
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStrong ion pair charge transfer interaction of 1,8-naphthalimide–bipyridinium conjugates with basic anions – towards the development of a new type of turn-on fluorescent anion sensors†
Zoltán
Szakács
 a,
Márton
Bojtár
bc,
Dóra
Hessz
ad,
Sylvia
Rousseva
a,
István
Bitter
b,
László
Drahos
c,
Michiel
Hilbers
e,
Hong
Zhang
e,
Mihály
Kállay
a and
Miklós
Kubinyi
*ad
a,
Márton
Bojtár
bc,
Dóra
Hessz
ad,
Sylvia
Rousseva
a,
István
Bitter
b,
László
Drahos
c,
Michiel
Hilbers
e,
Hong
Zhang
e,
Mihály
Kállay
a and
Miklós
Kubinyi
*ad
aDepartment of Physical Chemistry and Materials Science, Budapest University of Technology and Economics, 1521 Budapest, Hungary. E-mail: kubinyi@mail.bme.hu
bDepartment of Organic Chemistry and Technology, Budapest University of Technology and Economics, 1521 Budapest, Hungary
cInstitute of Organic Chemistry, Research Center for Natural Sciences, Hungarian Academy of Sciences, P. O. B. 286, 1519 Budapest, Hungary
dInstitute of Materials and Environmental Chemistry, Research Center for Natural Sciences, Hungarian Academy of Sciences, P. O. B. 286, 1519 Budapest, Hungary
eVan’t Hoff Institute for Molecular Sciences, University of Amsterdam, P. O. B. 94157, 1090 GD Amsterdam, The Netherlands
First published on 1st April 2019
Abstract
The electron transfer processes of two conjugates consisting of an N,N′-dimethyl-bipyridinium unit and an 1,8-naphthalimide or 4-piperidinyl-1,8-naphthalimide fluorophore have been investigated. The two dyads show only weak fluorescence due to the intramolecular photoinduced electron transfer (PET) from the naphthalimide to the bipyridinium moiety. The dyads form radical pairs with the Lewis-base anions such as fluoride, acetate and benzoate via intermolecular electron transfer from the anions to the bipyridinium units. The electron transfer from the anions is indicated by an intense coloration – due to the absorption of the bipyridinium radicals – and a dramatic enhancement of the fluorescence intensity due to the cancellation of intramolecular PET in the dyads. These results demonstrate the efficient operation of bipyridinium–naphthalimide conjugates as signal transduction units from which a new type of turn-on fluorescent anion sensors may be created.
Introduction
N,N′-Disubstituted bipyridinium dications (known also as viologens, V2+ in Fig. 1) are strong electron acceptors and they can be reduced to a radical cation V˙+ and a neutral form V0 in consecutive steps.1 Viologens form ion-pair charge transfer (IPCT) complexes with many inorganic and organic anions of electron donor nature. For example, N,N′-dimethyl-4,4′-bipyridinium (methyl viologen, MV2+) forms MV2+X− ion pairs with anions X− = F−, Cl−, Br−, I−, perchlorate, sulphate2 and tetraphenylborate,3–5 which have a specific charge transfer band in their absorption spectra. It falls between 350 and 500 nm, depending on the solvent and the counter anion.6 Exciting the ion pair at that transition, the excited species IPCT* converts into the biradical structure MV˙+X˙ via an electron transfer from the anion to the cation. The biradicaloid complexes of MV can be easily identified by transient absorption spectroscopy as the MV˙+ radical has a characteristic sharp peak around ∼395 nm and a broad structured band with a maximum at ∼605 nm in its absorption spectrum.7 The electron transfer reaction in the excited ion pairs takes place directly from the Franck–Condon state of the excited ion-pair.4,8 The lifetime of the biradical structures in solution falls in the fs or ps range.8,9 In a microcrystalline state or in a polymer film, their lifetimes are much longer, reaching up to several hours.10 These features make the IPCT complexes of bipyridinium derivatives attractive candidates for applications in optical storage and switching technologies.10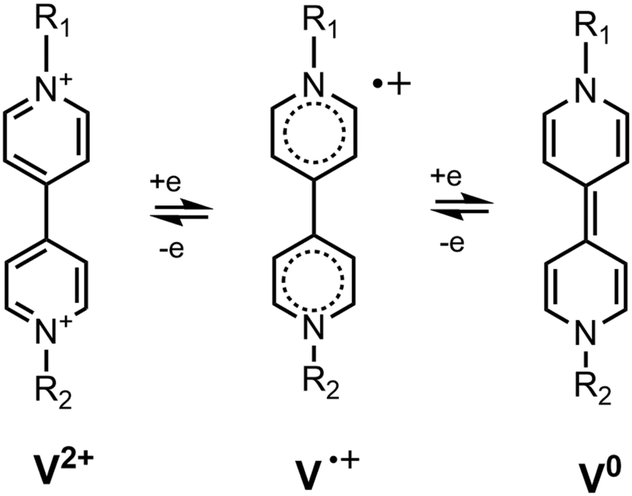 | ||
| Fig. 1 The two reduction steps of viologen derivatives. | ||
Attaching various coordination spheres to the viologen unit, colorimetric anion sensors have been constructed.11–15 They indicated the binding of the anion via a new absorption band that belonged to the IPCT transition. It was found in these studies that in polar aprotic solvents, the electron transfer between viologens and some basic anions took place directly – without photoexcitation – resulting in the formation of radicaloid species. The colourless samples turned yellow when weak IPCT complexes were formed and bright blue upon the formation of strong radicaloid complexes.
In the present work, we studied the optical responses of the naphthalimide–bipyridinium conjugates, NIV and PiNIV shown in Fig. 2, to the addition of Lewis base type anions which had been reported to form radicals directly with viologens.11–15 It was hoped that upon coupling a 1,8-naphthalimide (NI) fluorophore to the bipyridinium unit, the binding of anions will be indicated by fluorescence enhancement, in addition to coloration.
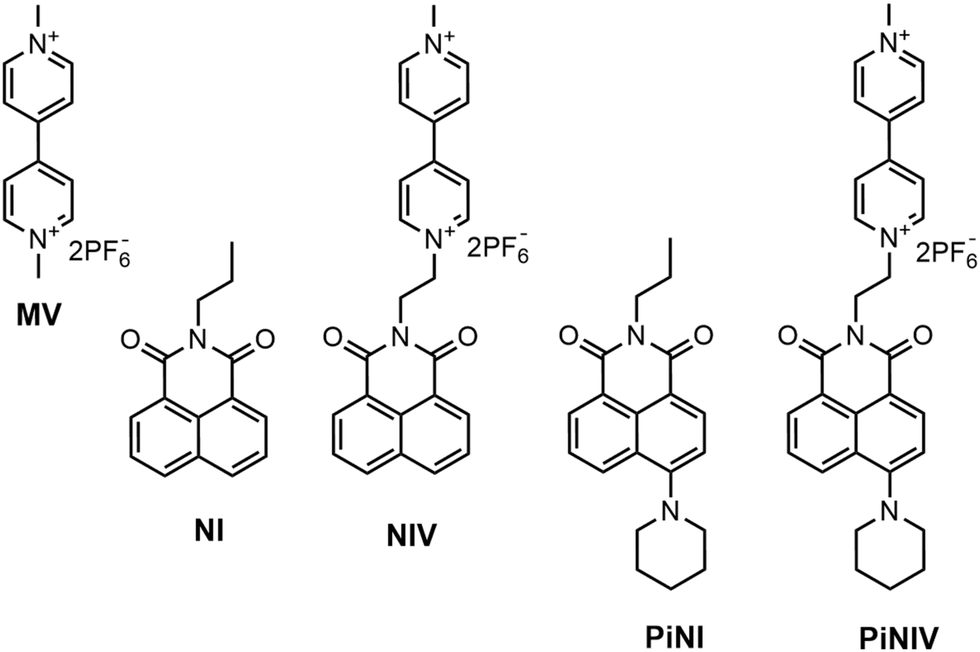 | ||
| Fig. 2 The investigated compounds, MV, NIV, and PiNIV and their reference compounds, NI and PiNI. | ||
A variety of ‘turn-on’ fluorescent chemosensors with NI fluorophore units have been reported.16–21 In general, the NI group is an electron acceptor in these sensors, and is linked to an electron donor substituent as the receptor site. The excitation of the NI group induces an electron transfer from the substituent to the NI moiety (photoinduced electron transfer, PET) resulting in quenched fluorescence. The binding of the analyte to the receptor site blocks the PET process which manifests in fluorescence enhancement. We note that in anion sensors of such types the naphthalimide signalling group is coupled most frequently to an urea or thiourea binding unit which coordinates the F−, AcO−, etc. analyte anions via hydrogen bonds.20 Thus, the analyte anions act as Brønsted bases in these sensors.
In contrast to these NI based sensors, in the presence of bipyridinium cations NIs act as electron donors in photoinduced electron transfer processes.22,23 This applies both for the mixtures of NI and viologen derivatives (intermolecular PET) and for covalently linked NI–viologen dyads (intramolecular PET), and the result is also fluorescence quenching – similar to that in NI–electron donor systems.22
PiNIV, the dyad substituted with a piperidine group in the 4-position is a new photoactive compound. It is expected that the introduction of the electron donor piperidine substituent will enhance the efficiency of the PET effect in the free sensor molecule and – as an additional advantage – will shift the absorption into the visible range.
Materials and methods
Preparation of the samples
The stock solutions of MV, NI, PiNI, NIV and PiNIV (c = 1 mM) for the investigation of the electron transfer and subsequent processes were made in anhydrous acetonitrile, and for the photophysical study in HPLC grade MeCN. Before the reactions, the solutions were deoxygenated by N2 bubbling for 10–15 min and the reactions were initiated by the addition of the anion using a N2 rinsed gastight Hamilton syringe. In this way, the reactants were kept water- and oxygen-free.The fluorescence quantum yields of the NIs were determined using quinine sulfate (in 0.1 M H2SO4) as reference.24,25
In the experiments on the intermolecular electron transfer, the tetramethylammonium (TMA) salts of the F−, Cl−, Br− and I− anions and the tetrabutylammonium (TBA) salts of the acetate (AcO−), benzoate (BzO−) and H2PO4− anions were used. The salts were purchased from Sigma-Aldrich and stored in a desiccator. The stock solutions of the anions (c = 0.5–0.25 mM depending on the solubility) were freshly prepared every day in a N2 atmosphere, using anhydrous acetonitrile and stored under a N2 atmosphere.
The NMR spectra were recorded in deoxygenated MeCN-d3 solutions.
Instrumentations
The absorption spectra were recorded using an Agilent 8453 spectrometer. The fluorescence emission spectra were recorded using a PerkinElmer LS-50B fluorescence spectrometer. The time-resolved fluorescence was measured using a lab-built time-correlated single photon counting (TCSPC) setup with a frequency-doubled Ti:sapphire laser (Chameleon Ultra, Coherent) as the light source. The IRF was of fwhm = 20–25 ps. The TCSPC system is described in detail in ref. 26.The NMR spectra were recorded using a 500 MHz Bruker Avance DRX-500 spectrometer.
The HR-MS spectra were recorded using a Q-TOF Premier mass spectrometer (Waters Corporation) using electrospray ionization in the positive mode.
The RP-HPLC-UV/Vis-MS measurements were performed using a Shimadzu LCMS-2020 instrument applying a Gemini C18 column. The chromatograms were detected using a UV–vis diode array (220–800 nm) and an MS system with electrospray ionization. More details on HPLC-MS experiments are presented in the ESI.†
The cyclic voltammetry (CV) measurements were performed using a Reference 600 potentiostat/galvanostat from Gamry Instruments. All the measurements were carried out in deoxygenated MeCN applying a continuous N2 flow over the cell, and the scan rate was 50 mV s−1. Pt electrodes were applied as the working and counter electrodes. The reduction potentials of MV, NIV and PiNIV were obtained using an Ag/AgCl (3 M aq. KCl) reference electrode and applying TBAPF6 at a concentration of 10 mM as the supporting electrolyte. The voltammograms of PiNIV were also recorded in the presence of the tetralkyammonium salts of F−, AcO− and BzO− anions. In these experiments an Ag/Ag+ (10 mM AgNO3 in MeCN) reference electrode was applied to avoid contact of the anhydrous reaction mixture with an aqueous phase.
Computational details
Quantum chemical calculations were performed to study the PET process of NIV using the Gaussian09 package of programs.27 The input geometry was created by conformational analysis using the Merck molecular force field (MMFF94) as implemented in the Marvin Beans program package.28 The minimal energy conformer was optimized at the density functional theory (DFT) level applying the PBE0 functional with the 6-311++G** basis set.29,30 This optimized ground-state geometry was applied as the initial structure in configuration interaction singles (CIS) level calculations using the same basis set to optimize the excited state geometry and calculate oscillator strengths.We note that we employed a relatively inaccurate CIS method instead of time-dependent DFT because the considered molecule has several low-lying CT states, and the latter method may yield very poor results for such states, while at least qualitatively correct results can be expected from CIS.
Results and discussion
Synthesis of naphthalimide–viologen conjugates
NI and PiNI were prepared as previously described.22,31 An overview of the synthesis of the conjugates NIV and PiNIV is given in Scheme 1. The initial 9-(2-bromoetyl)-naphthalimides 132 and 231 were first prepared according to the literature. Then, they were reacted with 4,4′-bipyridine, leading to N-ethyl-4,4′-bipyridyl-1,8-naphthalimide conjugates NIBP and PiNIBP. Methylation of these intermediates with methyl iodide yielded NIV and PiNIV. Hexafluorophosphate was chosen as the counter anion because of its solubility and non-coordinating properties. The details of the synthetic work, NMR and HRMS data are presented in the ESI.†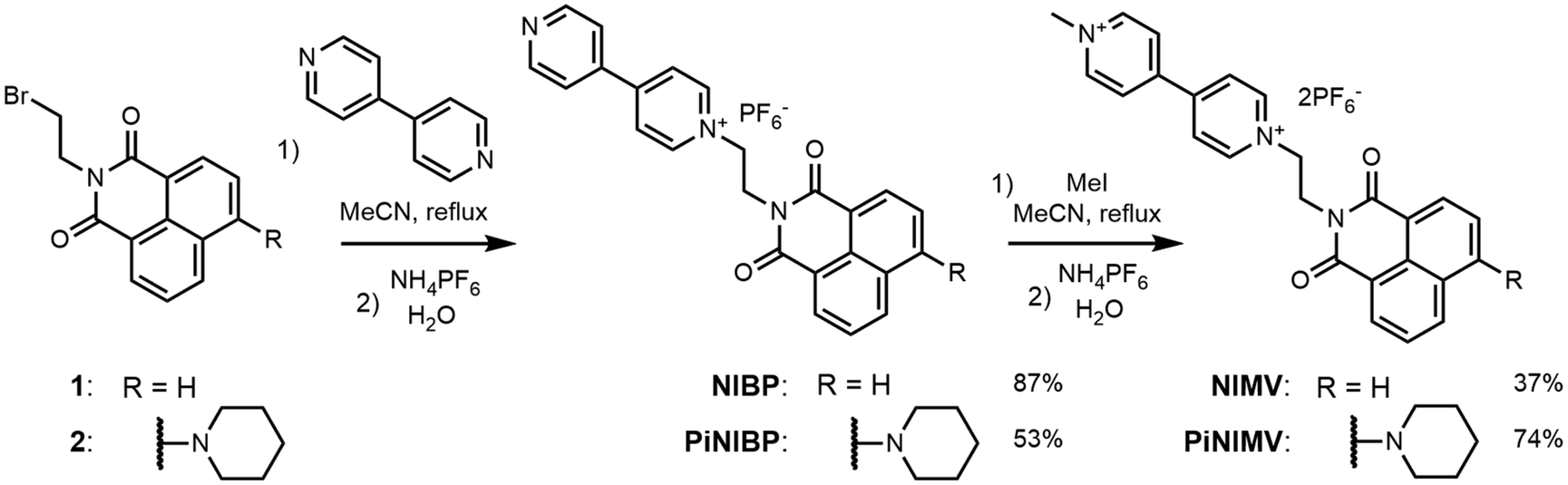 | ||
| Scheme 1 Synthesis of the naphthalimide–viologen dyads. | ||
PET in naphthalimide–viologen conjugates
The absorption and fluorescence spectra of NIV and PiNIV, together with the spectra of the NI, PiNI and MV reference compounds are displayed in Fig. 3. The photophysics of NI and PiNI was investigated in detail in a previous study.26 Here we only underline that the longest wavelength absorption band as well as the fluorescence band of NI show a substantial redshift in the spectra of PiNI: the absorption band from 331 nm to 405 nm and the fluorescence band from 375 to 535 nm. The absorption spectra of the dyads, NIV and PiNIV are almost the linear combinations of the spectra of MV and the respective naphthalimide reference compounds, NI and PiNI, which indicates only a weak coupling between the electronic structures of the donor and acceptor moieties of the conjugates. The longest wavelength bands of NI and PiNI show a minor redshift in the spectra of the dyads, which may be due to the dipole interactions with the viologen groups. The shapes of the emission bands of NIV and PiNIV are very similar to that of the bands of NI and PiNI, respectively, showing that there is only one radiative species, the naphthalimide unit in the conjugates. The fluorescence quantum yields of the viologen conjugates are lower, Φ = 0.0013 for NIV compared to 0.023 for NI and Φ = 0.00084 for PiNIV compared to 0.043 for PiNI. The reason for the weakened fluorescence is the PET from the naphthalimide to the viologen moiety that has been evidenced by flash photolysis and fluorescence quenching experiments.22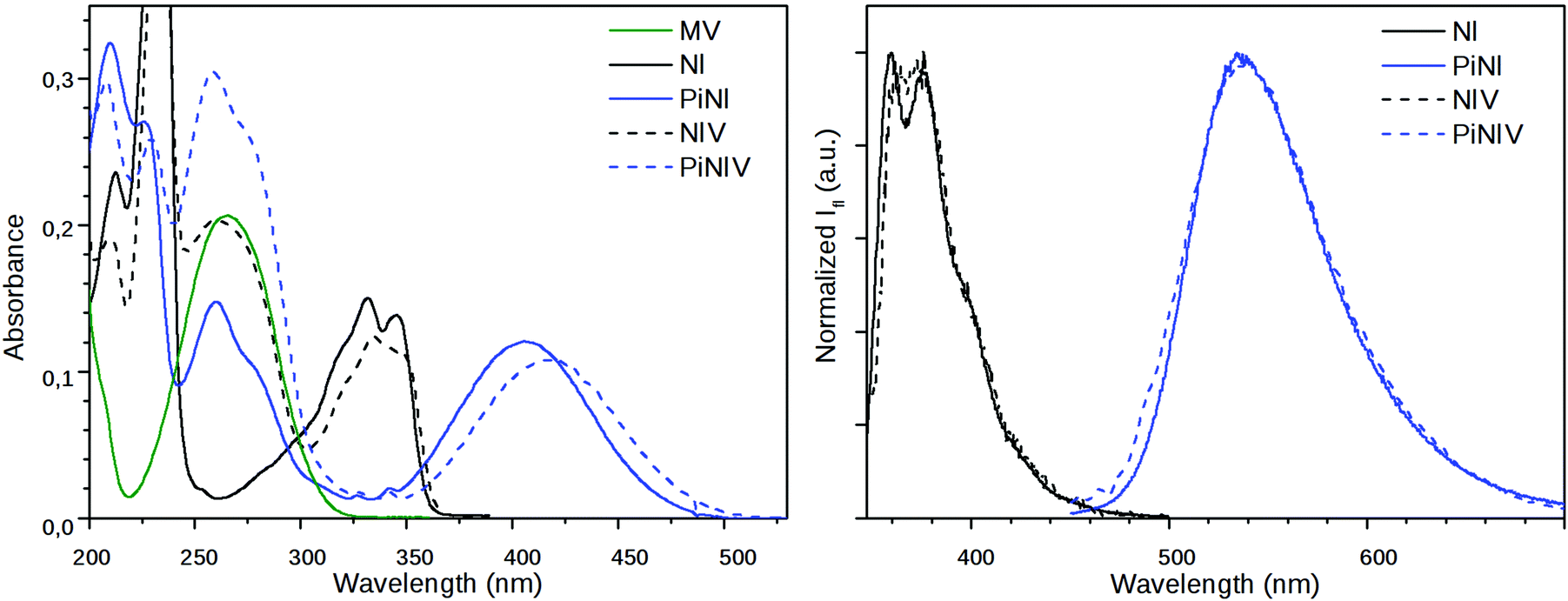 | ||
| Fig. 3 (Left) The absorption spectra of MV and the naphthalimide derivatives and (right) the emission spectra of the naphthalimides, c = 10 μM, λex = 340 nm (NI and NIV); λex = 410 nm (PiNI and PiNIV). | ||
The dynamics of the excited singlet species was investigated by fluorescence lifetime measurements. The decay curves of the two dyads are displayed in Fig. 4 and the fitted time constants are shown in Table 1. The reference compounds NI and PiNI show biexponential decays in MeCN.26 Presumably, the deactivation of NI involves an efficient S1–T2 intersystem crossing.33 The decay of PiNI is consistent with the conversion of the directly excited emissive molecule into a dark TICT species.26 The decays of the naphthalimide–viologen dyads, NIV and PiNIV, are triexponential, with two time constants close to the values of the time constants of the respective naphthalimides and an additional fast component (8 ps for NIV, at the limit of the temporal resolution of the TCSPC system, 55 ps for PiNIV) which are associated with the reversible formation of the biradical species from the singlet species. As these time constants are one and two orders of magnitude shorter than the other two time constants of the respective dyads, their reciprocals can be taken as the estimated values of the rate coefficients of the intramolecular electron transfer processes. This value is kET ∼ 1.25 × 1011 s−1 for NIV and kET ∼ 1.8 × 1010 s−1 for PiNIV. The kET values in the 109 to 1011 s−1 range were reported for the conjugates of viologens with various other electron acceptors.34–38
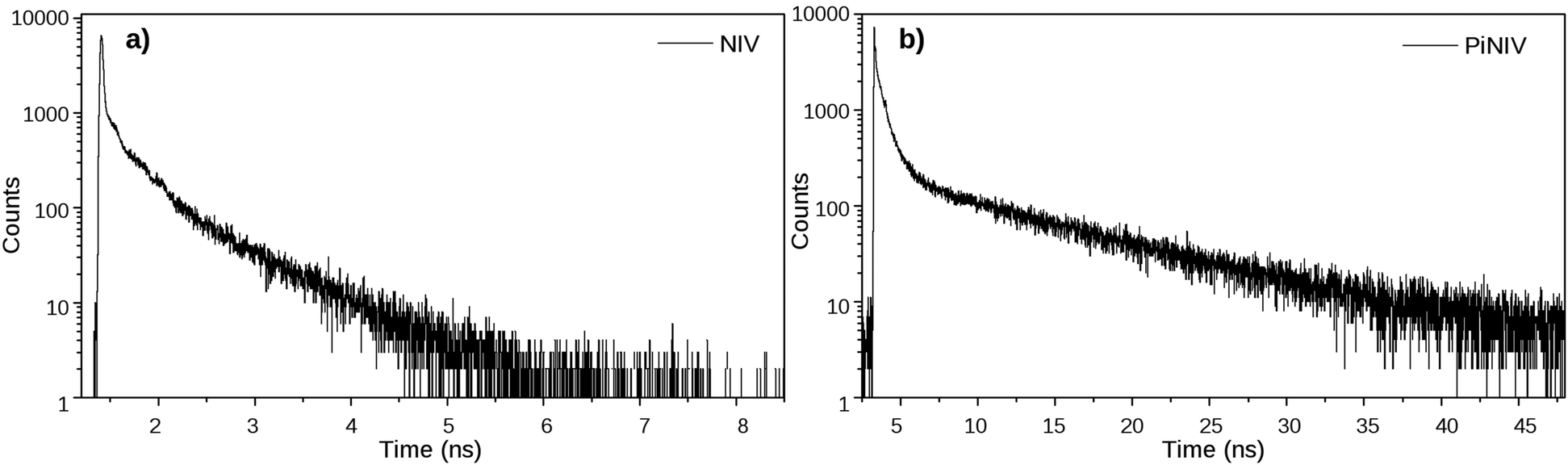 | ||
| Fig. 4 The fluorescence decay curves of NIV and PiNIV in MeCN, λex = 350 nm and λem = 370 nm (NIV); λex = 410 nm and λem = 540 nm (PiNIV). | ||
Theoretical calculations were performed to study the PET in the case of the simpler dyad, NIV. In the ground-state optimized structure, the dihedral angle between the two pyridinium rings was found to be 41°. Starting with the excited state CIS optimization from this geometry, the calculation resulted in a planar structure of the bipyridinium with an S1 → S0 transition probability of zero. We had attempted to find the radiative excited species of NIV applying very tight step sizes during the optimization and optimizing higher excited states as well (S2, S3, and S4) – assuming a crossing between the surfaces – but every calculation resulted in the same geometries and electronic structures. This suggests that the PET has a very small activation barrier or it is a barrierless process.
The biradical nature is represented well by the CIS calculations, and the dominant configuration (>98%) was found to be a determinant which corresponds to a HOMO → LUMO excitation, where the HOMO is a π orbital localized on the naphthalimide and the LUMO is a π* orbital of the bipyridinium. The schematic energy diagram of PET in NIV and the shapes of the Hartree–Fock orbitals contributing to the PET process can be seen in Fig. 5 (we note that in the dyad NIV, LUMO+8 corresponds to the LUMO of NI).
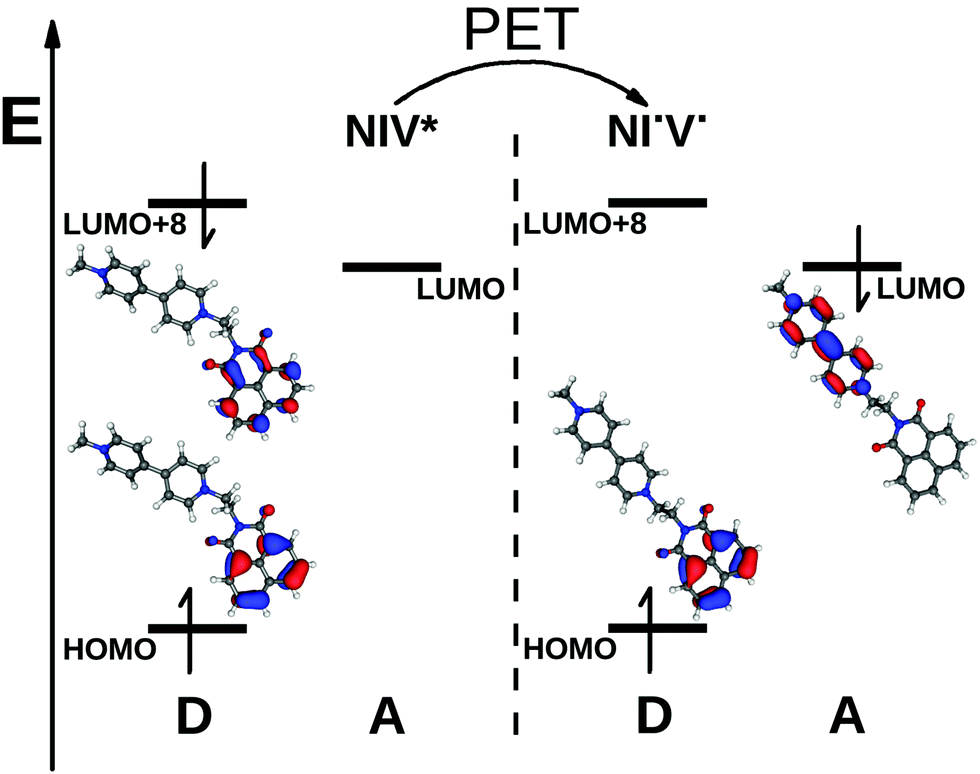 | ||
| Fig. 5 A schematic energy diagram of the PET in NIV and the shapes of the contributing MO's (NIV* corresponds to the Franck–Condon structure and NI˙V˙ to the biradical species with the excited state optimized structure). | ||
Strong charge transfer complexes with anions
The reactions of MV, NIV and PiNIV with anions were studied in anhydrous, deaerated acetonitrile solutions. It was found that under these conditions the reaction of the viologen derivatives with F−, AcO− and BzO− anions could be observed by the naked eye – the colourless samples of NIV turned to purple, and the yellow samples of PiNIV turned to green when the salts of these anions were added and their fluorescence became notably stronger (Fig. 6). The intermolecular electron transfer from these Lewis base type anions to the viologen units of the dyads was proved by the appearance of the characteristic absorption bands of the viologen radicals.7 The fluorescence intensity increased by ∼20 times when 5 equiv. of BzO− was added to the sample (10 and 15 times enhancements were observed in the case of F− and AcO−, respectively), demonstrating the chemosensing ability towards these anions. The addition of Cl−, I−, Br− and H2PO4− ions did not lead to detectable changes in the spectra, indicating the high selectivity.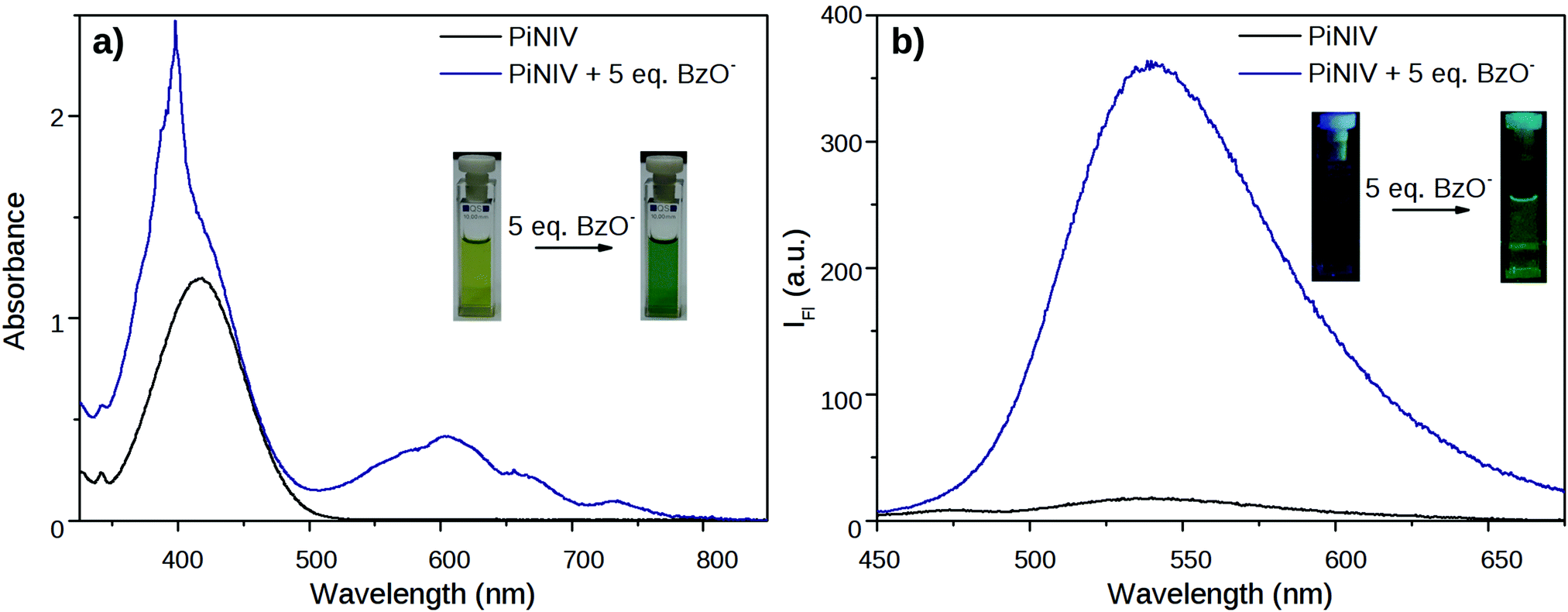 | ||
| Fig. 6 Changes in the absorption and fluorescence spectra of PiNIV 1 min after the addition of 5 equiv. of BzO− anions. The concentration of PiNIV was (a) 100 μM and (b) 10 μM. | ||
The 1H NMR spectra of the conjugates also changed spectacularly when F− or the other Lewis base type anions were added to the solution of the viologen derivatives. As shown in Fig. 7, the signals of the bipyridinium units and the ethylene bridges disappeared from the spectra, marking the region of the unpaired spin density.39
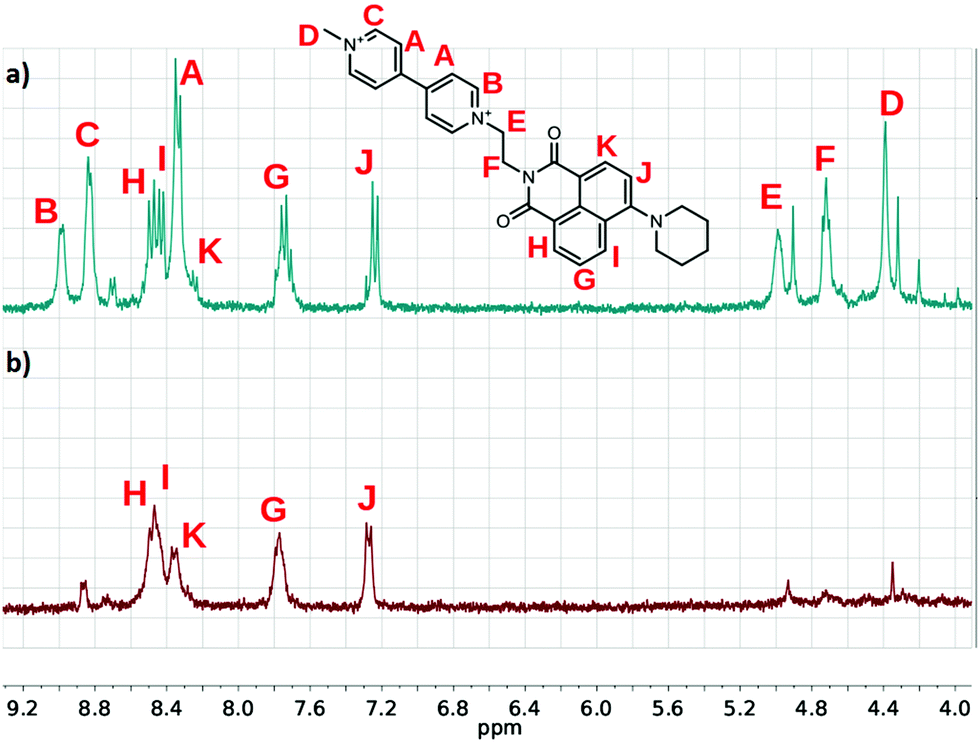 | ||
Fig. 7 Partial 1H NMR spectra of (a) PiNIV and (b) its 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture with TMAF (MeCN-d3, 0.5 mM). :1 mixture with TMAF (MeCN-d3, 0.5 mM). | ||
Degradation products
The coloration of the mixtures of NIV and PiNIV with the salts of F−, AcO− and BzO− anions attributed to the viologen radicals lasted for a long time – a few days or a few hours – depending on the temperature. Subsequently, the structured absorption bands of the radical cation around 400 and 600 nm decayed (see Fig. 8). A new absorption band around 370 nm, assignable to the doubly reduced viologen derivatives40 (V0 in Fig. 1) could not be observed. In contrast, the band of the naphthalimide unit around 420 nm became more pronounced.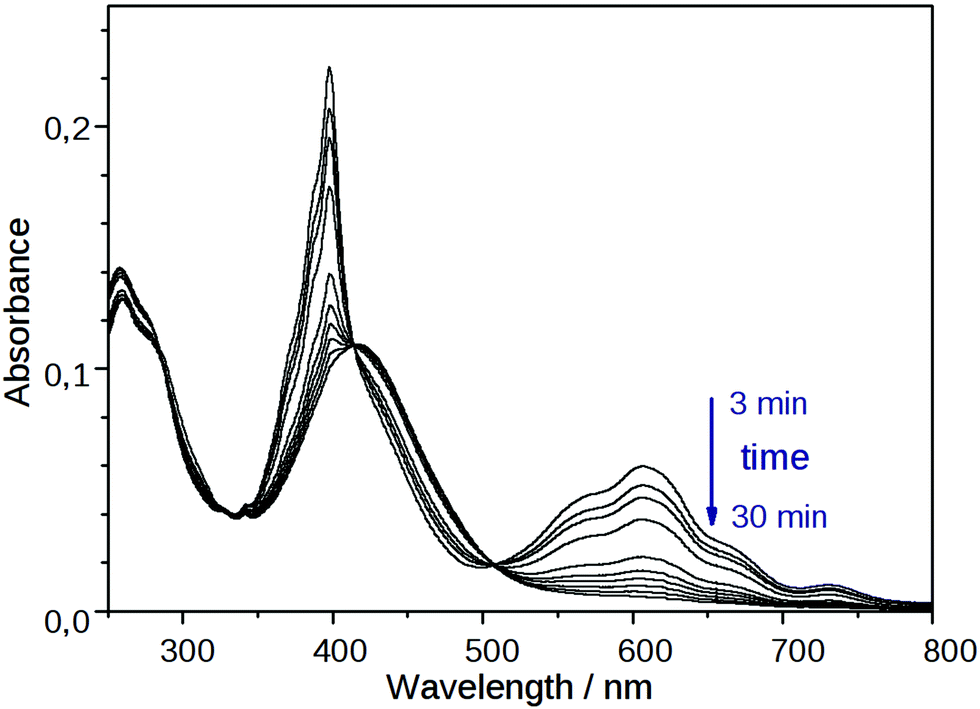 | ||
| Fig. 8 Temporal change in the absorption spectrum of PiNIV (10 μM) after the addition of 2 equiv. of F− anions. | ||
Control experiments were performed using more concentrated (c = 0.5 mM) deaerated solutions stored under N2 overpressure. The signal of the radical cation decayed in the concentrated solutions as well, confirming that a slow decomposition of the radical cations took place even when the ratio of the dissolved O2 was negligible.
The main degradation products of NIV and PiNIV in the presence of TMAF were identified by LC-MS analysis and HRMS. The results are illustrated in Fig. 9. It was found that the dominant degradation routes were the dealkylation of the pyridinium N-atoms, i.e., the demethylation of the pyridinium unit and the splitting of the dyad into an N-methylpyridinium cation and the respective 9-vinyl-naphthalimide. The reaction mixtures of NIV and PiNIV with the salts of AcO− and BzO− ions were also analysed and the respective demethylated molecules were identified as the main degradation products. Further details of the analysis are presented in the ESI.†
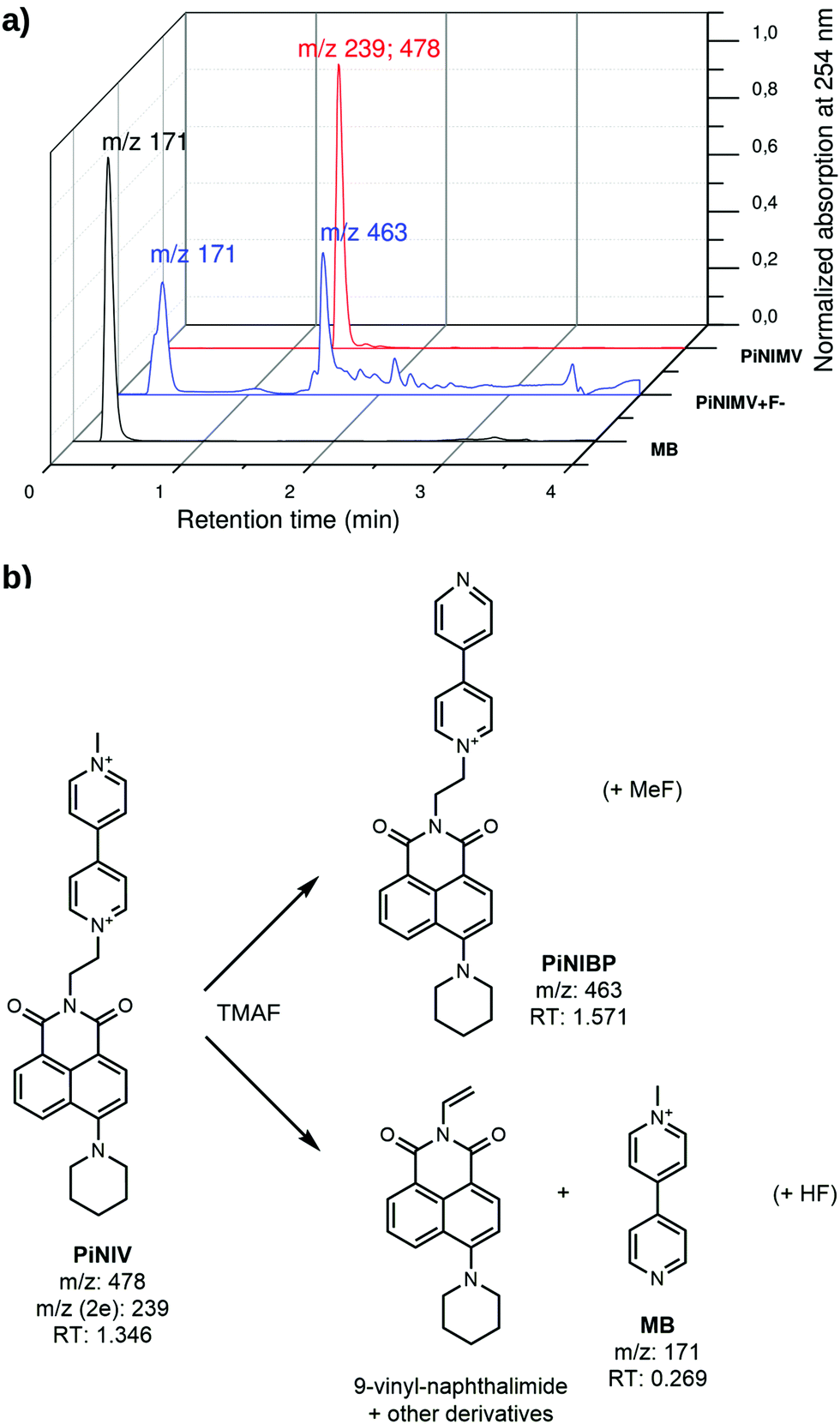 | ||
| Fig. 9 (a) LC-MS traces of PiNV, PiNIV + fluoride and MB and (b) degradation routes of PiNIV. | ||
It is known from the literature that in strongly basic solutions viologens undergo a dealkylation reaction,1 and our results suggest a reaction mechanism shown in Scheme S5 in the ESI,† in which there are radical intermediates formed via a complete electron transfer from the anion to the viologen.
Kinetics of anion binding
The kinetics of the reactions of MV, NIV and PiNIV with the anions F−, AcO− and BzO− was investigated by measuring the temporal variation of the absorption spectra of the reaction mixtures. The solvent used was anhydrous MeCN, and the solutions of the reactants were deoxygenated before mixing. The initial concentrations were [MV]0, [NIV]0, and [PiNIV]0 = 20 μM, [F−]0 and [AcO−]0 = 40 μM, and [BzO−]0 = 100 μM – and the reactions of the latter anion were slower.Preliminary experiments were carried out to check if weak IPCT complexes were present in a detectable concentration. The characteristic absorption band of the weak complexes of viologens around 400 nm did not appear at the early stage of the electron transfer reaction. However, the formation of weak IPCT complexes could not be excluded at later stages, and thus the spectra were recorded employing a 500 nm cut-off filter to avoid the photoinduced generation of the viologen radicals.
The concentrations of the biradical complexes were calculated from the absorbance values at 605 nm where the MV˙+ radical has a band maximum with an absorption coefficient of 13900 M−1 cm−1.41 To determine the activation barrier, experiments were carried out at five different temperatures between 283 and 323 K.
The concentration profiles of the viologen radical were analysed in terms of the reaction scheme shown in Scheme 2. In addition to the second-order electron transfer process, the scheme involved – as a simple approach – the recombination of the radical species and their decomposition as first-order reactions. The differential rate equations for this scheme are as follows:
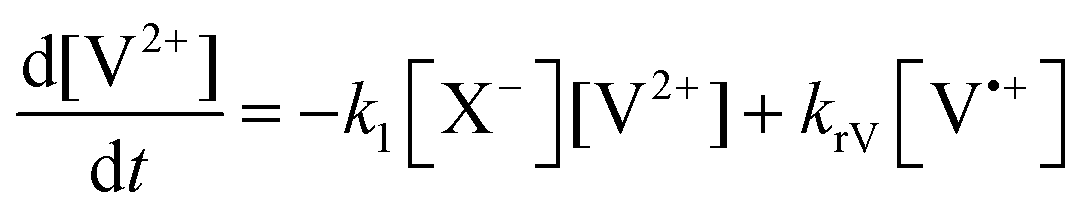 | (1) |
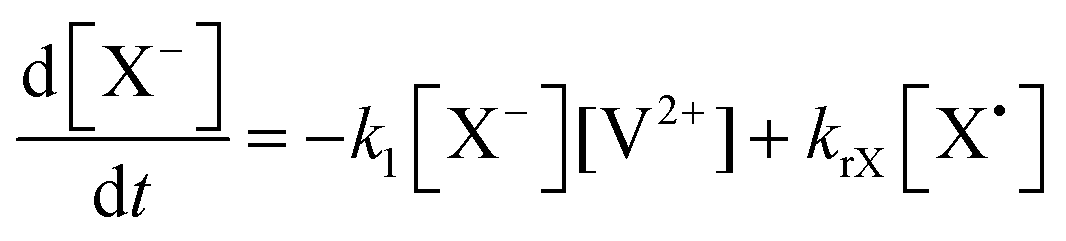 | (2) |
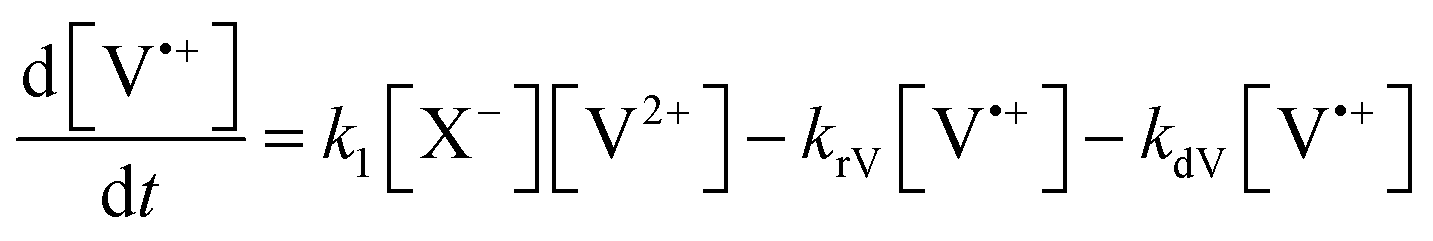 | (3) |
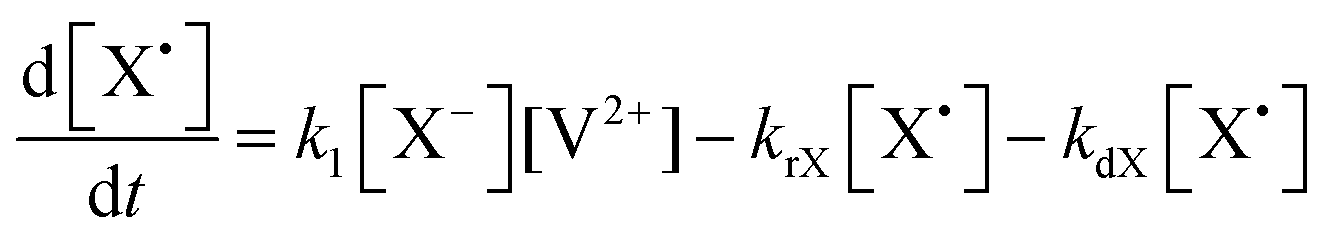 | (4) |
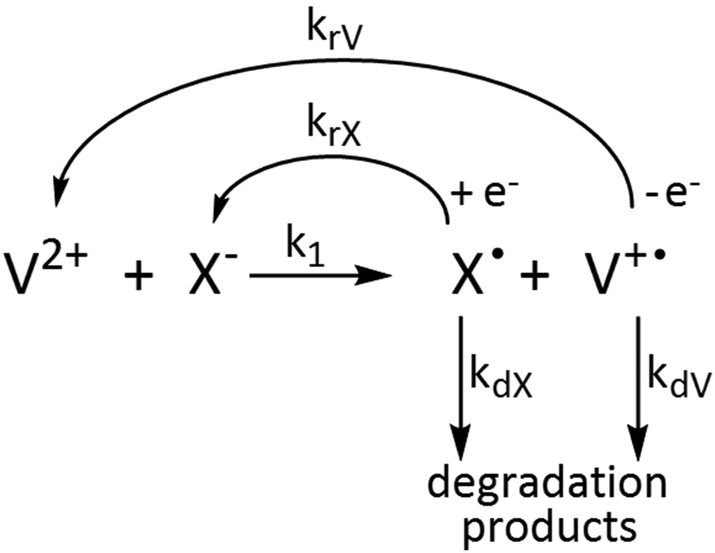 | ||
| Scheme 2 Kinetic scheme of the reactions of viologens with basic anions. | ||
The eqn (1)–(4) were solved numerically and the rate constants, k1, kr′s and kd′s for the mixtures of MV2+, NIV2+ and PiNIV2+ dications with F−, AcO− and BzO− anions were determined from the measured concentration profiles of the V˙+ radicals applying least square fittings. As can be seen in Fig. 10 (a typical result of the fitting), the scheme was suitable for the description of the kinetics.
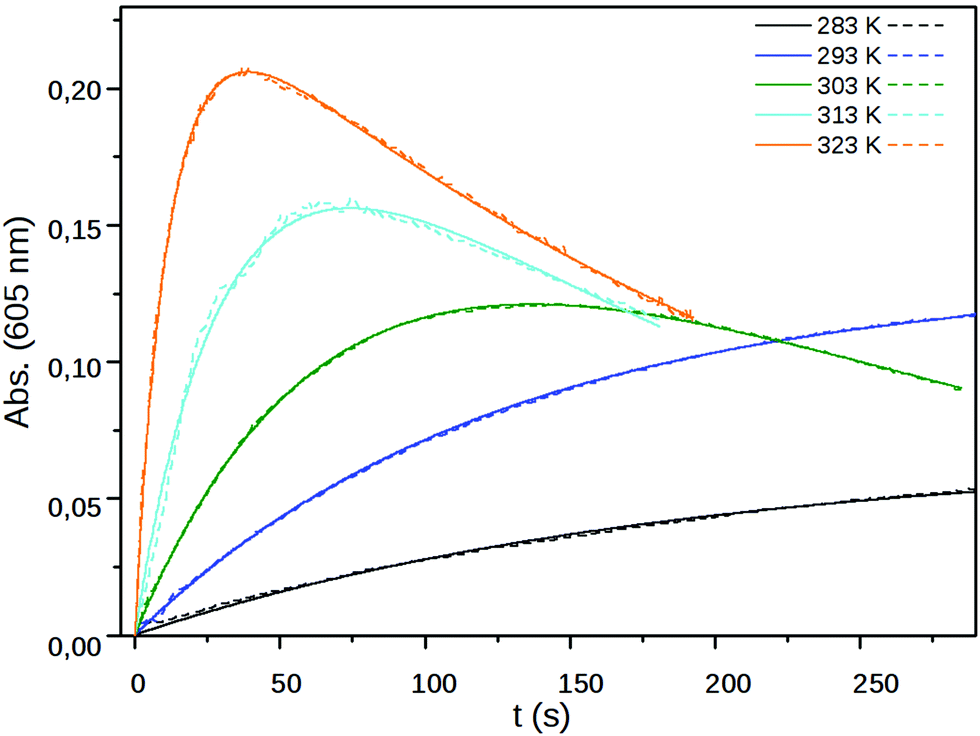 | ||
| Fig. 10 The measured (dashed line) and fitted (solid line) absorbance at 605 nm of PiNIV (c = 20 μM) after the addition (t = 0 s) of 100 μM BzO− as a function of time at five different temperatures. | ||
We present here the values of the rate coefficients of the intermolecular electron transfer, k1, only. In the initial period of the reactions where k1 is the dominant parameter, the effect of the remaining dissolved oxygen is negligible. The kd and kr parameters may have some contributions from unconsidered reactions, e.g. the reactions with traces of dissolved O2.
The fitted k1 rate constants are displayed in Table 2 and the parameters of the Arrhenius-plots (Fig. 11) are shown in Table 3. As can be seen in Table 2, the rate coefficient of the X− → V2+ electron transfer strongly depends on the applied anion and is much less sensitive to the substituent attached to the viologen unit. The electron transfer processes from BzO− have the lowest rate coefficients and have the highest activation barriers – this anion has the lowest surface charge density.
| Anion | Viologen | k 1/M−1 s−1 | ||||
|---|---|---|---|---|---|---|
| 283 K | 293 K | 303 K | 313 K | 323 K | ||
| F− | MV 2+ | 169 | 639 | 1624 | 3690 | 5200 |
| NIV 2+ | 149 | 268 | 681 | 944 | 1290 | |
| PiNIV 2+ | 161 | 380 | 729 | 1460 | 3740 | |
| AcO− | MV 2+ | 137 | 342 | 855 | 1790 | 3670 |
| NIV 2+ | 180 | 560 | 1310 | 2590 | 4290 | |
| PiNIV 2+ | 166 | 376 | 1080 | 2680 | 6660 | |
| BzO− | MV 2+ | 4.80 | 15.7 | 54.7 | 151 | 463 |
| NIV 2+ | 13.3 | 43.6 | 86.4 | 247 | 477 | |
| PiNIV 2+ | 12.9 | 37.7 | 93.6 | 253 | 765 | |
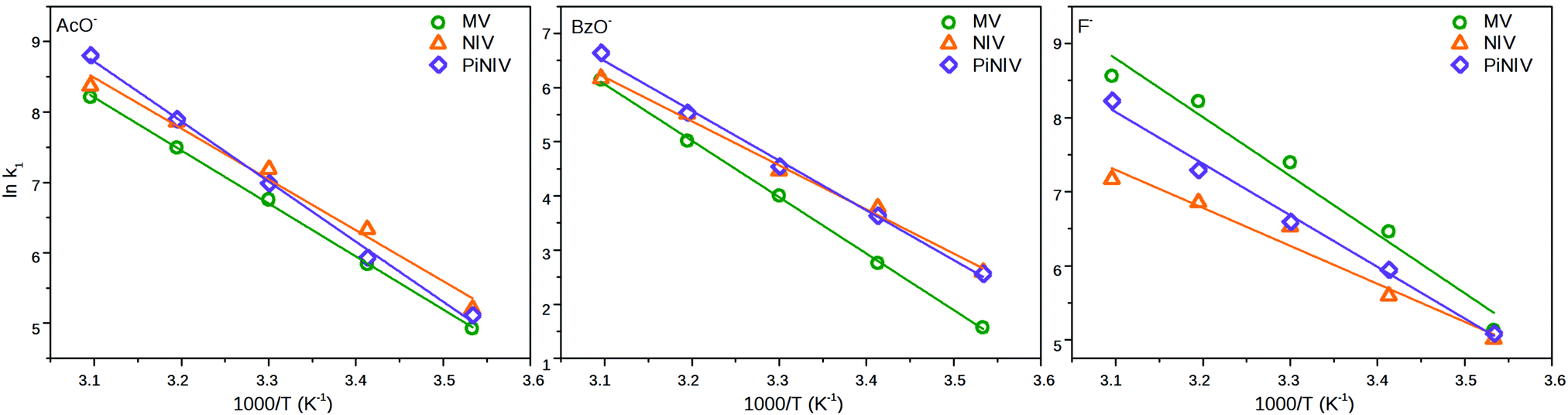 | ||
| Fig. 11 The Arrhenius plots of the rate coefficients of the electron transfer of MV2+, NIV2+ and PiNIV2+ with the anions, F−, AcO− and BzO−. | ||
| AcO− | BzO− | F− | ||||
|---|---|---|---|---|---|---|
| E a (kJ mol−1) | lnA |
E a (kJ mol−1) | lnA |
E a (kJ mol−1) | lnA |
|
| MV 2+ | 62.6 ± 0.9 | 31.6 ± 0.4 | 86.7 ± 1.3 | 38.4 ± 0,5 | 65.9 ± 6.4 | 33.3 ± 2.5 |
| NIV 2+ | 60.1 ± 4.0 | 30.9 ± 1.6 | 67.7 ± 2.9 | 31.4 ± 1.2 | 42.6 ± 4.5 | 23.2 ± 1.8 |
| PiNIV 2+ | 71.0 ± 2.2 | 35.2 ± 0.9 | 76.4 ± 2.6 | 35.0 ± 1.0 | 57.9 ± 2.8 | 29.7 ± 1.1 |
Cyclic voltammetry
The redox properties of the viologen derivatives were also characterized by cyclic voltammetry. An Ag|AgCl|KCl (aq., 3 M) reference electrode was chosen to allow a comparison of the reduction potentials obtained for MV2+ with the values reported in the literature.22,42 The reduction potentials obtained for MV2+, NIV2+ and PiNIV2+ are displayed in Table 4. The potentials E1/2(red1) belong to the V2+ + e− ⇌ V˙+ reduction step and the potentials E1/2(red2) to the V˙+ + e− ⇌ V0 process. The closeness of the E1/2(red1) and E1/2(red2) values for NIV2+ and PiNIV2+ to the respective values of MV2+ shows that the electronic systems of the bipyridinium moieties are not affected by the naphthalimide substituents. Waves corresponding to the oxidation of the naphthalimide units of NIV and PiNIV were not observed up to the oxidation potential of the MeCN solvent.| E 1/2(red1) (V) | E 1/2(red2) (V) | |
|---|---|---|
| MV 2+ | −0.340 | −0.784 |
| NIV 2+ | −0.315 | −0.769 |
| PiNIV 2+ | −0.329 | −0.816 |
To gain insight into the intermolecular electron transfer between viologens and Lewis base anions, the cyclic voltammograms of PiNIV were also recorded in the presence of 5 equiv. of F− and AcO− anions and of 15 equiv. of BzO− anion. As the radical ionic complexes are stable only in water-free solvents, an Ag|Ag+|AgNO3 (10 mM in MeCN) reference electrode was used in these experiments. The results are illustrated in Fig. 12, on the example of the voltammogram of PiNIV in the presence of F− ions. The wave belonging to the V2+ + e− ⇌ V˙+ step was flattened upon the addition of F− anions, whereas the wave of the second reduction step still appeared. The reduction of degradation products was also observed in the voltammograms with lower reduction potentials. A plausible explanation for this behavior is that the PiNIV2+ dications are reduced chemically by F− ions, forming the [PiNIV˙+–F˙]complex as an intermediate, from which the radical cations PiNIV˙+ are reduced further electrochemically into the neutral species, PiNIV0. The addition of the AcO− resulted in similar changes in the voltammogram, and the addition of BzO− caused only minor changes, in accord with its lower reactivity (see Fig. S7 in the ESI†).
 | ||
| Fig. 12 Cyclic voltammograms of 0.2 mM PiNIV in acetonitrile solution in the absence and in the presence of 5 equiv. TMAF. | ||
For comparison, the voltammograms of the tetraalkylammonium salts of the anions F−, AcO− and BzO− were also recorded (see Fig. S8 in the ESI†). They showed only an anodic peak which appeared between 1.1 and 1.3 V. The lack of the cathodic peak indicated that the three anions were oxidized irreversibly.43
Conclusion
In the present work it was shown that 1,8-naphthalimide–viologen conjugates can act as signal transducers in turn-on fluorescent chemosensors for Lewis base anions, like F−, AcO− and BzO−. Their operation is based upon a combination of intra- and intermolecular electron transfer: the intramolecular electron transfer from the naphthalimide to the viologen moiety of the unbound dyad quenches the fluorescence. The Lewis base type anions reduce the dyads into monocationic radicals, with the bipyridinium unit and the ethylene bridge as the region of unpaired spin density. The intramolecular PET is supressed in these radicals which results in a strong fluorescence enhancement. We hope that our results will contribute to the development of a new type of fluorescent sensor family which is based on the blocking of the PET by the intermolecular electron transfer with the basic anions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Higher Education Excellence Program of the Ministry of Human Capacities (BME FIKP-BIO) and by LASERLAB-EUROPE (grant no. LLAMS002347, European Union's Horizon 2020 research and innovation programme). M. Kállay is grateful for the financial support from the National Research, Development, and Innovation Office (NKFIH, Grant No. KKP126451). We are grateful to the MTA-BME “Lendület” Chemical Nanosensors Research Group for their help with the cyclic voltammetric measurements.Notes and references
- C. L. Bird and A. T. Kuhn, Electrochemistry of the Viologens, Chem. Soc. Rev., 1981, 10, 49–82 RSC.
- P. M. S. Monk, C. Turner and S. P. Akhtar, Electrochim. Acta, 1999, 44, 4817–4826 CrossRef CAS.
- Y. Isoda, H. Kawai, S. Muta and T. Nagamura, J. Photochem. Photobiol., A, 1996, 97, 113–120 CrossRef CAS.
- F. Ito and T. Nagamura, J. Photochem. Photobiol., C, 2007, 8, 174–190 CrossRef CAS.
- W. G. Santos, D. S. Budkina, V. M. Deflon, A. N. Tarnovsky, D. R. Cardoso and M. D. E. Forbes, J. Am. Chem. Soc., 2017, 139, 7681–7684 CrossRef CAS PubMed.
- M. S. Monk, N. M. Hodgkinson and R. D. Partridge, Dyes Pigm., 1999, 43, 241–251 CrossRef.
- T. W. Ebbesen, L. E. Manring and K. S. Peters, J. Am. Chem. Soc., 1984, 106, 7400–7404 CrossRef CAS.
- T. Nagamura, H. Sakaguchi, T. Ito and S. Muta, Mol. Cryst. Liq. Cryst., 1994, 247, 39–48 CrossRef CAS.
- W. Jarzeba, S. Pommeret and J. C. Mialocq, Chem. Phys. Lett., 2001, 333, 419–426 CrossRef CAS.
- T. Nagamura, Mol. Cryst. Liq. Cryst., 1993, 224, 75–83 CrossRef CAS.
- R. Kannappan, C. Bucher, E. Saint-Aman, J. C. Moutet, A. Milet, M. Oltean, E. Métay, S. Pellet-Rostaing, M. Lemaire and C. Chaix, New J. Chem., 2010, 34, 1373–1386 RSC.
- J. J. Park, Y. H. Kim, S. Rhim and J. Kang, Tetrahedron Lett., 2012, 53, 247–252 CrossRef CAS.
- Y. H. Kim, S. Rhim, J. J. Park and J. Kang, J. Inclusion Phenom. Macrocyclic Chem., 2012, 74, 317–323 CrossRef CAS.
- A. N. Swinburne, M. J. Paterson, K. H. Fischer, S. J. Dickson, E. V. B. Wallace, W. J. Belcher, A. Beeby and J. W. Steed, Chem. – Eur. J., 2010, 16, 1480–1492 CrossRef CAS.
- Z. Y. Dong, D. W. Zhang, X. Z. Jiang, H. Li and G. H. Gao, Chin. Chem. Lett., 2013, 24, 688–690 CrossRef CAS.
- A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef CAS PubMed.
- R. A. Bissell, A. P. de Silva, H. Q. N. Gunaratne, P. L. M. Lynch, G. E. M. Maguire, C. P. McCoy and K. R. A. S. Sandanayake, Top. Curr. Chem., 1993, 168, 223–264 CrossRef CAS.
- G. R. C. Hamilton, S. K. Sahoo, S. Kamila, N. Singh, N. Kaur, B. W. Hyland and J. F. Callan, Chem. Soc. Rev., 2015, 44, 4415–4432 RSC.
- A. P. de Silva, H. Q. N. Gunaratne and C. P. McCoy, Nature, 1993, 364, 42–44 CrossRef.
- R. M. Duke, E. B. Veale, F. M. Pfeffer, P. E. Kruger and T. Gunnlaugsson, Chem. Soc. Rev., 2010, 39, 3936–3953 RSC.
- J. B. Czirok, M. Bojtár, D. Hessz, P. Baranyai, L. Drahos, M. Kubinyi and I. Bitter, Sens. Actuators, B, 2013, 182, 280–287 CrossRef CAS.
- T. P. Le, J. E. Rogers and L. A. Kelly, J. Phys. Chem. A, 2000, 104, 6778–6785 CrossRef CAS.
- J. E. Rogers, T. P. Le and L. A. Kelly, Photochem. Photobiol., 2001, 73, 223–229 CrossRef CAS.
- J. B. Birks, J. Res. Natl. Bur. Stand., Sect. A, 1976, 80, 389–399 CrossRef.
- M. J. Adams, J. G. Highfield and G. F. Kirkbright, Anal. Chem., 1977, 49, 1850–1852 CrossRef CAS.
- Z. Szakács, S. Rousseva, M. Bojtár, D. Hessz, I. Bitter, M. Kállay, M. Hilbers, H. Zhang and M. Kubinyi, Phys. Chem. Chem. Phys., 2018, 20, 10155–10164 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- MarvinSketch 16.4.25, ChemAxon, 2016, http://chemaxon.com.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- D. Wang, X. Zhang, C. He and C. Y. Duan, Org. Biomol. Chem., 2010, 8, 2923–2925 RSC.
- U. Hossain, S. Sengupta and S. Bhattacharya, Bioorg. Med. Chem., 2005, 13, 5750–5758 CrossRef PubMed.
- V. Wintgens, P. Valat, J. Kossányi, L. Biczók, A. Demeter and T. Bérces, J. Chem. Soc., Faraday Trans., 1994, 90, 411–421 RSC.
- J. W. Park, B. A. Lee and S. Y. Lee, J. Phys. Chem. B, 1998, 102, 8209–8215 CrossRef CAS.
- J. Joseph, N. V. Eldho and D. Ramaiah, J. Phys. Chem. B, 2003, 107, 4444–4450 CrossRef CAS.
- M. Hariharan, J. Joseph and D. Ramaiah, J. Phys. Chem. B, 2006, 110, 24678–24686 CrossRef CAS PubMed.
- Y. Araki, H.-X. Luo, T. Nakamura, M. Fujitsuka, O. Ito, H. Kanato, Y. Aso and T. Otsubo, J. Phys. Chem. A, 2004, 108, 10649–10655 CrossRef CAS.
- D. Frath, J. E. Yarnell, G. Ulrich, F. N. Castellano and R. Ziessel, ChemPhysChem, 2013, 14, 3348–3354 CrossRef CAS PubMed.
- L. Chen, H. Willcock, C. J. Wedge, F. Hartl, H. M. Colquhoun and B. W. Greenland, Org. Biomol. Chem., 2016, 14, 980–988 RSC.
- M. Mohammad, J. Org. Chem., 1987, 52, 2779–2782 CrossRef CAS.
- T. Watanabe and K. Honda, J. Phys. Chem., 1982, 86, 2617–2619 CrossRef CAS.
- A. C. Benniston, A. Harriman, P.-Y. Li, J. P. Rostron, R. W. Harrington and W. Clegg, Chem. – Eur. J., 2007, 13, 7838–7851 CrossRef CAS PubMed.
- M. Galicia and F. J. Gonzalez, J. Electrochem. Soc., 2002, 149, D46–D50 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nj00382g |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |