
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oligofluorene with multiple spiro-connections: its and their use in blue and white OLEDs†
Debin
Xia‡
 a,
Chunbo
Duan‡
b,
Shihui
Liu
a,
Dongxue
Ding
b,
Martin
Baumgarten
*c,
Manfred
Wagner
c,
Dieter
Schollmeyer
d,
Hui
Xu
*b and
Klaus
Müllen
*d
a,
Chunbo
Duan‡
b,
Shihui
Liu
a,
Dongxue
Ding
b,
Martin
Baumgarten
*c,
Manfred
Wagner
c,
Dieter
Schollmeyer
d,
Hui
Xu
*b and
Klaus
Müllen
*d
aMIIT Key Laboratory of Critical Materials Technology for New Energy Conversion and Storage, School of Chemistry and Chemical Engineering, Harbin Institute of Technology, 150001 Harbin, China
bKey Laboratory of Functional Inorganic Material Chemistry, Ministry of Education, School of Chemistry and Material Science, Heilongjiang University, 74 Xuefu Road, 150080 Harbin, China. E-mail: hxu@hlju.edu.cn
cMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: martin.baumgarten@mpip-mainz.mpg.de
dJohannes Gutenberg-University Mainz, Duesbergweg 10-14, 55128 Mainz, Germany. E-mail: muellen@mpip-mainz.mpg.de
First published on 5th February 2019
Abstract
Bond rotation within molecules is regarded as one of the nonradiative decay pathways and is detrimental to high photoluminescence quantum yields. In this work, a bulky and rigid blue emitter (Spiro-F) with five spiro-carbon linkages is synthesized. Spiro-F can serve not only as an active component of blue organic light-emitting diodes (OLEDs), but also as a host and blue emitter of white OLEDs. The WOLED offers a low turn-on voltage of 3.5 V and a high current efficiency of 3.6 cd A−1, together with CIE coordinates of (0.29, 0.33). This work proves the potential of super-rigid oligofluorene emitters for OLEDs.
1. Introduction
Over the past three decades, numerous efforts have been devoted to organic light-emitting diodes (OLEDs) due to their potential applications in flat-panel displays and solid-state lightings.1–7 To achieve white OLEDs, a blue emitter is an essential component.6 However, compared to its red, green and orange counterparts, the development of blue OLEDs is less advanced. This is due to inefficient charge injection and unfavorable electrical properties, caused by the intrinsically wide energy gaps of blue emitters.8 Therefore, great efforts must be made to advance the area of high-performance blue-emitting materials to promote the commercialization of OLED technology.9–18Spirobifluorene19,20 is considered as one of the most promising building blocks of blue emitters due to its high photoluminescence quantum yield (PLQY) and good thermal stability.21–24 Also, spirobifluorene-based materials feature no excimer emission or keto defects, which often occur in fluorene-based emitters.25–27 Furthermore, spiro-connections would restrict intermolecular π–π interactions and thus facilitate an amorphous morphology.28 These features have prompted us to construct extended spirobifluorene-based blue emitters, which we expect to show high PLQY, high thermal stability and pure color emission.
According to the literature, linkages between spirobifluorenes in almost all the blue-emitting compounds are by single bonds or flexible segments.29–31 Consequently, bond rotations can occur, affecting the possibility of high PLQYs.32,33 Spirobifluorene-based large “super-rigid” molecules (with no bond rotation) are rarely reported. Only Cocherel et al. in 2008 developed an oligophenylene-based compound, dispirobifluorene–indenofluorene, which showed excellent thermal stability and color purity, together with high PLQY up to 90%.34 Therefore, it is essential to develop “super-rigid” blue emitters.
Precursor Spiro-4O was used to synthesize tetrafluorene-substituted tetraindeno-fused spirobifluorene (Spiro-F) with a “super-rigid”, bulky geometry extended into three directions (Scheme 1). We anticipate that this material holds promise for use in OLEDs based on the following considerations: (1) the orthogonal configuration between the backbone and the substituted fluorenes can prevent intermolecular aggregation in the solid state; (2) the geometry and tert-butyl groups can induce solubility and formation of amorphous thin films; (3) the “super-rigid” structure can reduce undesirable nonradiative decay.
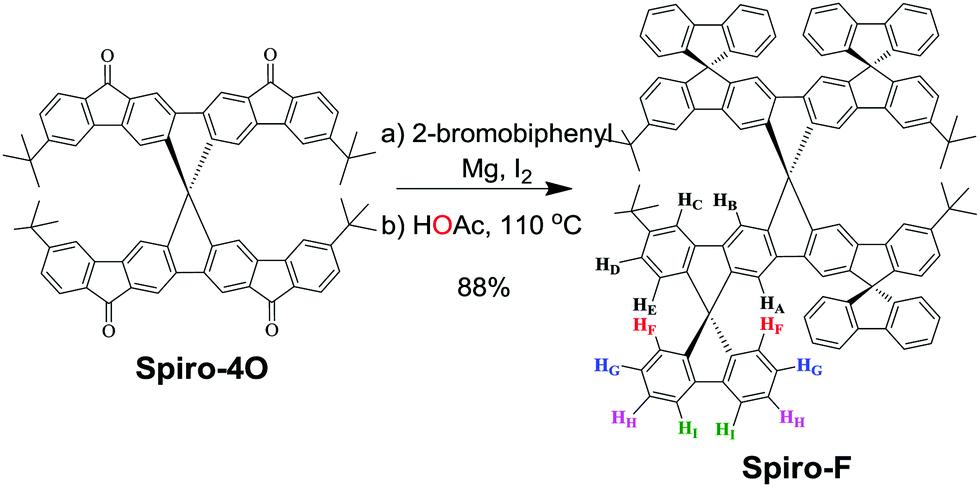 | ||
| Scheme 1 Synthesis of Spiro-F. | ||
Herein, we report the synthesis and characterization of the blue emitter Spiro-F with five spirobifluorene units. Its thermal stability, photophysical and electrochemical properties were analyzed. Finally, we investigated the performance of Spiro-F in blue and white organic light-emitting diodes (OLEDs).
2. Results and discussion
Synthesis
The facile synthetic route to Spiro-F is illustrated in Scheme 1. As we had previously reported, starting material tetraketone Spiro-4 was prepared on a multigram scale in a three-step synthesis.35,36 Firstly, excess fresh Grignard reagent, namely [1,1′-biphenyl]-2-ylmagnesium bromide, was prepared to ensure efficient nucleophilic attack upon tetraketone Spiro-4O. Then, the reaction was quenched with dilute hydrochloric acid to afford the tetra-alcohol intermediate, which was used directly in the next step without purification. A tetrafold intramolecular cyclization reaction was accomplished under reflux conditions using acetic acid as solvent. After purification by column chromatography, Spiro-F was obtained in 88% overall yield.The chemical structure of Spiro-F was unambiguously confirmed by NMR spectroscopy, high-resolution MALDI-TOF mass spectrometry, and single crystal X-ray diffraction. After 2D NMR analysis, which included H–H NOESY, H–H COSY and H–H TOCSY, it was possible to assign the aromatic protons, as shown in Fig. S1 (ESI†). The NOESY spectrum (Fig. S2, ESI†) allowed us to detect proton HF at 6.87 ppm, which is the only proton coupled through space with three other protons, namely HA, HE and HG. Based on the H–H COSY spectrum (Fig. S1, ESI†), HD (6.54 ppm), HH (7.36 ppm), and HI (7.84 ppm) were assigned. Thermogravimetric analysis (TGA) of Spiro-F revealed excellent thermal stability, with 5% weight loss occurring at 540 °C (Fig. S4, ESI†). The tert-butyl groups were completely cleaved upon increasing the temperature to 580 °C. The differential scanning calorimetry (DSC) curves of Spiro-F showed no phase transition in the range from 30 to 450 °C.
Crystallography
Suitable single crystals for X-ray diffraction analysis were obtained by slow evaporation of its pentane solution at room temperature. tert-Butyl groups played a significant role in inducing the solubility of Spiro-F and favored crystal formation in comparison to those with long alkyl chains.37,38 As shown in Fig. 1, the two diindeno-fused fluorene (DFF) backbones 15 Å in length are perpendicular to each other and the backbones are not completely flat with slight distortions on both sides. Annulated rigid and strained five-membered rings lead to the obvious backbone distortion, which is not observed in its analogues Spiro-4S, Spiro-4SO2 and 4Ph (chemical structures are shown in Fig. S5, ESI†).36,39 Two face-to-face fluorenes 8.76 Å in distance are quasi-parallel. This is one “super-rigid” structure characterized using X-ray crystallography. In the crystal-packing diagram (Fig. 1 bottom left) no intermolecular π–π interactions are detected between the two DFF π-conjugated systems, due to steric hindrance from perpendicular fluorene units.36 | ||
| Fig. 1 Crystal structure of Spiro-F. Top and bottom-left: A single molecule (tert-butyl groups and hydrogen atoms are omitted for clarity). Bottom-right: the unit cell. | ||
Optical and electrochemical properties
The electronic absorption and photoluminescence spectra of Spiro-F are illustrated in Fig. 2. From the UV-Vis absorption spectrum, we assign the four obvious bands to the conjugated DFF backbones since the characteristic peaks (312, 334, 360, 379 nm) are very similar to those of its analogues Spiro-4S, Spiro-4SO2 and 4Ph (chemical structures shown in Fig. S5, ESI†).36,39 | ||
| Fig. 2 Absorption (square) and photoluminescence (cycle) spectra of Spiro-F in p-xylene (10−5 mol L−1) (λExc = 360 nm) and thin film (λExc= 360 nm). | ||
Moreover, in the case of Spiro-F, two emission bands at 386 and 406 nm in CH2Cl2 were observed, similar to those in the above-mentioned model compounds, which further indicates that the spiro-carbon linkages disrupt intramolecular electronic coupling. Furthermore, the symmetric mirror images of the absorption and emission spectra, as well as the very small Stokes shift (4 nm), coincide with the super-rigid molecular structure of Spiro-F. It is noted that the relative emission intensity of the peaks at 379 and 383 is changed while increasing the solution concentration (Fig. S6, ESI†). When comparing the solid with the solution state, the maximum emission peak of Spiro-F presents a redshift, together with the appearance of a long tail in the range of λ = 470–620 nm, suggesting the existence of intermolecular aggregation. Using 9,10-diphenylanthracene (Φ = 0.9 in cyclohexane, λExc = 325 nm) as a standard, the photoluminescence quantum yield (PLQY) of Spiro-F was measured as high as 0.76 ± 0.03, which qualifies Spiro-F for use in OLEDs.
To obtain information on the frontier orbital energies of Spiro-F, its redox behavior was investigated by cyclic voltammetry (CV) with the ferrocene/ferrocenium (Fc/Fc+) couple as a reference. As shown in Fig. S7 (ESI†), during the anodic and cathodic sweep in CH2Cl2, only three oxidation waves were observed, with E1/2 potentials at 1.27, 1.40 and 1.48 V. The first wave is ascribed to the oxidation of the DFF backbones (Fig. S5, ESI†).39 This further illustrates that the spiro-carbon linkage maintains the properties of the conjugated DFF backbones. The energy of the highest occupied molecular orbital (HOMO) is estimated from the equation EHOMO = [Ered1/2 − EFc1/2 + 4.8] is −5.62 eV. According to the optical gap (Eg) determined from the absorption onset, the lowest unoccupied molecular orbital (LUMO) level of Spiro-F is −2.43 eV based on the equation ELUMO = EHOMO + Eg.
In order to obtain further insight into the optoelectronic characteristics of Spiro-F, density functional theory (DFT) calculations were performed. As shown in Fig. 3, the HOMO, HOMO−1, LUMO and LUMO−1 are largely localized on the DFF backbones, while the fluorene units are not involved in the corresponding frontier orbitals. In the case of HOMO+2 and LUMO−2, they are predominately contributed by the fluorene moieties, which is similar to the situation in ladder-type oligophenylene materials.34 Interestingly, reports have shown that the S1/T1 values of Spiro-4S and Spiro-4SO2 are the same.36 Thus, we can conclude that the optoelectronic properties of Spiro-F mainly arise from its DFF backbone.
 | ||
| Fig. 3 Frontier molecular orbitals calculated by DFT B3LYP/6-31G(d). | ||
Electroluminescent devices
A single-layer device was fabricated with the configuration: ITO/Spiro-F/LiF/Al (Fig. S8, ESI†). As shown in Fig. S9 (ESI†), the turn-on voltage of the blue-emitting device is around 4.7 V at 1 Cd m−2, and its maximum brightness of 240.7 Cd m−2 is achieved at 15 V with the current density of 179.0 mA cm−2. The maximum current efficiency is 1.31 Cd A−1. This represents a significant enhancement over ladder-type oligo- or polyphenylene-based devices.34 In order to further develop a Spiro-F-based white OLED, the yellow emitter 3,4,5,6-tetrakis(3,6-diphenyl-carbazol-9-yl)-1,2-dicyanobenzene (4CzPNPh) was employed as a dopant, since the emission spectrum of Spiro-F largely overlaps with the absorption spectrum of 4CzPNPh, thus allowing energy transfer. We fabricated the complementary emitting devices with a bi-EML configuration of ITO|MoO3 (6 nm)|NPB (70 nm)|mCP (5 nm)|Spiro-F:4CzPNPh(20 nm, 0.5 wt%)|Spiro-F (5 nm)|TPBI (30 nm)|LiF (1 nm)|Al (Fig. 4a). As expected, blue and orange emission peaks were observed, while the relative intensity of the orange component was voltage dependent (Fig. S10, ESI†). This can be ascribed to incomplete energy transfer from Spiro-F to 4CzPNPh, or partial carrier trapping at the 4CzPNPh site. Noticeably, our preliminary device studies present a low turn-on voltage (3.5 V) and white emission with CIE coordinates of (0.29, 0.33) at 8 V. Efficiencies have also been realized with maxima of 3.6 Cd A−1 for current efficiency, 1.4 lm W−1 for power efficiency, and 1.8% for external quantum efficiency, which are comparable to the well-known white OLEDs based upon phosphole derivatives (Fig. 4b and c).40,41 The peak positions for Spiro-F and 4CzPNPh emissions do not depend on 4CzPNPh doping concentration (Fig. S11, ESI†). The efficiencies can be dramatically enhanced to 11.3 Cd A−1, 4.3 lm W−1, and 3.9%, respectively when the doping ratio of 4CzPNPh increases to 5% (Fig. S12 and S13, ESI†). A “super-rigid” structure such as Spiro-F thus plays an important role in achieving highly efficient white OLEDs. | ||
| Fig. 4 (a) Device configuration and energy level diagram of Spiro-F and 4CzPNPh-based diodes, and chemical structures of the employed materials. (b) Luminance–current density and (J)–voltage characteristics. (c) Efficiency-luminance correlations of the devices. | ||
3. Conclusions
In conclusion, we have developed a rigid multiple spirobifluorene-based blue emitter with a high photoluminescence quantum yield. The rigid structure with spiro-carbon linkages proves beneficial for thermal stability and solution processability, respectively. High-quality amorphous films can be obtained, whilst avoiding keto defects. Due to fluorenes introducing a steric effect, no intermolecular π–π interactions are observed in the single crystals. Spiro-F based WOLEDs provide promising results, highlighting the great potential of the title structure as both host and blue emitter. This work not only supports the significance of extended rigid structures for emitting materials but also constitutes a feasible approach toward high molecular-weight 3D emitters with multiple spiro-connections.4. Experimental section
Synthesis and characterization of 3,3′,9,9′-tetra-tert-butyl-6,6′spirobi[cyclopenta[2,1-b:3,4-b′]tetrafluorene]12,12′,15,15′-spirotetra[fluorene] (Spiro-F)
2-Bromobiphenyl (1.2 mL, 7.1 mmol) in dry THF (25 mL) was added slowly to a flask charged with iodine (10 mg, 0.04 mmol) and magnesium (0.18 g, 7.5 mmol) to initiate the Grignard reaction under an inert atmosphere. The mixture was then refluxed for 5 h. Subsequently, the 25 mL THF solution of Spiro-4O (0.3 g, 0.32 mmol) was added all at once and refluxed overnight. After cooling to room temperature, the THF was evaporated under reduced pressure, followed by the addition of water (50 mL) and 1 M HCl (50 mL) to quench the reaction. The mixture was stirred for 3 h and then extracted with DCM (3 × 50 mL). The organic phases were combined, washed with brine (2 × 100 mL), dried over Na2SO4 and filtered. The solvent was removed by rotary evaporation. The resulting residue was dissolved in acetic acid (50 mL) and refluxed overnight. After cooling to room temperature, the mixture was then poured into water (100 mL). The crude product was precipitated and collected by filtration, washed with water and subjected to column chromatography, using hexane/DCM (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as eluent to obtain the desired pure compound as a white solid (420 mg, 88%). 1H NMR (500 MHz, C2D2Cl4): δ 7.84 (d, J = 7.7 Hz, 8H), 7.59 (d, J = 1.9 Hz, 4H), 7.36 (t, J = 7.5 Hz, 8H), 7.25 (s, 4H), 7.13 (t, J = 7.4 Hz, 8H), 7.04 (dd, J = 8.0, 1.9 Hz, 4H), 7.01 (s, 4H), 6.87 (d, J = 7.5 Hz, 8H), 6.54 (d, J = 8.0 Hz, 4H), 1.30 (s, 36H).13C NMR (126 MHz, C2D2Cl4): δ 151.02, 149.78, 149.52, 149.13, 146.54, 142.53, 141.91, 141.70, 141.34, 127.73, 127.54, 125.11, 124.40, 123.20, 119.95, 116.66, 115.76, 115.71, 65.85, 34.75, 31.58, 29.57. HR-MS (MALDI) m/z calculated for C117H88 [M+] 1492.6881. Found 1492.6874. Crystallographic data (CCDC deposition number): CCDC 1408340.† Melting point has not been observed below 300 °C.
:1) as eluent to obtain the desired pure compound as a white solid (420 mg, 88%). 1H NMR (500 MHz, C2D2Cl4): δ 7.84 (d, J = 7.7 Hz, 8H), 7.59 (d, J = 1.9 Hz, 4H), 7.36 (t, J = 7.5 Hz, 8H), 7.25 (s, 4H), 7.13 (t, J = 7.4 Hz, 8H), 7.04 (dd, J = 8.0, 1.9 Hz, 4H), 7.01 (s, 4H), 6.87 (d, J = 7.5 Hz, 8H), 6.54 (d, J = 8.0 Hz, 4H), 1.30 (s, 36H).13C NMR (126 MHz, C2D2Cl4): δ 151.02, 149.78, 149.52, 149.13, 146.54, 142.53, 141.91, 141.70, 141.34, 127.73, 127.54, 125.11, 124.40, 123.20, 119.95, 116.66, 115.76, 115.71, 65.85, 34.75, 31.58, 29.57. HR-MS (MALDI) m/z calculated for C117H88 [M+] 1492.6881. Found 1492.6874. Crystallographic data (CCDC deposition number): CCDC 1408340.† Melting point has not been observed below 300 °C.
Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 51603055), the Natural Science Foundation of Heilongjiang Province (Grant No. QC2017055), the China Postdoctoral Science Foundation (Grant No. 2016M601424, 2017T100236), the Postdoctoral Foundation of Heilongjiang Province (Grant No. LBH-Z16059), the Fundamental Research Funds for the Central Universities (Grant No. HIT.NSRIF.2019038), and the Key Laboratory of Functional Inorganic Material Chemistry (Heilongjiang University). Klaus Müllen thanks the Gutenberg Research College for their generous support. Open Access funding provided by the Max Planck Society.Notes and references
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913 CrossRef CAS.
- M. A. Baldo, M. E. Thompson and S. R. Forrest, Nature, 2000, 403, 750 CrossRef CAS PubMed.
- B. W. D'Andrade and S. R. Forrest, Adv. Mater., 2004, 16, 1585 CrossRef.
- J. Zhang, D. Ding, Y. Wei, F. Han, H. Xu and W. Huang, Adv. Mater., 2016, 28, 479 CrossRef CAS PubMed.
- H. Xu, R. Chen, Q. Sun, W. Lai, Q. Su, W. Huang and X. Liu, Chem. Soc. Rev., 2014, 43, 3259 RSC.
- J. Li, D. Ding, Y. Tao, Y. Wei, R. Chen, L. Xie, W. Huang and H. Xu, Adv. Mater., 2016, 28, 3122 CrossRef CAS PubMed.
- M. Zhu and C. Yang, Chem. Soc. Rev., 2013, 42, 4963 RSC.
- M.-T. Lee, H.-H. Chen, C.-H. Liao, C.-H. Tsai and C. H. Chen, Appl. Phys. Lett., 2004, 85, 3301 CrossRef CAS.
- D. Xia, B. Wang, B. Chen, S. Wang, B. Zhang, J. Ding, L. Wang, X. Jing and F. Wang, Angew. Chem., Int. Ed., 2014, 53, 1048 CrossRef CAS PubMed.
- M. Yu, S. Wang, S. Shao, J. Ding, L. Wang, X. Jing and F. Wang, J. Mater. Chem. C, 2015, 3, 861 RSC.
- L. Zhao, S. Wang, S. Shao, J. Ding, L. Wang, X. Jing and F. Wang, J. Mater. Chem. C, 2015, 3, 8895 RSC.
- M. Romain, D. Tondelier, J.-C. Vanel, B. Geffroy, O. Jeannin, J. Rault-Berthelot, R. Métivier and C. Poriel, Angew. Chem., Int. Ed., 2013, 125, 14397 CrossRef.
- C. Fan, L. Zhu, T. Liu, B. Jiang, D. Ma, J. Qin and C. Yang, Angew. Chem., Int. Ed., 2014, 53, 2147 CrossRef CAS.
- S. Zhang, L. Yao, Q. Peng, W. Li, Y. Pan, R. Xiao, Y. Gao, C. Gu, Z. Wang, P. Lu, F. Li, S. Su, B. Yang and Y. Ma, Adv. Funct. Mater., 2015, 25, 1755 CrossRef CAS.
- M. Romain, D. Tondelier, J.-C. Vanel, B. Geffroy, O. Jeannin, J. Rault-Berthelot, R. Métivier and C. Poriel, Angew. Chem., Int. Ed., 2013, 52, 14147 CrossRef CAS.
- J. Zhang, D. Ding, Y. Wei and H. Xu, Chem. Sci., 2016, 7, 2870 RSC.
- C. Fan, C. Duan, C. Han, B. Han and H. Xu, ACS Appl. Mater. Interfaces, 2016, 8, 27383 CrossRef CAS PubMed.
- C. Duan, J. Li, C. Han, D. Ding, H. Yang, Y. Wei and H. Xu, Chem. Mater., 2016, 28, 5667 CrossRef CAS.
- R. G. Clarkson and M. Gomberg, J. Am. Chem. Soc., 1930, 52, 2881 CrossRef CAS.
- T. P. I. Saragi, T. Spehr, A. Siebert, T. Fuhrmann-Lieker and J. Salbeck, Chem. Rev., 2007, 107, 1011 CrossRef CAS.
- D. Katsis, Y. H. Geng, J. J. Ou, S. W. Culligan, A. Trajkovska, S. H. Chen and L. J. Rothberg, Chem. Mater., 2002, 14, 1332 CrossRef CAS.
- L. Wang, B. Pan, L. Zhu, B. Wang, Y. Wang, Y. Liu, J. Jin, J. Chen and D. Ma, Dyes Pigm., 2015, 114, 222 CrossRef CAS.
- Y. Wang, W. Liu, J. Deng, G. Xie, Y. Liao, Z. Qu, H. Tan, Y. Liu and W. Zhu, Chem. – Asian J., 2016, 11, 2555 CrossRef CAS PubMed.
- Q. Dong, H. Lian, Z. Gao, Z. Guo, N. Xiang, Z. Zhong, H. Guo, J. Huang and W.-Y. Wong, Dyes Pigm., 2017, 137, 84 CrossRef CAS.
- X. Wang, L. Zhao, S. Shao, J. Ding, L. Wang, X. Jing and F. Wang, Polym. Chem., 2014, 5, 6444 RSC.
- L. Romaner, A. Pogantsch, P. Scandiucci de Freitas, U. Scherf, M. Gaal, E. Zojer and E. J. W. List, Adv. Funct. Mater., 2003, 13, 597 CrossRef CAS.
- M. Gaal, E. J. W. List and U. Scherf, Macromolecules, 2003, 36, 4236 CrossRef CAS.
- M. Zhu and C. Yang, Chem. Soc. Rev., 2013, 42, 4963 RSC.
- Y. H. Kim, D. C. Shin, S. H. Kim, C. H. Ko, H. S. Yu, Y. S. Chae and S. K. Kwon, Adv. Mater., 2001, 13, 1690 CrossRef CAS.
- J. Luo, Y. Zhou, Z.-Q. Niu, Q.-F. Zhou, Y. Ma and J. Pei, J. Am. Chem. Soc., 2007, 129, 11314 CrossRef CAS.
- Y.-I. Park, J.-H. Son, J.-S. Kang, S.-K. Kim, J.-H. Lee and J.-W. Park, Chem. Commun., 2008, 2143 RSC.
- K. Skonieczny, J. Yoo, J. M. Larsen, E. M. Espinoza, M. Barbasiewicz, V. I. Vullev, C.-H. Lee and D. T. Gryko, Chem. – Eur. J., 2016, 22, 7485 CrossRef CAS PubMed.
- H.-W. Tseng, J.-Q. Liu, Y.-A. Chen, C.-M. Chao, K.-M. Liu, C.-L. Chen, T.-C. Lin, C.-H. Hung, Y.-L. Chou, T.-C. Lin, T.-L. Wang and P.-T. Chou, J. Phys. Chem. Lett., 2015, 6, 1477 CrossRef CAS.
- N. Cocherel, C. Poriel, J. Rault-Berthelot, F. Barrière, N. Audebrand, A. M. Z. Slawin and L. Vignau, Chem. – Eur. J., 2008, 14, 11328 CrossRef CAS PubMed.
- D. Xia, X. Guo, M. Wagner, M. Baumgarten, D. Schollmeyer and K. Müllen, Cryst. Growth Des., 2017, 17, 2816 CrossRef CAS.
- J. Guan, F. Zhang, Y. Zhang, Z. Liu, Y. Wu and D. Xia, CrystEngComm, 2017, 19, 6752 RSC.
- J. Liu, B.-W. Li, Y.-Z. Tan, A. Giannakopoulos, C. Sanchez-Sanchez, D. Beljonne, P. Ruffieux, R. Fasel, X. Feng and K. Müllen, J. Am. Chem. Soc., 2015, 137, 6097 CrossRef CAS PubMed.
- B. Kohl, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2015, 54, 6051 CrossRef CAS PubMed.
- K.-T. Wong, L.-C. Chi, S.-C. Huang, Y.-L. Liao, Y.-H. Liu and Y. Wang, Org. Lett., 2006, 8, 5029 CrossRef CAS PubMed.
- O. Fadhel, M. Gras, N. Lemaitre, V. Deborde, M. Hissler, B. Geffroy and R. Réau, Adv. Mater., 2009, 21, 1261 CrossRef CAS.
- M. P. Duffy, W. Delaunay, P. A. Bouit and M. Hissler, Chem. Soc. Rev., 2016, 45, 5296 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1408340. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8nj06135a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |