
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Acylseleno- and acylthioureato complexes of gold(I) N-heterocyclic carbenes†
Julia
Kuchar
a,
Jörg
Rust
b,
Christian W.
Lehmann
b and
Fabian
Mohr
 *a
*a
aFakultät für Mathematik und Naturwissenschaften, Anorganische Chemie, Bergische Universität Wuppertal, Gaußstr. 20, 42119 Wuppertal, Germany. E-mail: fmohr@uni-wuppertal.de; Web: http://www.aoc.uni-wuppertal.de/mohr
bChemische Kristallographie und Elektronenmikroskopie, Max-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470 Mülheim an der Ruhr, Germany
First published on 14th June 2019
Abstract
A series of gold(I) N-heterocyclic carbene complexes containing monoanionic acylchalcogenourea (chalcogen = S, Se) ligands was prepared and characterised by various spectroscopic methods and X-ray diffraction. The compounds were conveniently accessible in good yields through a one-pot reaction involving [AuCl(tht)] (tht = tetrahydrothiophene), the imidazolium salt and the acylchalcogenourea in the presence of excess base. The X-ray diffraction experiments showed that in these complexes, the normally chelating ligands adopt the rare κS/Se-coordination mode in which only the chalcogen is bound to gold.
Introduction
Acylselenoureas are bidentate Se,O-ligands, which, upon deprotonation typically form six-membered chelate rings with a large variety of metals. Examples for such compounds include bis(chelates) with the divalent transition metal ions Cu2+, Ni2+, Pd2+, Pt2+, Zn2+ and Cd2+.1–10 There are also examples of tris(chelates) with the trivalent metal ions Fe3+, Co3+ and In3+.11–13 Amongst the main group elements, bis(chelates) containing lead(II) have been of recent interest as single-source precursors for various PbSe nanomaterials.14,15 However, examples of such species containing silver and gold are still quite rare. We have previously reported the first Ag+ and Au+ compounds containing acylselenoureato and acylthioureato ligands together with a detailed study of their biological activity.16–19 We also demonstrated that square planar gold(III) complexes with acylselenoureato ligands are accessible when a dicarbanionic co-ligand is present.20 This stabilizing co-ligand is essential to prevent reduction of gold by the acylselenourea during the reaction. Given our interest in gold complexes with selenium and sulfur ligands,21 we communicate here the synthesis of some gold(I) complexes containing acylselenoureas and acylthioureas with an N-heterocyclic carbene co-ligand by a simple one-pot process.Results and discussion
Initially, we examined three different routes towards the desired NHC-gold(I) acylchalcogenoureato complexes: (1) the reaction of the acylchalcogenoureas with [AuCl(IPr)] [IPr = 1,3-di(2,6-diisopropylphenyl)imidazolylidene] in the presence of base, (2) the reaction of the acylchalcogenoureas with [AuOH(IPr)] and (3) by transmetallation of [AuCl(IPr)] with the corresponding silver(I) acylchalcogenoureato complexes.17 To our surprise, the latter two routes afforded either no products at all or we observed formation of dark, inseparable mixtures. In particular, the lack of reactivity of the gold(I) hydroxy complex [AuOH(IPr)] in this system is somewhat unexpected. The Nolan group has previously demonstrated that this reagent readily reacts with a variety of species containing acidic protons to form the corresponding gold(I) complexes.22 Given that the reaction between the acylchalcogenoureas and [AuCl(IPr)] showed the most promising results, we planned to further simplify and generalise the synthesis route by preparing [AuCl(IPr)] in situ directly from the imidazolium salt and [AuCl(tht)] (tht = tetrahydrothiophene) in the presence of base and the acylchalcogenoureas. Gratifyingly, the one-pot reaction of [AuCl(tht)] with the acylchalcogenoureas and the imidazolium salt in the presence of two equivalents of KOtBu afforded the desired complexes in two hours at room temperature in pure form and good yields (Scheme 1).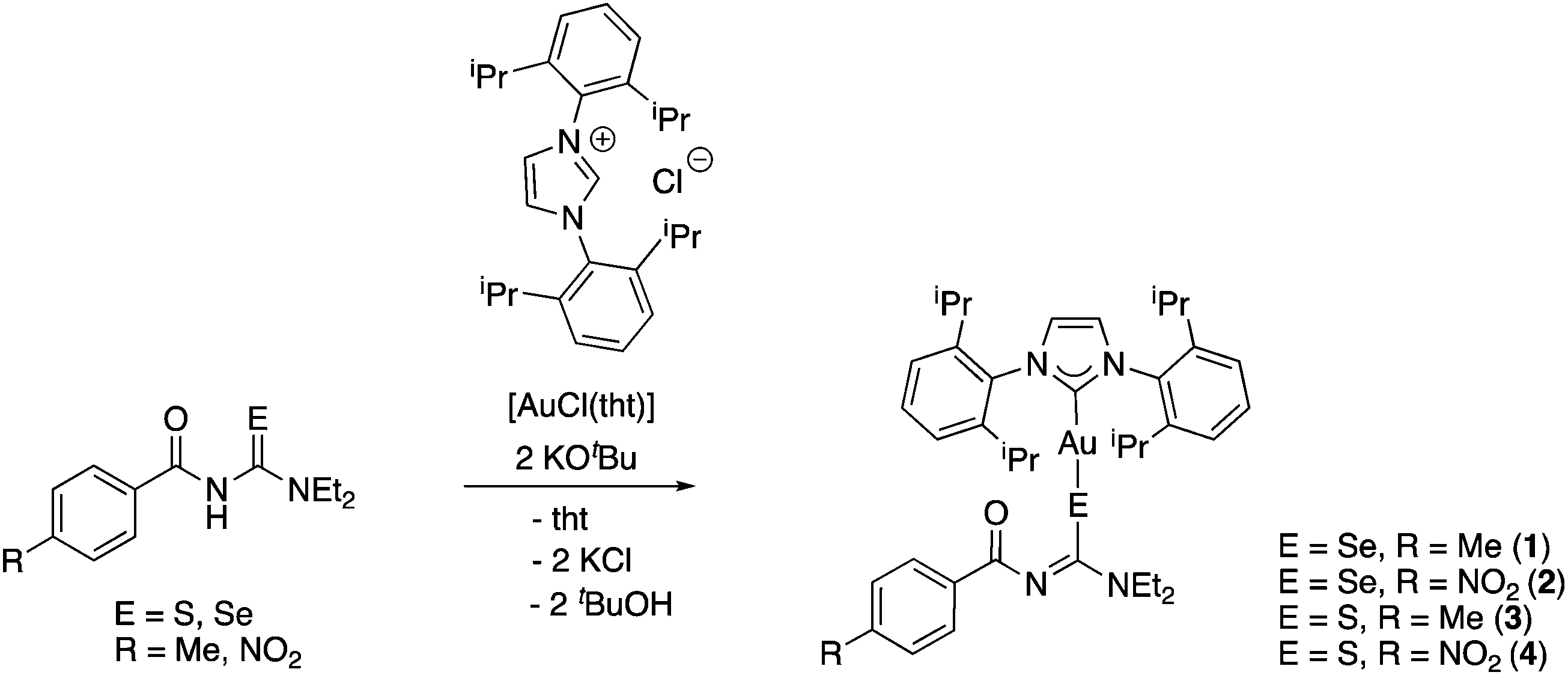 | ||
| Scheme 1 | ||
Complexes 1–4 were isolated as air-, light- and moisture-stable colourless or yellow compounds in yields greater than 60%. The proton NMR spectra of the compounds lack the signal of the NH-proton, consistent with the presence of the deprotonated acylchalcogenourea ligands. Furthermore, the signal due to the carbenic carbon-atom bound to gold can be observed at around 186 ppm (in selenium compounds 1 and 2) or 183 ppm (in sulfur compounds 3 and 4) in their 13C NMR spectra. These values are higher than that found in the starting material [AuCl(IPr)] (175 ppm),23 confirming that substitution of the chlorido ligand has indeed occurred. Similar chemical shifts for the carbene–carbon-atom have been observed in other gold(I) NHC-complexes containing anionic chalcogen ligands.24,25 For the selenium compounds we were able to record 77Se NMR spectra, which showed singlet resonances shifted upfield by 371 ppm (1) and 380 ppm (2), relative to those of the acylselenoureas themselves.18 For comparison, the singlet resonances of the triphenylphosphine counterparts of 1 and 2 are shifted upfield by 279 ppm and 266 ppm, respectively.16 This nicely illustrates the electronic differences that phosphine (Ph3P) or carbene (IPr) ligands trans to the selenium atom have on its 77Se chemical shift.
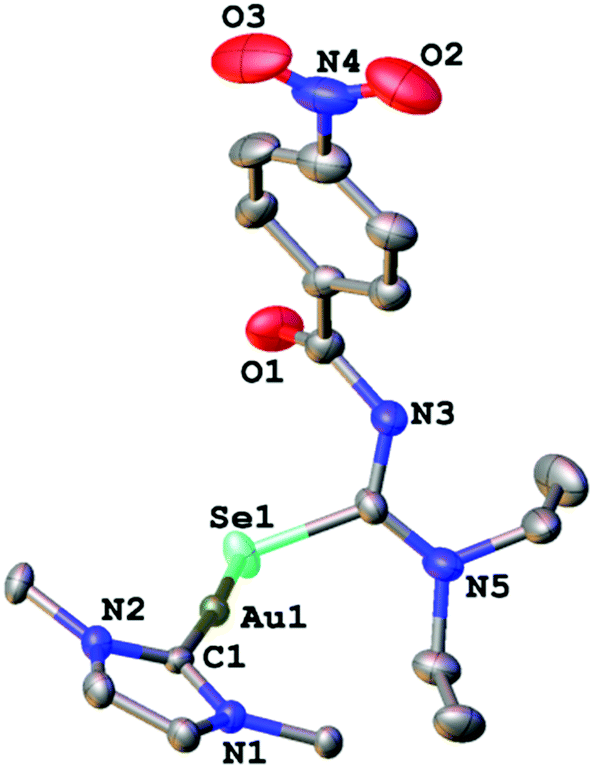
We managed to grow crystals of complexes 1, 2 and 4 suitable for an X-ray diffraction experiment. The compounds crystallise in the monoclinic crystal system in the space groups P21/n, P21/c and C2/c, respectively. The asymmetric unit of 1 contains a single molecule of the complex together with one molecule of co-crystallised ethanol. Compounds 2 and 4 contain four and three independent molecules, respectively in their asymmetric units. The differences in geometric parameters (bond distances and angles) of the independent molecules are however insignificant. In all three complexes the gold atom is bound to the carbon-atom of the IPr-ligand and a selenium or sulfur atom from the deprotonated acylchalcogenourea ligands (Fig. 1–3).
 | ||
| Fig. 1 Molecular structure of 1. Ellipsoids show 30% probability levels. Hydrogen atoms as well as the co-crystallised ethanol of solvation have been omitted for clarity. Only the ipso carbon-atoms of the 2,6-iPr2C6H3 groups are shown. | ||
 | ||
| Fig. 2 Molecular structure of one of the independent molecules of 2. Ellipsoids show 30% probability levels. Hydrogen atoms have been omitted for clarity. Only the ipso carbon-atoms of the 2,6-iPr2C6H3 groups are shown. | ||
 | ||
| Fig. 3 Molecular structure of one of the independent molecules of 4. Ellipsoids show 30% probability levels. Hydrogen atoms have been omitted for clarity. Only the ipso carbon-atoms of the 2,6-iPr2C6H3 groups are shown. | ||
The metal centre is two-coordinate linear with C–Au–Se/S angles ranging from 174° to 177°, as expected for gold(I) compounds. The angles at the chalcogen atoms are around 100°, similar to what is observed in the solid-state structures of gold(I) compounds with anionic Se/S-ligands. Furthermore, the gold–carbon bond distances of approximately 2.0 Å are typical values for gold(I) carbenes. There is only one other example of an NHC-Au(I) complex containing an anionic selenium ligand (−SeCN) which has been structurally characterised.26 In this compound the Au–Se bond length of 2.414 Å is slightly longer than those found in complexes 1 and 2 (around 2.39 Å).
Thus, in these compounds the typically bidentate chelating (via Se/S and O-atoms) acylchalcogenoureato ligands behave as simple monodentate Se− or S− ligands. The ArC(O) units are rotated by almost 90° away from the CSe- and CS-groups which are bound to the gold centre. There is therefore no interaction whatsoever between the oxygen atom of the acylchalcogenoureato ligand and the metal. We may invoke the Pearson hard/soft principle to account for this observation: the soft nature of the gold(I) centre combined with the soft Se- and S-ligand results in a favourable interaction, whereas the hard-soft mismatch between gold and oxygen disfavours any contact between those atoms. This κSe,S-coordination mode of acylchalcogenoureato ligands is very rare, apart from our previously reported examples containing gold or rhenium,27,40 there are no other examples of structurally characterised metal complexes containing Se-bound monoanionic acylselenourea ligands. In the case of the sulfur counterparts, there are three reports in the literature dealing with κS-coordinated acylthioureato ligands (Fig. 4).
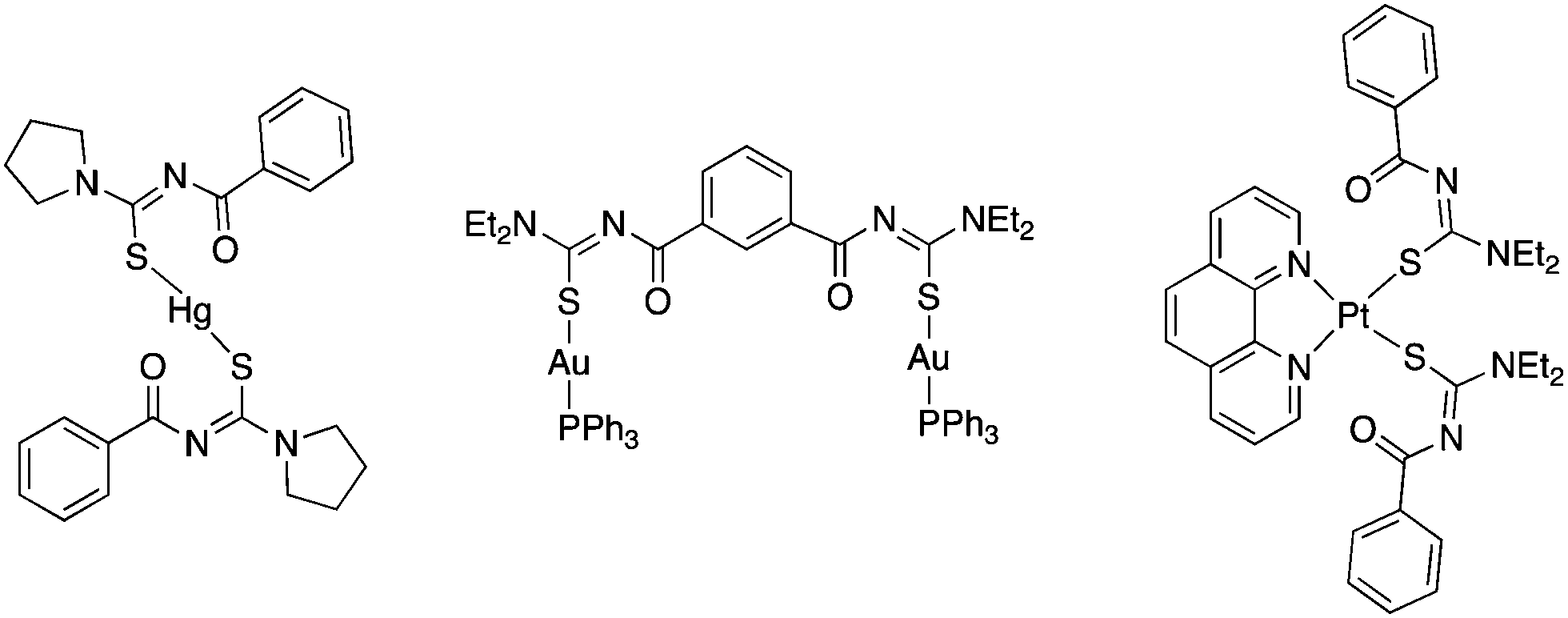 | ||
| Fig. 4 Known complexes featuring κS-coordination of acylthiourea ligands. | ||
An early publication from Richter describes the mercury(II) complexes [Hg{PhC(O)NC(S)NR2}2] (NR2 = morpholine, pyrrolidine) featuring κS-bound ligands (Fig. 4 left).28 Although the oxygen atoms are directed towards the metal, the Hg–O distance of 2.52 Å was deemed too large for the S,O-chelating mode. The bis(acylthiourea) derived from isophthaloyl dichloride 1,3-{Et2NC(S)NHC(O)}2C6H4 reacts with two equivalents of [AuCl(PPh3)] in the presence of base to afford the corresponding bimetallic gold(I) complex (Fig. 4 centre).29 More recently, Koch reported the platinum(II) complexes [Pt(phen){ArC(O)NC(S)NR2}2] (Ar = 1-nap, R = nBu; Ar = Ph, R = Et), in which two deprotonated acylthiourea ligands are κS-bound to the metal centre (Fig. 4 right).30
In summary we disclose here the first examples of gold(I) carbene complexes containing Se- or S-bound acylchalcogenoureato ligands. The compounds are readily accessible through a one-pot method starting from readily available materials. We are currently exploring the scope of this method with respect to the carbene co-ligand and potential biological activity of this new class of substances.
Experimental
General
Reactions were carried out under aerobic conditions using HPLC grade solvents. The acylchalcogenoureas,31–33 [AuCl(tht)]34 and the imidazolium salt IPrHCl35 were prepared by reported procedures. All other reagents and solvents were procured from commercial suppliers and were used as received. NMR spectra were recorded on Bruker Avance 400 or Bruker Avance III 600 instruments. Spectra were referenced externally to Me4Si (1H, 13C) and Me2Se (77Se). Elemental analyses were performed by staff of the in-house elemental analysis facility using an Elementar Vario EL system.Preparation of the complexes
To a solution containing the respective acylchalcogenourea, [AuCl(tht)] (1 equiv.) and IPrHCl (1 equiv.) in MeOH (15 mL) was added KOtBu (2.2 equiv.). After ca. 2 h at room temperature, the mixture was evaporated to dryness and the residue was extracted into CH2Cl2 (3 × 10 mL). The combined extracts were filtered through Celite and the filtrate was subsequently dried in vacuum.X-ray crystallography
Diffraction data for complexes 2 and 4 were collected using an Enraf-Nonius KappaCCD system located in front of a FR591 rotating anode equipped with an Incoatec focusing Montel optic for Mo radiation and an Oxford Cryosystems Cryostream 700. For data integration the EVAL-14 package36 was employed. For data scaling and absorption correction (combination of Gaussian and multiscan corrections) SADABS was used. Diffraction data for complex 1 was collected using a Rigaku Oxford Diffraction Gemini E Ultra diffractometer, equipped with an EOS CCD area detector and a four-circle kappa goniometer. Data integration, scaling and empirical absorption correction was carried out using the CrysAlis Pro program package.37 All crystal structures were solved using SHELXT38 and refined using SHELXL.38 The Olex2 graphical user interface39 was used for all structure manipulations and for molecular graphics. Crystallographic and refinement details are collected in Table 1.| 1 EtOH | 2 | 4 | |
|---|---|---|---|
| CCDC code | 1907692 | 1907635 | 1907634 |
| Empirical formula | C42H59AuN4O2Se | C39H50AuN5O3Se | C39H50AuN5O3S |
| Colour | Colourless | Yellow | Colourless |
| Formula weight (g mol−1) | 927.85 | 912.76 | 865.86 |
| Temperature (K) | 150(2) | 100(2) | 100(2) |
| Wavelength (Å) | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/n | P21/c | C2/c |
| Unit cell dimensions | a = 14.1899(3) Å | a = 19.003(7) Å | a = 33.860(7) Å |
| b = 16.2803(4) Å | b = 21.806(3) Å | b = 36.211(7) Å | |
| c = 18.4131(5) Å | c = 37.911(6) Å | c = 19.899(3) Å | |
| β = 101.693(2)° | β = 91.39(2)° | β = 107.145(18)° | |
| Volume | 4165.47(18) Å3 | 15705(7) Å3 | 23314(8) Å3 |
| Z | 4 | 16 | 24 |
| ρ calc | 1.480 g cm−3 | 1.544 g cm−3 | 1.480 g cm−3 |
| μ | 4.444 mm−1 | 4.716 mm−1 | 3.882 mm−1 |
| F(000) | 1872.0 | 7296.0 | 10512 |
| Crystal size | 0.03 × 0.06 × 0.12 mm | 0.05 × 0.12 × 0.18 mm | 0.04 × 0.05 × 0.10 mm |
| θ range for data collection | 4.758 to 59.144° | 2.599 to 32.974° | 2.646 to 28.300° |
| Reflections collected | 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 188 188 |
405585 |
133811 |
| Independent reflections | 9924 | 58942 |
28915 |
| Parameters | 464 | 1805 | 1350 |
| Goodness-of-fit on F2 | 1.100 | 1.029 | 1.007 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0388 | R 1 = 0.0614 | R1 = 0.0659 |
| wR2 = 0.0784 | wR2 = 0.1157 | wR2 = 0.1048 | |
| Largest difference peak/hole | 3.06/−1.38 e Å−3 | 6.7/−3.6 e Å−3 | 1.4/−2.0 e Å−3 |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank RETORTE GmbH for a generous gift of selenium metal.References
- L. Beyer, R. Kirmse and E. Hoyer, Z. Chem., 1975, 15, 197 CrossRef CAS.
- R. Kirmse, L. Beyer and E. Hoyer, Z. Chem., 1975, 15, 454–455 CrossRef CAS.
- R. Kirmse, L. Beyer and E. Hoyer, Chem. Phys. Lett., 1977, 49, 544–546 CrossRef CAS.
- J. Stach, R. Kirmse, A. Heinrich, W. Dietzsch, J. Hartung and L. Beyer, Z. Chem., 1983, 23, 453–454 CrossRef CAS.
- M. Kampf, R. Richter, S. Gerber and R. Kirmse, Z. Anorg. Allg. Chem., 2004, 630, 1437–1443 CrossRef CAS.
- M. Kampf, R. Richter, L. Hennig, A. Eidner, J. Baldamus and R. Kirmse, Z. Anorg. Allg. Chem., 2004, 630, 2677–2686 CrossRef CAS.
- M. Kampf, R. Richter, J. Griebel, A. Weller and R. Kirmse, Z. Anorg. Allg. Chem., 2005, 631, 698–708 CrossRef CAS.
- J. C. Bruce and K. R. Koch, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2008, 64, m1–m4 CrossRef CAS.
- W. Bensch and M. Schuster, Z. Anorg. Allg. Chem., 1993, 619, 786–790 CrossRef CAS.
- W. Bensch and M. Schuster, Z. Anorg. Allg. Chem., 1993, 619, 791–795 CrossRef CAS.
- M. Akhtar, J. Akhtar, M. A. Malik, F. Tuna, M. Helliwell and P. O'Brien, J. Mater. Chem., 2012, 22, 14970–14975 RSC.
- W. Bensch and M. Schuster, Z. Anorg. Allg. Chem., 1994, 620, 1479–1482 CrossRef CAS.
- M. Schuster and W. Bensch, Z. Anorg. Allg. Chem., 1994, 620, 737–742 CrossRef CAS.
- J. Akhtar, M. A. Malik, S. K. Stubbs, P. O'Brien, M. Helliwell and D. J. Binks, Eur. J. Inorg. Chem., 2011, 2984–2990 CrossRef CAS.
- J. Akhtar, M. Akhtar, M. A. Malik, P. O'Brien and J. Raftery, J. Am. Chem. Soc., 2012, 134, 2485–2487 CrossRef CAS PubMed.
- A. Molter, J. Rust, C. W. Lehmann and F. Mohr, Tetrahedron, 2012, 68, 10586–10591 CrossRef CAS.
- M. Dörner, J. M. Rautiainen, J. Rust, C. W. Lehmann and F. Mohr, Eur. J. Inorg. Chem., 2017, 789–797 CrossRef.
- A. Molter, S. Kathrein, B. Kircher and F. Mohr, Dalton Trans., 2018, 47, 5055–5064 RSC.
- J. Kuchar, J. Rust, C. W. Lehmann and F. Mohr, Eur. J. Inorg. Chem., 2018, 5215–5222 CrossRef CAS.
- B. David, U. Monkowius, J. Rust, C. W. Lehmann, L. Hyzak and F. Mohr, Dalton Trans., 2014, 43, 11059–11066 RSC.
- A. Molter and F. Mohr, Coord. Chem. Rev., 2010, 254, 19–45 CrossRef CAS.
- S. Gaillard, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2010, 46, 2742–2744 RSC.
- P. de Frémont, N. M. Scott, E. D. Stevens and S. P. Nolan, Organometallics, 2005, 24, 2411–2418 CrossRef.
- E. Schuh, C. Pflüger, A. Citta, A. Folda, M. P. Rigobello, A. Bindoli, A. Casini and F. Mohr, J. Med. Chem., 2012, 55, 5518–5528 CrossRef CAS PubMed.
- R. Rubbiani, E. Schuh, A. Meyer, J. Lemke, J. Wimberg, N. Metzler-Nolte, F. Meyer, F. Mohr and I. Ott, MedChemComm, 2013, 4, 942–948 RSC.
- M. V. Baker, P. J. Barnard, S. K. Brayshaw, J. L. Hickey, B. W. Skelton and A. H. White, Dalton Trans., 2005, 37–43 RSC.
- A. Molter, J. Rust, C. W. Lehmann and F. Mohr, Tetrahedron, 2012, 68, 10586–10591 CrossRef CAS.
- R. Richter, J. Seiler, L. Beyer, O. Lindqvist and L. Andersen, Z. Anorg. Allg. Chem., 1985, 522, 171–183 CrossRef CAS.
- V. D. Schwade, L. Kirsten, A. Hagenbach, E. Schulz Lang and U. Abram, Polyhedron, 2013, 55, 155–161 CrossRef CAS.
- I. A. Kotzé, V. J. Smith, E. F. Kangara and K. R. Koch, New J. Chem., 2017, 41, 14995–15002 RSC.
- I. B. Douglass, J. Am. Chem. Soc., 1937, 59, 740–742 CrossRef CAS.
- A. Molter, J. Rust, C. W. Lehmann and F. Mohr, ARKIVOC, 2011, vi, 10–17 Search PubMed.
- K. H. König, L. Kaul, M. Kuge and M. Schuster, Liebigs Ann. Chem., 1987, 1115–1116 CrossRef.
- R. Usón, A. Laguna and M. Laguna, Inorg. Synth., 1989, 26, 85–91 Search PubMed.
- L. Hintermann, Beilstein J. Org. Chem., 2007, 3, 1–5 CrossRef PubMed.
- A. J. M. Duisenberg, L. M. J. Kroon-Batenburg and A. M. M. Schreurs, J. Appl. Crystallogr., 2003, 36, 220–229 CrossRef CAS.
- CrysAlisPro, Rigaku Oxford Diffraction Ltd, Oxford, UK, 2016.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- V. D. Schwade, A. Hagenbach, E. S. Lang, K. Klauke, F. Mohr and U. Abram, Eur. J. Inorg. Chem., 2014, 1949–1954 CrossRef CAS.
Footnote |
| † CCDC 1907692, 1907635 and 1907634. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c9nj02229e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |