
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New enantiopure binaphthyl-cinchona thiosquaramides: synthesis and application for enantioselective organocatalysis†
Sándor
Nagy
 a,
Gyula
Dargó
a,
Péter
Kisszékelyi
a,
Zsuzsanna
Fehér
a,
András
Simon
b,
Júlia
Barabás
b,
Tibor
Höltzl
bc,
Béla
Mátravölgyi
a,
Levente
Kárpáti
d,
László
Drahos
e,
Péter
Huszthy
a and
József
Kupai
*a
a,
Gyula
Dargó
a,
Péter
Kisszékelyi
a,
Zsuzsanna
Fehér
a,
András
Simon
b,
Júlia
Barabás
b,
Tibor
Höltzl
bc,
Béla
Mátravölgyi
a,
Levente
Kárpáti
d,
László
Drahos
e,
Péter
Huszthy
a and
József
Kupai
*a
aDepartment of Organic Chemistry & Technology, Budapest University of Technology & Economics, Szent Gellért tér 4, Budapest, 1111, Hungary. E-mail: jkupai@mail.bme.hu; Tel: +36-1463-2229
bDepartment of Inorganic & Analytical Chemistry, Budapest University of Technology & Economics, Szent Gellért tér 4, Budapest, 1111, Hungary
cFurukawa Electric Institute of Technology, Késmárk utca 28/A, Budapest, 1157, Hungary
dLaboratory of Plastics & Rubber Technology, Budapest University of Technology & Economics, Műegyetem rkp. 3., Budapest, 1111, Hungary
eMS Proteomics Research Group, Research Centre for Natural Sciences, Hungarian Academy of Sciences, Magyar Tudósok körútja 2, Budapest, 1117, Hungary
First published on 21st March 2019
Abstract
This work presents the first successful applications of cinchona-thiosquaramides in asymmetric reactions. Binaphthyl-cinchona squaramides and thiosquaramides were synthesised, and then used as organocatalysts to promote the catalytic enantioselective Michael addition reaction of pentane-2,4-dione to trans-β-nitrostyrene with excellent yields (up to 99%) and enantioselectivities (up to 99% ee) at as low as 0.2 mol% catalyst loadings. Thiosquaramides gave higher enantioselectivities (up to 92% ee) in conjugate addition reaction of lawsone to β,γ-unsaturated α-keto ester than its oxo analogue, with high yields (up to 100%). Also, only thiosquaramide could catalyse the aza-Diels–Alder addition reaction of 2-siloxydiene to benzylideneacetone. Furthermore, quantum chemical computations showed that the geometrical structure of binaphthyl-cinchona thiosquaramide is similar to that of squaramide.
Introduction
Over the last 20 years an explosive growth of research in the field of asymmetric synthesis has taken place.1–5 The aim of enantioselective synthesis or catalysis is to produce enantiopure products (a single enantiomer as the ultimate goal) starting from achiral substrates by chiral reagents, catalysts or auxiliaries. The role of these chiral reagents or catalysts is to generate diastereomeric transition states leading to the formation of two enantiomers so that one of them is preferentially formed. In recent years binaphthyl compounds have found frequent applications in the design of various asymmetric organocatalysts,6–12 since the binaphthyl structure is an attractive platform for catalyst development, particularly in light of their axially chiral characteristic. The enantiomeric atropisomers of 2,2′-substituted binaphthyls have been developed to exploit the axial dissymmetry induced by the restricted rotation about the biaryl bond.8 Application of binaphthyl unit as a chiral scaffold is well-established in asymmetric catalysis, especially the binaphthyl-based bifunctional mono-, bisthioureas, and cinchona-squaramides (Fig. 1) have been found beneficial in asymmetric Michael addition.13–16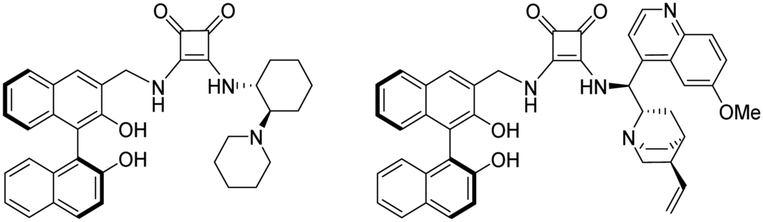 | ||
| Fig. 1 Binaphthyl-based bifunctional squaramides. | ||
Nowadays, squaramides have become a dominant core among hydrogen bond catalysts,17–21 thanks to their rigid four-membered ring, the appropriate distance between the donor hydrogens, and higher acidity. Very recently, Rawal et al. published22 the first application of thiosquaramides, the thio analogues of squaramides, as asymmetric catalysts. In a recent review, the main benefits of thio analogues compared to those of squaramides, like increased aromaticity, higher acidity and greater solubility in non-polar solvents, were summarised.23 Bifunctional thiosquaramides are a promising new class of catalysts, and their simple preparation procedure is expected to further expand the scope of reactions in hydrogen bonding catalysis.24
Stereodivergence in cooperative asymmetric catalysis with simultaneous involvement of two chiral catalysts is a persistent challenge in asymmetric catalysis. Depending on whether the orientation of the two chiral attachments is complimentary or opposed, stereochemical enhancement or stereochemically destructive interference might occur. The question of how two chiral catalytic units work in concert, surmounting the chiral match–mismatch issue, is profoundly important.25,26 Chirality transfer from two catalysts to the product can be particularly complex. Sunoj et al. revealed the origin of high enantioselectivities in a reaction catalysed by axially chiral binaphthyl and cinchona units through transition-state modelling.26 Besides applying theoretical studies, it can be proved experimentally if stereodivergence could be achieved by inverting the configuration of the chiral catalysts that are involved in the activation of the reactants.
Herein we report the synthesis of new binaphthyl-cinchona squaramides and their thio analogues, which are good multiple hydrogen bond donor organocatalysts, and the comparison of their application in asymmetric Michael addition of pentane-2,4-dione to trans-β-nitrostyrene, in asymmetric conjugate addition reaction of lawsone and β,γ-unsaturated α-keto ester and, moreover, in aza-Diels–Alder addition reaction of 2-siloxydiene and benzylideneacetone is presented. Furthermore, the effect of the configuration of the axially chiral binaphthyl unit and match–mismatch of chiralities with the cinchona unit was studied.
Results and discussion
Planning the synthesis of catalysts 1a and 1b has begun with a retrosynthetic analysis (Scheme 1). Based on this, a convergent synthetic route starting from commercially available 2,2′-dimethyl-1,1′-binaphthalene [(R)-3], squaric esters (Sq-M, Sq-B) and cinchona alkaloids (4a and 4b) was designed for the preparation of catalysts 1a and 1b.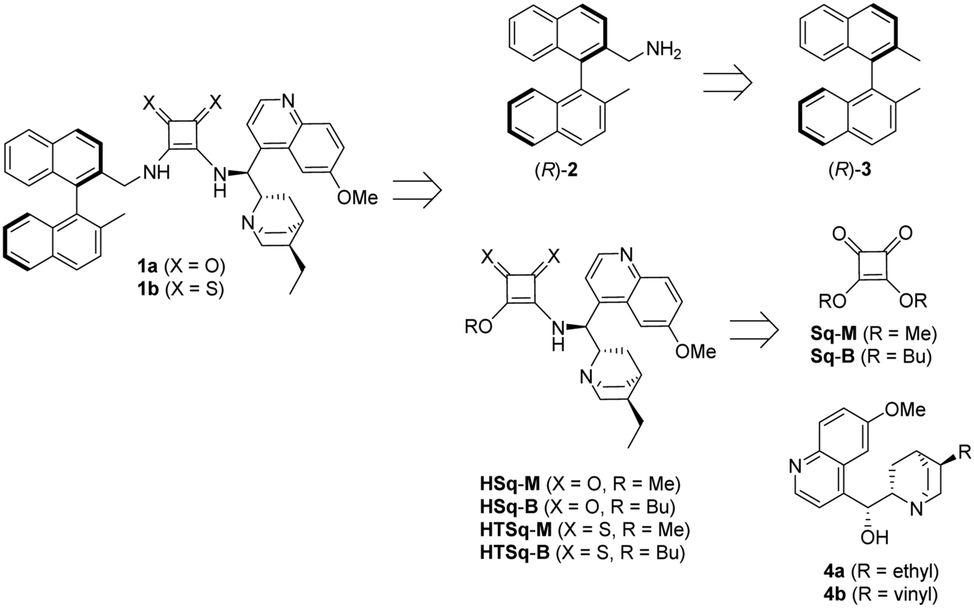 | ||
| Scheme 1 Retrosynthetic analysis of 1a and 1b. | ||
Amine fragment (R)-2 was prepared through bromination of dimethyl binaphthalene followed by substitution (Scheme 2). The monobromo derivative (R)-5 was synthesised using N-bromosuccinimide (NBS) as brominating agent and benzoyl peroxide (BPO) as initiator.27 This product was converted into amine (R)-2 in two different ways (Scheme 2): reacting with sodium azide, and reducing the corresponding azide by catalytic hydrogenation (Method A), or applying large excess of aqueous ammonia (Method B). The latter was the preferred procedure, because it is a one-step, easily implemented method providing amine (R)-2 with the same yield. Also, in comparison to Pd/C and H2; aqueous ammonia solution is an easy to handle reagent.
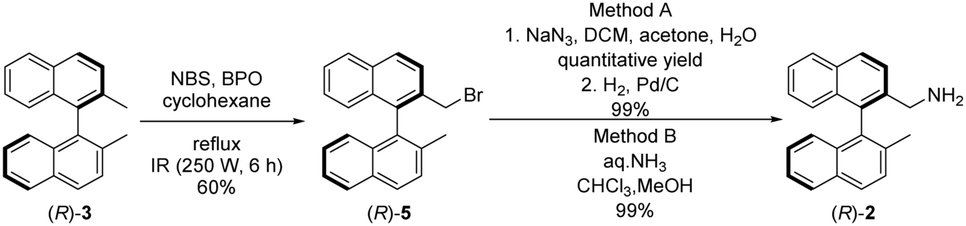 | ||
| Scheme 2 Synthesis of the axially chiral binaphthalene precursor (R)-2. | ||
In our recent publications,28,29 we used hydroquinine (4a) as starting material to form its amino derivative (6),30 however quinine (4b) is a cheaper source of the cinchona scaffold. Considering that this method contained a catalytic hydrogenation step, the ethyl derivative 6 was synthesized by reducing the vinyl and the azide group in the same step (Scheme 3). Applying the latter procedure, the intermediates were used without any purification.
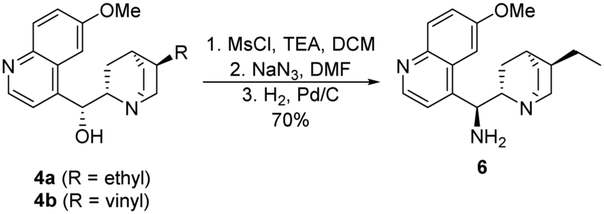 | ||
| Scheme 3 Synthesis of cinchona amine 6. | ||
The binaphthyl-cinchona squaramide 1a was gained by reacting half-squaramides HSq-M or HSq-B with the aminomethyl binaphthalene (R)-2. Application of methyl ester HSq-M as reagent is advantageous due to the higher yield (91% vs. 66%), and the easier separation of 1a from the excess of HSq-M, than from HSq-B during column chromatography.
The synthesis of binaphthyl-cinchona thiosquaramide 1b was attempted in many ways (see Scheme 4 and Scheme S1, ESI†), but it was successful only by thionation of its dioxo form 1a using P4S10·pyridine complex.31,32 In this synthetic route, the purification of the intermediates is easier than in those that apply thionated starting materials or intermediates, due to decomposition of thiosquaramide-derivatives on silica gel. We note here that all compounds were characterised by well-established methods including low- and high-resolution MS, IR, 1D and 2D NMR (see Experimental section and ESI†). The binaphthyl-cinchona squaramide 1a was gained by reacting half-squaramides HSq-M or HSq-B with the aminomethyl binaphthalene (R)-2.
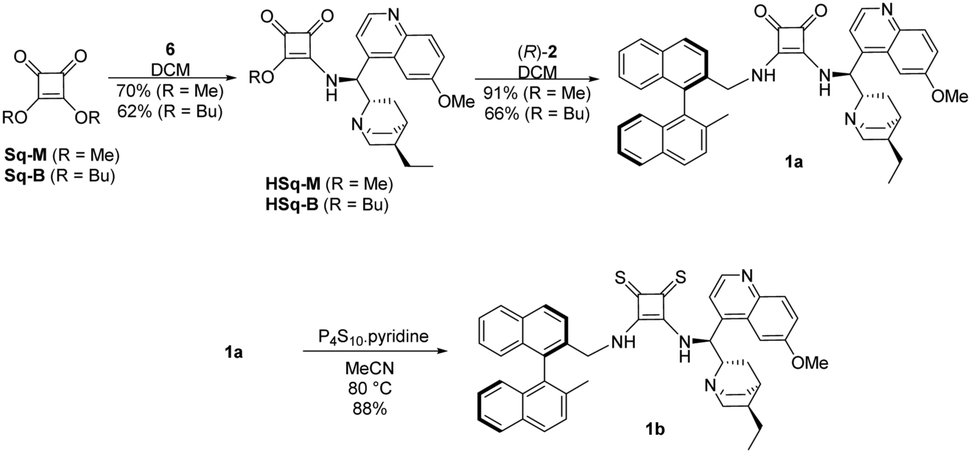 | ||
| Scheme 4 Synthesis of binaphthyl-cinchona squaramide 1a and thiosquaramide 1b organocatalysts. | ||
The catalytic activity of binaphthyl-cinchona (thio)squaramides 1a and 1b was tested in Michael addition reaction using trans-β-nitrostyrene (7) and pentane-2,4-dione (8) (Table 1).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Yieldb [%] | eec [%] |
| a Reaction conditions: pentane-2,4-dione (8) (0.41 mmol) was added to the solution of trans-β-nitrostyrene (7) (0.16 mmol) and 5 mol% of catalyst 1a or 1b in 1 mL of solvent, then the resulting mixture was stirred at room temperature for 48 hours. b Isolated yields. c Determined by chiral HPLC (S enantiomer). | ||||
| 1 | 1a | DCM | 56 | 95 |
| 2 | 1a | Toluene | 80 | 96 |
| 3 | 1a | EtOAc | 83 | 98 |
| 4 | 1a | MTBE | 70 | 97 |
| 5 | 1a | MeOH | 52 | 61 |
| 6 | 1a | H2O | 42 | 93 |
| 7 | 1a | Neat | 59 | 98 |
| 8 | 1b | DCM | 44 | 80 |
| 9 | 1b | Toluene | 85 | 96 |
| 10 | 1b | EtOAc | 76 | 98 |
| 11 | 1b | MTBE | 63 | 98 |
| 12 | 1b | MeOH | 51 | 67 |
| 13 | 1b | H2O | 71 | 98 |
| 14 | 1b | Neat | 52 | 100 |
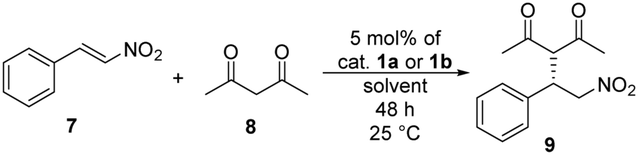
The highest yields and enantiomeric excesses were reached in ethyl acetate and toluene. Following solvent selection guidelines33–35 – based on properties of solvents such as boiling point, health hazard, aquatic and air impact – ethyl acetate was chosen to optimise the amount of catalysts and to decrease the reaction time of the Michael addition reaction from 48 hours to 24 hours. The results are shown in Table 2.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Amount of catalyst [mol%] | Yieldb [%] | eec [%] |
| a Reaction conditions: pentane-2,4-dione (8) (0.41 mmol) was added to the solution of trans-β-nitrostyrene (7) (0.16 mmol) and catalyst 1a or 1b in 1 mL of ethyl acetate, then the reaction mixture was stirred at room temperature for 24 hours. b Isolated yields. c Determined by chiral HPLC (S enantiomer). | ||||
| 1 | 1a | 5 | 85 | 98 |
| 2 | 1a | 1 | 93 | 98 |
| 3 | 1a | 0.5 | 93 | 96 |
| 4 | 1a | 0.2 | 90 | 93 |
| 5 | 1a | 0.1 | 88 | 88 |
| 6 | 1b | 5 | 81 | 98 |
| 7 | 1b | 1 | 92 | 97 |
| 8 | 1b | 0.5 | 90 | 97 |
| 9 | 1b | 0.2 | 91 | 98 |
| 10 | 1b | 0.1 | 88 | 77 |
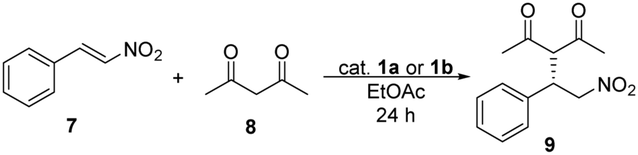
The yields and enantiomeric excesses have not changed significantly by decreasing the reaction time from 48 hours to 24 hours in the presence of 5 mol% of catalysts 1a or 1b. Comparing these two catalysts (1a and 1b), the differences between the yields and enantiomeric excesses were only relevant, when less than 0.5 mol% of catalysts were applied. However, in the presence of 0.1 mol% catalyst, in both cases, a decrease was noticed in these values, mainly in enantiomeric excesses. No reaction took place in the absence of a catalyst. For thiosquaramide 1b 0.2 mol% was the minimum catalyst loading, that gave the Michael adduct with high yield and enantiomeric excess, however the ee was only 5% lower when its dioxo analogue (1a) was used.
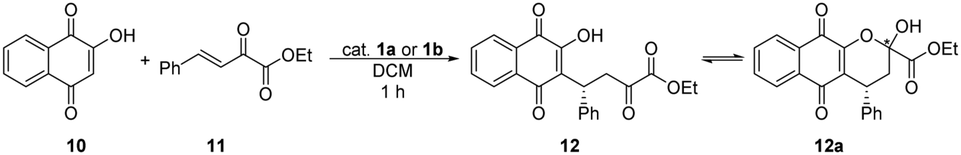
The first, preliminary studies of bifunctional aryl thiosquaramides have been reported by Rawal et al.22 They applied and compared oxo- and the corresponding thiosquaramides in the conjugate addition reaction of lawsone (10) to β,γ-unsaturated α-keto ester (11), and they achieved 9–22% higher enantiomeric excesses when thiosquaramides were used, demonstrating the superior performance of this new type of bifunctional catalysts. Also, the higher acidity of aryl thiosquaramides allowed their application as Brønsted acids in aza-Diels–Alder reaction. In this reaction, the corresponding thiourea, oxo-, and thiosquaramides were applied. Product was only afforded, when thiosquaramide was used.
Thus, we applied 1a and 1b organocatalysts in the aforementioned two asymmetric reactions (Tables 3 and 4).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Amount of catalyst [mol%] | Yieldb [%] | eec [%] |
| a Reaction conditions: β,γ-unsaturated α-keto ester (11) (0.11 mmol) was added to the solution of lawsone (10) (0.10 mmol) and catalyst 1a or 1b or DBU in 0.5 mL of DCM, then stirred at room temperature for 1 hour. b Isolated yields. c Determined by chiral HPLC (R enantiomer). | ||||
| 1 | 1a | 10 | 93 | 79 |
| 2 | 1a | 5 | 98 | 83 |
| 3 | 1a | 1 | 74 | 83 |
| 4 | 1b | 10 | 89 | 91 |
| 5 | 1b | 5 | 93 | 92 |
| 6 | 1b | 1 | 46 | 88 |
| 7 | DBU | 10 | 44 | 0 |
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Yieldb [%] | drb,c [%] |
| a Reaction conditions: 2-siloxydiene 13 (0.20 mmol) was added to the solution of N-benzylideneaniline (14) (0.24 mmol) and 20 mol% of catalyst 1a or 1b in 1 mL of toluene, stirred at room temperature for 12 hours. b Isolated yields. c Determined by 1H NMR. | |||
| 1 | 1a | 0 | — |
| 2 | 1b | 80 | 5.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
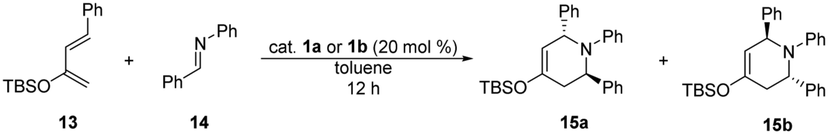
In the conjugate addition (Table 3), high yields were achieved by the application of catalysts 1a and 1b, even with 5% catalyst loading. In all cases, thiosquaramide (1b) gave higher enantioselectivity than its dioxo analogue (1a). These results also prove the noted high catalytic activity of thiosquaramides.
Following the preliminary studies of Rawal et al.,22 both organocatalysts (1a and 1b) were also applied in aza-Diels–Alder reaction (Table 4). Similarly to their observation, only thiosquaramide (1b) was able to act as Brønsted acid, therefore adduct 15 was only afforded in the reaction when it was catalysed by 1b. Adduct 15 was obtained with 80% yield in a diastereomeric ratio of 5.4 to 1 (major diastereomer shown in Table 4). Using catalyst 1a, only the unreacted starting materials were detected.
Containing two chiral fragments, namely the cinchona and the binaphthyl unit, 1a and 1b could either act as a cooperating (matched pair) or as an uncooperating (mismatched pair) multistereogenic catalyst system. Hence, after the successful utilization of catalysts 1a and 1b, the application of the “the opposite” catalysts (1c and 1d) synthesised from (S)-3, the enantiomer pair of (R)-dimethyl binaphthalene [(R)-3], was investigated in the same asymmetric transformations (Scheme 5).
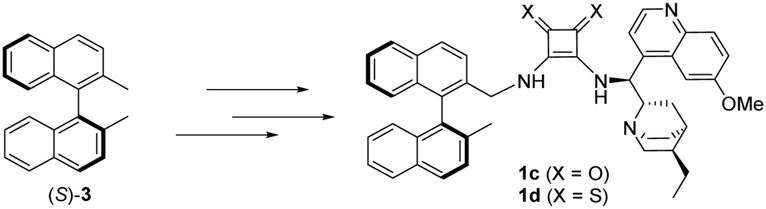 | ||
| Scheme 5 Synthesis of the opposite catalysts 1c and 1d. | ||
The synthesis of the “opposite” catalysts (1c and 1d) was accomplished by the same route (Scheme S2, ESI†) as catalysts 1a and 1b.
First, we tested them in Michael addition reaction using trans-β-nitrostyrene (7) and pentane-2,4-dione (8) (Table 5).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Amount of catalyst [mol%] | Yieldb [%] | eec [%] |
| a Reaction conditions: pentane-2,4-dione (8) (0.41 mmol) was added to the solution of trans-β-nitrostyrene (7) (0.16 mmol) and catalyst 1c or 1d in 1 mL of ethyl acetate, then the reaction mixture was stirred at room temperature for 24 hours. b Isolated yields. c Determined by chiral HPLC (S enantiomer). | ||||
| 1 | 1c | 5 | 96 | 96 |
| 2 | 1c | 1 | 93 | 94 |
| 3 | 1c | 0.5 | 83 | 95 |
| 4 | 1c | 0.2 | 62 | 90 |
| 5 | 1c | 0.1 | 56 | 81 |
| 6 | 1d | 5 | 93 | 98 |
| 7 | 1d | 1 | 96 | 97 |
| 8 | 1d | 0.5 | 91 | 97 |
| 9 | 1d | 0.2 | 86 | 91 |
| 10 | 1d | 0.1 | 83 | 84 |
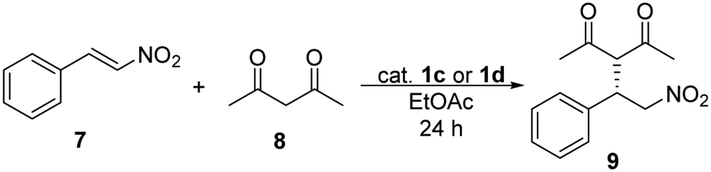
In this Michael addition 1c and 1d gave the same enantiomer [(S)] of 9 in excess as was given by using 1a and 1b. Comparing oxo-catalysts 1a and 1c, the yields were significantly lower (up to 32% difference) when 1c was used, in cases of 0.5 mol% or lower catalyst loadings, but the enantiomeric excesses have not changed considerably. Notable difference in the efficiency and selectivity between thio-analogues 1b and 1d was not noticed. Between 1c and 1d, the yields were up to 27% higher when thio-squaramide (1d) was used, but the selectivity of these catalysts is comparable.
The conjugate addition of lawsone (10) to β,γ-unsaturated α-keto ester (11) proved the noted high catalytic activity of thiosquaramides22 (Table 3), therefore we tested the catalysts 1c and 1d in this reaction as well. The results are shown in Table 6. In this conjugate addition, both the yield and selectivity were higher in the presence of only 1% of thiosquaramide (1d). Comparing these results to the outcomes of conjugate addition catalysed by 1a and 1b, the same tendency was observed: the enantiomeric excess is approximately 5–10% higher when thiosquaramide is applied. By applying 1a and 1b or 1c and 1d, the same enantiomer [(R)] of the adduct 12 was obtained.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Amount of catalyst [mol%] | Yieldb [%] | eec [%] |
| a Reaction conditions: β,γ-unsaturated α-keto ester 11 (0.11 mmol) was added to the solution of lawsone (10) (0.10 mmol) and catalyst 1c or 1d in 0.5 mL of DCM, stirred at room temperature for 1 hour. b Isolated yields. c Determined by chiral HPLC (R enantiomer). | ||||
| 1 | 1c | 10 | 100 | 83 |
| 2 | 1c | 5 | 100 | 84 |
| 3 | 1c | 1 | 73 | 79 |
| 4 | 1d | 10 | 100 | 88 |
| 5 | 1d | 5 | 100 | 89 |
| 6 | 1d | 1 | 100 | 90 |
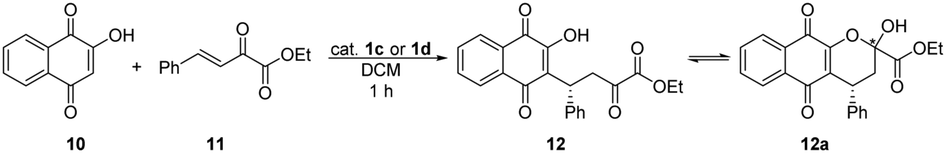
Finally we tested 1c and 1d catalysts in aza-Diels–Alder reaction, although, none of them gave product; however the reaction has taken place when thiosquaramide 1b was used. Therefore, the optimisation of the reaction conditions is still in progress.
Theoretical calculations
The distance between the two NH groups of the squaramide or thiosquaramide unit and the H-bond angle has a significant effect on the enantioselectivity of the reaction.36,37 Therefore, we analysed the differences in the 3D geometric structure of the binaphthyl-cinchona squaramide (1a) and thiosquaramide (1b) catalysts (Fig. 2) using quantum chemical calculations.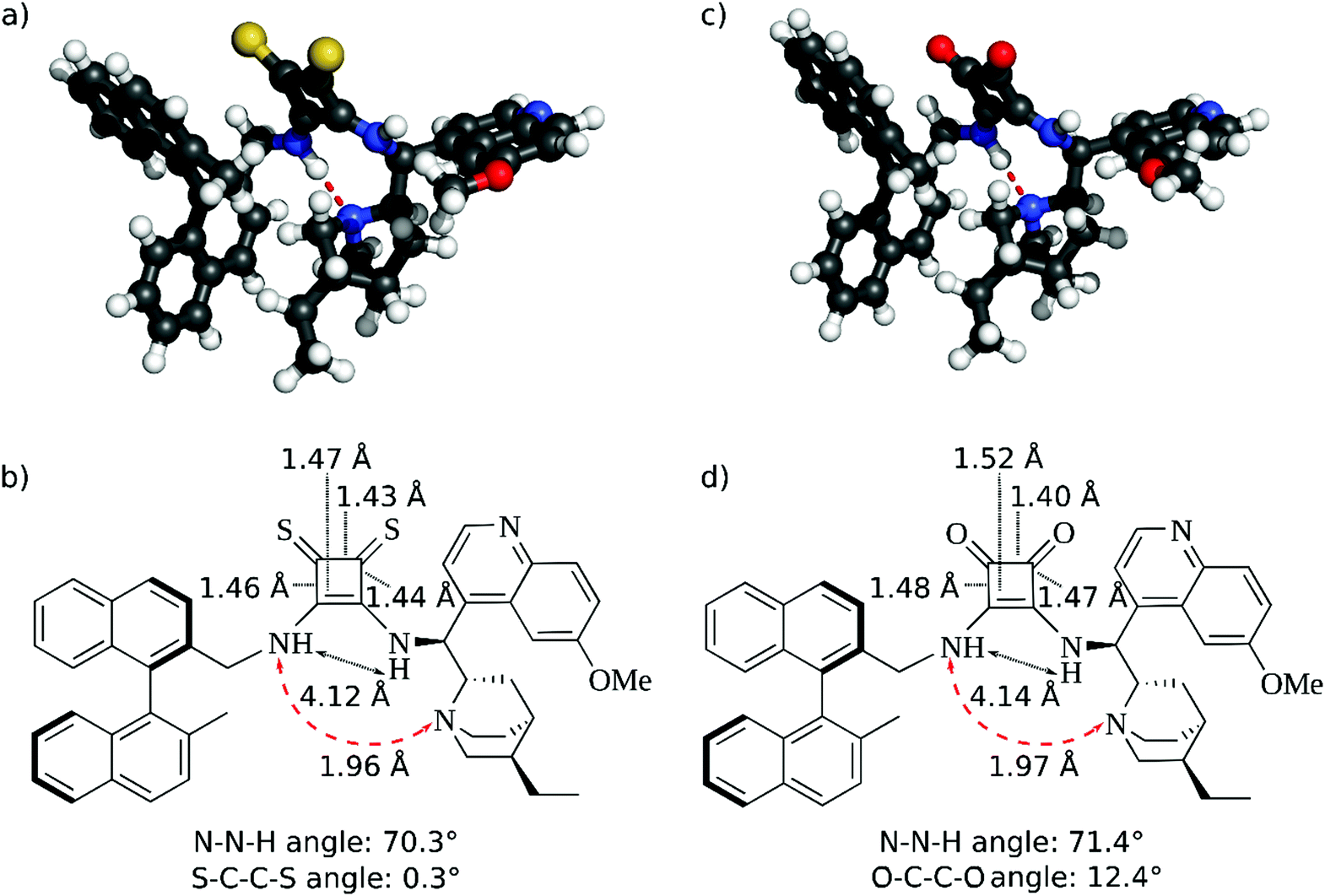 | ||
| Fig. 2 Optimised geometries and schematic structures of binaphthyl-cinchona thiosquaramide 1b (a and b) and binaphthyl-cinchona squaramide 1a (c and d) catalysts with the calculated distances and angles. The hydrogen bond between the NH group and the quinuclidine part is shown by red dashed arrows. | ||
The calculated distances between the NH groups (4.12 Å and 4.14 Å) and also the N–N–H angles (70.3° and 71.4°) are very similar in both the thiosquaramide and in squaramide catalysts (Fig. 2). Furthermore, a hydrogen bond is formed between the quinuclidine and the squaramide part. Also, only minor differences are observed in the geometry of the four membered rings.
Thus, the molecular geometries of thiosquaramide and squaramide do not imply a significant difference in their enantioselectivities.
Conclusions
In conclusion, the synthesis of a new class of chiral cinchona (thio)squaramide organocatalysts containing axially chiral binaphthyl moiety was demonstrated. We have addressed a fundamental problem of chiral induction in asymmetric catalysis when two chiral catalytic units act together in a reaction. Cinchona-thiosquaramides (1b and 1d) were first applied as enantioselective organocatalysts in asymmetric Michael addition of pentane-2,4-dione (8) to trans-β-nitrostyrene (7) performing a thorough solvent screening and parameter optimisation. Compared to their oxo analogues, thiosquaramide catalyst 1b gave Michael adduct 9 with almost the same yield (90% and 91%) and 1d gave the adduct with significantly higher yield, than its oxo analogue (86% vs. 62%). Thiosquaramide 1b gave slightly higher enantioselectivity (98% ee vs. 93% ee) with even 0.2% of catalyst loading in ethyl acetate. In the conjugate addition reaction of lawsone (10) to β,γ-unsaturated α-keto ester 11, the adduct 12 was gained with high yields (up to 100%), even when 1% of catalyst loading was applied. In this conjugate addition, the application of 1b and 1d thio derivatives resulted in up to 12% higher enantioselectivities (up to 92%) than by using the corresponding oxosquaramides 1a or 1c. Our results demonstrated clearly that there is no considerable effect of the match–mismatch pair of (R)- and (S)-binaphthyl cinchona (thio)squaramide diastereomers (1avs.1c and 1bvs.1d) on the selectivity. The Brønsted acid activity of the squaramides and their thio analogues were tested in aza-Diels–Alder addition of 2-siloxydiene 13 to N-benzylideneaniline (14). In this reaction the adduct (15) was only obtained when thiosquaramide 1b was used, with dr of 5.4 (trans):1 (cis). Quantum chemical computations showed that the geometric structures of binaphthyl-cinchona thiosquaramide and squaramide are similar, in line with the experimentally observed similar yield and ee values. These findings significantly expand the scope of applications of thiosquaramide derivatives in asymmetric syntheses.
Experimental
General
Infrared spectra were recorded on a Bruker Alpha-T FT-IR spectrometer. Optical rotations were measured on a PerkinElmer 241 polarimeter that was calibrated by measuring the optical rotations of both enantiomers of menthol. NMR spectra were recorded at Directorate of Drug Substance Development, Egis Pharmaceuticals Plc., on a Bruker Avance III HD (at 600 MHz for 1H and at 150 MHz for 13C spectra) or at Department of Inorganic & Analytical Chemistry, Budapest University of Technology and Economics, on a Bruker DRX-500 Avance spectrometer (at 500 MHz for 1H and at 125 MHz for 13C spectra) or on a Bruker 300 Avance spectrometer (at 300 MHz for 1H and at 75 MHz for 13C spectra) at temperatures given. Mass spectra were recorded on CAMAG LCMS Interface (HPLC pump: Shimadzu LC-20AD Prominence SQ MS: Shimadzu LCMS-2020 MS settings: detector voltage: 1.10 kV, m/z: 105–1000, scan speed: 1075 μ s−1, DL temperature: 250 °C, nebulizing gas flow: 1.5 L min−1, drying gas flow: 15 L min−1. Eluent: acetonitrile: 0.1% (v/v) formic acid 95:5, 1.5 mL min−1). The exact mass measurements were performed using Q-TOF Premier mass spectrometer (Waters Corporation, 34 Maple St, Milford, MA, USA) in positive electrospray ionisation mode. The enantiomeric ratios of the samples were determined by chiral high-performance liquid chromatography (HPLC) measurements using reversed phase mode (Thermo Finnigan Surveyor LC System). Elemental analyses were performed in the Microanalytical Laboratory of the Department of Organic Chemistry, Institute for Chemistry, Eötvös Loránd University, Budapest, Hungary. Melting points were taken on a Boetius micro-melting point apparatus and they were uncorrected. Starting materials were purchased from Aldrich Chemical Company unless otherwise noted. Silica gel 60 F254 (Merck) plates and aluminium oxide 60 F254 (Merck) were used for TLC. The spots of materials on TLC plates were visualised by UV light at 254 nm. Silica gel 60 (70–230 mesh, Merck) was used for column chromatography. Silica gel 60 with particle size of 0.063 mm was used for dry column vacuum chromatography (DCVC).38 Ratios of solvents for the eluents are given in milliliters. PerkinElmer TGA 6 was used to determine the thermal stability of the catalysts with nitrogen purging. The beginning of thermal degradation was determined at 95% (w/w) of the measured sample. A 10 °C min−1 heating rate was used from 30 to 700 °C, then the sample was kept at 700 °C for 10 min. PerkinElmer DSC 7 was used to measure melting point of the catalysts with nitrogen purging.
The computations were carried out using density functional theory applying the ωB97X-D functional39 and 6-31G* basis set,40 as it is implemented in the Q-Chem 5.2. quantum chemical software package.41 This functional includes long range and dispersion corrections and the accuracy of this method has been tested for similar systems in our previous studies.29,42,43 The (75302) integration grid was applied in all cases. The geometries of the catalysts were optimised both in the gas phase and in ethyl acetate solvent using SM8 continuum solvation method.44 Molecules were visualised using the PyMol program.45
(R)-2-(Bromomethyl)-2′-methyl-1,1′-binaphthalene [(R)-5]. White waxy oil (383 mg, 60%). TLC (SiO2; hexane
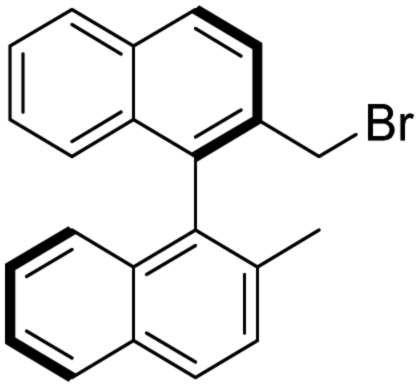
:DCM = 2:1, Rf = 0.67). [α]D20 +121.2 (c 1.00, CHCl3); IR νmax 3046, 1506, 1212, 812, 751 cm−1; 1H NMR δ (ppm, 300 MHz, DMSO-d6, 25 °C) 1.96 (3 H, s), 4.04 (1 H, d, JH,H 14.0 Hz), 4.13 (1 H, d, JH,H 14.0 Hz), 6.85 (1 H, d, JH,H 8.5 Hz), 6.86 (1 H, d, JH,H 8.3 Hz), 7.23 (1 H, overlapped), 7.26 (1 H, overlapped), 7.42 (1 H, overlapped), 7.45 (1 H, overlapped), 7.58 (1 H, d, JH,H 8.4 Hz), 7.90 (1 H, d, JH,H 8.6 Hz), 7.97 (2 H, overlapped), 8.00 (1 H, overlapped), 8.07 (1 H, d, JH,H 8.5 Hz); 13C NMR δ (ppm, 75 MHz, DMSO-d6, 25 °C) 19.8, 60.6, 124.8, 125.0, 125.0, 125.4, 126.3, 127.6, 127.7, 128.0, 128.2, 128.7, 131.6, 131.8, 132.1, 132.4, 132.4, 133.2, 134.0, 138.0; MS-ESI+ (m/z): the ionization was not feasible of this compound under the circumstances of the applied methods (ESI H+ addition or Na+ addition). Anal. calc. for C22H17Br (%): C, 73.14; H, 4.74; Br, 22.12. Found: C, 73.11; H, 4.77; Br, 22.09.
To the best of our knowledge the synthesis of (R)-5 has not been reported.
(S)-2-(Bromomethyl)-2′-methyl-1,1′-binaphthalene [(S)-5]. White waxy oil (396 mg, 62%). [α]D20 −121.2 (c 1.00, CHCl3), the other spectroscopic data of this product are the same as those of (R)-5.
To the best of our knowledge the synthesis of (S)-5 has not been reported.
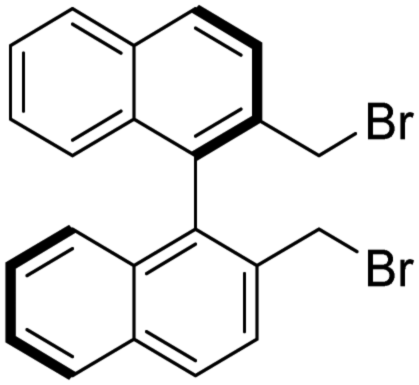
(R)-2,2′-Bis(bromomethyl)-1,1′-binaphthalene [(R)-5b] (the by-product of the preparation of (R)-5). White crystals (157 mg, 20%). TLC (SiO2; hexane
:DCM = 2:1, Rf = 0.74). M.p. 161–165 °C. [α]D20 +161.9 (c 1.00, CHCl3); IR νmax 3048, 3010, 2966, 2917, 2849, 1912, 1773, 1719, 1507, 1463, 1212, 1025, 821, 756, 722, 686 cm−1; 1H NMR δ (ppm, 600 MHz, DMSO-d6, 25 °C) 4.33 (2 H, d, JH,H 10.0 Hz), 4.37 (2 H, d, JH,H 10.0 Hz), 6.89 (2 H, d, JH,H 8.6 Hz), 7.33 (2 H, t, JH,H 7.5 Hz), 7.55 (2 H, t, JH,H 7.5 Hz), 7.84 (2 H, d, JH,H 8.6 Hz), 8.06 (2 H, d, JH,H 8.3 Hz), 8.17 (2 H, d, JH,H 8.6 Hz); 13C NMR δ (ppm, 75 MHz, DMSO-d6, 25 °C) 33.2, 126.4, 127.1, 127.2, 128.2, 128.4, 129.5, 132.0, 133.1, 133.8, 134.2; MS-ESI+ (m/z): the ionization was not feasible of this compound under the circumstances of the applied methods (ESI H+ addition or Na+ addition).
To the best of our knowledge the NMR assignment of (R)-5b has not been reported.
(S)-2,2′-Bis(bromomethyl)-1,1′-binaphthalene [(S)-5b] (the by-product of the preparation of (S)-5). White crystals (151 mg, 19%). M.p. 161–165 °C. [α]D20 −161.9 (c 1.00, CHCl3), the other spectroscopic data of this product are the same as those of (R)-5b.
To the best of our knowledge the NMR assignment of (S)-5b has not been reported.
(R)-2-(Azidomethyl)-2′-methyl-1,1′-binaphthalene [(R)-16]. (R)-2-Bromomethyl-2′-methyl-1,1′-binaphthalene [(R)-5] (200 mg, 0.556 mmol), was dissolved in DCM (1 mL), then acetone (4 mL), sodium azide (72.2 mg, 1.11 mmol) and water (10 μL) was added. The mixture was stirred until the reaction was completed. The acetone was removed under reduced pressure, and the remaining material was dissolved in a mixture of DCM and water (2 mL of each). The phases were shaken well and separated. The organic phase was dried over anhydrous MgSO4 and the solvent was removed under reduced pressure. Further purification was not necessary to afford the pure product as white waxy oil (179 mg, quantitative yield). TLC (SiO2 TLC; hexane
:DCM = 2:1, Rf = 0.49). [α]D20 +60.5 (c 1.00, CHCl3); IR νmax 3053, 3010, 2918, 2805, 2090, 810, 742 cm−1; 1H NMR δ (ppm, 300 MHz, DMSO-d6, 25 °C) 1.99 (3 H, s), 4.07 (1 H, d, JH,H 13.5 Hz), 4.12 (1 H, d, JH,H 13.6 Hz), 6.83 (1 H, d, JH,H 8.4 Hz), 6.92 (1 H, d, JH,H 8.5 Hz), 7.25 (1 H, ddd, J1,H,H 8.4 Hz, J2,H,H 6.9 Hz, J3,H,H 1.4 Hz), 7.31 (1 H, ddd, J1,H,H 8.5 Hz, J2,H,H 6.9 Hz, J3,H,H 1.3 Hz), 7.44 (1 H, ddd, J1,H,H 8.2 Hz, J2,H,H 6.9 Hz, J3,H,H 1.4 Hz), 7.525 (1 H, ddd, J1,H,H 8.2 Hz, J2,H,H 6.9 Hz, J3,H,H 1.3 Hz), 7.61 (1 H, d, JH,H 8.5 Hz), 7.77 (1 H, d, JH,H 8.5 Hz), 7.99 (1 H, overlapped), 8.02 (1 H, overlapped), 8.06 (1 H, d, JH,H 8.4 Hz), 8.13 (1 H, d, JH,H 8.5 Hz); 13C NMR δ (ppm, 125 MHz, DMSO-d6, 25 °C) 19.8, 52.1, 124.7, 125.2, 125.3, 126.4, 126.4, 126.8, 126.9, 128.1, 128.1, 128.3, 128.3, 128.6, 131.7, 131.8, 132.3, 132.5, 133.0, 134.5, 135.6; MS-ESI+ (m/z): [M + H+–N2] calcd for C22H18N: 296.14, found: 296.20. Anal. calc. for C22H17N3 (%): C, 81.71; H, 5.30; N, 12.99. Found: C, 81.70; H, 5.33; N, 12.96.
To the best of our knowledge the synthesis of (R)-16 has not been reported.
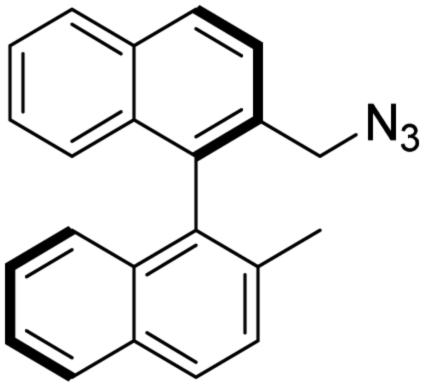
Method B: 2-bromomethyl-2′-methyl-1,1′-binaphthalene [(R)-5 or (S)-5] (200 mg, 0.556 mmol), was dissolved in a mixture of chloroform and methanol (2 mL of each), then it was added dropwise to a solution of ammonia (4.3 mL of methanol in 8.6 mL 25% aq. NH3), and this reaction mixture was stirred for 12 h. After the reaction was completed, the organic solvents were removed under reduced pressure. The remaining aqueous mixture was extracted using chloroform (5 mL). The organic phase was dried over MgSO4 and the solvent was removed under reduced pressure. Further purification was not necessary to obtain the pure product.
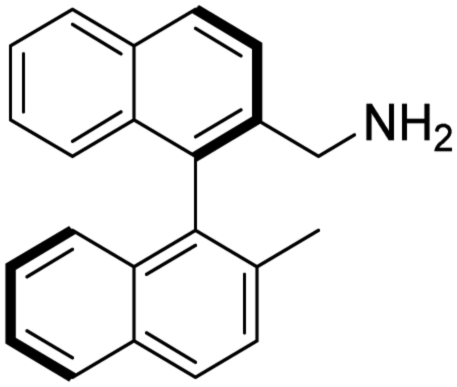
(R)-2-(Aminomethyl)-2′-methyl-1,1′-binaphthalene [(R)-2]. Pale-yellow crystals (165 mg, 99%). TLC (SiO2 TLC; hexane
:DCM = 2:1, Rf = 0.77). M.p. 69–71 °C; [α]D20 +12.7 (c 0.99, CHCl3); IR νmax 3371, 3051, 3007, 2917, 2856, 1593, 1506, 812, 744 cm−1; 1H NMR δ (600 MHz; DMSO-d6) 1.97 (3 H, s), 3.28 (1 H, d, JH,H 14.8 Hz), 3.37 (1 H, d, JH,H 14.9 Hz), 6.85 (1 H, d, JH,H 8.6 Hz), 6.87 (1 H, d, JH,H 8.6 Hz), 7.24 (1 H, overlapped), 7.25 (1 H, overlapped), 7.42 (1 H, overlapped), 7.44 (1 H, overlapped), 7.59 (1 H, d, JH,H 8.5 Hz), 7.92 (1 H, d, JH,H 8.6 Hz), 7.97 (1 H, overlapped), 7.99 (1 H, overlapped), 8.00 (1 H, overlapped), 8.06 (1 H, d, JH,H 8.6 Hz); 13C NMR δ (ppm, 150 MHz, DMSO-d6, 25 °C) 20.1, 43.4, 125.1, 125.1, 125.3, 125.5, 126.3, 126.5, 126.6, 127.8, 127.9, 128.3, 128.3, 128.9, 131.9, 132.0, 132.4, 132.5, 133.2, 133.9, 134.3, 139.5; MS-ESI+ (m/z): [M + H+] calcd for C22H20N: 298.16, found: 298.20. Anal. calc. (%): C, 88.85; H, 6.44; N, 4.71. Found: C, 88.84; H, 6.45; N, 4.71.
To the best of our knowledge the synthesis of (R)-2 has not been reported.
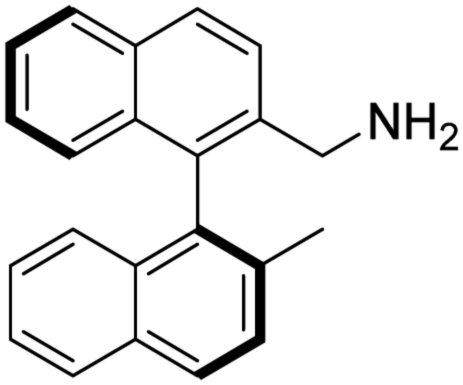
(S)-2-(Aminomethyl)-2′-methyl-1,1′-binaphthalene [(S)-2]. Pale-yellow crystals (165 mg, 99%). [α]D20 −12.7 (c 0.99, CHCl3), the other spectroscopic data of this product are the same as those of (R)-2.
To the best of our knowledge the synthesis of (S)-2 has not been reported.
(1S)-[((2S,4S,5R)-5-Ethylquinuclidin-2-yl)(6-methoxyquinolin-4-yl)]methanamine (6). This compound was synthesized as described in the literature46 with some minor modifications. To a solution of triethylamine (4.1 mL, 2.97 g, 29.4 mmol), and quinine (4b) (2.00 g, 6.2 mmol) in THF (35 mL) mesyl chloride (1.9 mL, 2.81 g, 24.5 mmol) was added at 0 °C. The reaction mixture was stirred at room temperature for 4 h. The solvent was removed under reduced pressure, subsequently DCM (50 mL) and saturated aq. NaHCO3 solution (50 mL) were added to the residue. The phases were shaken well and separated. The organic phase was dried over anhydrous MgSO4 and the solvent was removed. This mesylate intermediate was dissolved in DMF (50 mL), and sodium azide (1.6 g, 24.7 mmol) was added. The reaction mixture was stirred overnight. After the reaction was completed, the solvent was removed under reduced pressure. The crude product was dissolved in methanol (50 mL). 20% Pd/C catalyst (200 mg) was used for the catalytic hydrogenation of the azide intermediate. The hydrogenation was carried out at room temperature in atmospheric pressure using hydrogen gas. After the reaction was completed, the catalyst was filtered through a pad of Celite®, then the solvent was removed under reduced pressure. Further purification was not necessary to obtain the pure product as a yellow oil (1.61 g, 70% overall yield). Spectral data were fully consistent with those reported in the literature.46 TLC (SiO2 TLC; DCM
:methanol:NH3 (25% aq. solution) = 10:1:0.01, Rf = 0.19). [α]D20 +63.6 (c 0.97, CHCl3), lit.: [α]D20 +71.8 (c 0.97, CHCl3);47 IR νmax 3371, 3299, 2926, 2859, 2119, 1918, 1620, 1588, 1506, 1473, 1454, 1431, 1355, 1317, 1259, 1228, 1174, 1133, 1076, 1028, 977, 916, 851, 826, 741, 712, 635 cm−1; 1H NMR δ (ppm, 500 MHz, DMSO-d6, 80 °C) 0.71 (1 H, dd, J1,H,H 7.6, J1,H,H 13.2), 0.81 (3 H, t, JH,H 7.3 Hz), 1.23 (1 H, m, overlapped), 1.27 (2 H, m, overlapped), 1.40 (1 H, m, overlapped), 1.44 (1 H, m, overlapped), 1.52 (1 H, m, overlapped), 1.54 (1 H, m, overlapped), 2.45 (1 H, m), 2.68 (1 H, m, overlapped), 3.00 (1 H, m, overlapped), 3.20 (1 H, m, overlapped), 3.24 (1 H, m, overlapped), 3.96 (3 H, s), 4.59 (1 H, d, JH,H 10.1 Hz), 7.41 (1 H, dd, J1,H,H 2.5, J2,H,H 9.2), 7.56 (1 H, d, JH,H 4.5 Hz), 7.83 (1 H, s), 7.95 (1 H, d, JH,H 9.2 Hz), 8.70 (1 H, d, JH,H 4.5 Hz); 13C NMR δ (ppm, 125 MHz, DMSO-d6, 80 °C) 11.4, 24.8, 25.1, 26.6, 28.0, 36.8, 40.1, 52.5, 55.2, 57.1, 60.9, 102.9, 119.9, 120.4, 128.2, 130.8, 143.9, 147.2, 147.4, 156.6; MS-ESI+ (m/z): [M + H+] calcd for C20H28N3O: 326.22, found: 326.22.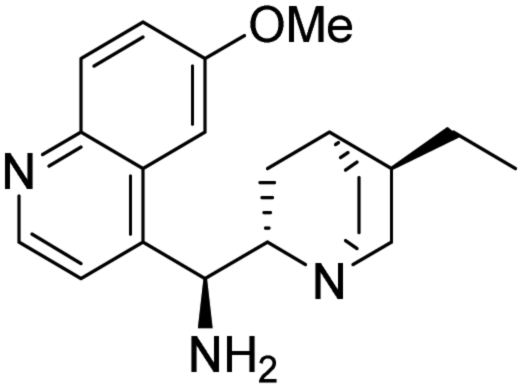 :methanol = 10:1) to obtain HSq-M or HSq-B.
:methanol = 10:1) to obtain HSq-M or HSq-B.
(((1S)-((2S,4S,5R)-5-Ethylquinuclidin-2-yl)(6-methoxyquinolin-4-yl)methyl)amino)-4-methoxycyclobut-3-ene-1,2-dione (HSq-M). Pale-yellow crystals (375 mg, 70%). TLC (SiO2 TLC; DCM
:methanol = 10:1, Rf = 0.28). M.p. 120 °C (decomposed) [α]D20 −60 (c 1.00, chloroform); IR νmax 3186, 3078, 2954, 2932, 2870, 1802, 1705, 1667, 1622, 1606, 1552, 1509, 1475, 1434, 1392, 1224 cm−1; 1H NMR δ (ppm, 500 MHz, DMSO-d6, 80 °C) 0.61 (1 H, m), 0.79 (3 H, t, JH,H 12.0 Hz), 1.27 (2 H, overlapped), 1.31 (1 H, overlapped), 1.41 (1 H, overlapped), 1.43 (1 H, overlapped), 1.53 (1 H, overlapped), 1.56 (1 H, overlapped), 2.46 (1 H, d, JH,H 13.5 Hz), 2.66 (1 H, m), 3.15 (1 H, overlapped), 3.19 (1 H, overlapped), 3.39 (1 H, overlapped), 3.95 (3 H, s), 4.25 (3 H, s), 5.75 (1 H, broad), 7.45 (1 H, d, JH,H 9.0 Hz), 7.64 (1 H, d, JH,H 4.0 Hz, overlapped), 7.66 (1 H, overlapped), 7.97 (1 H, d, JH,H 9.0 Hz), 8.77 (1 H, d, JH,H 4.0 Hz); 13C NMR δ (ppm, 125 MHz, DMSO-d6, 80 °C) 12.0, 24.8, 25.6, 27.0, 28.0, 36.7, 40.2, 52.0, 55.6, 57.1, 58.5, 60.1, 101.1, 119.9, 121.9, 127.3, 131.6, 142.9, 144.2, 147.8, 157.9, 170.8, 177.8, 181.4, 189.9; MS-ESI+ (m/z): [M + H+] calcd for C25H30N3O4: 436.22, found: 436.30. Anal. calc. (%): C, 68.95; H, 6.71; N, 9.65. Found: C, 68.92; H, 6.74; N, 9.65.
To the best of our knowledge the synthesis of HSq-M has not been reported.
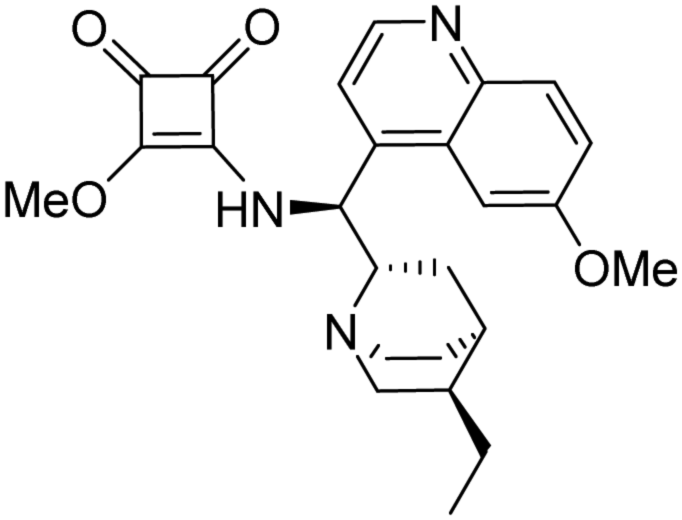
3-(((1S)-((2S,4S,5R)-5-Ethylquinuclidin-2-yl)(6-methoxyquinolin-4-yl)methyl)amino)-4-butoxycyclobut-3-ene-1,2-dione (HSq-B). White crystals (287 mg, 62%). TLC (SiO2 TLC; DCM
:methanol = 10:1, Rf = 0.38). M.p. 120 °C (decomposed) [α]D20 −129 (c 1.00, chloroform); IR νmax 3234, 3075, 3034, 2956, 2932, 2870, 1802, 1706, 1606, 1509, 1475, 1433, 1359, 1297, 1259, 1229, 1171, 1084, 1029, 919, 853 cm−1; 1H NMR δ (600 MHz; DMSO-d6, 60 °C) 0.80 (3 H, t, JH,H 7.4), 0.85 (3 H, overlapped), 1.29 (2 H, broad), 1.33 (2 H, overlapped), 1.39 (1 H, overlapped), 1.42 (1 H, overlapped), 1.52 (1 H, overlapped), 1.54 (1 H, overlapped), 1.67 (1 H, broad), 2.43 (1 H, broad), 2.64 (1 H, broad), 3.14 (1 H, overlapped), 3.17 (1 H, overlapped), 3.35 (1 H, broad), 3.96 (3 H, s), 4.56 (2 H, broad), 7.46 (1 H, d, JH,H 9.1), 7.63 (1 H, d, JH,H 4.5 Hz, overlapped), 7.72 (1 H, broad), 7.98 (1 H, d, JH,H 9.1), 8.77 (1 H, d, JH,H 4.5), 9.02 (1 H, broad); 13C NMR δ (150 MHz; DMSO-d6, 60 °C) 11.8, 13.2, 17.9, 24.9, 25.5, 26.9, 28.1, 31.3, 36.9, 40.3, 53.3, 55.6, 57.0, 58.7, 72.6, 101.1, 120.1, 121.4, 127.4, 131.5, 143.2, 144.2, 147.7, 157.8, 171.2, 177.3, 181.6, 189.5; MS-ESI+ (m/z): [M + H+] calcd for C28H36N3O4: 478.27, found: 478.30. Anal. calc. (%): C, 70.42; H, 7.39; N, 8.80. Found: C, 70.40; H, 7.40; N, 8.79.
To the best of our knowledge the synthesis of HSq-B has not been reported.
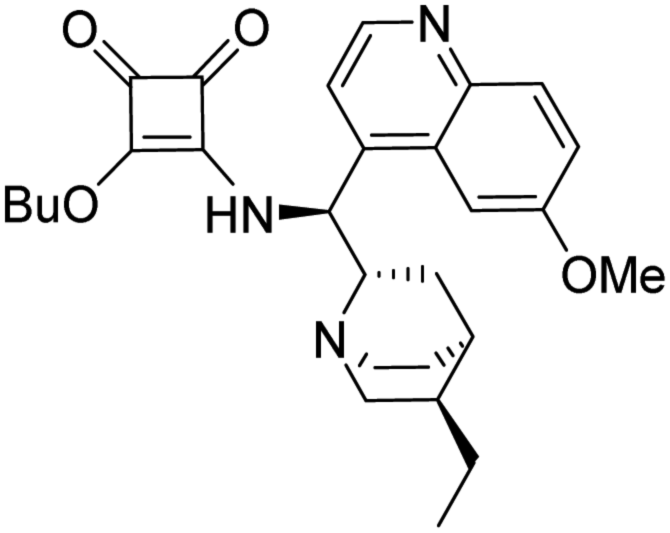 :methanol:NH3 (40:1:0.01) mixture as an eluent to obtain the pure product.
:methanol:NH3 (40:1:0.01) mixture as an eluent to obtain the pure product.
3-(((1S)-((2S,4S,5R)-5-Ethylquinuclidin-2-yl)(6-methoxyquinolin-4-yl)methyl)amino)-4-(R)-(((2′-methyl-[1,1′-binaphthalen]-2-yl)methyl)amino)cyclobut-3-ene-1,2-dione (1a). Pale-yellow crystals (427 mg, 91% with reagent HSq-M and 310 mg, 66% with reagent HSq-B). M.p. 170 °C (DSC), TLC (SiO2 TLC; DCM
:methanol:NH3 = 40:1:0.01, Rf = 0.31). [α]D20 −44,7 (c 1.00, CHCl3); IR νmax 3234, 3051, 3004, 2930, 2860, 1794, 1676, 1621, 2589, 1530, 1508, 1458, 1359, 1260, 1241, 1224 cm−1; 1H NMR δ (ppm, 500 MHz, DMSO-d6, 60 °C) 0.70 (1 H, m), 0.81 (3 H, t, JH,H 7.4 Hz), 1.36 (2 H, overlapped), 1.53 (1 H, overlapped), 1.54 (2 H, overlapped), 1.64 (1 H, overlapped), 1.65 (1 H, overlapped), 1.75 (3 H, s), 2.57 (1 H, m), 2.77 (1 H, wide), 3.28 (1 H, overlapped), 3.45 (1 H, overlapped), 3.66 (1 H, overlapped), 3.96 (3 H, s), 4.31 (1 H, dd, J1,H,H 5.2 Hz, J2,H,H 14.9 Hz), 4.40 (1 H, dd, J1,H,H 6.8 Hz, J2,H,H 14.9 Hz), 5.98 (1 H, wide), 7.19 (1 H, ddd, J1,H,H 1.3 Hz, J2,H,H 6.7 Hz, J3,H,H 14.9 Hz), 7.26 (2 H, overlapped), 7.37 (1 H, ddd, J1,H,H 1.0 Hz, J2,H,H 6.7 Hz, J3,H,H 14.9 Hz), 7.46 (1 H, overlapped), 7.48 (1 H, overlapped), 7.59 (1 H, d, JH,H 4.6 Hz), 7.69 (1 H, d, JH,H 8.6 Hz), 7.78 (1 H, d, JH,H 2.7 Hz), 7.84 (1 H, d, JH,H 7.2 Hz), 7.90 (1 H, d, JH,H 8.1 Hz), 7.99 (2 H, overlapped), 8.04 (1 H, d, JH,H 8.6 Hz), 8.78 (1 H, d, JH,H 4.6 Hz); 13C NMR δ (ppm, 125 MHz, DMSO-d6, 60 °C) 11.4, 19.4, 24.5, 25.1, 26.1, 26.7, 35.9, 40.2, 45.1, 53.0, 55.6, 56.5, 58.7, 101.6, 121.6, 124.3, 124.7, 124.9, 125.8, 125.8, 126.2, 126.5, 127.2, 127.6, 127.8, 127.9, 128.0, 128.1, 131.3, 131.6, 131.7, 132.0, 132.3, 132.5, 133.5, 134.0, 134.5, 144.2, 147.4, 157.8, 165.9, 167.6, 181.3, 182.2; HRMS-ESI+ (m/z): [M + H+] calcd for C46H45N4O3: 701.3492, found: 701.3501.
To the best of our knowledge the synthesis of 1a has not been reported.
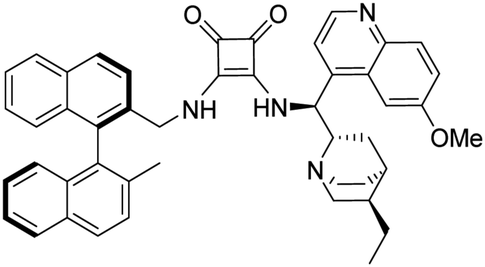
3-(((1S)-((2S,4S,5R)-5-Ethylquinuclidin-2-yl)(6-methoxyquinolin-4-yl)methyl)amino)-4-(S)-(((2′-methyl-[1,1′-binaphthalen]-2-yl)methyl)amino)cyclobut-3-ene-1,2-dione (1c). Pale-yellow crystals (413 mg, 88% with reagent HSq-M). M.p. 204–206 °C, TLC (SiO2 TLC; DCM
:methanol:NH3 = 40:1:0.01, Rf = 0.29). [α]D20 −76,6 (c 1.00, CHCl3); IR νmax 3425, 2931, 1796, 1677, 1621, 1590, 1530, 1509, 1460, 1360, 1260, 1242, 1028, 813, 687 cm−1; 1H NMR δ (ppm, 400 MHz, DMSO-d6, 117 °C) 0.76 (1 H, b), 0.83 (3 H, t, JH,H 7.4 Hz), 1.07 (2 H, t, JH,H 7 Hz), 1.37 (2 H, q, JH,H 8 Hz), 1.53 (1 H, b), 1.65 (1 H, b), 1.69 (1 H, b), 1.94 (3 H, s), 2.67 (1 H, b), 3.76 (1 H, b), 3.37 (1 H, m), 3.47 (2 H, q, JH,H = 7 Hz), 3.94 (3 H, s), 4.39 (2 H, d, JH,H = 5.2 Hz), 5.97 (1 H, d, JH,H = 10.6 Hz), 6.79 (1 H, d, JH,H = 8.6 Hz), 6.88 (1 H, d, JH,H = 8.6 Hz), 6.93 (1 H, t, JH,H 7 Hz), 7.04 (1 H, t, JH,H 7.6 Hz), 7.23 (1 H, t, JH,H 7.6 Hz), 7.44 (1 H, m), 7.45 (1 H, m), 7.46 (1 H, m), 7.54 (1 H, d, JH,H 4.6 Hz), 7.67 (1 H, d, JH,H 8.6 Hz), 7.73 (1 H, m), 7.74 (1 H, m), 7.84 (1 H, d, JH,H 8.4 Hz), 7.96 (1 H, m), 7.98 (1 H, m), 8.00 (1 H, m), 8.75 (1 H, d, JH,H = 4.6 Hz); 13C NMR δ (ppm, 100 MHz, DMSO-d6, 117 °C) 10.9, 17.9, 19.2, 24.3, 24.5, 25.7, 35.5, 45.1, 52.9, 55.5, 55.7, 56.3, 59.3, 101.9, 119.3, 121.4, 124.2, 124.3, 124.8, 125.5, 125.5, 125.6, 126.1, 127.0, 127.3, 127.4, 127.6, 127.7, 128.1, 131.1, 131.4, 131.6, 131.7, 132.0, 132.3, 132.5, 133.4, 133.8, 134.6, 144.2, 147.2, 157.8, 165.7, 168.0, 181.5, 182.3; HRMS-ESI+ (m/z): [M + H+] calcd for C46H45N4O3: 701.3492, found: 701.3501.
To the best of our knowledge the synthesis of 1c has not been reported.
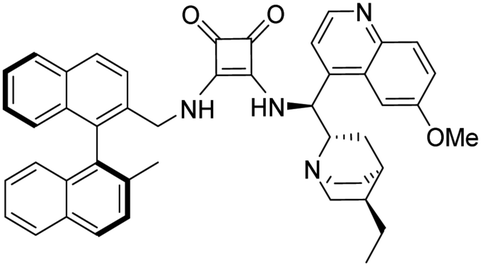
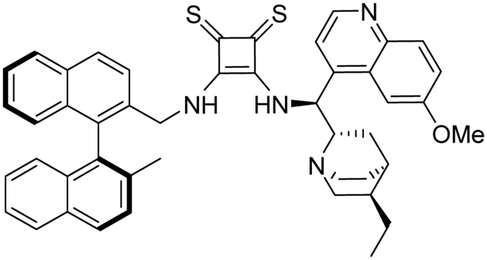
3-(((1S)-((2S,4S,5R)-5-Ethylquinuclidin-2-yl)(6-methoxyquinolin-4-yl)methyl)amino)-4-(R)-(((2′-methyl-[1,1′-binaphthalen]-2-yl)methyl)amino)cyclobut-3-ene-1,2-dithione (1b). Orange crystals (74 mg, 88%). TLC (aluminium oxide TLC; chloroform
:methanol:NH3 = 40:1:0.01) Rf = 0.69; (SiO2 TLC; chloroform:methanol = 40:1:0.01 NH3) Rf = 0.23. M.p. 237 °C (DSC), [α]D20 −44.6 (c 0.25, CHCl3); IR νmax 3436, 3169, 3051, 2999, 2930, 2872, 1917, 1762, 1698, 1620, 1588, 1507, 1474, 1432, 1357, 1243, 1143, 1085, 1028, 812, 748 cm−1; 1H NMR δ (ppm, 600 MHz, DMSO-d6, 60 °C) 0.74 (3 H, t, JH,H 7.4 Hz), 0.94 (1 H, m, JH,H 13.0 Hz), 1.28 (1 H, t, JH,H 7.4 Hz), 1.60 (1 H, t, JH,H 12.0 Hz), 1.74 (3 H, s), 1.81 (1 H, overlapped), 1.83 (3 H, overlapped), 3.09 (1 H, overlapped), 3.12 (1 H, overlapped), 3.57 (1 H, wide), 4.03 (1 H, s), 4.12 (1 H, overlapped), 4.43 (1 H, wide), 4.76 (3 H, d, JH,H 15.4 Hz), 5.16 (1 H, d, JH,H 15.4 Hz), 6.84 (2 H, d, JH,H 8.4 Hz), 7.16 (1 H, wide), 7.20 (2 H, overlapped), 7.24 (1 H, t, JH,H 7.6 Hz), 7.35 (1 H, t, JH,H 7.2 Hz), 7.45 (1 H, t, JH,H 7.1 Hz), 7.49 (1 H, dd, J1,H,H 2.7 Hz, J2,H,H 9.2 Hz), 7.82 (1 H, d, JH,H 8.4 Hz), 7.85 (1 H, overlapped), 7.86 (1 H, overlapped), 7.92 (1 H, wide), 7.97 (1 H, d, JH,H 8.3 Hz), 8.00 (1 H, overlapped), 8.02 (1 H, overlapped), 8.04 (1 H, d, JH,H 8.7 Hz), 8.86 (1 H, d, JH,H 4.6 Hz); 13C NMR δ (ppm, 150 MHz, DMSO-d6, 60 °C) 11.2, 19.8, 23.3, 23.9, 24.5, 25.2, 34.5, 41.2, 44.9, 51.8, 55.2, 56.3, 59.8, 102.2, 120.7, 122.4, 124.6, 124.9, 124.9, 125.7, 125.9, 126.4, 126.6, 126.6, 127.9, 128.0, 128.1, 128.2, 128.3, 131.7, 131.8, 131.9, 132.1, 132.3, 132.6, 132.7, 134.2, 134.4, 141.1, 144.4, 147.8, 158.2, 169.6, 170.9, 202.4, 204.9; HRMS-ESI+ (m/z): [M + H+] calcd for C46H45N4S2O: 733.3035, found: 733.3043.
To the best of our knowledge the synthesis of 1b has not been reported.
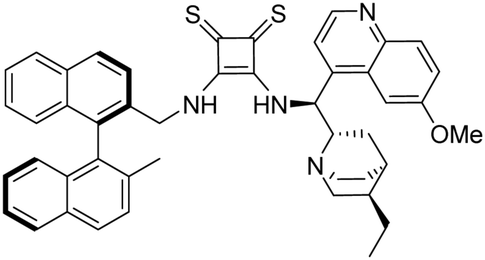
3-(((1S)-((2S,4S,5R)-5-Ethylquinuclidin-2-yl)(6-methoxyquinolin-4-yl)methyl)amino)-4-(S)-(((2′-methyl-[1,1′-binaphthalen]-2-yl)methyl)amino)cyclobut-3-ene-1,2-dithione (1d). Orange crystals (72 mg, 86%). TLC (aluminium oxide TLC; chloroform
:methanol:NH3 = 40:1:0.01) Rf = 0.66; (SiO2 TLC; chloroform:methanol = 40:1:0.01 NH3) Rf = 0.25. M.p. 208–210 °C, [α]D20 −93.3 (c 1.00, CHCl3); IR νmax 3431, 2929, 1698, 1566, 1508, 1474, 1244, 1145, 1086, 1029, 813, 746 cm−1; 1H NMR δ (ppm, 600 MHz, DMSO-d6, 22 °C) 0.76 (3 H, t, JH,H 7.3 Hz), 1.25 (2 H, b), 1.35 (2 H, m), 1.44 (1 H, b), 1.47 (1 H, b), 1.56 (1 H, b), 1.98 (3 H, s), 1.81 (1 H, b), 2.43 (1 H, b), 2.60 (1 H, b), 3.16 (1 H, b), 3.67 (1 H, b), 4.00 (1 H, b), 4.02 (3 H, s), 4.89 (1 H, d, JH,H 14.6 Hz), 5.13 (1 H, d, JH,H 14.6 Hz), 6.73 (1 H, b), 6.82 (1 H, b), 6.86 (1 H, d, JH,H 8.5 Hz), 6.96 (1 H, b), 7.28 (1 H, m), 7.48 (1 H, m), 7.49 (1 H, m), 7.50 (1 H, m), 7.61 (1 H, b), 7.72 (1 H, b), 7.79 (1 H, d, JH,H 8.5 Hz), 7.86 (1 H, d, JH,H 8.4 Hz), 8.00 (1 H, m), 8.01 (1 H, m), 8.02 (1 H, m), 8.10 (1 H, d, JH,H 8.5 Hz), 8.83 (1 H, d, JH,H 4.3 Hz); 13C NMR δ (ppm, 150 MHz, DMSO-d6, 22 °C) 12.0, 20.2, 22.3, 24.0, 25.4, 27.6, 36.5, 41.1, 44.9, 56.0, 56.4, 57.0, 102.7, 120.2, 122.5, 124.7, 124.7, 125.2, 125.4, 126.3, 126.6, 127.1, 127.3, 128.0, 128.2, 128.4, 128.6, 129.0, 131.6, 131.9, 132.1, 132.4, 132.5, 133.0, 134.4, 134.8, 135.5, 144.5, 147.9, 158.0, 168.8, 171.2, 201.0, 204.3; HRMS-ESI+ (m/z): [M + H+] calcd for C46H45N4S2O: 733.3035, found: 733.3043.
To the best of our knowledge the synthesis of 1d has not been reported.
 :ethyl acetate 2:1 mixture (Rf = 0.36) as eluent to obtain Michael adduct as pale-yellow crystals. Yields and enantiomeric excess (ee) values can be seen in Tables 1, 2 and 5. These products had the same spectroscopic data than those of reported (the absolute configuration was determined by the optical rotation of the products).29 HPLC: Phenomonex Lux Cellulose-3 column (3 μm, 250 × 4.6 mm), eluent CH3CN/20 mM NH4OAc in H2O = 40/60, isocratic mode; 0.6 mL min−1; UV detector 222 nm, 5 μL or 10 μL injection, 20 °C. Retention time for (S)-9: 11.94 min, for (R)-9: 14.20 min. The applied solvents, the amounts of the catalysts and reaction times are shown in Tables 1, 2 and 5.
:ethyl acetate 2:1 mixture (Rf = 0.30) as eluent to obtain adduct 12 as yellow crystals. Yields and enantiomeric excess (ee) values can be seen in Tables 3 and 6. These products had the same spectroscopic data than those of reported (the absolute configuration was determined by the optical rotation of the products).48 HPLC: Phenomonex Lux Cellulose-1 column (3 μm, 250 × 4.6 mm); eluent CH3CN/0.1% AcOH in H2O and = 40:60, isocratic mode; 0.8 mL min−1; UV detector 222 nm; 5 μL or 10 μL injection, 25 °C. Retention time for (R)-12: 9.16 min, for (S)-12: 10.87 min. The amounts of the catalysts are shown in Tables 3 and 6.
:ethyl acetate 2:1 mixture (Rf = 0.36) as eluent to obtain Michael adduct as pale-yellow crystals. Yields and enantiomeric excess (ee) values can be seen in Tables 1, 2 and 5. These products had the same spectroscopic data than those of reported (the absolute configuration was determined by the optical rotation of the products).29 HPLC: Phenomonex Lux Cellulose-3 column (3 μm, 250 × 4.6 mm), eluent CH3CN/20 mM NH4OAc in H2O = 40/60, isocratic mode; 0.6 mL min−1; UV detector 222 nm, 5 μL or 10 μL injection, 20 °C. Retention time for (S)-9: 11.94 min, for (R)-9: 14.20 min. The applied solvents, the amounts of the catalysts and reaction times are shown in Tables 1, 2 and 5.
:ethyl acetate 2:1 mixture (Rf = 0.30) as eluent to obtain adduct 12 as yellow crystals. Yields and enantiomeric excess (ee) values can be seen in Tables 3 and 6. These products had the same spectroscopic data than those of reported (the absolute configuration was determined by the optical rotation of the products).48 HPLC: Phenomonex Lux Cellulose-1 column (3 μm, 250 × 4.6 mm); eluent CH3CN/0.1% AcOH in H2O and = 40:60, isocratic mode; 0.8 mL min−1; UV detector 222 nm; 5 μL or 10 μL injection, 25 °C. Retention time for (R)-12: 9.16 min, for (S)-12: 10.87 min. The amounts of the catalysts are shown in Tables 3 and 6.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the New National Excellence Program of the Ministry of Human Capacities, grant numbers ÚNKP-18-4-BME-270 and ÚNKP-18-2-II-BME-243, and the Janos Bolyai Research Scholarship of the Hungarian Academy of Sciences. It was also supported by the National Research, Development and Innovation office (former OTKA, grant number K128473), the Servier-Beregi PhD Research Fellowship, and the Gedeon Richter's Talentum Foundation. The authors are grateful to Zsófia Garádi from Budapest University of Technology and Economics and to Dr András Dancsó from Egis Pharmaceuticals Plc., Directorate of Drug Substance Development for their assistance with the NMR measurements.Notes and references
- P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2001, 40, 3726–3748 CrossRef CAS PubMed.
- K. N. Houk and B. List, Acc. Chem. Res., 2004, 37, 487 CrossRef CAS.
- D. W. C. MacMillan, Nature, 2008, 455, 304–308 CrossRef CAS PubMed.
- G. Zhan, W. Du and Y. C. Chen, Chem. Soc. Rev., 2017, 46, 1675–1692 RSC.
- D. L. Hughes, Org. Process Res. Dev., 2018, 22, 574–584 CrossRef CAS.
- Y. Chen, S. Yekta and A. K. Yudin, Chem. Rev., 2003, 103, 3155–3211 CrossRef CAS PubMed.
- P. Kocovsky, S. Vyskocil and M. Smrcina, Chem. Rev., 2003, 103, 3213–3245 CrossRef CAS PubMed.
- J. M. Brunel, Chem. Rev., 2005, 105, 857–897 CrossRef CAS PubMed.
- D. Kampen, C. M. Reisinger and B. List, Top. Curr. Chem., 2009, 291, 395–456 CrossRef.
- S. Schenker, A. Zamfir, M. Freund and S. B. Tsogoeva, Eur. J. Org. Chem., 2011, 2209–2222 CrossRef CAS.
- D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed.
- R. B. Sunoj, Acc. Chem. Res., 2016, 49, 1019–1028 CrossRef CAS PubMed.
- B. Liu, X. Han, Z. Dong, H. Lv, H. B. Zhou and C. N. Dong, Tetrahedron: Asymmetry, 2013, 24, 1276–1280 CrossRef CAS.
- E. S. Zhou, B. Liu and C. N. Dong, Tetrahedron: Asymmetry, 2014, 25, 181–186 CrossRef CAS.
- S. Tang, Z. Y. Wang, B. Liu and C. E. Dong, Chin. Chem. Lett., 2015, 26, 744–748 CrossRef CAS.
- V. Modrocka, E. Veverkova, R. Baran and R. Sebesta, ChemistrySelect, 2018, 3, 1466–1471 CrossRef CAS.
- C. Jin, M. Zhang, L. Wu, Y. F. Guan, Y. Pan, J. L. Jiang, C. Lin and L. Y. Wang, Chem. Commun., 2013, 49, 2025–2027 RSC.
- X. Han, H. B. Zhou and C. E. Dong, Chem. Rec., 2016, 16, 897–906 CrossRef CAS PubMed.
- M. Chennapuram, U. V. S. Reddy, C. Seki, Y. Okuyama, E. Kwon, K. Uwai, M. Tokiwa, M. Takeshita and H. Nakano, Eur. J. Org. Chem., 2017, 1638–1646 CrossRef CAS.
- B. L. Zhao, J. H. Li and D. M. Du, Chem. Rec., 2017, 17, 994–1018 CrossRef CAS PubMed.
- L. J. Yan, G. X. Huang, H. F. Wang, F. J. Xiong, H. H. Peng and F. Chen, Eur. J. Org. Chem., 2018, 99–103 CrossRef CAS.
- M. Rombola and V. H. Rawal, Org. Lett., 2018, 20, 514–517 CrossRef CAS PubMed.
- S. Nagy, P. Kisszekelyi and J. Kupai, Period. Polytech., Chem. Eng., 2018, 62, 467–475 Search PubMed.
- T. X. Lu and S. E. Wheeler, Chem. – Eur. J., 2013, 19, 15141–15147 CrossRef CAS PubMed.
- M. P. Doyle, J. P. Morgan, J. C. Fettinger, P. Y. Zavalij, J. T. Colyer, D. J. Timmons and M. D. Carducci, J. Org. Chem., 2005, 70, 5291–5301 CrossRef CAS PubMed.
- B. Bhaskararao and R. B. Sunoj, J. Am. Chem. Soc., 2015, 137, 15712–15722 CrossRef CAS PubMed.
- P. C. B. Page, B. R. Buckley and A. J. Blacker, Org. Lett., 2004, 6, 1543–1546 CrossRef PubMed.
- S. Nagy, Z. Feher, P. Kisszekelyi, P. Huszthy and J. Kupai, New J. Chem., 2018, 42, 8596–8602 RSC.
- C. Didaskalou, J. Kupai, L. Cseri, J. Barabas, E. Vass, T. Holtzl and G. Szekely, ACS Catal., 2018, 8, 7430–7438 CrossRef CAS.
- S. H. McCooey and S. J. Connon, Org. Lett., 2007, 9, 599–602 CrossRef CAS PubMed.
- R. B. P. Elmes, N. Busschaert, D. D. Czech, P. A. Gale and K. A. Jolliffe, Chem. Commun., 2015, 51, 10107–10110 RSC.
- J. Bergman, B. Pettersson, V. Hasimbegovic and P. H. Svensson, J. Org. Chem., 2011, 76, 1546–1553 CrossRef CAS PubMed.
- D. Prat, J. Hayler and A. Wells, Green Chem., 2014, 16, 4546–4551 RSC.
- T. Welton, Proc. R. Soc. A, 2015, 471, 20150502 CrossRef PubMed.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC.
- J. P. Malerich, K. Hagihara and V. H. Rawal, J. Am. Chem. Soc., 2008, 130, 14416–14417 CrossRef CAS PubMed.
- J. Aleman, A. Parra, H. Jiang and K. A. Jorgensen, Chem. – Eur. J., 2011, 17, 6890–6899 CrossRef CAS PubMed.
- D. S. Pedersen and C. Rosenbohm, Synthesis, 2001, 2431–2434 CAS.
- J. D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- Y. H. Shao, Z. T. Gan, E. Epifanovsky, A. T. B. Gilbert, M. Wormit, J. Kussmann, A. W. Lange, A. Behn, J. Deng, X. T. Feng, D. Ghosh, M. Goldey, P. R. Horn, L. D. Jacobson, I. Kaliman, R. Z. Khaliullin, T. Kus, A. Landau, J. Liu, E. I. Proynov, Y. M. Rhee, R. M. Richard, M. A. Rohrdanz, R. P. Steele, E. J. Sundstrom, H. L. Woodcock, P. M. Zimmerman, D. Zuev, B. Albrecht, E. Alguire, B. Austin, G. J. O. Beran, Y. A. Bernard, E. Berquist, K. Brandhorst, K. B. Bravaya, S. T. Brown, D. Casanova, C. M. Chang, Y. Q. Chen, S. H. Chien, K. D. Closser, D. L. Crittenden, M. Diedenhofen, R. A. DiStasio, H. Do, A. D. Dutoi, R. G. Edgar, S. Fatehi, L. Fusti-Molnar, A. Ghysels, A. Golubeva-Zadorozhnaya, J. Gomes, M. W. D. Hanson-Heine, P. H. P. Harbach, A. W. Hauser, E. G. Hohenstein, Z. C. Holden, T. C. Jagau, H. J. Ji, B. Kaduk, K. Khistyaev, J. Kim, J. Kim, R. A. King, P. Klunzinger, D. Kosenkov, T. Kowalczyk, C. M. Krauter, K. U. Lao, A. D. Laurent, K. V. Lawler, S. V. Levchenko, C. Y. Lin, F. Liu, E. Livshits, R. C. Lochan, A. Luenser, P. Manohar, S. F. Manzer, S. P. Mao, N. Mardirossian, A. V. Marenich, S. A. Maurer, N. J. Mayhall, E. Neuscamman, C. M. Oana, R. Olivares-Amaya, D. P. O’Neill, J. A. Parkhill, T. M. Perrine, R. Peverati, A. Prociuk, D. R. Rehn, E. Rosta, N. J. Russ, S. M. Sharada, S. Sharma, D. W. Small, A. Sodt, T. Stein, D. Stuck, Y. C. Su, A. J. W. Thom, T. Tsuchimochi, V. Vanovschi, L. Vogt, O. Vydrov, T. Wang, M. A. Watson, J. Wenzel, A. White, C. F. Williams, J. Yang, S. Yeganeh, S. R. Yost, Z. Q. You, I. Y. Zhang, X. Zhang, Y. Zhao, B. R. Brooks, G. K. L. Chan, D. M. Chipman, C. J. Cramer, W. A. Goddard, M. S. Gordon, W. J. Hehre, A. Klamt, H. F. Schaefer, M. W. Schmidt, C. D. Sherrill, D. G. Truhlar, A. Warshel, X. Xu, A. Aspuru-Guzik, R. Baer, A. T. Bell, N. A. Besley, J. D. Chai, A. Dreuw, B. D. Dunietz, T. R. Furlani, S. R. Gwaltney, C. P. Hsu, Y. S. Jung, J. Kong, D. S. Lambrecht, W. Z. Liang, C. Ochsenfeld, V. A. Rassolov, L. V. Slipchenko, J. E. Subotnik, T. Van Voorhis, J. M. Herbert, A. I. Krylov, P. M. W. Gill and M. Head-Gordon, Mol. Phys., 2015, 113, 184–215 CrossRef CAS.
- W. Yang, Y. M. Cui, J. F. Zou, C. Y. Wang, W. S. Huang, J. Zhang and L. W. Xu, Synth. Commun., 2014, 44, 3173–3182 CrossRef CAS.
- P. Kisszekelyi, A. Alammar, J. Kupai, P. Huszthy, J. Barabas, T. Holtzl, L. Szente, C. Bawn, R. Adams and G. Szekely, J. Catal., 2019, 371, 255–261 CrossRef CAS.
- C. J. Cramer and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 760–768 CrossRef CAS PubMed.
- The PyMOL Molecular Graphics System, V. S., LLC.
- C. Cassani, R. Martin-Rapun, E. Arceo, F. Bravo and P. Melchiorre, Nat. Protoc., 2013, 8, 325–344 CrossRef CAS PubMed.
- B. Vakulya, S. Varga, A. Csampai and T. Soos, Org. Lett., 2005, 7, 1967–1969 CrossRef CAS PubMed.
- Y. J. Gao, Q. Ren, S. M. Ang and J. Wang, Org. Biomol. Chem., 2011, 9, 3691–3697 RSC.
- K. Takasu, N. Shindoh, H. Tokuyama and M. Ihara, Tetrahedron, 2006, 62, 11900–11907 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: FT-IR, 1H, 13C, 2D NMR and MS spectra of new compounds, thermoanalytical measurements (TGA, DSC) of catalysts 1a and 1b, chiral HPLC profiles of Michael and conjugate adducts and data of theoretical calculations. See DOI: 10.1039/c8nj06451b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |