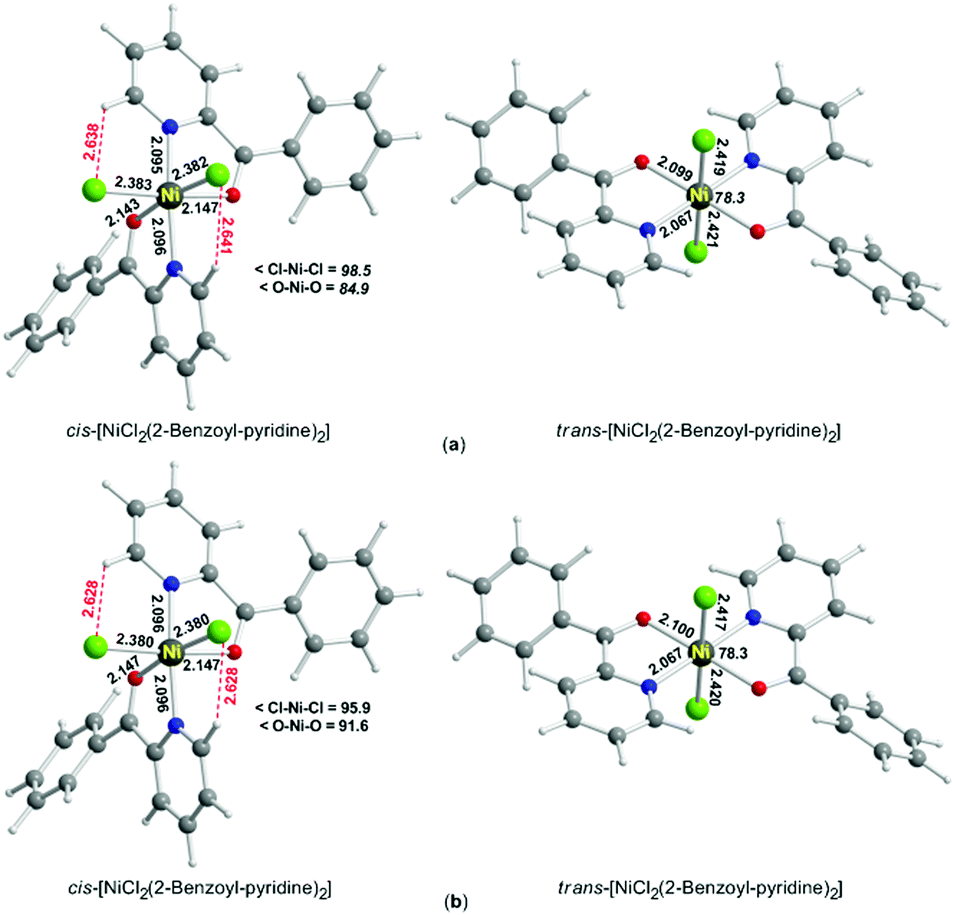 | ||
| Fig. 3 Equilibrium geometries of the molecules cis,cis,trans-[NiCl2{(py)(ph)CO}2] (2) and trans,trans,trans-[NiCl2{(py)(ph)CO}2] (1) in MeCN (a) and EtOH (b) solutions calculated at the PBE0/SDD(Ni)∪6-31G(d,p)(E) level of theory. | ||
Fig. 3 shows that the solvent has a marginal effect on the structural parameters of the two diastereoisomers. The calculated Ni–Cl, Ni–O and Ni–N bond lengths for the two diastereoisomers in solution are in good agreement with the experimental data in the solid state. The minor elongations in most of the calculated Ni–Cl, Ni–O and Ni–N bond lengths, relative to the corresponding ones in the solid-state structures, are expected on the ground that solvation effects play a role in the solution structures.
Interestingly, the cis,cis,trans-[NiCl2{(py)(ph)CO}2] diastereoisomer is more stable than the trans,trans,trans one in solutions of both MeCN and EtOH by 2.0 and 1.7 kcal mol−1, respectively. This could be due to the intramolecular C–H⋯Cl hydrogen bond formation for the cis,cis,trans molecule in solution (Fig. 3), which does not occur in the trans,trans,trans isomer; these H bonds are also observed in the solid-state structure of 2 (vide supra).
However, two distinct diastereoisomers have been crystallized. The orange-red isomer 1 is precipitated from EtOH and this can be attributed to the lattice EtOH and H2O molecules that are present in its crystal structure, and the stabilizing effect of intermolecular O(H2O)–H⋯Cl and O(EtOH)–H⋯Cl H bonds, which exist in the solid-state structure (see the ESI†). The lack of solvents in the crystal lattice of 2 results in the absence of extra intermolecular H bonds, favouring the formation of the stabilizing intramolecular C–H⋯Cl H bonds and the isolation of this isomer.
In conclusion, two of the five possible diastereoisomers of [NiCl2{(py)(ph)CO}2] have been isolated and studied. DFT calculations reveal that the cis,cis,trans isomer 2 is more stable than the trans,trans,trans isomer 1 in both MeCN and EtOH solutions; this is proven by the green colour of the reaction solutions that lead to the two complexes (vide supra) and the fact that the electronic spectra of both reaction solutions are very similar with the solid-state spectrum of 2 (the spectrum of the reaction solution that leads to 1 indicates the presence of a very small percentage of the trans,trans,trans isomer). Work is in progress in our laboratories to isolate other diastereoisomers of [NiCl2{(py)(ph)CO}2] (including the two enantiomers of the all cis diastereoisomer) and to study the effect of the halogenido ligand (Br− and I−vs. Cl−) on the diastereoisomerism of the interesting [NiX2{(ph)(py)CO}2] system.
Experimental
Syntheses and characterization
All manipulations were performed under aerobic conditions using materials (reagent grade) and solvents as received.Preparation of trans,trans,trans-[NiCl2{(py)(ph)CO}2]·0.4H2O·0.6EtOH (1·0.4H2O·0.6EtOH)
To a pale green solution of NiCl2·6H2O (0.059 g, 0.25 mmol) in EtOH (6 mL) was added solid (py)(ph)CO (0.092 g, 0.50 mmol). The solid was immediately dissolved and the colour of the solution turned to olive green. The reaction solution was stirred for a further 15 min and filtered. Vapour diffusion of Et2O into the reaction solution gave X-ray quality, dark orange-red crystals of the product after 21 d. The crystals were collected by filtration, washed with ice-cold EtOH (2 × 0.5 mL) and Et2O (3 × 2 mL), and dried in vacuo over P4O10 overnight. Typical yields were in the range 50–55%. The product was analyzed satisfactorily as lattice solvent-free, i.e.1. Anal. calcd for C24H18N2O2Cl2Ni: C, 58.11; H, 3.67; N, 5.65%. Found: C, 57.92; H, 3.73; N, 5.33%. Diagnostic IR (KBr, cm−1) band: 1624 (s). Solid-state electronic spectrum (λ/nm): 405, 696, 810 (sh), 1080.Preparation of cis,cis,trans-[NiCl2{(py)(ph)CO}2] (2)
To a pale green solution of NiCl2·6H2O (0.059 g, 0.25 mmol) in MeCN (15 mL) was added solid (py)(ph)CO (0.092 g, 0.50 mmol). The solid was soon dissolved, and the resultant green solution was stirred for a further 15 min. The reaction solution was layered with Et2O (20 mL). Slow mixing gave X-ray quality, green crystals of the product over a period of 10 d. The crystals were collected by filtration, washed with Et2O (4 × 2 mL), and dried in air. The yield was 67%. Anal. calcd for C24H18N2O2Cl2Ni: C, 58.11, H, 3.67, N, 5.65%. Found: C, 58.27; H, 3.80; N, 5.47%. Diagnostic IR (KBr, cm−1) band: 1622 (s). Solid-state electronic spectrum (λ/nm): 397, 425, 710 (sh), 1010.XRD data for 1·0.4H2O·0.6EtOH: monoclinic, P21/n, a = 10.5379(2) Å, b = 9.1919(2) Å, c = 25.3211(5) Å, β = 95.362(1)°, V = 2441.95(9) Å3, Z = 4, R1[I > 2σ(I)] = 0.0449, wR2 (all data) = 0.1441. XRD data for 2: orthorhombic, Pcan, a = 7.9540(1) Å, b = 13.9375(3) Å, c = 16.6479(4) Å, V = 2178.14(7) Å3, Z = 4, R1[I > 2σ(I)] = 0.0306, wR2 (all data) = 0.0819.
Conversion of 1 into 2
A green solution of 1 (0.099 g, 0.20 mmol) in MeCN (20 mL) was stirred overnight. The solution was layered with Et2O (25 mL). Slow mixing gave X-ray quality, green crystals in a period of one week; the unit cell of a crystal was almost identical with that of the authentic 2 prepared as described above. The crystals were collected by filtration, washed with Et2O (4 × 2 mL), and dried in air. The yield was ∼55%. Analytical data calcd for C24H18NiN2O2Cl2 (found values are in parentheses): C 58.11 (58.43), H 3.67 (3.51), N 5.65 (5.70)%. The IR and diffuse reflectance spectra of this sample are superimposable with the corresponding spectra of the authentic complex 2. Effective magnetic moment (μeff) at 22 °C: 3.36 BM. The complex can be isolated (albeit in a rather poor crystal quality and a lower yield of ∼20%) upon slow solvent evaporation of the green solution at room temperature over a period of 1 week.All DFT calculations were performed using the Gaussian program suite employing the 1999 hybrid functional of Perdew, Burke and Ernzerhof (denoted as PBE0). For Ni, the relativistic effective core potentials (RECP) were used in conjunction with the SDD basis set to describe the metal valence electrons, and two sets of f polarization functions were added; the 6-31G(d,p) basis set was used for all other main group elements, E. The method used is abbreviated as PBE0/SDD(Ni)∪6-31G(d,p)(E).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
V. P. would like to thank the Special Account of NCSR “Demokritos” for financial support concerning the operation of the X-ray facilities at INN through the internal program entitled “Structural study and characterization of crystalline materials” (NCSR Demokritos, ELKE #10813).Notes and references
- C. E. Housecroft and A. G. Sharpe, Inorganic Chemistry, Pearson, Harlow, UK, 5th edn, 2018, pp. 676–679 Search PubMed.
- G. A. Lawrance, Introduction to Coordination Chemistry, Wiley, Chichester, UK, 2010, pp. 105–114 Search PubMed.
- G. L. Miessler, P. J. Fischer and D. A. Tarr, Inorganic Chemistry, Pearson, Boston, USA, 5th edn, 2014, pp. 322–336 Search PubMed.
- J. R. Gispert, Coordination Chemistry, Wiley-VCH, Weinheim, Germany, 2008, pp. 95–124 Search PubMed.
- F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bochmann, Advanced Inorganic Chemistry, Wiley, New York, 6th edn, 1999, pp. 16–18, 839 Search PubMed.
- S. J. Lippard and J. M. Berg, Principles of Bioinorganic Chemistry, University Science Books, Mill Valley, CA, USA, 1994, pp. 16–18 Search PubMed.
- Y. Rojas Pérez, L. D. Slep and R. Etchenique, Inorg. Chem., 2019, 58, 11606–11613 CrossRef PubMed.
- C. D. Polyzou, Z. G. Lada, A. Terzis, C. P. Raptopoulou, V. Psycharis and S. P. Perlepes, Polyhedron, 2014, 79, 29–36 CrossRef CAS.
- For example, see: B. Machura, I. Nawrot, K. Michalik and Z. Drzazga, Polyhedron, 2011, 30, 2294–2302 CrossRef CAS.
- For example, see: J. M. Casas, L. R. Falvello, J. Forniés and A. Martin, Inorg. Chem., 1996, 35, 56–62 CrossRef CAS PubMed.
- J. G. Malecki and A. Maroń, Transition Met. Chem., 2011, 36, 297–305 CrossRef CAS.
- D. Kovala-Demertzi, A. Michaelides and A. Aubry, Inorg. Chim. Acta, 1992, 194, 189–194 CrossRef CAS.
- Y. Fuchita, H. Ieda, Y. Tsunemune, J. Kinoshita-Nagaoka and H. Kawano, J. Chem. Soc., Dalton Trans., 1998, 791–796 RSC.
- M.-L. Hsueh and C.-H. Yang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m269 CrossRef CAS PubMed.
- A. A. Kitos, D. P. Giannopoulos, C. Papatriantafyllopoulou, L. Cunha-Silva and S. P. Perlepes, Inorg. Chem. Commun., 2016, 64, 53–55 CrossRef CAS.
- A. A. Kitos, C. G. Efthymiou, M. J. Manos, A. J. Tasiopoulos, V. Nastopoulos, A. Escuer and S. P. Perlepes, Dalton Trans., 2016, 45, 1063–1077 RSC.
- Z. G. Lada, A. Soto Beobide, A. Savvidou, C. P. Raptopoulou, V. Psycharis, G. A. Voyiatzis, M. M. Turnbull and S. P. Perlepes, Dalton Trans., 2017, 46, 260–274 RSC.
- N. C. Anastasiadis, C. M. Granadeiro, N. Klouras, L. Cunha-Silva, C. P. Raptopoulou, V. Psycharis, V. Bekiari, S. S. Balula, A. Escuer and S. P. Perlepes, Inorg. Chem., 2013, 52, 4145–4147 CrossRef CAS PubMed.
- T. C. Stamatatos, S. P. Perlepes, C. P. Raptopoulou, A. Terzis, C. S. Patrickios, A. J. Tasiopoulos and A. K. Boudalis, Dalton Trans., 2009, 3351–3362 Search PubMed.
- J. G. Malecki, B. Machura, A. Świtlicka and J. Kusz, Polyhedron, 2011, 30, 410–416 CrossRef CAS.
- P. Chakraborty, S. Purkait, S. Mondal, A. Bouzá, A. Frontera, C. Massera and D. Das, CrystEngComm, 2015, 17, 4680–4690 RSC.
- T. Ghosh, T. Chattopadhyay, S. Das, S. Mondal, E. Suresh, E. Zangrando and D. Das, Cryst. Growth Des., 2011, 11, 3198–3205 CrossRef CAS.
- J. G. Malecki and R. Kruszynski, Polyhedron, 2007, 26, 2686–2694 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full synthetic and characterization (spectroscopic, theoretical and structural) details for the two complexes (text, figures, tables). Cifs and checkcifs for the two compounds. CCDC 1949392 and 1949393. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9nj04876f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |