
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A dry chemical method for dispersing Ir nanoparticles in the pores of activated carbon and their X-ray absorption spectroscopy analysis†
Hiroyuki
Itoi
 *a,
Takashi
Tachikawa
b,
Ryutaro
Suzuki
a,
Hideyuki
Hasegawa
a,
Hiroyuki
Iwata
c,
Yoshimi
Ohzawa
a,
Atsushi
Beniya
b and
Shougo
Higashi
*b
*a,
Takashi
Tachikawa
b,
Ryutaro
Suzuki
a,
Hideyuki
Hasegawa
a,
Hiroyuki
Iwata
c,
Yoshimi
Ohzawa
a,
Atsushi
Beniya
b and
Shougo
Higashi
*b
aDepartment of Applied Chemistry, Aichi Institute of Technology, Yachigusa 1247, Yakusa-cho, Toyota 470-0392, Japan. E-mail: itoi-hiroyuki@aitech.ac.jp
bFrontier Research-Domain, Toyota Central R&D Labs, Inc., Yokomichi 41-1, Nagakute, 480-1192, Japan. E-mail: shigashi@mosk.tytlabs.co.jp
cDepartment of Electrical and Electronics Engineering, Aichi Institute of Technology, Yachigusa 1247, Yakusa-cho, Toyota 470-0392, Japan
First published on 29th October 2019
Abstract
Ir nanoparticles are finely dispersed over whole particles of activated carbon (AC) via the gas phase adsorption of an organoiridium complex in the AC and subsequent heat treatment. Structural analysis of the supported Ir is performed in combination with X-ray absorption spectroscopy.
Metal nanoparticles have been widely studied for their use in catalysts, electronics, sensors, and many other fields. In particular, precious metals, such as platinum, iridium, and rhodium, have high catalytic activity and selectivity and have been used in industry for catalytic applications. The amount used can be easily reduced by decreasing the size of the metal nanoparticles, which increases their active surface areas per unit mass. Moreover, decreasing the particle size demonstrates an enhancement of the catalytic activity per unit surface area of the metals.1–3 The resulting synergetic effect, which is also affected by the kind of support, has been of significant interest.4
For catalyst applications, metal nanoparticles are generally deposited or grown on thermally and mechanically stable metal oxides (e.g., silica and alumina), which serve as catalyst supports. Normally, supported metal nanoparticles are prepared via a wet chemical method using relatively inexpensive inorganic metal complexes compared to organometallic complexes, whereby an inorganic metal complex is mixed with a support in the liquid phase and reduced with a reducing reagent. In addition to metal oxide supports, carbon-based materials, such as carbon black, have also been used as catalyst supports, due to their high electrical conductivity and chemical stability. As a result, fuel cell catalysts using carbon-based supports have undergone intense study to demonstrate their early practical applications.5–7 Carbon materials have versatile structures, which include fullerenes,8 carbon nanotubes,9,10 graphenes,11etc. In addition to these nanostructured carbons, activated carbon (AC) has a large surface area and a strong adsorption property, and its surface area and pore size can be precisely controlled by appropriate synthetic methods and conditions. Such nanosized pore spaces confine the materials adsorbed or synthesized in the pores, and thus can be used as a template for synthesizing polymers12–16 or nanoparticles17,18 inside them.
Here, we demonstrate the fine dispersion of Ir nanoparticles over AC using an organoiridium complex and perform their structural analysis using X-ray absorption spectroscopy. In comparison to inorganic metal complexes, organometallic complexes have relatively high vapor pressure and can be reduced just through heat treatment, without requiring reducing reagents or gases.19,20 For the preparation of the AC-supported Ir nanoparticles, we examine the gas phase adsorption of an organoiridium complex in AC to synthesize AC/organoiridium complex hybrids and their subsequent heat treatment (i.e., a dry chemical method). This method does not need any special apparatus, organic solvents, cumbersome chemical reactions, or the synthesis of nanostructured carbons via a multi-step process. Although any porous carbon can be used in this method, the superiority of this method is the availability of AC, which is commercially available at high volumes. The advantage of this method over a wet chemical method is the controllability of the accurate weight ratios of the AC and organometallic complex in the AC/organometallic complex hybrids, unless the amount of an organometallic complex exceeds the saturation amount of the AC used.15,21,22 The accurate adjustment of the weight ratio enables reproducible dispersion of Ir nanoparticles with the aid of the pore confinement of the AC, which controls the size of the Ir nanoparticles to below the pore size of the AC used. Our strategy establishes the methodology for systematically and reproducibly synthesizing metal nanoparticles without the formation of undesired large nanoparticles on porous carbons. We believe that our method facilitates the optimization of the size of nanoparticles for the synthetic catalysts and electrocatalysts.
Fig. 1 illustrates an experimental schematic of the gas phase adsorption of an Ir complex, (1,5-cyclooctadiene)-η5-indenyl iridium(I) (IrIndCOD), in activated carbon (AC) and its subsequent thermal decomposition. The AC used in this study is KOH-activated carbon with a surface area of 3160 m2 g−1 and pore sizes less than 4 nm (Fig. S1, ESI†). AC was dried under vacuum using a glass ampoule (Fig. 1a), and the amount of dried AC was determined by the weight difference between the empty ampoule and the one containing the dried AC. IrIndCOD was precisely weighed to create accurate AC/Ir weight ratios of (100 − X)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :X (X = 0.5–10), and added to the ampoule with a magnetic stir bar, followed by sealing the ampoule under vacuum. The ampoule was then heated at a ramp rate of 1 °C min−1 up to 100 °C and kept at this temperature for 24 h with stirring to adsorb IrIndCOD uniformly over the entirety of the AC particles (Fig. 1b). Because IrIndCOD crystals are larger than the pore size of AC, vaporized IrIndCOD molecules may be adsorbed in the AC pores preferentially in the vicinity of the IrIndCOD crystals unless the mixture of AC and IrIndCOD is stirred. The decomposition of adsorbed IrIndCOD in the resulting AC/IrIndCOD samples was caused by heating the ampoule at 400 °C for 3 h (Fig. 1c). The resulting samples are denoted AC/Ir (X%).
:X (X = 0.5–10), and added to the ampoule with a magnetic stir bar, followed by sealing the ampoule under vacuum. The ampoule was then heated at a ramp rate of 1 °C min−1 up to 100 °C and kept at this temperature for 24 h with stirring to adsorb IrIndCOD uniformly over the entirety of the AC particles (Fig. 1b). Because IrIndCOD crystals are larger than the pore size of AC, vaporized IrIndCOD molecules may be adsorbed in the AC pores preferentially in the vicinity of the IrIndCOD crystals unless the mixture of AC and IrIndCOD is stirred. The decomposition of adsorbed IrIndCOD in the resulting AC/IrIndCOD samples was caused by heating the ampoule at 400 °C for 3 h (Fig. 1c). The resulting samples are denoted AC/Ir (X%).
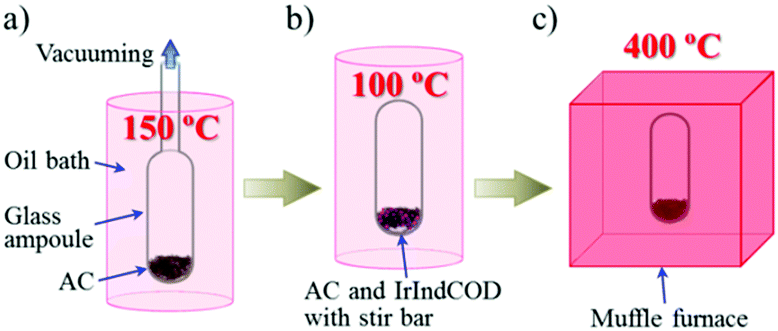 | ||
| Fig. 1 Schematic of the sample preparation. (a) Vacuum drying of AC at 150 °C. (b) Adsorption of IrIndCOD in AC at 100 °C with stirring. (c) Heat treatment at 400 °C. | ||
Fig. 2 shows the X-ray diffraction (XRD) patterns of IrIndCOD, AC, and the AC/Ir samples. In addition, the XRD pattern of AC/IrIndCOD (10%), which is the precursor of AC/Ir (10%) and has the maximum Ir content among all the AC/Ir precursors, is also plotted for comparison. As shown in the figure, AC shows a carbon (10) diffraction at 44°, but does not show a carbon (002) diffraction at ca. 26° because the carbon structure is highly activated by KOH and few stacked graphene sheets remain.23 Meanwhile, IrIndCOD shows many distinct peaks derived from its crystalline structure. Although the weight percent of IrIndCOD in AC/IrIndCOD (10%) is as high as 19 wt%, the sample does not show any distinct peaks of IrIndCOD. Instead, AC/IrIndCOD (10%) shows a broad peak at ca. 15° because IrIndCOD is adsorbed inside the nanosized pores of AC and does not form sufficiently crystalline structures in such constrained spaces to display distinct peaks;21,22i.e., IrIndCOD exists in an amorphous phase in the AC pores. It has been reported that the amorphous phases of adsorbed metallocenes (e.g., ferrocene and cobaltocene) in the constrained spaces of carbon nanotubes (CNTs) can be identified by transmittance electron microscopy (TEM) imaging.24,25 This observation was realized due to the fact that the highly crystalline structures of CNTs made it possible to distinguish the amorphous metallocenes from the highly crystalline CNT wall. If IrIndCOD is not completely adsorbed in the AC pores, the sample should show distinct peaks originating from the remaining IrIndCOD crystals on the AC particles. Therefore, the IrIndCOD in AC/IrIndCOD (10%) is finely dispersed inside the AC pores, suggesting that IrIndCOD is completely adsorbed for the other AC/Ir precursors (i.e., the AC/IrIndCOD samples). On the other hand, the AC/Ir samples do not visibly show the broad peak at 15° in their XRD patterns, suggesting the decomposition of IrIndCOD during the heat treatment at 400 °C. Nevertheless, the AC/Ir samples also do not show any distinct peaks derived from the face-centered cubic (fcc) structure of the Ir nanoparticles,26 even for AC/Ir (10%) with the highest Ir loading among all the AC/Ir samples. These results suggest that the AC/Ir samples contain small Ir nanoparticles. However, there remain undecomposed IrIndCOD structures, and the size of the Ir nanoparticles cannot be estimated from such a broad peak in their XRD patterns. These issues are discussed in the following sections.
 | ||
| Fig. 2 XRD patterns of AC, IrIndCOD, AC/IrIndCOD (10%), and the AC/Ir samples. | ||
The TEM images of AC, AC/IrIndCOD (10%), AC/Ir (0.5%), and AC/Ir (10%) are shown in Fig. 3. Since the IrIndCOD in AC/IrIndCOD (10%) is finely dispersed inside the amorphous AC pores, no difference was observed between AC and AC/IrIndCOD (10%) (Fig. 3a and b), i.e., the IrIndCOD is adsorbed as a complex without decomposition. In contrast, nanoparticles are observed in AC/Ir (0.5%) and AC/Ir (10%) (Fig. 3c and d). Although the Ir content largely differs depending on the samples, the size of the Ir nanoparticles is controlled to below ca. 4 nm due to the pore confinement effect of the AC, of which the maximum pore size is ca. 4 nm (Fig. S1, ESI†). AC/Ir (10%) contains a large amount of Ir, but we confirmed very fine dispersion of Ir nanoparticles over the whole AC particles. By considering the least Ir loading in AC/Ir (0.5%) among all the AC/Ir samples, the amount of the observed Ir nanoparticles in Fig. 3c is relatively large, because the Ir nanoparticles in AC/Ir (0.5%) exist preferentially near the particle surface of AC, which was further confirmed from the TEM and X-ray photoelectron spectroscopy analyses (for details, see the ESI†). We have previously reported the dispersion of Ni subnanoclusters on zeolite-templated carbon (ZTC) via a dry chemical method.27 ZTC is prepared using zeolite as a template and has a uniform pore size of 1.2 nm.28 However, in addition to the subnanoclusters, nanoparticles larger than the pore size of ZTC also form on the particle surface of ZTC. The undesired formation of the large nanoparticles, which hampers the rigorous study of the size-dependent performances of nanoparticles, is considered to proceed during the adsorption procedure. In the previous method, an organonickel complex was separated from ZTC in a glass ampoule during the adsorption, and decomposed on the ZTC particles before adsorption. In the present study, we modified the adsorption method by directly mixing IrIndCOD and AC with stirring and gradually increasing the temperature so that IrIndCOD can be completely adsorbed in the pores of AC without decomposition. AC/Ir (0.5%) contains the smallest amount of Ir among all the AC/Ir samples, but the maximum size of the Ir nanoparticles is almost the same as that in AC/Ir (10%). This result implies that Ir atoms resulting from the thermally decomposed IrIndCOD rapidly diffuse inside the AC pores to grow into nanoparticles during the heat treatment. However, the TEM observation and XRD analysis cannot conclude that all the adsorbed IrIndCOD molecules were completely decomposed by the heat treatment.
 | ||
| Fig. 3 TEM images of (a) AC, (b) AC/IrIndCOD (10%), (c) AC/Ir (0.5%), and (d) AC/Ir (10%). | ||
Fig. 4 shows the X-ray absorption near edge structure (XANES) spectra of the Ir L3 edge for the AC/Ir samples. The XANES spectra of Ir, IrIndCOD, and IrO2 are also shown in the figure. The spectrum shapes of the AC/Ir samples do not match with either that of individual Ir, IrO2, and IrIndCOD spectra, so it is natural to think that those obtained spectra are the mixture of the individual spectra of Ir, IrO2, and IrIndCOD. Thus, we tried to fit the obtained XANES spectra by linearly combining the individual Ir, IrO2, and IrIndCOD spectra. The spectra were successfully fitted well with the parameters as summarized in Table 1. We determined the spectrum agreement using R-factors.29–31 Note that the ratios used for the linear combination fitting are equal to the molar ratios (for details, see the ESI†). In the TEM analysis, the formation of Ir nanoparticles was confirmed, but no IrO2 was detected; however our XANES analyses revealed the existence of IrO2 in all five samples due to the oxidation of the outer surfaces of the Ir nanoparticles. The reason why clear evidence of IrO2 was absent in the XRD patterns for the AC/Ir samples is explained by the very thin IrO2 layers formed on the outer surface of the Ir nanoparticles. Since the sizes of the Ir nanoparticles were controlled below the pore sizes of AC, it was difficult to detect such very thin IrO2 layers clearly with our laboratory XRD instrument. Meanwhile, the content of IrIndCOD is low compared to those of Ir and IrO2 for all the samples, but we see a tendency for it to decrease with decreasing IrIndCOD precursor loading. A good R-factor of 0.004 in the linear fitting was achieved for the simulated spectrum of AC/Ir (0.5%) (i.e., the minimum IrIndCOD loaded sample), based on the assumption that IrIndCOD was completely decomposed, suggesting that the content of IrIndCOD was negligible (Fig. S2, ESI†). However, the results obtained from the XANES spectra revealed that the other AC/Ir samples contain undecomposed IrIndCOD.
 | ||
| Fig. 4 XANES spectra of the samples. Solid lines correspond to the fitting results. | ||
| Total Ir content (wt%) | Molar ratio | R-Factor | ||
|---|---|---|---|---|
| IrIndCOD | IrO2 | Ir | ||
| 0.5 | 0.054 ± 0.024 | 0.464 ± 0.012 | 0.482 ± 0.012 | 0.0037 |
| 2 | 0.143 ± 0.022 | 0.331 ± 0.010 | 0.527 ± 0.010 | 0.0030 |
| 3 | 0.247 ± 0.020 | 0.410 ± 0.008 | 0.343 ± 0.008 | 0.0017 |
| 5 | 0.235 ± 0.020 | 0.382 ± 0.008 | 0.383 ± 0.008 | 0.0017 |
| 10 | 0.281 ± 0.024 | 0.380 ± 0.012 | 0.338 ± 0.012 | 0.0037 |
In practical applications, the challenge is to reduce the amount of undecomposed metal precursors and maximize the nanoparticle yield with controlled sizes. There are two options for this purpose: increasing the temperature and/or the duration of the heat treatment, both of which help the decomposition of the remaining IrIndCOD. In fact, we confirmed an increase in the number of nanoparticles with sizes of ∼4 nm by simply increasing the heat treatment duration from 3 to 24 h or the heat treatment temperature from 400 to 500 °C for AC/IrIndCOD (10%) (Fig. S3 and S4, ESI†). The optimization for completely decomposing the adsorbed IrIndCOD for each sample is beyond the scope of the current study. The purpose of this study was to focus on the synthesis and structural analysis of the Ir nanoparticles below the pore size of AC by taking advantage of the pore confinement.
The remaining concern is the porosity of the AC/Ir samples necessary for the accessibility of substrates to the surface of the Ir nanoparticles within the AC pores when they are used as catalysts. The nitrogen adsorption/desorption measurement of the AC/Ir samples demonstrated that AC/Ir (10%) had a surface area of as high as 2310 m2 g−1 and micro- and mesopore volumes of 0.75 and 0.43 cm3 g−1, respectively (Table S1, ESI†). The other AC/Ir samples have larger surface areas and pore volumes than AC/Ir (10%) (for details, see the ESI†). Therefore, such large surface areas and pore volumes of the AC/Ir samples enable the access of substrates to the Ir nanoparticles when they are used as catalysts. In addition, the pore size distributions of the AC/Ir samples revealed that both micro- and mesopore volumes decreased by the Ir loading (Fig. S1, ESI†), indicating that Ir nanoparticles exist not only in the micropores but also in the mesopores, irrespective of the amount of Ir.
The proposed dry chemical method is obviously not limited to Ir metal nanoparticle synthesis and should be applicable to make other metal nanoparticles by selecting appropriate organometallic complexes and porous carbon supports. The size of the metal nanoparticles can be adjusted by using AC with appropriate pore sizes, and attention should be paid not to overlook the remaining complexes. Our XANES analyses identified the remaining metal precursor after the synthesis for high mass loading samples, indicating that XANES can be used for identifying the remaining precursors. This method would be desirable to systematically synthesize the nanoparticles without the undesirable formation of nanoparticles larger than the pore size of porous carbons and to discuss the correlation between the particle sizes and their performances as electrocatalysts, for example.
In summary, Ir nanoparticles with sizes less than the pore sizes of AC were finely dispersed over the whole AC particles via the dry chemical method. Undecomposed organometallic complexes in AC cannot be identified by TEM imaging and XRD analyses, while X-ray absorption analysis provided the information on the state of the supported Ir. This methodology can provide facile synthesis of the metal nanoparticles with sizes less than the pore sizes of porous carbons, by using appropriate organometallic complexes and porous carbons. The proposed dry chemical method is expected to be a facile method for optimizing the performances as electrocatalysts and other devices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Number 17K06034.Notes and references
- K. Judai, S. Abbet, A. S. Wörz, U. Heiz and C. R. Henry, J. Am. Chem. Soc., 2004, 126, 2732–2737 CrossRef CAS PubMed.
- A. A. Herzing, C. J. Kiely, A. F. Carley, P. Landon and G. J. Hutchings, Science, 2008, 321, 1331–1335 CrossRef CAS PubMed.
- S. Vajda, M. J. Pellin, J. P. Greeley, C. L. Marshall, L. A. Curtiss, G. A. Ballentine, J. W. Elam, S. Catillon-Mucherie, P. C. Redfern, F. Mehmood and P. Zapol, Nat. Mater., 2009, 8, 213–216 CrossRef CAS PubMed.
- A. Beniya and S. Higashi, Nat. Catal., 2019, 2, 590–602 CrossRef.
- Y. Holade, E. N. Sahin, K. Servat, W. T. Napporn and B. K. Kokoh, Catalysts, 2015, 5, 310–348 CrossRef CAS.
- F. Raimondi, G. G. Scherer, R. Kötz and A. Wokaun, Angew. Chem., Int. Ed., 2005, 44, 2190–2209 CrossRef CAS PubMed.
- B. Fang, N. K. Chaudhari, M.-S. Kim, J. H. Kim and J.-S. Yu, J. Am. Chem. Soc., 2009, 131, 15330–15338 CrossRef CAS PubMed.
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- S. Iijima and T. Ichihashi, Nature, 1993, 363, 603–605 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- D. Salinas-Torres, J. M. Sieben, D. Lozano-Castello, E. Morallón, M. Burghammer, C. Riekel and D. Cazorla-Amorós, Carbon, 2012, 50, 1051–1056 CrossRef CAS.
- D. Salinas-Torres, J. M. Sieben, D. Lozano-Castelló, D. Cazorla-Amorós and E. Morallón, Electrochim. Acta, 2013, 89, 326–333 CrossRef CAS.
- H. Itoi, S. Hayashi, H. Matsufusa and Y. Ohzawa, Chem. Commun., 2017, 53, 3201–3204 RSC.
- H. Itoi, S. Maki, T. Ninomiya, H. Hasegawa, H. Matsufusa, S. Hayashi, H. Iwata and Y. Ohzawa, Nanoscale, 2018, 10, 9760–9772 RSC.
- H. Itoi, K. Shimomura, H. Hasegawa, N. Nomura, Y. Ohta, H. Iwata, Y. Hoshikawa and Y. Ohzawa, Adv. Mater. Interfaces, 2019, 6, 1801799 CrossRef CAS.
- C. D. Saquing, T.-T. Cheng, M. Aindow and C. Erkey, J. Phys. Chem. B, 2004, 108, 7716–7722 CrossRef CAS.
- C. Galeano, J. C. Meier, V. Peinecke, H. Bongard, I. Katsounaros, A. A. Topalov, A. Lu, K. J. J. Mayrhofer and F. Schüth, J. Am. Chem. Soc., 2012, 134, 20457–20465 CrossRef CAS PubMed.
- S. Mayavan, J.-B. Sim and S.-M. Choi, J. Mater. Chem., 2012, 22, 6953–6958 RSC.
- H. Itoi, H. Nishihara, S. Kobayashi, S. Ittisanronnachai, T. Ishii, R. Berenguer, M. Ito, D. Matsumura and T. Kyotani, J. Phys. Chem. C, 2017, 121, 7892–7902 CrossRef.
- H. Itoi, Y. Yasue, K. Suda, S. Katoh, H. Hasegawa, S. Hayashi, M. Mitsuoka, H. Iwata and Y. Ohzawa, ACS Sustainable Chem. Eng., 2017, 5, 556–562 CrossRef CAS.
- H. Itoi, H. Hasegawa, H. Iwata and Y. Ohzawa, Sustainable Energy Fuels, 2018, 2, 558–565 RSC.
- H. Nishihara, H. Fujimoto, H. Itoi, K. Nomura, H. Tanaka, M. T. Miyahara, P. A. Bonnaud, R. Miura, A. Suzuki, N. Miyamoto, N. Hatakeyama, A. Miyamoto, K. Ikeda, T. Otomo and T. Kyotani, Carbon, 2018, 129, 854–862 CrossRef CAS.
- Y. F. Li, R. Hatakeyama, T. Kaneko, T. Izumida, T. Okada and T. Kato, Nanotechnology, 2006, 17, 4143–4147 CrossRef CAS PubMed.
- L.-J. Li, A. N. Khlobystov, J. G. Wiltshire, G. A. D. Briggs and R. J. Nicholas, Nat. Mater., 2005, 4, 481–485 CrossRef CAS PubMed.
- H. Kobayashi, M. Yamauchi and H. Kitagawa, J. Am. Chem. Soc., 2012, 134, 6893–6895 CrossRef CAS PubMed.
- H. Nishihara, F. Ohtake, A. Castro-Muniz, H. Itoi, M. Ito, Y. Hayasaka, J. Maruyama, J. N. Kondo, R. Osuga and T. Kyotani, J. Mater. Chem. A, 2018, 6, 12523–12531 RSC.
- H. Itoi, H. Nishihara, T. Kogure and T. Kyotani, J. Am. Chem. Soc., 2011, 133, 1165–1167 CrossRef CAS PubMed.
- S. Kelly, R. Ingalls, F. Wang, B. Ravel and D. Haskel, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 7543–7550 CrossRef CAS.
- J. W. Sobczak, E. Sobczak, A. Drelinkiewicz, M. Hasik and E. Wenda, J. Alloys Compd., 2004, 362, 162–166 CrossRef CAS.
- M. Takaoka, A. Shiono, K. Nishimura, T. Yamamoto, T. Uruga, N. Takeda, T. Tanaka, K. Oshita, T. Matsumoto and H. Harada, Environ. Sci. Technol., 2005, 39, 5878–5884 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nj04659c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |