
Interaction of 5-S-cysteinyl-dopamine with graphene oxide: an experimental and theoretical study for the detection of a Parkinson's disease biomarker†

Abstract
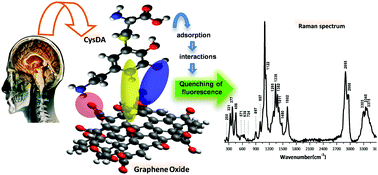
5-S-Cysteinyl-dopamine (CysDA) is a metabolite from dopamine oxidation found in patients with Parkinson's disease (PD), and has been suggested to be a biomarker for PD diagnosis. Therefore, the development of methodologies for its detection and identification is a matter of continuous interest for clinical neurology. Graphene oxide (GO) is an efficient 2D material known for its molecular adsorption capabilities, which also improves the spectroscopic signal and quenches the fluorescence of molecules adsorbed on it. For this reason, in the present work we examine the interaction of CysDA with GO. First, we demonstrate that Raman spectroscopy is a viable alternative analytical approach for the identification of CysDA, particulary due to the capacity of GO to quench the molecular fluorescence of the analyte. In order to enhance the interaction of CysDA with GO and to improve the detection of CysDA by vibrational spectroscopy, we studied the physicochemical processes involved in the adsorption of CysDA on GO through methods of Fourier transform infrared spectroscopy and quantum chemistry. These combined strategies revealed a wide variety of molecular interactions involved in the adsorption process. The stabilization of CysDA with GO is achieved primarily by hydrogen bonds and polarization effects, with a minor role of dispersion forces. In addition, we found that solvent molecules strengthens the adsorption of CysDA over GO. This work is the first reported strategy, as an alternative approach to the standard chromatographic methods, in the detection of CysDA, based on GO enhanced vibrational spectroscopy. The combination of Raman spectroscopy with graphenic substrates paves new ground for the future development of selective analytical platforms, which may ultimately enhance the detection of CysDA as a biomarker for PD.
 Please wait while we load your content...
Please wait while we load your content...

