
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRelease of reactive selenium species from phthalic selenoanhydride in the presence of hydrogen sulfide and glutathione with implications for cancer research†
Ammar
Kharma
 ab,
Anton
Misak
a,
Marian
Grman
a,
Vlasta
Brezova
c,
Lucia
Kurakova
d,
Peter
Baráth
e,
Claus
Jacob
b,
Miroslav
Chovanec
f,
Karol
Ondrias
a and
Enrique
Domínguez-Álvarez
*g
ab,
Anton
Misak
a,
Marian
Grman
a,
Vlasta
Brezova
c,
Lucia
Kurakova
d,
Peter
Baráth
e,
Claus
Jacob
b,
Miroslav
Chovanec
f,
Karol
Ondrias
a and
Enrique
Domínguez-Álvarez
*g
aInstitute of Clinical and Translational Research, Biomedical Research Center, University Science Park for Medicine, Slovak Academy of Sciences, Dúbravská cesta 9, 845 05 Bratislava, Slovak Republic
bDivision of Bioorganic Chemistry, School of Pharmacy, Saarland University, Campus B2 1, D-66123 Saarbruecken, Germany
cFaculty of Chemical and Food Technology, Slovak University of Technology, Radlinskeho 9, 812 37 Bratislava, Slovak Republic
dDepartment of Pharmacology and Toxicology, Faculty of Pharmacy, Comenius University, Ulica Odbojárov 10, 832 32 Bratislava, Slovak Republic
eInstitute of Chemistry, Slovak Academy of Sciences, Dúbravská cesta 9, 845 38 Bratislava, Slovak Republic
fCancer Research Institute, Biomedical Research Center, University Science Park for Medicine, Slovak Academy of Sciences, Dúbravská cesta 9, 845 05 Bratislava, Slovak Republic
gInstituto de Química Orgánica General, Consejo Superior de Investigaciones Científicas (IQOG, CSIC), Juan de la Cierva 3, 28006 Madrid, Spain. E-mail: e.dominguez.alvarez@csic.es
First published on 1st July 2019
Abstract
The last decade has witnessed a renewed interest in selenium (Se) as an element able to prevent a range of illnesses in humans, mainly through supplementation. However, such supplementation relies on species such as sodium selenite or selenomethionine, which proved to have limited solubility and bioavailability, thus leading to limited activity. To overcome this limitation, other selenium species need to be explored, such as phthalic selenoanhydride (R-Se), which is soluble in physiological media. R-Se releases various reactive selenium species (RSeS), including hydrogen selenide (H2Se), that can interact with cellular components, such as glutathione (GSH) and hydrogen sulfide (H2S). This interplay between R-Se and the cellular components provides a sophisticated biochemical release mechanism that could be behind the noteworthy biological activities observed for this compound. In order to investigate the interactions of phthalic chalcogen anhydrides with H2S or GSH, we have employed UV-vis spectrophotometry, electron paramagnetic resonance spectroscopy (EPR) and plasmid DNA (pDNA) cleavage assay. We found that apart from R-Se, the other analogues do not have the ability to scavenge the ˙cPTIO radical or to cleave pDNA on their own. In contrast, the scavenging potency for the ˙cPTIO radical and for the O2˙− radical exerted by R-Se and its sulfur analogue (R-S) significantly increased when they were evaluated in the presence of H2S. However, GSH only changed the radical scavenging activity of R-Se. These new discoveries may explain some of the biological activities associated with this class of compounds and open a new approach to ascertain the possible mechanisms underlying their biological actions.
Introduction
Selenium (Se) is an essential element for human health and its deficiency causes severe disorders, such as Keshan or Kashin-Beck diseases, which are endemic in farming self-sufficient regions with low levels of Se in the soil.1,2 Both Se-containing compounds and the Se atom present in the active sites of 25 mammalian selenoproteins identified so far participate in key cellular and physiological processes, as well as diseases such as cancer, inflammation, immunity, type 2 diabetes or liver diseases.3–8 However, the molecular mechanism of action is not fully understood yet for some of these selenoproteins.During recent years, Se-compounds have gained substantial interest as H2Se donors, as potential anti-cancer agents or as selenocompounds with potential use in selenium supplementation.8–11 Among other effects of Se-compounds related to cancer, they could induce apoptosis in cancer cells through the production of Reactive Oxygen Species (ROS), thus inducing oxidative stress (OS).12,13 Furthermore, certain Se-compounds can damage DNA. Then, they may not protect against cancer and other chronic diseases, and can even cause or enhance some types of cancer. These facts indicate that Se may exert a broad pattern of toxic effects.14,15 The exact mechanisms underlying the beneficial and toxic effects of Se-compounds are not fully understood yet and they are still under intensive investigation, due to the interest in the potential applications of this dual behaviour: these pro-oxidant/antioxidant Se-compounds could act as novel cellular redox modulators.
In this context, the accessibility and reactivity of the selenols (mainly deprotonated at biological pH values) and selenol-derived compounds, such as Se-methyl selenocysteine and diselenides,16–19 may be behind the reported chemopreventive activity of different Se-containing compounds, which has been reviewed extensively by numerous authors.11,20–25 Several mechanisms have been proposed to explain the chemopreventive activity, such as the direct scavenging of free radicals,26,27 the amelioration of the toxic effects of anticancer drugs,28 the glutathione peroxidase (GPx)-like activity,29,30 the protection from toxic elements such as arsenic by protecting PC12 cells from arsenic induced oxidative stress31 or the protection from radiotherapy,32 and the modulation of the intracellular redox homeostasis33 and of the protein kinases.34
In the last twenty years, H2S (H2S/HS−/S2−) has been emerging as a new gaseous signalling molecule besides nitric oxide (˙NO) and carbon monoxide (CO). H2S is produced endogenously in almost all mammalian cells and affects many physiological and pathological processes.35–37 H2S has mostly beneficial effects under conditions of oxidative stress by reacting with reactive oxygen and nitrogen species.38–42 It has both pro- and anti-cancer effects depending on the cell type, concentration and interaction with other cellular molecules.43–45 Glutathione (GSH) is another intracellular natural antioxidant, which has many biological roles including modulation of cellular redox homeostasis and protection against reactive oxygen and nitrogen species.46,47 As has been determined in previous work, sodium selenite (Na2SeO3) and selenium tetrachloride (SeCl4) have the ability to interact separately with GSH and H2S. This fact could be behind their biological effects.48 However, as mentioned above, sodium selenite can have reduced bioavailability and can also exert toxic effects.14,15 Thus, it is desirable to move towards novel Se-containing compounds that retain this observed ability to interact with relevant sulfur compounds (herein, GSH and H2S) and, at the same time, show reduced toxicity in comparison with sodium selenite. This would improve the applicability of these new selenium-based redox modulators and simultaneously opens a new approach in Se-supplementation, by finding redox-active derivatives with less toxicity.
In this context, our previous data showed promising chemopreventive, antiproliferative, cytotoxic, free radical scavenging, pro-apoptotic and multidrug-resistance (MDR) reversing activity of phthalic selenoanhydride (R-Se, Fig. 1), the Se-analogue of phthalic-anhydride.49–52 Probably, these reported biological activities are directly related to the Se atom, as no relevant biological effects were displayed by its oxygen analogue, phthalic anhydride (R-O).51 A hypothesis which may explain the amazing biological properties of R-Se draws attention to the lability of the CO–Se chemical bond.49 This lability suggests the interesting possibility that R-Se behaves in the organism as a prodrug: it enables the internalization of the compound in the cells and once inside it can release selenium slowly inside the cell, in the form of H2Se, of other uncharged Se-species such as nanoparticles of selenium or of charged Se-anions able to interact swiftly with cellular components.49,51 Due to their particular chemical affinity towards thiol-containing agents, such as hydrogen sulfide (H2S) or glutathione (GSH), and with the enzymes and proteins which are components of the cell thiolstat, such reactivity may initiate pronounced biological responses. In this way, R-Se could be a very simple, elegant and straightforward method to transport selenium inside cells and, at the same time, it would enable its release in an “activated” form as a reactive selenium species, ready to exert immediately a wide variety of biological effects. Besides, these compounds have been proven in previous studies to exert a selective action to be less toxic in non-tumour cells than in cancer cells.49,51 Thus, they could be used as safer redox modulators.
 | ||
| Fig. 1 (A) Chemical structure of the chalcogen derivatives of phthalic anhydride. X = Se → R-Se; S → R-S; O → R-O. Synthetic procedure for R-Se and R-S. (B) Hypothesized reactions that would lead to the release of hydrogen selenide (H2Se) from R-Se in physiological media. | ||
Herein, we have studied further the possible mechanism(s) underlying these initial promising biological activities of R-Se, and we will ascertain if these hypothesized interactions with H2S and GSH take place effectively. The activities of R-O and phthalic thioanhydride (R-S) were examined in parallel for comparison. Since H2S and GSH interact with several biologically active molecules, modulating their activities,42,46–48,53 the interactions of H2S and GSH with these three phthalic anhydride derivatives and their molecular consequences were also analysed, expecting that in these instances R-Se would be the most reactive one thanks to its higher expected reactivity. The reduction of the ˙cPTIO and superoxide (O2˙−) radicals, and plasmid DNA (pDNA) cleavage assay were employed to monitor these consequences. We show that only R-Se displays any significant biological activity on its own. This activity was augmented considerably when tested in combination with H2S or GSH. In contrast, GSH showed no impact on the activities of R-S or of R-O, whilst H2S was efficient to some extent in this context. Hence, the products of the H2S or GSH interaction with R-Se and R-S show free radical scavenging properties and appear to cleave pDNA, potentially explaining some of their biological activities.
Results and discussion
To unveil the possible mechanisms underlying the reported activities of R-Se against cancer, we have designed different experiments with R-Se, R-S and R-O, such as mass spectrometry (ESI-MS), spectrophotometrically-monitored radical scavenging and electron paramagnetic resonance (EPR), in an attempt to ascertain how this promising Se-containing compound can interact with different cellular redox targets.Chemistry
In the present work, we have evaluated three chalcogen phthalic anhydrides, as shown in Fig. 1: phthalic selenoanhydride (X = Se → R-Se), phthalic thioanhydride (X = S → R-S) and phthalic anhydride (X = O → R-O), as well as phthalic acid (R-OH). R-O and R-OH were commercially available, whereas R-Se and R-S were synthesized following an adaption of the procedure previously described for the synthesis of R-Se (Fig. 1).49In brief, elemental selenium or sulfur is reduced with lithium aluminium hydride, and the in situ formed hydrogen chalcogenide attacks the phthaloyl chloride to form a reactive intermediate. Sulphuric acid is added to form the desired final R-Se or R-S. Compounds were isolated in the form of stable solids, whose purity was assessed through NMR and LC-MS.
ESI-MS
R-Se showed a complex pattern of fragmentation (Fig. 2) in the ESI-MS (electrospray ionization mass spectrometry) spectrum taken in a 50% methanol/H2O solution (Fig. 3). The possible fragments corresponding to the main peaks observed in ESI are suggested in Fig. 2. | ||
| Fig. 2 Hypothesized fragmentation pattern for protonated R-Se in ESI-MS. | ||
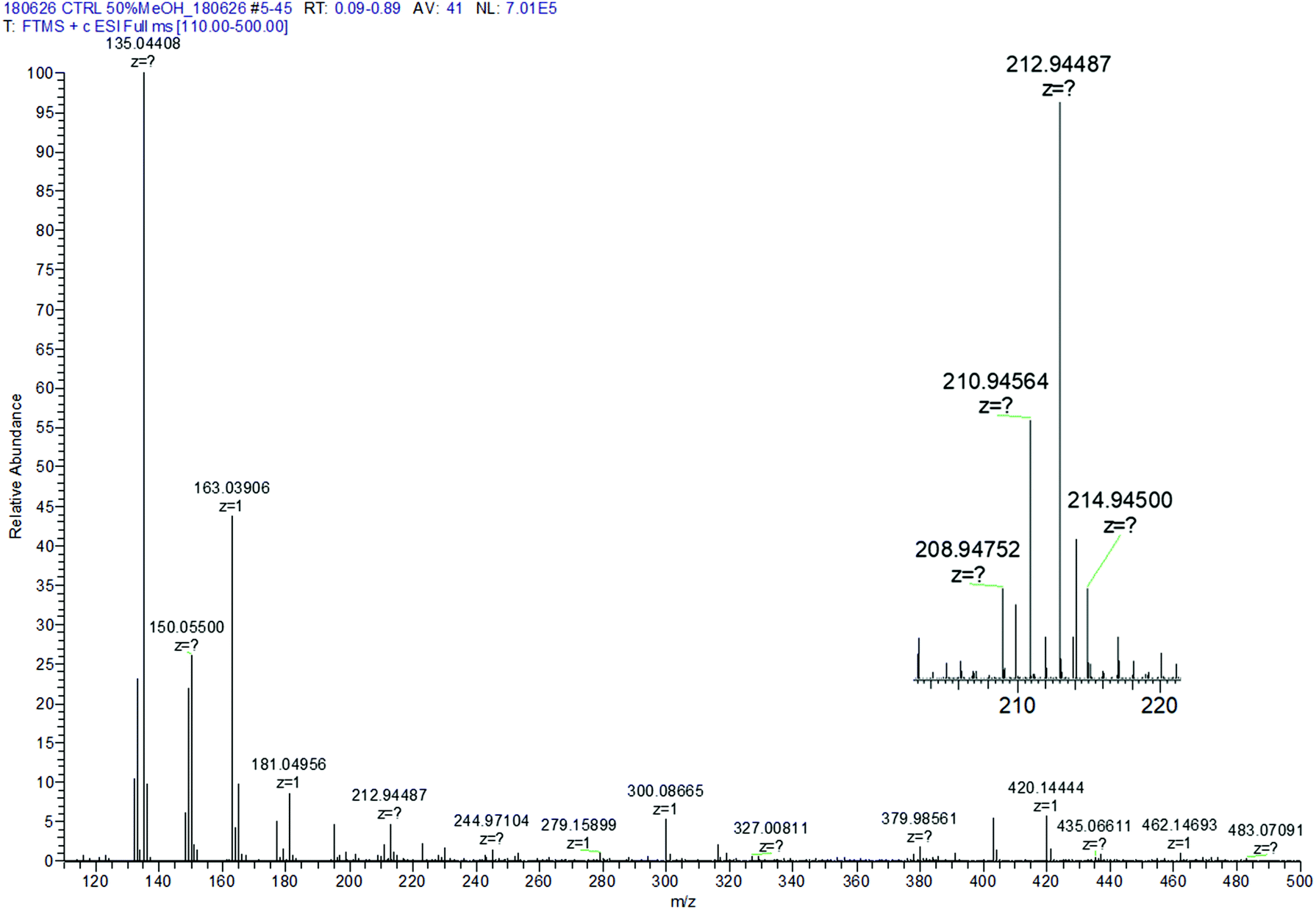 | ||
| Fig. 3 Control R-Se ESI spectrum in 50% methanol/H2O. Inset: Zoom of the protonated molecular peak showing the characteristic isotopic pattern of Se. | ||
The M+H molecular peak of R-Se is easily recognisable thanks to the characteristic isotopic pattern of the Se-containing fragments, and the remaining peaks of low m/z can be assigned to specific fragments (Fig. 2). Briefly, the protonated molecular peak (m/z = 212.94487) is attacked by methanol to generate the fragment with m/z = 244.97104 (also with the characteristic isotopic pattern of Se). This fragment loses a molecule of hydrogen selenide, leading to m/z = 163.03906, which can suffer additional fragmentation (release of CO, m/z = 135.04408) or can incorporate a molecule of water to later generate protonated R-O (m/z = 181.04956). The majority of these peaks can be seen when R-Se is analysed together with Na2S (Fig. 4). The remaining peaks, especially those with m/z > 245, are the result of complex couplings with other R-Se molecules/fragments, and with water or with the solvent (methanol). The low abundance of the protonated molecular peak in the ESI-MS spectrum may be an indicator of the readiness of the R-Se compound to react with different compounds, and this reactivity can explain the biological activities found so far for this bioactive compound.
 | ||
| Fig. 4 Experimental ESI-MS spectrum of R-Se + Na2S in 50% methanol/H2O. Inset: Structures suggested to explain the differential peaks with respect to the R-Se ESI-MS spectrum. | ||
When Na2S is added to the R-Se solution in 50% methanol/H2O (Fig. 4), the protonated molecular peak of R-Se (m/z = 212.94487) disappears, although m/z = 244.97110 (the result of methanol addition to this peak and also with the Se isotopic pattern) can be observed, but with a significant lower abundance than in the spectrum of R-Se alone. It is also possible to observe the peaks of lower m/z that were hypothesized above in Fig. 2: m/z = 135.04414, m/z = 149.02, m/z = 163.03908 and m/z = 181.04958. Besides, new peaks appear. The most relevant of them is m/z = 197.02664, which is the equivalent of m/z = 244.97110 but replacing the Se atom by sulfur (Fig. 4, inset). This peak can be formed through the coupling of H2S (generated in situ from Na2S) with m/z = 163.03908. Finally, two peaks (m/z = 266.95303 and m/z = 219.00859) with a difference of 48 Da are found, a difference that can be attributed to a Se–S change, taking into account that the first peak presents the characteristic isotopic pattern of Se. Tentative structures that could explain these two peaks are the polyhydroxy-containing compounds drawn in the Fig. 4 inset. In any case, the absence of the R-Se protonated molecular peak and the two sulfur-containing peaks (m/z = 197.02664 and m/z = 219.00859) together is indicative of an interaction between R-Se and Na2S.
According to the data obtained, ESI-MS experiments show how this Se-compound, in the electrospray ionization conditions, suffers specific fragmentation (Fig. 2) and how it forms complex couplings with the solvent and other R-Se molecules/fragments (Fig. 3), indicating that this compound has high reactivity. This fact could be indicative of potential interactions of R-Se with reactive species present in the cell, such as ROS, Reactive Nitrogen Species (RNS) and the sulfur compounds of the redox thiolstat. To prove this hypothetical interaction between R-Se and sulfur species present in cells such as H2S, we also applied the ESI-MS methodology to a mixture of R-Se and Na2S (Fig. 4). In this second ESI-MS spectrum, it is observed how the peaks of the initial R-Se are practically irrelevant, whereas its fragment peaks are the main peaks. Additionally, new peaks related to coupling of its fragments with the added sulfur atoms start to be observed, proving also the reaction between the two chalcogen compounds.
Results of the reduction of ˙cPTIO by phthalic-anhydride derivatives and H2S
Since H2S is endogenously produced in living organisms, we studied the interaction of H2S with R-Se. We observed that H2S interacts with R-Se. Since H2S and Se derivatives were reported to interact with radicals,54 it was of high interest to reveal whether the products of the H2S/R-Se reaction interact with radicals and how this interaction is unique in comparison to other phthalic-anhydride derivatives. Therefore, we studied the potency of H2S, R-Se, R-S, R-O and R-OH and the interaction products of H2S/R-Se, H2S/R-S, H2S/R-O and H2S/R-OH to reduce the ˙cPTIO radical. | ||
| Fig. 5 Reduction of ˙cPTIO by H2S, R-Se, R-S, RO and the R-Se/H2S mixture. (A) Time resolved UV-vis spectra of 100 μM ˙cPTIO after addition of 100 μM R-S. Spectra were recorded every 30 s for 20 min. The first spectrum was recorded 15 s after R-S addition. Arrows indicate the decrease of ABS at 356 and 560 nm. Inset: Kinetics of changes in absorbance at 560 nm of 100 μM ˙cPTIO after addition (indicated by an arrow) of: 100 μM H2S (black); 50 and 100 μM R-Se (red and pink); 50 and 100 μM R-S (blue and cyan); 50 and 100 μM R-O (green and dark green) and a mixture of 100 μM H2S with 100 μM R-O (dark yellow). Means ± SEM, n = 2–4. (B) Time resolved UV-vis spectra of the interaction of 100 μM ˙cPTIO with 100 μM H2S (3 times repeated every 30 s, black) and subsequent addition of 12.5 μM R-Se. Spectra were recorded every 30 s for 20 min, the first spectrum, indicated by the red line, was measured 15 s after addition of R-Se. Inset: Details of the time resolved spectra of the ˙cPTIO/H2S (100/100 μM/μM) interaction before (black) and after addition of R-Se (12.5 μM, the first spectrum is indicated by the solid red line, which is followed each 30 s by: long dashed red, medium dashed red, short dashed red, dotted red, solid blue, long dashed blue, medium dashed blue, etc.). | ||
Notably, 6.25, 12.5 and 25 μM of R-Se in the presence of 100 μM H2S had two time-dependent phases of decreasing the concentration of 100 μM ˙cPTIO. The first was a fast decrease (in ≤2 min) followed by a second gradual decrease (Fig. 6A). On the other hand, 6.25, 12.5 and 25 μM of R-S decreased ˙cPTIO in the first phase slower than what was observed when R-Se was employed (≤5 min), but later the ˙cPTIO concentration did not decrease significantly (Fig. 6C). This indicates that the molecular mechanism of ˙cPTIO reduction by H2S/R-Se and H2S/R-S is different. The ˙cPTIO reduction potency of the H2S/R-Se mixture was several fold higher than (≥5×) that of H2S/R-S (Fig. 6E). The reduction of ˙cPTIO strongly depended on the H2S/R-Se molar ratio: it was low at 0.5 and 1 H2S/R-Se molar ratios but increased significantly at 2 and 4 molar ratios (Fig. 6B and F). On the other hand, the reduction of ˙cPTIO gradually increased with the H2S/R-Se molar ratio (Fig. 6D and F). The results show that H2S interacting with R-Se and R-S (but not with R-O or R-OH) forms reactive products which reduce the ˙cPTIO radical. The order of potency is as follows: H2S/R-Se > H2S/R-S ≫ H2S/R-O ∼ R-OH = 0.
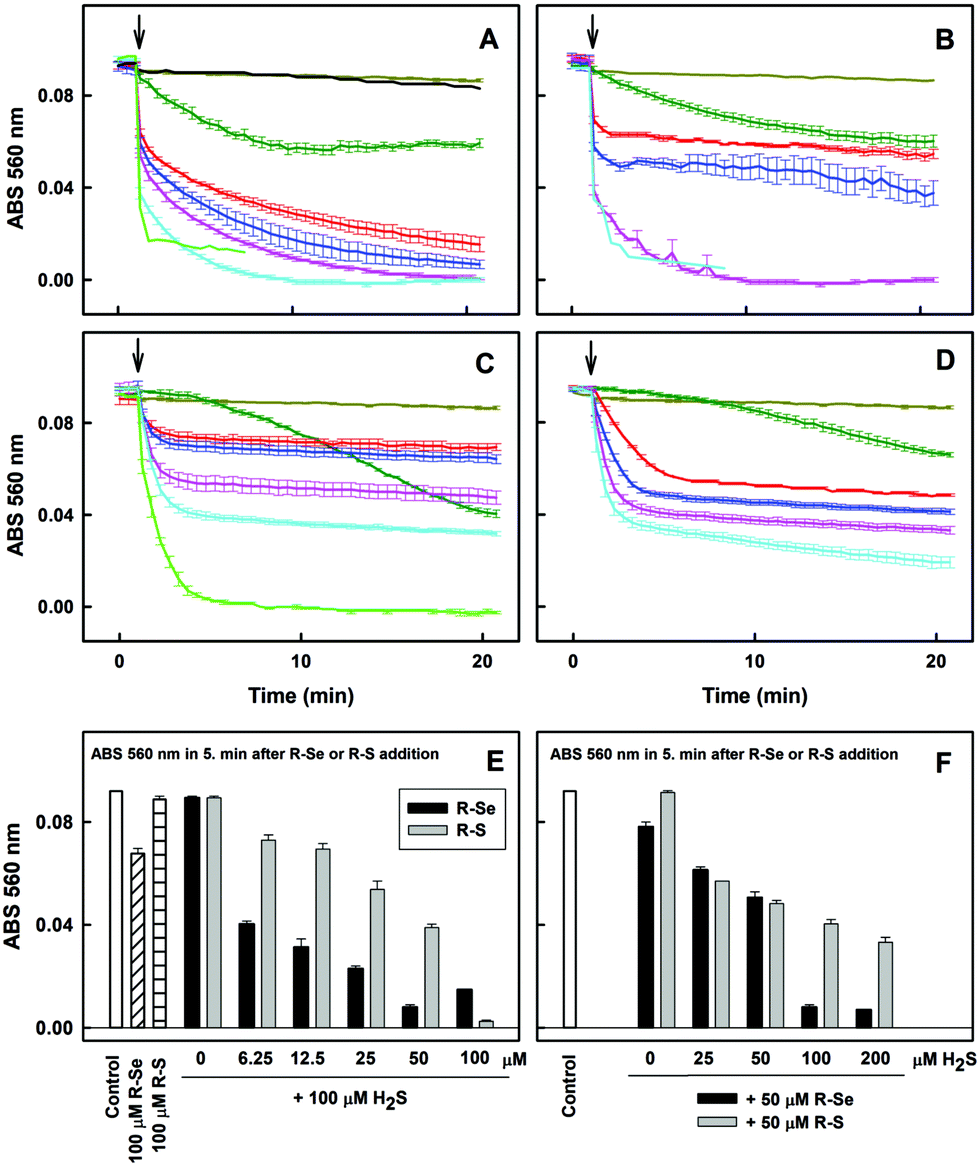 | ||
| Fig. 6 The effect of R-Se and R-S on the kinetics of ˙cPTIO reduction in the absence and presence of H2S. (A) The effect of R-Se and R-OH on the time-dependent reduction of ˙cPTIO/H2S. Kinetics of changes in absorbance at 560 nm of 100 μM ˙cPTIO after addition (indicated by an arrow) of 100 μM R-Se alone (dark green) and compared to the addition of 0 (dark yellow), 6.25 (red), 12.5 (blue), 25 (pink), 50 (cyan) and 100 μM (green) R-Se to ˙cPTIO/H2S (100/100 μM/μM). 50 μM R-OH added to ˙cPTIO/H2S (black; 100/100 μM/μM). Data were collected from UV-vis spectra every 30 s for 20 min. (B) The effect of H2S on the kinetics of reduction of ˙cPTIO in the presence of R-Se. Kinetics of changes in absorbance at 560 nm of 100 μM ˙cPTIO after addition (indicated by an arrow) of 100 μM H2S alone (dark yellow) and after addition of 50 μM R-Se to 0 μM (dark green), 25 μM (red), 50 μM (blue), 100 μM (pink) and 200 μM H2S (cyan). (C) The effect of R-S on the kinetics of ˙cPTIO reduction in the presence of H2S. Kinetics of changes in absorbance at 560 nm of 100 μM ˙cPTIO after addition (indicated by an arrow) of 100 μM R-S alone (dark green) and compared to the addition of 0 (dark yellow), 6.25 (red), 12.5 (blue), 25 (pink), 50 (cyan) and 100 μM (green) R-S to ˙cPTIO/H2S (100/100 μM/μM). (D) The effect of H2S on the kinetics of ˙cPTIO reduction in the presence of R-S. Kinetics of changes in absorbance at 560 nm of 100 μM ˙cPTIO after addition (indicated by an arrow) of 100 μM H2S alone (dark yellow) and after addition of 50 μM R-S to 0 μM (dark green), 25 μM H2S (red), 50 μM (blue), 100 μM (pink) and 200 μM H2S (cyan). (E) Comparison of the potency of R-Se and R-S to reduce ˙cPTIO in the presence of H2S. Reduction of ˙cPTIO (100 μM) by R-Se and R-S (100 μM) and reduction of ˙cPTIO in the presence of H2S (100 μM) after addition of 0, 6.12, 12.5, 25, 50 and 100 μM R-Se or R-S. The changes in absorbance at 560 nm of 100 μM ˙cPTIO were taken from (A and C) at the 5th min after the addition of the compound. (F) Comparison of the potency of R-Se and R-S to reduce ˙cPTIO in the presence of different concentrations of H2S. Reduction of ˙cPTIO (100 μM) by R-Se and R-S (50 μM) in the presence of 0, 25, 50, 100 and 200 μM H2S. The changes in absorbance at 560 nm of 100 μM ˙cPTIO were taken from (B and D) at the 5th min after the addition of the compound. Mean ± SEM, n = 2–4. | ||
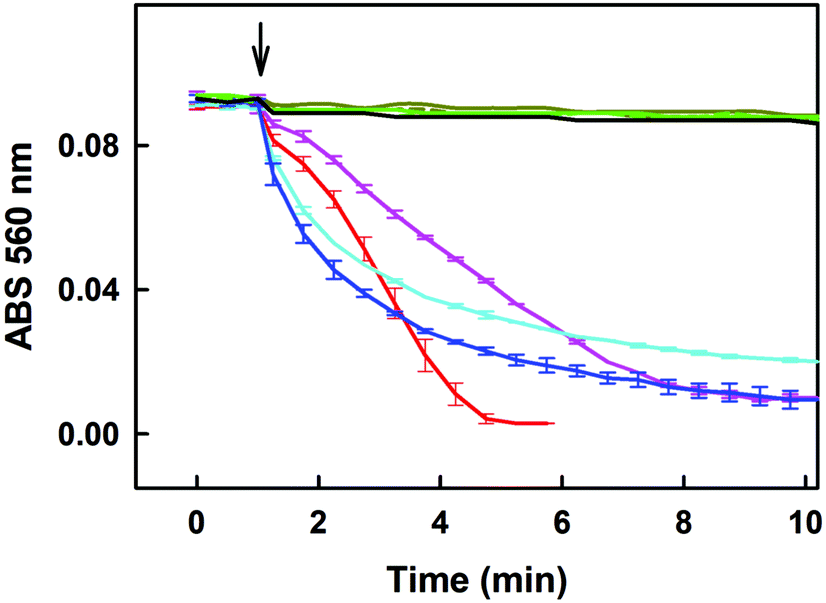 | ||
| Fig. 7 Effect of GSH on the ˙cPTIO reduction kinetics in the presence of R-Se, R-S, R-O and R-OH. Kinetics of the changes in absorbance at 560 nm of 100 μM ˙cPTIO after addition (indicated by an arrow) of 100 (dashed dark-yellow) and 500 μM GSH (solid dark yellow); after addition of 50 μM R-Se to ˙cPTIO/GSH (100/200 μM/μM; pink), and to ˙cPTIO/GSH (100/500 μM/μM; red); after addition of 50 μM R-S to ˙cPTIO/GSH (100/200 μM/μM; cyan), and to ˙cPTIO/GSH (100/500 μM/μM; blue); after addition of 50 μM R-O to ˙cPTIO/GSH (100/200 μM/μM; dashed green), and to ˙cPTIO/GSH (100/500 μM/μM; solid green); and after addition of 50 μM R-OH to ˙cPTIO/GSH (100/500 μM/μM; black). Mean ± SEM, n = 2–3. | ||
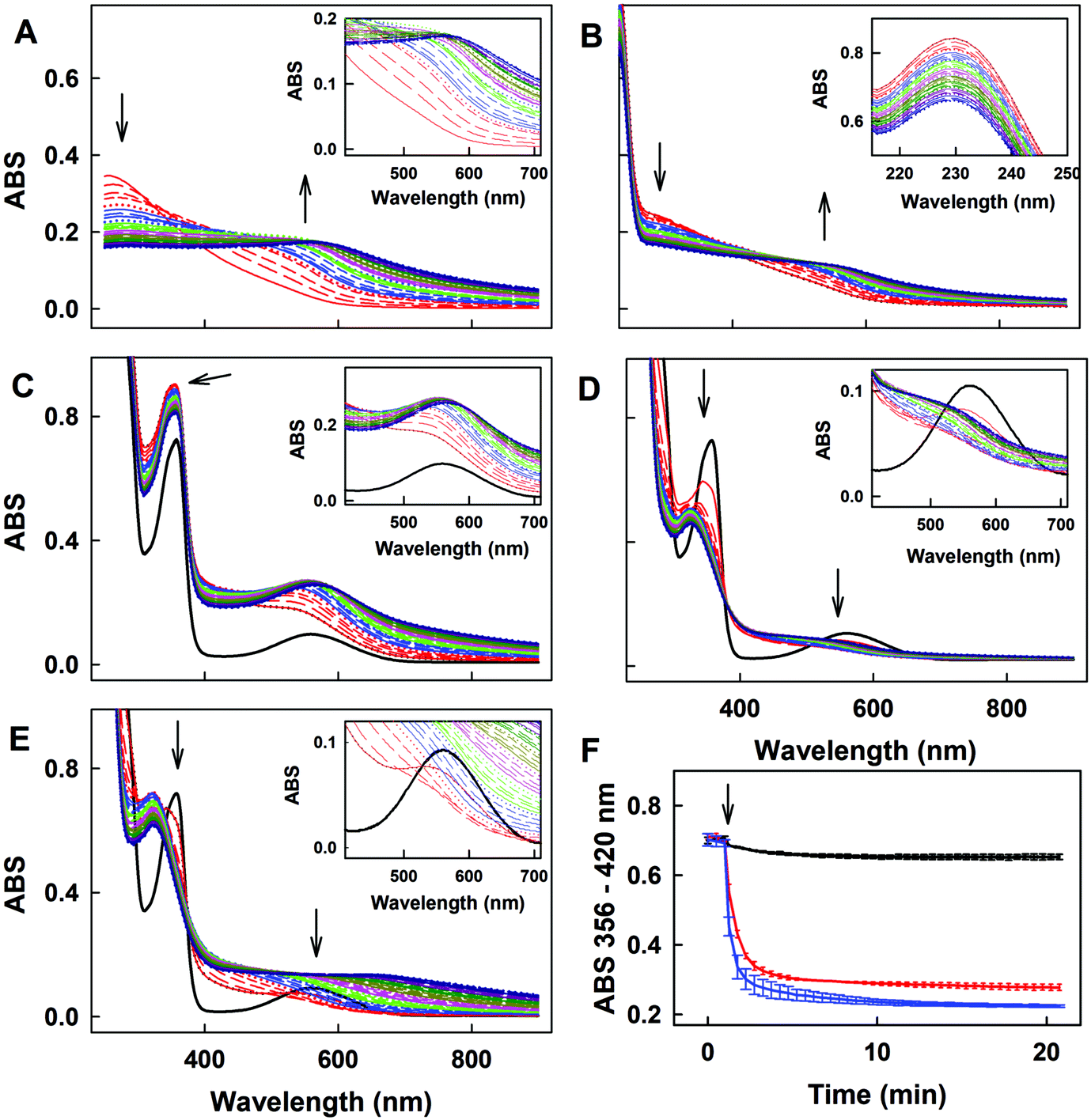 | ||
| Fig. 8 UV-vis spectra of the interaction of Na2Se with ˙cPTIO/H2S and ˙cPTIO/GSH. (A) Time resolved UV-vis spectra of 100 μM Na2Se in 100 mM sodium phosphate, 100 μM DTPA, pH 7.4 buffer at 37 °C. Spectra were collected every 30 s for 20 min, the first spectrum was measured 15 s after addition of Na2Se. Insets are details. The first spectrum is indicated by the solid red line, which is followed each 30 s by: long dashed red, medium dashed red, short dashed red, dotted red, solid blue, long dashed blue, medium dashed blue, etc. (B) Time resolved UV-vis spectra of the interaction of 100 μM Na2Se with 100 μM H2S. Spectra were collected every 30 s for 20 min, the first spectrum, indicated by the solid red line, was measured 15 s after addition of Na2Se to H2S. Inset: Details of the time resolved spectra of HS−, peak at 230 nm. (C) Time resolved UV-vis spectra of the interaction of 100 μM ˙cPTIO with 100 μM Na2Se (˙cPTIO – 3 times repeated every 30 s, black) and subsequent addition of 100 μM Na2Se. Spectra were collected every 30 s for 20 min, the first spectrum, indicated by the solid red line, was measured 15 s after addition of Na2Se. The arrow indicates ABS at 356 nm. Inset: Details of the time resolved spectra of the ˙cPTIO/Na2Se (100/100 μM/μM) interaction before (black) and after addition of Na2Se (100 μM). (D) Time resolved UV-vis spectra of the interaction of Na2Se with ˙cPTIO/H2S. Control ˙cPTIO/H2S (100/100 μM/μM; 3 times repeated every 30 s, black), and subsequent addition of 100 μM Na2Se. Spectra were collected every 30 s for 20 min, the first spectrum, indicated by the red line, was measured 15 s after addition of Na2Se. The arrows indicate the decrease of ABS at 356 and 560 nm. Inset: Details of the time resolved spectra of the ˙cPTIO/H2S (100/100 μM/μM) interaction before (black) and after addition of Na2Se (100 μM). (E) Time resolved UV-vis spectra of the interaction of Na2Se with ˙cPTIO/GSH. Control ˙cPTIO/GSH (100/500 μM/μM; 3 times repeated every 30 s, black), and subsequent addition of 100 μM Na2Se. Spectra were collected every 30 s for 20 min, the first spectrum, indicated by the solid red line, was measured 15 s after addition of Na2Se. The arrows indicate the decrease of ABS at 356 and 560 nm. Inset: Details of the time resolved spectra of the ˙cPTIO/GSH (100/500 μM/μM) interaction before (black) and after addition of Na2Se (100 μM). (F) Kinetics of the interaction of Na2Se (100 μM, marked by an arrow) with ˙cPTIO (100 μM, black), ˙cPTIO/H2S (100/100 μM/μM, blue) and ˙cPTIO/GSH (100/500 μM/μM, red) monitored as changes of ABS at 356 nm with correction to 420 nm; mean ± SEM, n = 2–3. | ||
The time resolved UV-vis spectra of ˙cPTIO/Na2Se (100/100 μM/μM) show no ˙cPTIO reduction by Na2Se alone, since ABS at 356 nm (marked by an arrow) did not decrease over the time (Fig. 8C and F), nor ABS at 560 nm (Fig. 8C, inset). H2S (100 μM) or GSH (500 μM) had only minor effects (≤7%) on their own in terms of ˙cPTIO (100 μM) reduction within 20 min. However, addition of freshly prepared Na2Se (100 μM) to the H2S/˙cPTIO (100/100 μM/μM) or GSH/˙cPTIO (500/100 μM/μM) mixture reduced ˙cPTIO (decreased ABS at 356 and 560 nm) in <1 min (Fig. 8D–F), indicating formation of reducing species during the interaction of Na2Se-derivatives with H2S and GSH which reduce the ˙cPTIO radical. The results support the suggestion that H2S and GSH significantly potentiated the reducing properties of Se derivatives which are released from R-Se.
Discussion of the reduction of ˙cPTIO by phthalic-anhydride derivatives and H2S
Regarding the reduction of the ˙cPTIO radical, R-S and R-Se showed a higher capacity to reduce this radical in comparison with H2S. But H2S was most effective in the reduction of the ˙cPTIO free radical compared to phthalic acid and phthalic anhydride. This fact suggests that the mechanism that explains this reduction must be related to a characteristic reaction of R-S and R-Se that is not demonstrated by R-O or by R-OH. A potential candidate for the reaction that led to this observation could be then the release of sulfide or selenide anions, respectively, as the release of O2− is non-existent. All sulfide or selenide anions would probably be partially protonated in buffered solution. H2S is produced endogenously and exerts relevant biological effects and functions. It can be found in tissue cells in non-negligible physiological concentrations that reach even higher than 1 μM.55 Besides, its local space–time concentration in microenvironments can be even several times higher. This fact indicates that the H2S/R-Se interaction may be involved also in the biological activities of R-Se. Thus, we have evaluated how it interacts with these chalcogen phthalic derivatives. The results were in line with the previous observations and supported our hypothesis of the S2− and Se2− release: the addition of H2S to R-S and R-Se potentiated the above mentioned reduction of the ˙cPTIO radical. It is noteworthy that the addition of H2S promoted the reduction exerted by R-Se more than the one induced by R-S. In contrast, the addition of H2S to R-O and R-OH did not exert any effect. Interestingly, the interaction of H2S with R-Se is dependent on the molar ratio between the Se analogue and H2S: when R-Se is predominant or when both are equimolar, a low reduction is observed. However, when the molar ratio H2S/R-Se is 2 to 4, the detected reduction increased significantly. This fact could suggest that a reaction between H2S and R-Se takes place and that the concentration of the first potentiates this reaction, although the R-Se is also crucial as its replacement by R-S reduced the observed potentiation effect.Another sulfur-containing biogenic compound with crucial functions is GSH, which can reach intracellular concentrations in the range 0.5–10 mM. Thus, we have also evaluated its interaction with the phthalic anhydride derivatives, as this interaction may be involved in the R-Se biological effects. And effectively similar results of potentiation of the R-Se and R-S ability to reduce the ˙cPTIO radical were obtained, being then again more significant for R-Se than for R-S. The phthalic anhydride derivatives could interact also with other components of the cellular thiolstat, or even with other different enzymes. For example, they could be activated through different cellular enzymes, such as disulfide reductases and esterases. However, further research needs to be conducted in future work to confirm this extent. In this study we have selected GSH and H2S as representative compounds of the sulfur containing components involved in the redox thiolstat. The enhancement of the R-Se activity with respect to the potentiation observed for R-S by the two thiols evaluated (H2S and GSH) was observed and therefore we tested how H2S and GSH reduced the ˙cPTIO radical in the absence of R-Se, finding that in this case there was no interaction. This confirms the key role of R-Se in this interaction, and the requirement of having both Se and S species to have a more effective interaction.
Cleavage of pDNA
We wanted to ascertain whether the products of the H2S/R-Se and/or GSH/R-Se interaction can directly attack pDNA without the contribution of other (unknown) biologically important molecules and/or pathways. Briefly, the pDNA cleavage assay can detect any activity that attacks and disrupts the sugar-phosphate backbone of DNA (e.g., reactive oxygen species, free radicals etc.).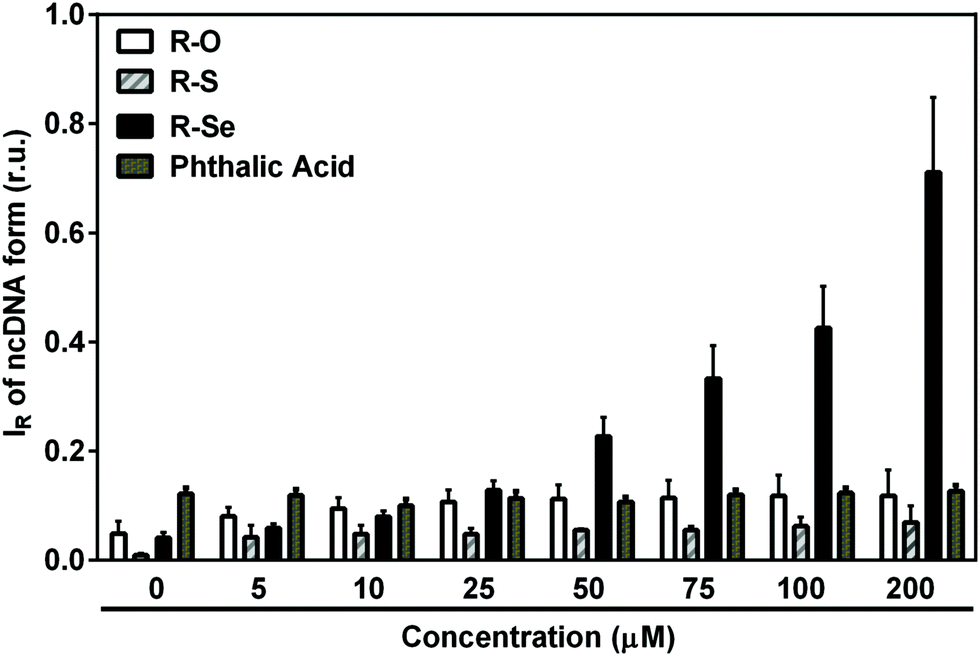 | ||
| Fig. 9 The effect of anhydride-derivatives on pDNA integrity. Increasing concentrations of R-Se, R-S, R-O or phthalic acid were incubated with pDNA for 30 min at 37 °C and the resulting pDNA forms were resolved using agarose gel electrophoresis. Mean ± SEM, n = 3. | ||
Notably, H2S modulated the pDNA cleavage activity of the anhydride derivatives depending on the H2S/anhydride derivative molar ratio. In the presence of 50 μM of R-Se, increasing concentrations of H2S caused bell-shaped effects with a maximum level being reached at 100/50 and 200/50 μM/μM H2S/R-Se molar ratios. The effect of 50 μM of R-S, R-O and R-OH increased with increasing H2S concentrations (Fig. 10A). In the presence of 100 μM H2S, the pDNA cleavage activity of R-Se was several times higher in comparison to R-S, R-O or R-OH. Also GSH modulated the pDNA cleavage activity of the anhydride derivatives. In the presence of 50 μM R-Se, increasing concentrations of GSH mediated bell-shaped effects with a maximum level being seen at a 100/50 μM/μM GSH/R-Se molar ratio. In the presence of 50 μM R-S, R-O or R-OH, increasing concentrations of GSH had no effects on the pDNA cleavage (Fig. 10B).
 | ||
| Fig. 10 The effect of increasing concentrations of H2S (A) and GSH (B) on the pDNA integrity in the presence of the anhydride-derivatives R-Se, R-S, R-O and R-OH at 50 μM concentrations. Mean ± SEM, n = 3–5. | ||
As we detected relatively similar pDNA cleavage efficiencies at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:3 molar ratios of R-Se:H2S, we checked whether there was a difference in kinetics between the two reactions. However, the two reactions displayed the same (linear) time-dependent pDNA cleavage efficiency, suggesting that within a 1:1–1:3 (R-Se:H2S) molar ratio window, R-Se is a rate-limiting factor in the reaction (Fig. 11).
:1 and 1:3 molar ratios of R-Se:H2S, we checked whether there was a difference in kinetics between the two reactions. However, the two reactions displayed the same (linear) time-dependent pDNA cleavage efficiency, suggesting that within a 1:1–1:3 (R-Se:H2S) molar ratio window, R-Se is a rate-limiting factor in the reaction (Fig. 11).
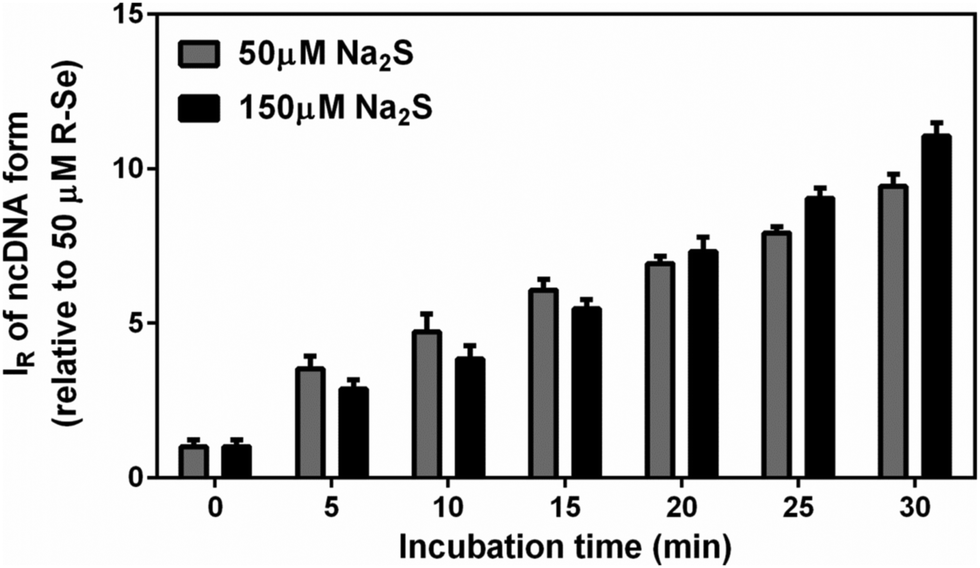 | ||
| Fig. 11 Comparison of time-dependent pDNA cleavage by H2S/R-Se at molar ratios 50/50 and 150/50 μM/μM. The H2S/R-Se mixture was incubated with pDNA for the given time. IR of ncDNA was normalized relative to 50 μM R-Se. Mean ± SEM, n = 3. | ||
Summing up, we have observed that R-S, R-O and R-OH only exerted limited pDNA cleavage activity, whereas this activity increased significantly when R-Se was employed in a concentration-dependent manner. Interestingly, the addition of H2S and GSH modulated strongly this pDNA cleavage action, and showed a bell-shaped effect, suggesting that the kinetics may be a rate-limiting factor in this action. In this case, the pDNA cleavage exerted by the remaining phthalic derivatives tested in the presence of H2S increased in a concentration dependent manner with H2S, which may be caused by the H2S-related pDNA cleavage effect. Thus, it is noteworthy that the pDNA cleavage in the absence of H2S and the bell-shaped modulation in the presence of H2S and GSH are only observed when R-Se is employed, which underlines the unique redox-modulating properties of R-Se and indicates the possibility of the formation of a S–Se intermediate when H2S interacts with R-Se.
Under these conditions, the relative intensity of the ˙BMPO-OOH adducts decreased slowly over the time and was comparable to the values reported under physiological conditions (Fig. 12A1–A3).56 The addition of R-Se or R-O (25 μM) had a minor effect on the ˙BMPO-OOH adducts formation, their concentration or rate of decay (Fig. 12B1–B3, D1–D3, 13A and B). In contrast, R-S (25 μM) significantly decreased the quantity of the ˙BMPO-adducts (Fig. 12C1–C3, 13A and B) and from the decreased ratio of the ˙BMPO-OOH/˙BMPO-adducts (Fig. 12C and D), a superposition of at least two radicals, ˙BMPO-OOH and ˙BMPO-OH, was recognized. H2S (50 μM) had similar effects to R-S, however its potency to decrease the quantity of the ˙BMPO-adducts and ratio of ˙BMPO-OOH/˙BMPO-OH was lower in comparison to R-S (Fig. 12E1–E3 and 13).
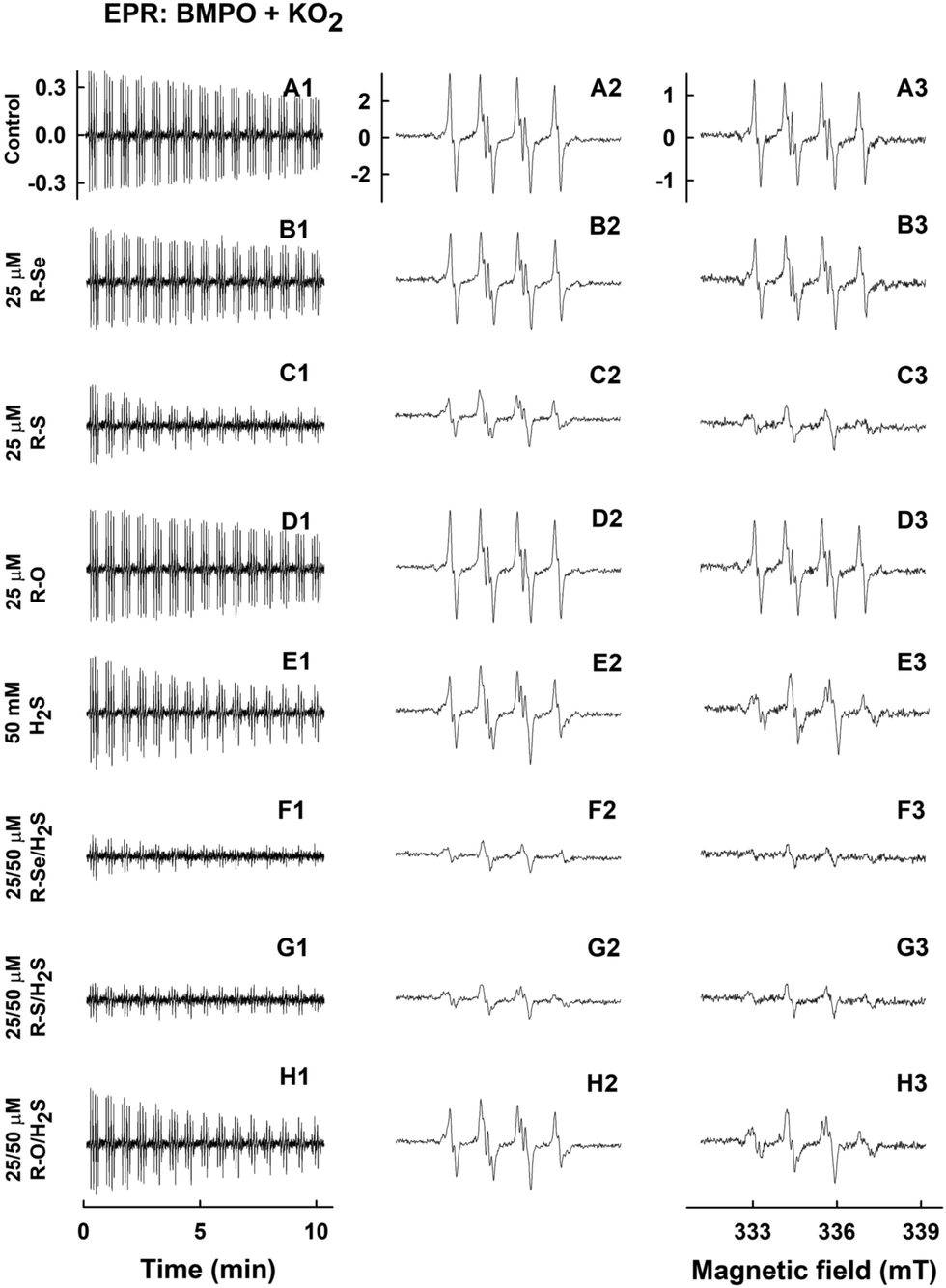 | ||
| Fig. 12 EPR spectra of ˙BMPO in the presence of O2˙− and modulated by R-Se, R-S and R-O without and with H2S. Representative EPR spectra of the ˙BMPO-adducts were monitored in 10% v/v saturated KO2/DMSO solution in 50 mM sodium phosphate buffer, 0.1 mM DTPA, pH 7.4, 37 °C in the presence of the studied species investigated and 20 mM BMPO. Sets of individual EPR spectra of the ˙BMPO-adducts monitored with 15 sequential scans, each 42 s (A1–H1), starting acquisition 2 min after sample preparation in: control 10% v/v KO2/DMSO in the buffer (A1); KO2/DMSO in the presence of 25 μM R-Se (B1); 25 μM R-S (C1); 25 μM R-O (D1); 50 μM H2S (E1); a mixture of 25/50 μM/μM R-Se/H2S (F1); a mixture of 25/50 μM/μM R-S/H2S (G1) and a mixture of 25/50 μM/μM R-O/H2S (H1). The spectra A2–H2 show details of the accumulated first ten A1–H1 spectra. The spectra A3–H3 show details of the accumulated last five A1–H1 spectra. The intensities of the time-dependent EPR spectra (A1–H1) and detailed spectra (A2–H2 and A3–H3) are comparable, as they were measured under identical EPR settings. | ||
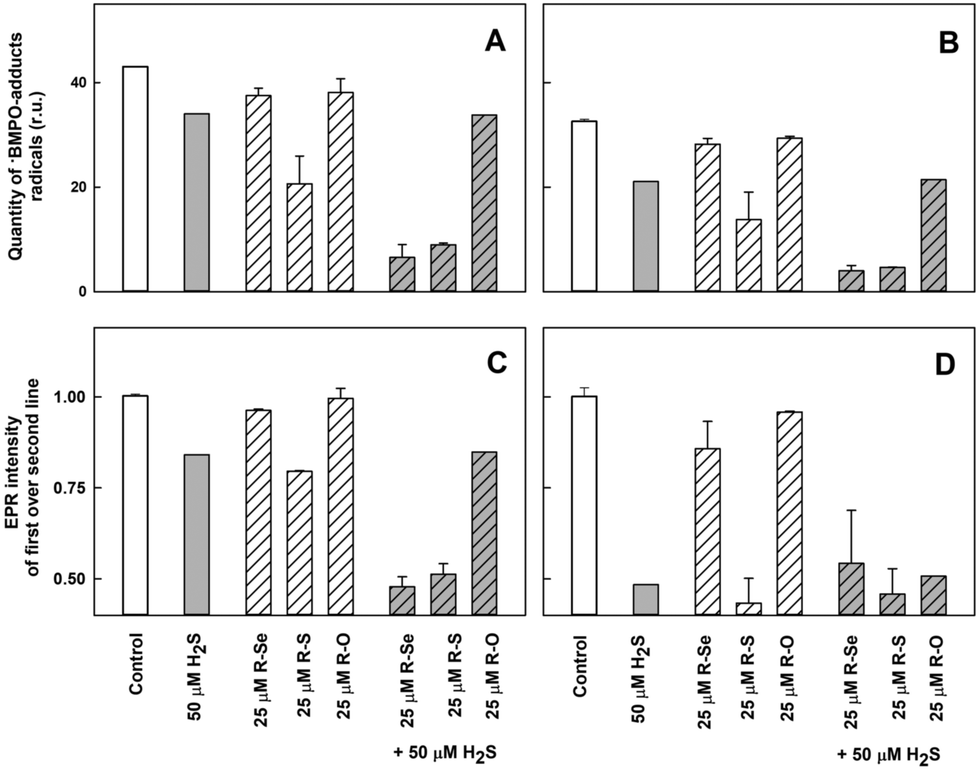 | ||
| Fig. 13 The effects of the compounds on the ˙BMPO-adduct radicals. The effects of the compounds (25 μM) and their mixture with H2S (50 μM) on the quantity of the ˙BMPO-adduct radicals (double integral of the EPR spectra from Fig. 12) in the presence of 10% v/v saturated KO2/DMSO solution. Average radical quantity during 2–9 (A, from Fig. 12A2–H2) and 10–13 (B, from Fig. 12A3–H3) min after sample preparation. The effects of the compounds (25 μM) and their mixture with H2S (50 μM) on the ratio of the EPR intensity of the first over the second line spectra of the ˙BMPO-adduct radicals (data from Fig. 12). Average ratio during 2–9 (C, from Fig. 12A2–H2) and 10–13 (D, from Fig. 12A3–H3) min after sample preparation. Buffer: 50 mM sodium phosphate, 0.1 mM DTPA, pH 7.4, 37 °C. Mean ± SEM, n = 2. | ||
The presence of H2S (50 μM) in the R-Se or R-S (25 μM) solution significantly decreased the quantity of the ˙BMPO-adducts (Fig. 12F1–F3, G1–G3, 13A and B) and significantly decreased the ratio of the ˙BMPO-OOH/˙BMPO-adducts (Fig. 13C and D), where a superposition of at least two radicals, ˙BMPO-OOH and ˙BMPO-OH, was recognized. Alternatively, a mixture of H2S (50 μM) with R-O (25 μM) caused a similar effect to H2S alone (Fig. 12H1–H3 and 13). Based on the decreasing quantity of the ˙BMPO-adducts, we suggest that R-S, H2S/R-Se and H2S/R-S scavenge the ˙BMPO-OOH/OH adducts, which may include direct scavenging of O2˙− or its derivatives. The decreasing ratio of the ˙BMPO-OOH/˙BMPO-adducts indicates that the compounds cause the decomposition of ˙BMPO-OOH to ˙BMPO-OH and scavenge both ˙BMPO-adducts. However, we cannot exclude the possibility of trapping an unknown radical by BMPO which decomposed to ˙BMPO-OH before measurement of the sample. Our data suggest that R-S, H2S/R-Se and H2S/R-S have high potency to scavenge different radicals.
To summarize this section, taking into account the ability of the phthalic derivatives to reduce the ˙cPTIO radical, we have studied also how these derivatives interact with O2˙−, observing how they interfere with the formation of the ˙BMPO-OOH adduct in the presence of KO2 and BMPO. In this experiment, R-Se and R-O showed a minor interaction with the O2˙− radical, whereas R-S significantly decreased the formation of the ˙BMPO-OOH adducts. Interestingly, the addition of H2S to R-Se and to R-S significantly enhances their capacity to decrease the formation of the ˙BMPO-OOH adduct, again highlighting the enhanced activity of the products of the interaction between H2S and R-Se: the presence of both Se and S atoms seems to be crucial for all these activities.
Final discussion
At the sight of the results presented herein, we suggest that the products of the H2S or GSH interaction with R-Se, having free radical scavenging and pDNA cleavage activities, can also affect intracellular molecules other than DNA. Based on the well-known consequences of oxidative stress, protein oxidation is also highly expected. We are aware of the fact that many more effects should be examined to obtain a more complete picture of the action of the products of the H2S or GSH interaction with R-Se and our intention is to present these promising initial results.In summary, the results confirm our initial hypothesis: selenoanhydride (R-Se) can act as a H2Se donor, serving as a prodrug that enables the internalisation of selenium into cells, and the subsequent release of H2Se and ionic species of Se inside the cell. Besides, we have proven that H2S and GSH interact with R-Se, and that the intermediates and/or products of this interaction have significant properties to reduce (scavenge) the ˙cPTIO and superoxide (O2˙−) radicals or their derivatives and to cleave pDNA. The antioxidant (reducing) properties observed of the intermediates and/or products of the H2S/R-Se and GSH/R-Se interaction to reduce ˙cPTIO, scavenge O2˙− and decompose ˙BMPO-OOH to ˙BMPO-OH, indicate that they may modulate redox properties and free radical signalling. However, qualifying the significance of these observations is a challenge for future research.
Experimental
Chemical synthesis of the anhydride-derivatives
R-Se and R-S (Fig. 1) were synthetized according to a procedure based on the one previously described in the literature,49 with minor modifications, whereas R-O was commercially available. Briefly, a suspension of grey selenium (for R-Se) or elemental sulfur (for R-S) in water-free tetrahydrofuran is reduced by dropwise addition of lithium aluminium hydride. Once the reaction is completed (visible by the ceasing of the generation of molecular hydrogen), phthaloyl chloride is added to the reaction and left reacting till the end of the reaction (usually 1 h). Then, the solution is filtered to eliminate the metallic salts generated during the process, and over the filtrate, 10 ml of concentrated sulfuric acid is added dropwise. The mixture is left reacting and the solid formed is filtered and washed with chloroform. The product isolated from the organic fraction is recrystallized in hexane.The structure of the compounds R-Se and R-S was confirmed by 1H-NMR, and their purity by LC-MS. Spectra are provided in the ESI† and they are in accordance with the literature. The purity of both compounds was 100% according to LC-MS (see data in the ESI†), so both derivatives were suitable for biological evaluation as they accomplished the 95% purity considered as the minimum threshold purity value required for biological assays. The chemical reactions of this synthetic procedure are shown in Fig. 1. It is quite interesting to see how the reaction normally used to get oxygen anhydrides (dehydration of phthalic acid) also serves to synthesize the sulfur and selenium anhydride analogues. What is more, it is noteworthy to point out the selectivity of the formation of the respective thio- and selenoanhydride when an oxygen atom is bound to the second carbonyl of the intermediate chalcogen phthalate.
In previous work in a PhD dissertation,57 to learn more about the reactivity of different phthalic derivatives to form phthalic selenoanhydride (R-Se) following this procedure, a synthetic study was performed. R-Se was synthesized according to the procedure mentioned above, departing from phthaloyl chloride and using lithium aluminium hydride for the reaction. The yield before recrystallization in this case was 94%. When the reaction was carried out using water as a solvent and employing sodium borohydride as a reducing agent, the yield before recrystallization was 47%. The reaction could also use different phthalic derivatives as substrates (always employing lithium aluminium hydride), in this case with different yields. We explored phthalic anhydride and N-hydroxyphthalimide, achieving yields of 16% and 62%, respectively. Interestingly (unpublished results), phthalimide did not render R-Se after dehydration with sulfuric acid. It seems that a selenazine is formed instead of the selenoanhydride, obtaining 1H-benzo[d][1,2]selenazine-1,4(3H)-dione. Unfortunately, this compound could not be obtained with a satisfactory purity and more research needs to be done to isolate and characterize this compound.
For the pDNA cleavage assay, the anhydride-derivatives were dissolved in ultrapure deionized water at a 1 mM final concentration by vortexing and 1 min water bath sonication, subsequently aliquoted and stored at −80 °C before their use. For UV-vis, EPR and ESI studies, the anhydride-derivatives were dissolved in anhydrous DMSO at a 50 mM concentration, aliquoted and stored at −80 °C before being used.
ESI-MS measurement
Saturated R-Se was prepared in 50% methanol/H2O, vortexed for 2–3 min, sonicated in a water bath for 1–2 min and centrifuged for 2 min. The sample without and with 7 mM Na2S (∼9 pH) was incubated for 1 min at 37 °C and 55 μl of the supernatant was used to measure ESI-MS spectra (Orbitrap Elite, ThermoScientific).Chemicals for UV-vis and EPR measurements
The studied compounds R-Se, R-S and R-O in DMSO (50 mM) were used after thawing. The spin trap 5-tert-butoxycarbonyl-5-methyl-1-pyrroline-N-oxide (BMPO, 100 mM, ENZO Life Sciences AG, Switzerland) was prepared in deionized H2O, stored at −80 °C and used after thawing. The radical 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (˙cPTIO, 10 mM, Cayman 81540 or Sigma C221) in deionized H2O was stored at −20 °C for several weeks. Na2S as a source of H2S (100 mM; SB01, DoJindo, Japan) was prepared in deionized H2O, stored at −80 °C and used after thawing. Na2S dissociates in solution and reacts with H+ to yield H2S, HS− and a trace of S2−. We use the term H2S to encompass the total mixture of H2S, HS− and S2−. To Na2Se powder (Alfa Aesar, 36187, stored under argon) H2O was added, and in 10 s an aliquot of the stock Na2Se solution (10 mM) was added to a UV-vis cuvette containing the studied compounds. 100 mM sodium phosphate buffer supplemented with 100 μM DTPA, pH 7.4, 37 °C, was employed for UV-vis experiments. 50 and 25 mM sodium phosphate buffer, supplemented with 100 and 50 μM DTPA (diethylenetriaminepentaacetic acid), pH 7.4, 37 °C was used for electron paramagnetic resonance (EPR) studies.UV-vis of ˙cPTIO
To a basic 900–990 μl solution of 100 mM sodium phosphate, 100 μM DTPA buffer (pH 7.4, 37 °C) the required aliquots of 100 μM ˙cPTIO and Na2S were added to obtain the desired final concentrations of ˙cPTIO and Na2S. Then, the UV-vis spectra (900–190 nm) were recorded, 3 × 30 s. The studied compounds R-Se, R-S and R-O (50 mM in DMSO), firstly dissolved in 50 μl buffer and vortexed for 3 s, were added and the spectra were recorded every 30 s for 20 min using a Shimadzu 1800 (Kyoto, Japan) spectrometer at 37 °C (the blank was H2O). For our study, the ˙cPTIO extinction coefficient at 560 nm of 920 M−1 cm−1 was used. Scavenging of the ˙cPTIO radical by Na2S (H2S) or GSH and its mixture with the studied compounds R-Se, R-S and R-O was determined as a decrease of absorbance at 356 and 560 nm (the absorption maximum of ˙cPTIO) after subtraction of the baseline absorbance, which was determined at 730 or 420 nm, respectively.42Plasmid DNA cleavage
The pDNA cleavage assay, which detects a disruption of the sugar-phosphate backbone of DNA, was used to study if the products of the H2S/R-Se and/or GSH/R-Se interaction can directly attack pDNA. In this assay, even a single hit is trapped, as it converts the circular supercoiled DNA molecule into its nicked relaxed circular form. These two forms display distinct mobility in agarose gels, and therefore they can easily be distinguished and quantified.The pBR322 vector (4.361 kb, New England Biolabs, N3033L) was used in the pDNA cleavage assay. In this assay, all samples contained 200 ng of pDNA in a final volume of 20 μl of buffer composed of 25 mM sodium phosphate and 50 μM DTPA (pH 7.4). Three different assay conditions were used: (i) pDNA per se (control), (ii) pDNA + phthalic-anhydride derivatives, and (iii) pDNA + anhydride-derivative + Na2S or GSH. The resulting mixtures were incubated for 30 min at 37 °C. Afterwards, the reaction mixtures were subjected to 0.6% agarose gel electrophoresis. The samples were electrophoresed in TBE buffer (89 mM Tris, 89 mM boric acid, 2 mM EDTA, pH 8.0) at 5.5 V cm−1 for 2 h. The gel was stained with Gel Red™ nucleic acid gel stain. Finally, the gels were photographed using a UV transilluminator. To quantify the pDNA cleavage efficiency, the integrated densities of two identified pBR322 forms (a supercoiled and a nicked circular form) in each lane were quantified using Total Lab TL100 image analysis software (Nonlinear Dynamic Ltd, USA).
EPR of ˙BMPO-adducts
To study the ability of R-Se, R-S and R-O without and with H2S to scavenge the O2˙− radical produced in DMSO/KO2 solution, sample preparation and EPR measurements were conducted in accordance with previously reported protocols.42 A solution (final concentrations) of BMPO (20 mM) and DTPA (100 μM) in sodium phosphate buffer (50 mM, pH 7.4) was incubated for 1 min at 37 °C. An aliquot of the compounds studied was added, followed by the addition of Na2S in 3 s and saturated KO2/DMSO solution (10% v/v DMSO/final buffer) 3 s later. The sample was mixed for 5 s and the first EPR spectrum was recorded 2 min after the addition of KO2/DMSO solution at 37 °C. Sets of individual EPR spectra of the ˙BMPO spin-adducts were recorded as 15 sequential scans, each 42 s, with a total time of 11 min. Each experiment was repeated at least twice. EPR spectra of the ˙BMPO spin-adducts were measured on a Bruker EMX spectrometer, X-band ∼9.4 GHz, 335.15 mT central field, 8 mT scan range, 20 mW microwave power, 0.1 mT modulation amplitude, 42 s sweep time, 20.48 ms time constant, and 20.48 ms conversion time at 37 °C.The relative quantity of the ˙BMPO-adduct radicals was calculated as a double integral of the EPR spectra. Since the EPR spectra were mostly low intensity, which did not permit spectral simulation, to quantify the relative ratio of the ˙BMPO-OOH/˙BMPO-adducts, the ratio of the EPR intensity of the first line over the second line was used. The ratio is ∼1 at ∼100% of ˙BMPO-OOH (Fig. 12A2) and ∼0.5 at ∼0% of ˙BMPO-OOH.42 A lower ratio (lower than 1) indicates a higher concentration of other ˙BMPO-adducts, in which mostly ˙BMPO-OH radicals are present.
Conclusions
Understanding the molecular mechanism of the biological effects of phthalic-anhydride derivatives could lead to development of more efficient drugs for treatment of cancer and ROS related diseases. To achieve this, we found that phthalic-anhydride derivatives R-Se, R-S, R-O and R-OH (≤50 μM) on their own have minor potency to reduce/scavenge radicals or cleave pDNA. However, the potency of R-Se and R-S, but not R-O or R-OH, significantly increased after interacting with H2S and GSH.Our in vitro data revealed unique properties of the H2S/R-Se, GSH/R-Se and H2S/R-S mixtures to reduce the ˙cPTIO and superoxide radicals. The unique potency of the H2S/R-Se mixture to cleave pDNA has a bell-shaped dependence on the H2S and GSH concentrations, whereas the potency of H2S/R-S increased linearly with H2S, but did not increase with the GSH concentration. The results underline that the interactions of R-Se and R-S with H2S and GSH enhanced significantly the different activities monitored, thus indicating that the intermediates and/or the products of the interaction of R-Se and R-S with endogenous H2S and GSH, which appear to include reactive selenium species such as H2Se, have significant antioxidant properties and that they can damage DNA. These findings may contribute to a more-in-depth understanding of the unique biological effects reported so far for R-Se and R-S. Besides, these findings open a new so far unexplored approach to study the action of Se-containing compounds. These experiments, for example, can be applied to the different selenium species that have been used until now in supplementation, to ascertain which ones have more ability to interact with GSH and H2S. This is of crucial importance, as it would enable detecting new compounds that could behave as Se-based redox modulators in potential Se supplementation. An example would be the phthalic selenoanhydride (R-Se) reported in this work, which retains the capacity of sodium selenite to react with key components of the redox thiolstat (such as GSH and H2S) and simultaneously, according to previous work, shows lower toxicity against non-tumour cells.
Author contributions
Conceptualization, E. D.-A., C. J., M. C., and K. O.; methodology, K. O., M. C., and V. B.; validation, K. O., M. C., V. B., A. M., M. G., and E. D.-A.; formal analysis, A. M., and K. O.; investigation, A. M., M. G., A. K., V. B., L. K., P. B., M. C., K. O., and E. D.-A.; resources, A. K., C. J., and E. D.-A.; writing – original draft preparation, K. O., M. C., and E. D.-A.; visualization, K. O., A. M., M. G., and E. D.-A.; supervision, K. O., C. J., M. C., and E. D.-A.; project administration, K. O., C. J., M. C., and E. D.-A.; funding acquisition, K. O.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Slovak Research & Development Agency, grant numbers APVV-15-0371, 15-0565 and 17-0384, and the Scientific Grant Agency of the Slovak Republic, grant numbers VEGA 1/0026/18, 2/0079/19, 2/0014/17 and 2/0053/19. The authors would also like to acknowledge the financial support of the Agencia Estatal Consejo Superior de Investigaciones Científicas (Spain, project 201780I027) and of the University of Saarland (through the Landesforschungsförderungsprogramm (LFPP) of the state of Saarland, Grant No. WT/2-LFFP 16/01). We also acknowledge the INTERREG-VA GR program (BIOVAL, Grant No. 4-09-21) and the NutRedOx (COST Project CA16112), as well as the support of the Erasmus+ program. We also thank Prof. Dr Anna K. H. Hirsch and Dr Eleonora Diamanti from Helmholtz Institute for Pharmaceutical Research in Saarland (HIPS) for helping with MS spectra measurements.Notes and references
- S. J. Fairweather-Tait and K. Cashman, in Nutrition for the Primary Care Provider. World Rev. Nutr. Diet., ed. D. M. Bier, J. Mann, D. H. Alpers, H. H. E. Vorster and M. J. Gibney, Karger AG, Basel, Switzerland, 2015, ch. 8, vol. 111, pp. 45–52 Search PubMed.
- S. Li, T. Xiao and B. Zheng, Sci. Total Environ, 2012, 421–422, 31–40 CrossRef CAS PubMed.
- S. Stranges, J. R. Marshall, R. Natarajan, R. P. Donahue, M. Trevisan, G. F. Combs, F. P. Cappuccio, A. Ceriello and M. E. Reid, Ann. Intern. Med., 2007, 147, 217–223 CrossRef PubMed.
- D. L. Hatfield, M. H. Yoo, B. A. Carlson and V. N. Gladyshev, Biochim. Biophys. Acta, Gen. Subj., 2009, 1790, 1541–1545 CrossRef CAS PubMed.
- J. C. Avery and P. R. Hoffmann, Nutrients, 2018, 10, 1203 CrossRef.
- N. Shang, X. Wang, Q. Shu, H. Wang and L. Zhao, J. Nanosci. Nanotechnol., 2019, 19, 1875–1888 CrossRef CAS PubMed.
- K. M. Peters, B. A. Carlson, V. N. Gladyshev and P. A. Tsuji, Free Radical Biol. Med., 2018, 127, 14–25 CrossRef CAS PubMed.
- V. Gandin, P. Khalkar, J. Braude and A. P. Fernandes, Free Radical Biol. Med., 2018, 127, 80–97 CrossRef CAS.
- M. Bodnar, M. Szczyglowska, P. Konieczka and J. Namiesnik, Crit. Rev. Food Sci. Nutr., 2016, 56, 36–55 CrossRef CAS.
- A. Tarze, M. Dauplais, I. Grigoras, M. Lazard, N. T. Ha Duong, F. Barbier, S. Blanquet and P. Plateau, J. Biol. Chem., 2007, 282, 8759–8767 CrossRef CAS.
- A. P. Fernandes and V. Gandin, Biochim. Biophys. Acta, Gen. Subj., 2015, 1850, 1642–1660 CrossRef CAS PubMed.
- J. J. An, K. J. Shi, W. Wei, F. Y. Hua, Y. L. Ci, Q. Jiang, F. Li, P. Wu, K. Y. Hui, Y. Yang and C. M. Xu, Cell Death Dis., 2013, 4, e973 CrossRef CAS PubMed.
- G. Nilsonne, X. Sun, C. Nyström, A. K. Rundlöf, A. Potamitou Fernandes, M. Björnstedt and K. Dobra, Free Radical Biol. Med., 2006, 41, 874–885 CrossRef CAS.
- J. Brozmanová, D. Mániková, V. Vlčková and M. Chovanec, Arch. Toxicol., 2010, 84, 919–938 CrossRef PubMed.
- E. Jablonska and M. Vinceti, J. Environ. Sci. Health, Part C: Environ. Carcinog. Ecotoxicol. Rev., 2015, 33, 328–368 CrossRef CAS.
- H. J. Reich and R. J. Hondal, ACS Chem. Biol., 2016, 11, 821–841 CrossRef CAS.
- R. Mousa, R. Notis Dardashti and N. Metanis, Angew. Chem., Int. Ed., 2017, 56, 15818–15827 CrossRef CAS PubMed.
- C. Jacob, G. I. Giles, N. M. Giles and H. Sies, Angew. Chem., Int. Ed., 2003, 42, 4742–4758 CrossRef CAS.
- L. Sancineto, M. Palomba, L. Bagnoli, F. Marini and C. Santi, Curr. Org. Chem., 2016, 20, 122–135 CrossRef CAS.
- D. Bartolini, L. Sancineto, A. Fabro de Bem, K. D. Tew, C. Santi, R. Radi, P. Toquato and F. Galli, in Advances in Cancer Research, ed. K. D. Tew and F. Galli, Academic Press Inc., Cambridge, USA, 2017, ch. 10, vol. 136, pp. 259–302 Search PubMed.
- C. Santi, C. Tomassini and L. Sancineto, Chimia, 2017, 71, 592–595 CrossRef CAS.
- M. Álvarez-Pérez, W. Ali, M. A. Marć, J. Handzlik and E. Domínguez-Álvarez, Molecules, 2018, 23, 628 CrossRef PubMed.
- W. Ali, M. Álvarez-Pérez, M. A. Marć, N. Salardón-Jiménez, J. Handzlik and E. Domínguez-Álvarez, Curr. Pharmacol. Rep., 2018, 4, 468–481 CrossRef CAS.
- S. Misra, M. Boylan, A. Selvam, J. E. Spallholz and M. Björnstedt, Nutrients, 2015, 7, 3536–3556 CrossRef CAS PubMed.
- C. M. Weekley and H. H. Harris, Chem. Soc. Rev., 2013, 42, 8870–8894 RSC.
- R. Terazawa, D. R. Garud, N. Hamada, Y. Fujita, T. Itoh, Y. Nozawa, K. Nakane, T. Deguchi, M. Koketsu and M. Ito, Bioorg. Med. Chem., 2010, 18, 7001–7008 CrossRef CAS PubMed.
- B. Romano, D. Plano, I. Encío, J. A. Palop and C. Sanmartín, Bioorg. Med. Chem., 2015, 23, 1716–1727 CrossRef CAS.
- Y. Zakharia, A. Bhattacharya and Y. M. Rustum, Oncotarget, 2018, 9, 10765–10783 CrossRef PubMed.
- C. Santi, C. Tidei, C. C. Scalera, M. Piroddi and F. Galli, Curr. Chem. Biol., 2013, 7, 25–36 CrossRef CAS.
- D. Bartolini, M. Piroddi, C. Tidei, S. Giovagnoli, D. Pietrella, Y. Manevich, K. D. Tew, D. Giustarini, R. Rossi, D. M. Townsend, C. Santi and F. Galli, Free Radical Biol. Med., 2015, 78, 56–65 CrossRef CAS PubMed.
- M. M. Rahman, R. A. Uson-Lopez, M. T. Sikder, G. Tan, T. Hosokawa, T. Saito and M. Kurasaki, Chemosphere, 2018, 196, 453–466 CrossRef CAS PubMed.
- M. Mix, N. Ramnath, J. Gomez, C. De Groot, S. Rajan, S. Dibaj, W. Tan, Y. Rustum, M. B. Jameson and A. K. Singh, World J. Clin. Oncol., 2015, 6, 156–165 CrossRef PubMed.
- D. Mániková, L. M. Letavayová, D. Vlasáková, P. Košík, E. C. Estevam, M. J. Nasim, M. Gruhlke, A. Slusarenko, T. Burkholz, C. Jacob and M. Chovanec, Molecules, 2014, 19, 12258–12279 CrossRef PubMed.
- C. Sanmartín, D. Plano, M. Font and J. A. Palop, Curr. Cancer Drug Targets, 2011, 11, 496–523 CrossRef.
- R. Wang, Physiol. Rev., 2012, 92, 791–896 CrossRef CAS.
- C. Szabo and A. Papapetropoulos, Pharmacol. Rev., 2017, 69, 497–564 CrossRef CAS.
- H. Kimura, Antioxid. Redox Signaling, 2015, 22, 362–376 CrossRef CAS.
- M. Whiteman, J. S. Armstrong, S. H. Chu, S. Jia-Ling, B. S. Wong, N. S. Cheung, B. Halliwell and P. K. Moore, J. Neurochem., 2004, 90, 765–768 CrossRef CAS.
- M. Whiteman, N. S. Cheung, Y. Z. Zhu, S. H. Chu, J. L. Siau, B. S. Wong, J. S. Armstrong and P. K. Moore, Biochem. Biophys. Res. Commun., 2005, 326, 794–798 CrossRef CAS PubMed.
- A. Staško, V. Brezová, M. Zalibera, S. Biskupič and K. Ondriaš, Free Radical Res., 2009, 43, 581–593 CrossRef PubMed.
- B. Olas, Chem.-Biol. Interact., 2014, 217, 46–56 CrossRef.
- A. Misak, M. Grman, Z. Bacova, I. Rezuchova, S. Hudecova, E. Ondriasova, O. Krizanova, V. Brezova, M. Chovanec and K. Ondrias, Nitric oxide, 2018, 76, 136–151 CrossRef CAS.
- C. Szabo, C. Coletta, C. Chao, K. Módis, B. Szczesny, A. Papapetropoulos and M. R. Hellmich, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 12474–12479 CrossRef CAS PubMed.
- D. Wu, W. Si, M. Wang, S. Lv, A. Ji and Y. Li, Nitric oxide, 2015, 50, 38–45 CrossRef CAS PubMed.
- J. Breza Jr., A. Soltysova, S. Hudecova, A. Penesova, I. Szadvari, P. Babula, B. Chovancova, L. Lencesova, O. Pos, J. Breza, K. Ondrias and O. Krizanova, BMC Cancer, 2018, 18, 591 CrossRef PubMed.
- V. I. Lushchak, J. Amino Acids, 2012, 736837 Search PubMed.
- C. Gaucher, A. Boudier, J. Bonetti, I. Clarot, P. Leroy and M. Parent, Antioxidants, 2018, 7, 62 CrossRef CAS PubMed.
- A. Kharma, M. Grman, A. Misak, E. Domínguez-Álvarez, M. J. Nasim, K. Ondrias, M. Chovanec and C. Jacob, Molecules, 2019, 24, 1359 CrossRef PubMed.
- E. Domínguez-Álvarez, D. Plano, M. Font, A. Calvo, C. Prior, C. Jacob, J. A. Palop and C. Sanmartín, Eur. J. Med. Chem., 2014, 73, 153–166 CrossRef PubMed.
- M. J. Nasim, W. Ali, E. Domínguez-Álvarez, E. N. da Silva Júnior, R. S. Z. Saleem and C. Jacob, in Organoselenium Compounds in Biology and Medicine: Synthesis, Biological and Therapeutic Treatments, ed. V. K. Jain and K. I. Priyadarsini, The Royal Society of Chemistry, Croydon, UK, 2018, ch. 10, pp. 277–302 Search PubMed.
- M. Gajdács, G. Spengler, C. Sanmartín., M. A. Marć, J. Handzlik and E. Domínguez-Álvarez, Bioorg. Med. Chem. Lett., 2017, 27, 797–802 CrossRef PubMed.
- E. Domínguez-Álvarez, M. Gajdács, G. Spengler, J. A. Palop, M. A. Marć, K. Kieć-Kononowicz, L. Amaral, J. Molnár, C. Jacob, J. Handzlik and C. Sanmartín, Bioorg. Med. Chem. Lett., 2016, 26, 2821–2824 CrossRef PubMed.
- M. M. Cortese-Krott, G. G. C. Kuhnle, A. Dyson, B. O. Fernandez, M. Grman, J. F. DuMond, M. P. Barrow, G. McLeod, H. Nakagawa, K. Ondrias, P. Nagy, S. B. King, J. E. Saavedra, L. K. Keefer, M. Singer, M. Kelm, A. R. Butler and M. Feelisch, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E4651–E4660 CrossRef CAS.
- M. T. Zimmerman, C. A. Bayse, R. R. Ramoutar and J. L. Brumaghim, J. Inorg. Biochem., 2015, 145, 30–40 CrossRef CAS.
- Y. H. Liu, M. Lu, L. F. Hu, P. T. H. Wong, G. D. Webb and J. S. Bian, Antioxid. Redox Signaling, 2012, 17, 141–185 CrossRef CAS PubMed.
- H. Zhao, J. Joseph, H. Zhang, H. Karoui and B. Kalyanaraman, Free Radical Biol. Med., 2001, 31, 599–606 CrossRef CAS.
- E. Domínguez-Álvarez, PhD Dissertation, University of Navarra, Spain, 2012.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nj02245g |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |